
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular recognition of planar and non-planar aromatic hydrocarbons through multipoint Ag–π bonding in a dinuclear metallo-macrocycle †
Kenichiro
Omoto
 ,
Shohei
Tashiro
and
Mitsuhiko
Shionoya
*
,
Shohei
Tashiro
and
Mitsuhiko
Shionoya
*
Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: shionoya@chem.s.u-tokyo.ac.jp
First published on 27th June 2019
Abstract
Exploration of a novel structural motif of host–guest interactions is one of the most fundamental topics to develop macrocycle-based host–guest/supramolecular systems. Herein, we present an unprecedented mode of inclusion of aromatic hydrocarbons into a macrocyclic cavity via multipoint Ag–π bonding as a driving force. A dinuclear AgI-macrocycle encapsulated one molecule of anthracene, a typical planar aromatic hydrocarbon, in solution and in the solid state. Single-crystal X-ray diffraction analysis of the host–guest inclusion complex revealed the binding of anthracene via multipoint Ag–π bonding to both AgI ions arranged within the open-ended nano-cavity of the dinuclear AgI-macrocycle. Notably, this binding motif based on Ag–π bonding was also applied to the inclusion of triptycene, a non-planar aromatic hydrocarbon with a steric tripodal structure, to evaluate the rotational motion of the molecular paddle-wheel in the AgI-macrocycle.
Introduction
Since the discovery of crown ether by Pedersen, a great number of excellent examples of functional macrocycles have been reported which can bind guest molecules or ions within their inner spaces.1 Guest binding abilities of macrocycles play vital roles in providing various implications to advanced functions in host–guest/supramolecular systems, such as molecular recognition,2 activation,3 transportation4 and fabrication of specific supramolecular architectures (i.e. rotaxanes and catenanes).5 In general, a variety of non-covalent interactions including hydrogen bonding, coulombic interactions, van der Waals forces and solvophobic effects have been utilised as driving forces for guest uptake of macrocycles.6 Besides, metal coordination capable of forming stable, reversible and directional bonds is another binding mode between host and guest compounds, and therefore macrocycles with coordinatively labile metal centres in the cavity have received a lot of attention in recent years in the field of supramolecular chemistry.7Aromatic hydrocarbons have been widely investigated as one of the most important classes of guest molecules for macrocycles owing to their ubiquitous structure and electronic properties related to π-conjugation.8 In the past few decades, a variety of macrocyclic receptors for aromatic molecules have been reported which can realise recognition,8 separation8c and regulation8d of their properties via host–guest interactions. However, in spite of the diversity of macrocyclic structures, molecular recognition of non-substituted, structurally simple aromatic hydrocarbons is still challenging because such hydrocarbons without polar functionalities are difficult to uptake by forming distinct chemical bonds (i.e. hydrogen bonds and coordination bonds) with a receptor, and alternatively vast-area contacts through a few weak interactions such as π–π, CH–π and van der Waals interactions are required to encapsulate a pristine aromatic hydrocarbon in a host framework (Fig. 1a). Such vast-area contacts generally need shape complementarity in the host–guest inclusion structure. Therefore, there are limitations in the diversity of host–guest structures, which make it difficult to rationally design a platform for the molecular recognition of non-planar aromatic hydrocarbons with a three-dimensional structure.
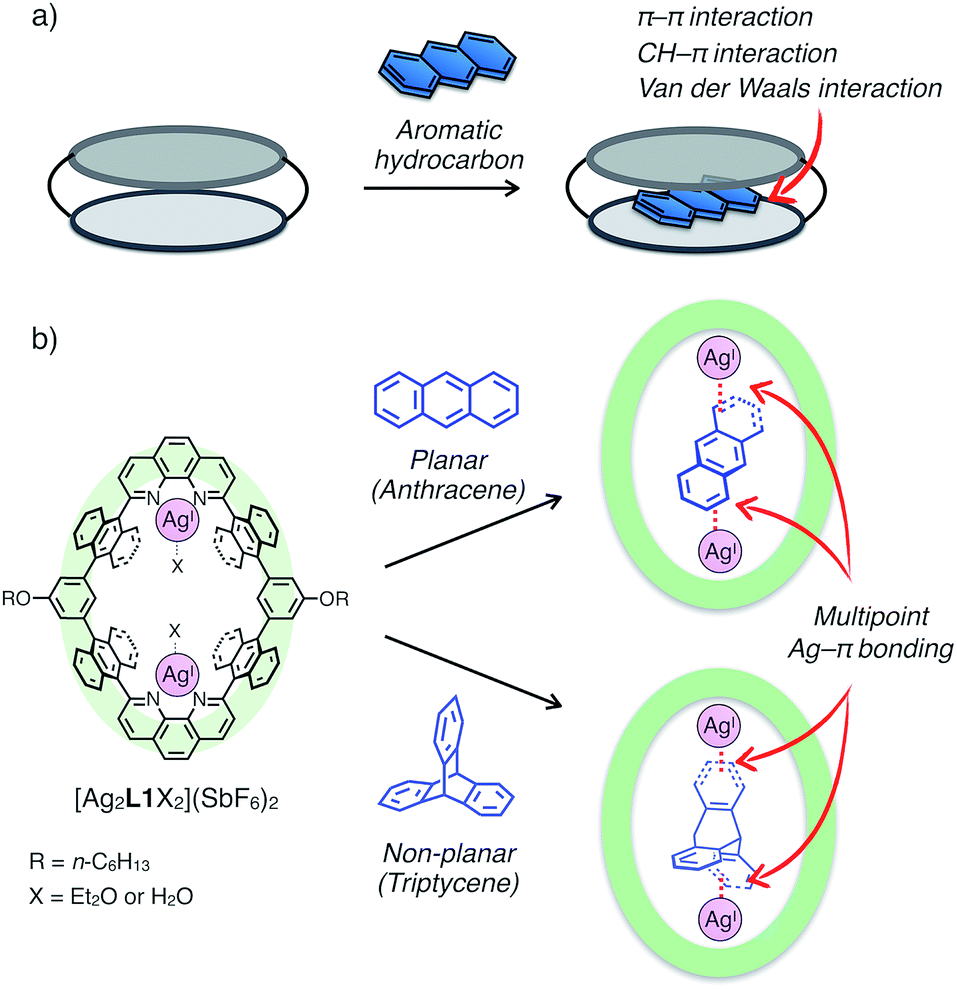 | ||
| Fig. 1 (a) Schematic representation of the binding mode of a non-substituted aromatic hydrocarbon within a macrocyclic host via vast-area contacts. (b) The chemical structure of a dinuclear AgI-macrocycle [Ag2L1X2](SbF6)2 and the schematic representation of the binding modes of planar and non-planar aromatic hydrocarbons via multipoint Ag–π bonding. | ||
Herein, we present an unprecedented mode of inclusion of non-substituted aromatic hydrocarbons into a metallo-macrocycle utilising M–π bonding as a driving force. M–π bonding is a kind of non-Werner type coordination bonding mainly originating from the interactions among d orbitals of metals and π orbitals of aromatic hydrocarbons.9–11 In general, M–π bonding has higher directionality and site-specificity than the above-mentioned conventional intermolecular interactions, and the resulting M–π complexes have the potential to exhibit unique functions based on the chemical and physical properties of the metal centres. Therefore, metallo-macrocycles possessing a nano-space with multiple metal centres for M–π bonding are expected to provide a novel structural motif to recognise aromatic guest molecules, in which the inclusion structure and the guest selectivity can be affected by the type, number, and arrangement of the inner metal ions.12 Based on this concept, we have recently developed a dinuclear AgI-macrocycle [Ag2L1X2](SbF6)2 (L1 = macrocyclic ligand and X = Et2O or H2O) which possesses an anthracene-based rhombic structure with a nano-space with two AgI centres (Fig. 1b). In this system, the AgI-macrocycle can accommodate a ditopic aromatic molecule such as cyclophanes and ferrocene utilising multiple Ag–π bonds as a main driving force.12a,c Herein we report that the AgI-macrocycle effectively recognised an anthracene molecule, a planar aromatic hydrocarbon, in the open-ended cavity through multiple Ag–π bonds with both ends of anthracene (Fig. 1b). Moreover, we found that this guest-binding motif based on Ag–π bonding does not require vast-area contacts between the host and guest, which achieved the binding of a non-planar aromatic hydrocarbon, triptycene, in a similar manner.
Results and discussion
The binding of anthracene (Ant) to a dinuclear AgI-macrocycle [Ag2L1X2](SbF6)2 was first investigated by 1H NMR titration experiments at 220 or 300 K and mass spectrometry. Upon the addition of Ant to a solution of [Ag2L1X2](SbF6)2 (0.07 mM) in CDCl3, the 1H NMR signals of [Ag2L1X2](SbF6)2 at 220 K were gradually replaced by a new set of signals with an increased amount of Ant (Fig. 2a–c). In the presence of more than 1.0 eq. of Ant, the original signals were almost completely replaced by a new set of signals, and the signals of free Ant (Aout, Bout and Cout) appeared at around 7.5–8.6 ppm, suggesting the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest inclusion complex, Ant⊂[Ag2L1](SbF6)2 (Fig. 2c–e). The new signals observed around the higher magnetic field region (Antin around 6.0–6.2 ppm) can be assigned to the included Ant, which are significantly up field shifted (|Δδ| = ca. 1.4–2.4 ppm) from their original positions due to the shielding effect from the anthracene walls of [Ag2L1]2+.13
:1 host–guest inclusion complex, Ant⊂[Ag2L1](SbF6)2 (Fig. 2c–e). The new signals observed around the higher magnetic field region (Antin around 6.0–6.2 ppm) can be assigned to the included Ant, which are significantly up field shifted (|Δδ| = ca. 1.4–2.4 ppm) from their original positions due to the shielding effect from the anthracene walls of [Ag2L1]2+.13
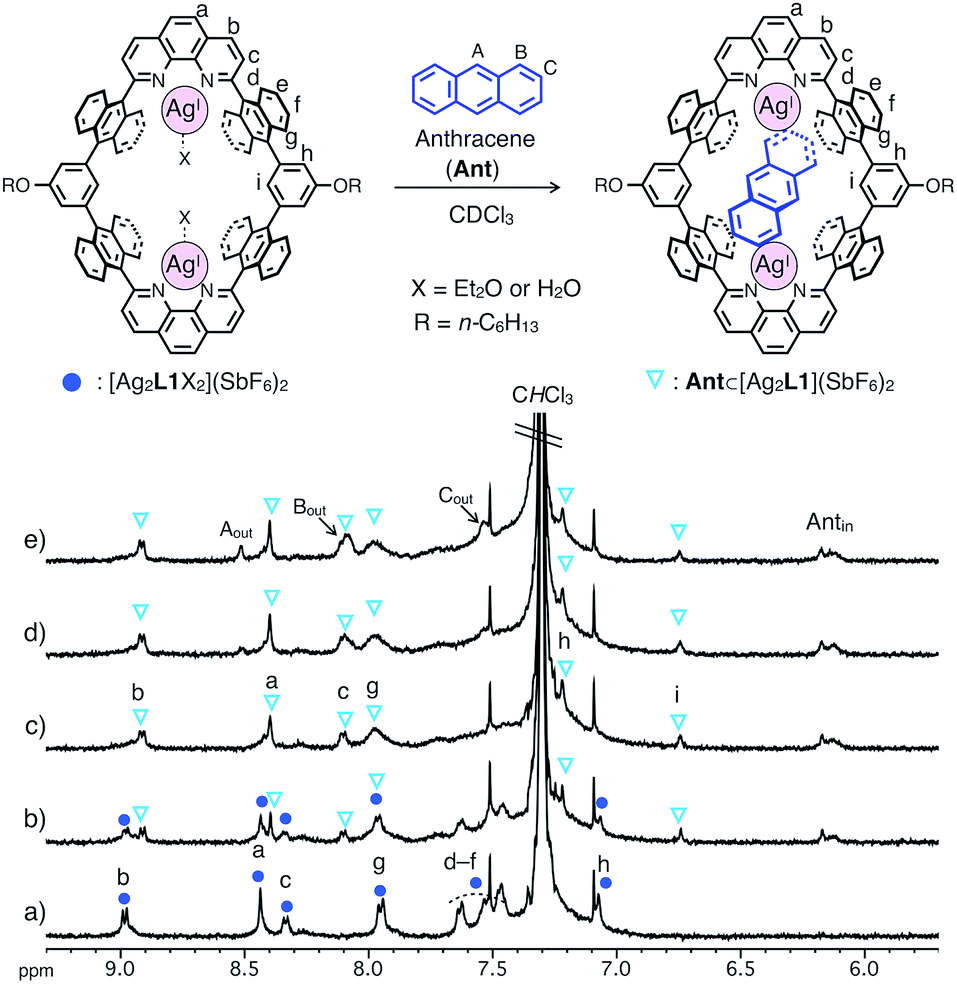 | ||
| Fig. 2 Partial 1H NMR spectra of [Ag2L1X2](SbF6)2 (0.07 mM) in the presence of (a) 0.0, (b) 0.5, (c) 1.0, (d) 1.5 and (e) 2.0 eq. of anthracene (Ant) (500 MHz, CDCl3, 220 K). Antin represents the signals of the included Ant. | ||
Notably, at a temperature as high as 300 K, as the intermolecular exchange between bound and free Ant molecules is faster than the timescale of 1H NMR spectroscopy, 1H NMR titration experiments at 300 K resulted in the sequential shift of the signals of [Ag2L1X2](SbF6)2 due to host–guest interactions (Fig. S1 and S2†). From the curve-fitting of the 1H NMR data at 300 K based on the formation of a 1:1 host–guest complex, the binding constant between Ant and the AgI-macrocycle (Ka(Ant) = [Ant⊂[Ag2L1]2+]/([[Ag2L1X2]2+][Ant]) M−1) was determined to be (3.0 ± 0.4) × 104 M−1 in CDCl3 at 300 K (Fig. S3†), which is comparable to those of already reported examples of macrocyclic receptors for aromatic hydrocarbons.8c,f,14 The formation of a 1:1 host–guest complex was also supported by ESI-TOF mass measurements (m/z = 903.72 as Ant⊂[Ag2L1]2+, Fig. S6†).
The structure and binding mode of the resulting complex were finally determined by single-crystal X-ray analysis (Fig. 3). By slow n-pentane vapour diffusion into a mixture of L1, AgSbF6 (4.2 eq.) and Ant (10 eq.) in CH2Cl2 in the dark at room temperature, yellow block crystals suitable for single-crystal X-ray analysis were obtained. In the resulting crystal structure, one molecule of Ant was included in the cavity of [Ag2L1]2+via η2-type Ag–π bonding at both terminal edges of the elongated π-surface in an anti-manner (Fig. 3a). These AgI ions were in a distorted square pyramidal five-coordinate geometry with two N-atoms of phenanthroline, two C-atoms of the included Ant, and one Cl-atom of a coordinating solvent: CH2Cl2 (Ag–N1 2.280(6) Å; Ag–N2 2.356(5) Å; Ag–C55 2.454(8) Å; Ag–C56 2.449(8) Å; Ag–Cl3 2.984(2) Å, Fig. 3b). These observations suggest that multipoint Ag–π bonding between Ant and AgI ions, which are precisely arranged on the inner surface of the nano-space, works as an effective driving force to bind Ant. Multipoint CH–π interactions between the H-atoms of the included Ant and the π-surface of anthracene-walls of the macrocycle may also contribute to stabilise the resulting complex as well. Notably, Ant⊂[Ag2L1(CH2Cl2)2]2+ possesses a Ci-symmetrical structure in the crystal due to the inclination of the included Ant. On the other hand, the 1H NMR spectrum of Ant⊂[Ag2L1](SbF6)2 showed a simple set of 1H NMR signals (Fig. 2b–e) suggesting a more symmetrical (D2h) structure with successive fluxional movement of Ant in the nano-cavity via intramolecular Ag–π exchange.
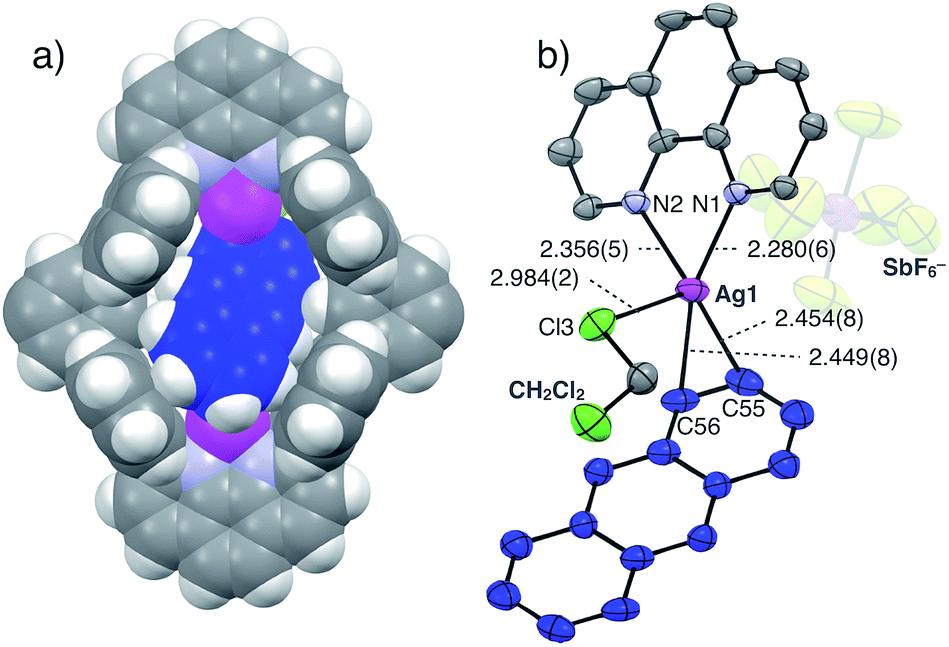 | ||
| Fig. 3 Crystal structure of Ant⊂[Ag2L1(CH2Cl2)2](SbF6)2. (a) Space filling model and (b) ORTEP view (50% probability level) of the partial structure (solvents, side-alkyloxy chains and counter anions in (a) are omitted for clarity). Ag: magenta, C: grey, C of Ant: blue, Cl: pale green, F: yellow, H: white, N: light blue and Sb: purple. | ||
Based on these results, the binding properties of [Ag2L1X2](SbF6)2 to other aromatic hydrocarbons of different sizes were examined. We previously reported that a smaller aromatic hydrocarbon, p-xylene, formed not a 1:1 but a 2:1 complex, (p-xylene)2⊂[Ag2L1]2+, in the crystalline state.12a On the other hand, a 1H NMR titration experiment using p-xylene showed negligible shift in the host signals (|Δδ| < 0.04 ppm) even after adding 4 eq. of p-xylene, suggesting a lower affinity to [Ag2L1X2](SbF6)2 or a different binding mode from that of Ant⊂[Ag2L1](SbF6)2 in solution (Fig. S22 and S24†). Similarly, naphthalene, which has an intermediate size between p-xylene and Ant, showed a smaller shift in the host signals (|Δδ| < 0.02 ppm) even after adding 4 eq. of naphthalene in the 1H NMR titration experiment at 300 K (Fig. S23† and S24†). These results may reflect the insufficiency of the molecular size of p-xylene and naphthalene to simultaneously form Ag–π bonds at both AgI centres arranged in the macrocyclic skeleton. These results suggest the importance of the arrangement mode of AgI ions in the macrocycle in controlling the stability, selectivity and structure of the resulting host–guest complex.
In contrast, triptycene (Trip), which is a non-planar aromatic hydrocarbon with a steric tripodal structure, was found to be effectively included within the cavity of [Ag2L1X2](SbF6)2. The inclusion behavior of Trip was revealed by a 1H NMR titration experiment in CDCl3 at 220 K, in which the host signals were replaced by a new set of signals in the presence of more than 1.0 eq. of Trip to form an 1:1 host–guest complex Trip⊂[Ag2L1](SbF6)2 (Fig. 4).13 The signals of the included Trip (Bin) showed a strong rotating frame Overhauser effect (ROE) correlation with the proton inside [Ag2L1]2+ (Hi), which suggests the existence of Trip within the nano-cavity of the macrocycle (Fig. S18†). The formation of Trip⊂[Ag2L1](SbF6)2 was also supported by ESI-TOF mass analysis (m/z = 941.21 as Trip⊂[Ag2L1]2+, Fig. S20†).
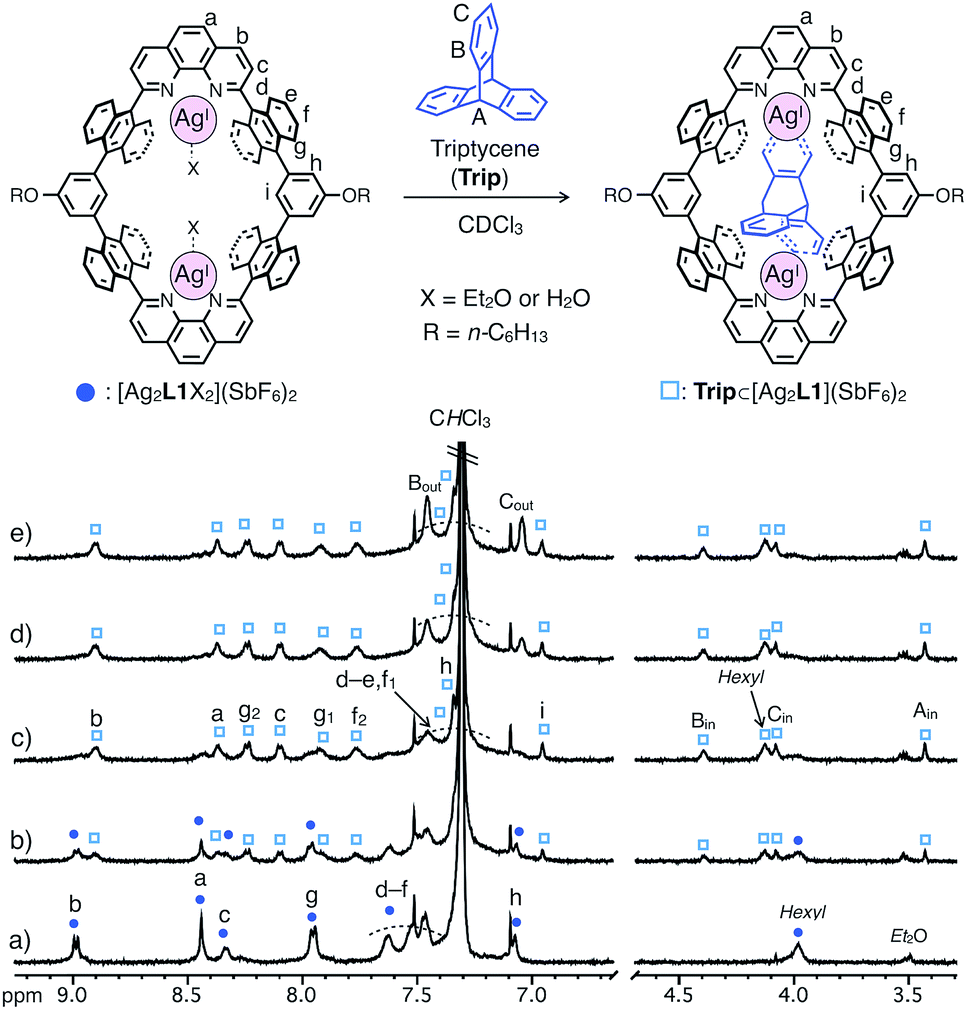 | ||
| Fig. 4 Partial 1H NMR spectra (500 MHz, CDCl3, 220 K) of [Ag2L1X2](SbF6)2 (0.11 mM) in the presence of (a) 0.0, (b) 0.5, (c) 1.0, (d) 1.5 and (e) 2.0 eq. of triptycene (Trip). Hexyl represents the signal of side-alkyloxy chains of L1. | ||
Similar to the case of Ant, the rates of intermolecular exchange of bound and free Trip at a higher temperature (300 K) became faster than the timescale of 1H NMR, which resulted in the observation of a dynamically averaged 1H NMR spectrum (Fig. S9 and S10†). From the curve-fitting of the 1H NMR data of the guest titration experiment at 300 K based on the formation of a 1:1 host–guest complex, the binding constant between Trip and the AgI-macrocycle (Ka(Trip) = [Trip⊂[Ag2L1]2+]/([[Ag2L1X2]2+][Trip]) M−1) was determined to be (3.1 ± 0.2) × 104 M−1 in CDCl3 at 300 K (Fig. S11†), which is comparable to that of Ant (Ka(Ant) = (3.0 ± 0.4) × 104 M−1).14 This result suggests host–guest interactions between Trip and AgI-macrocycle similar to that of Ant and AgI-macrocycle, where multipoint Ag–π bonding simultaneously formed at both ends of the aromatic molecule works as a dominant driving force for guest binding. Molecular modeling of Trip⊂[Ag2L1]2+ based on the crystal structure of the aforementioned Ant⊂[Ag2L1](SbF6)2 (Fig. 3) and that of a previously reported AgI complex of Trip supports the multipoint binding of Trip, where AgI ions coordinate to the π-planes of Trip in the anti or syn form (Fig. 5a and S21†).10d,g,h Such a binding motif of Trip suggests that the open-ended nano-cavity of [Ag2L1X2](SbF6)2 with two AgI ions would work as an effective receptor for not only planar but also non-planar aromatic molecules which allows multipoint Ag–π bonding.
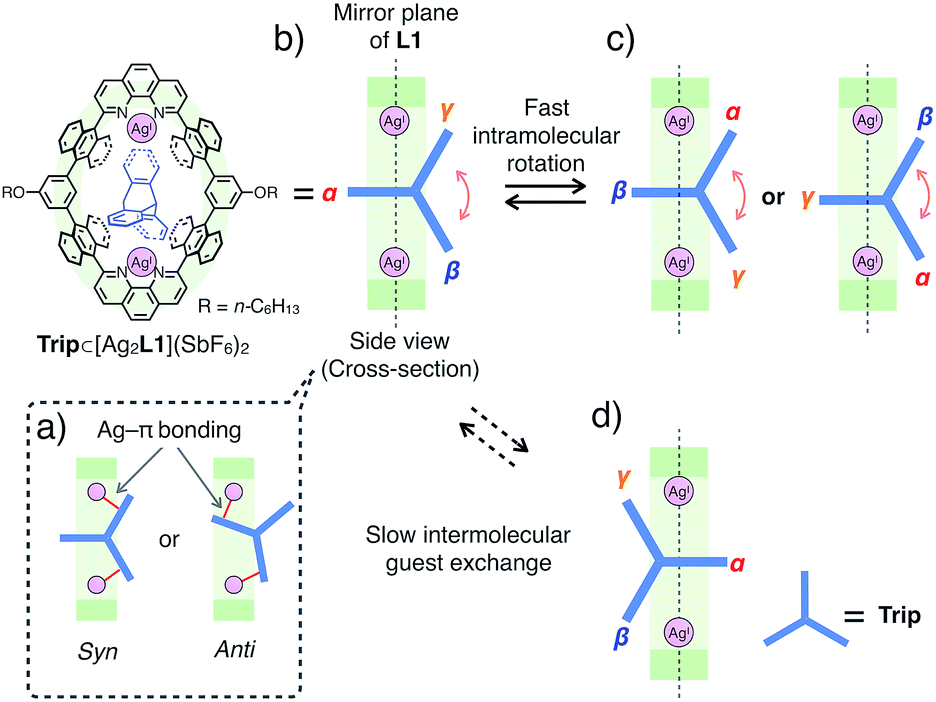 | ||
| Fig. 5 Schematic representation of the plausible binding modes and molecular dynamics of Trip⊂[Ag2L1](SbF6)2. (a) Plausible side cross-sectional views of Trip⊂[Ag2L1](SbF6)2 where AgI ions coordinate to the π-planes of Trip in an anti or a syn manner. (b–d) Plausible side cross-sectional views of the rotational motion of Trip, where Trip exhibits faster intramolecular rotational motion (b and c) and a slower intermolecular guest exchange (d) than the timescale of 1H NMR observation. | ||
Notably, the included Trip exhibits rotational motion at 220 K, which affects the symmetry of the resulting host–guest complex, Trip⊂[Ag2L1]2+ (Fig. 5b–d).15 Upon binding of Trip, the 1H NMR signals of the protons of anthracene walls (Hg in Fig. 4a) of the macrocycle split into two separate signals (Hg1 and Hg2 in Fig. 4c), suggesting the desymmetrisation of the host skeleton from D2h-symmetry to C2v-symmetry (loss of the mirror plane on the cyclic skeleton of L1). On the other hand, the three aromatic panels of the included Trip (α, β and γ in Fig. 5b and c) were observed as two identical signals in the higher magnetic field region (Bin and Cin at 4.4 and 4.1 ppm in Fig. 4c), suggesting conservation of the D3h-symmetry of the pristine tripodal structure of Trip even within the nano-cavity of the macrocycle. Considering such a respective loss and conservation of the symmetry of the host and the guest, the included Trip was supposed to be deviated from the mirror plane of the cyclic skeleton of the macrocycle L1 without fast intermolecular guest exchange to desymmetrise the host skeleton (Fig. 5d). Besides, the included Trip was supposed to exhibit fast intramolecular rotational movement around the 3-fold axis of the tripodal structure to average the magnetic environment of the three aromatic panels in the timescale of 1H NMR observation (α, β and γ in Fig. 5b and c). Such a specific rotational motion of Trip bound to two AgI ions suggests that the open-ended nano-cavity of [Ag2L1X2](SbF6)2 is suitable to bind aromatic guest molecules and to maintain and regulate the degree of freedom of their molecular motion, which potentially plays a key role as functional molecular machines.
Conclusions
In this study, host–guest interactions between a dinuclear AgI-macrocycle [Ag2L1X2](SbF6)2 and aromatic hydrocarbons were investigated. [Ag2L1X2](SbF6)2 has an ability to strongly include not only a planar (Ant) but also a non-planar (Trip) aromatic hydrocarbon which allows multipoint Ag–π bonding at both AgI centres simultaneously. Such a AgI-based specific binding motif is quite distinct from those of conventional macrocyclic receptors for aromatic molecules which require close and vast-area contacts between the aromatic guest and the nano-cavity of the macrocycle. Such a unique binding motif utilising non-Werner type M–π coordination would provide us an important clue to the rational design of the structures, properties and metal-centred functions of the resulting host–guest complexes. This finding would help to develop a new dimension to macrocycle-based supramolecular chemistry and molecular machines based on the selectivity, reactivity and dynamics of M–π bonding within a confined space of metallo-macrocycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI grant numbers JP16H06509 (Coordination Asymmetry) to M. S. and JP16H00956 (Molecular Architectonics) to S. T. K. O. acknowledges the Advanced Leading Graduate Course for Photon Science (ALPS).Notes and references
- (a) C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021–1027 CrossRef; (b) E. Kimura, Tetrahedron, 1992, 48, 6175–6217 CrossRef CAS; (c) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed; (d) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley, New York, 2nd edn, 2009 Search PubMed; (e) F. Davis and S. Higson, Macrocycles, John Wiley & Sons, Ltd, Chichester, 2011 CrossRef; (f) G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed; (g) Y. Segawa, A. Yagi, K. Matsui and K. Itami, Angew. Chem., Int. Ed., 2016, 55, 5136–5158 CrossRef CAS PubMed; (h) Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC; (i) T. Kakuta, T. Yamagishi and T. Ogoshi, Acc. Chem. Res., 2018, 51, 1656–1666 CrossRef CAS PubMed.
- (a) J. J. Christensen, D. J. Eatough and R. M. Izatt, Chem. Rev., 1974, 74, 351–384 CrossRef PubMed; (b) J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1988, 27, 89–112 CrossRef; (c) D. J. Cram, Angew. Chem., Int. Ed. Engl., 1988, 27, 1009–1020 CrossRef; (d) D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed; (e) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (f) K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800–5835 RSC; (g) J. Cai and J. L. Sessler, Chem. Soc. Rev., 2014, 43, 6198–6213 RSC.
- (a) Z. Clyde-Watson, A. Vidal-Ferran, L. J. Twyman, C. J. Walter, D. W. J. McCallien, S. Fanni, N. Bampos, R. S. Wylie and J. K. M. Sanders, New J. Chem., 1998, 22, 493–502 RSC; (b) R. Breslow and S. D. Dong, Chem. Rev., 1998, 98, 1997–2011 CrossRef CAS PubMed; (c) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- (a) M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed; (b) M. Lisbjerg, H. Valkenier, B. M. Jessen, H. Al-Kerdi, A. P. Davis and M. Pittelkow, J. Am. Chem. Soc., 2015, 137, 4948–4951 CrossRef CAS PubMed.
- (a) C. O. Dietrich-Buchecker, J.-P. Sauvage and J. M. Lern, J. Am. Chem. Soc., 1984, 106, 3043–3045 CrossRef CAS; (b) K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS PubMed; (c) N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed; (d) J.-F. Ayme, J. E. Beves, D. A. Leigh, R. T. McBurney, K. Rissanen and D. Schultz, Nat. Chem., 2012, 4, 15–20 CrossRef CAS PubMed.
- (a) E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210–1250 CrossRef CAS PubMed; (b) F. Biedermann and H.-J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS PubMed.
- (a) A. W. Maverick, S. C. Buckingham, Q. Yao, J. R. Bradbury and G. G. Stanley, J. Am. Chem. Soc., 1986, 108, 7430–7431 CrossRef CAS; (b) S. Anderson, H. L. Anderson, A. Bashall, M. McPartlin and J. K. M. Sanders, Angew. Chem., Int. Ed. Engl., 1995, 34, 1096–1099 CrossRef CAS; (c) R. V. Slone and J. T. Hupp, Inorg. Chem., 1997, 36, 5422–5423 CrossRef CAS; (d) J. A. Whiteford, P. J. Stang and S. D. Huang, Inorg. Chem., 1998, 37, 5595–5601 CrossRef CAS PubMed; (e) V. Amendola, L. Fabbrizzi, C. Mangano, P. Pallavicini, A. Poggi and A. Taglietti, Coord. Chem. Rev., 2001, 219–221, 821–837 CrossRef CAS; (f) M. Yanagisawa, K. Tashiro, M. Yamasaki and T. Aida, J. Am. Chem. Soc., 2007, 129, 11912–11913 CrossRef CAS PubMed; (g) P. D. Frischmann, A. J. Gallant, J. H. Chong and M. J. MacLachlan, Inorg. Chem., 2008, 47, 101–112 CrossRef CAS PubMed; (h) B. Kersting and U. Lehmann, Adv. Inorg. Chem., 2009, 61, 407–470 CrossRef CAS; (i) A. M. J. Devoille, P. Richardson, N. L. Bill, J. L. Sessler and J. B. Love, Inorg. Chem., 2011, 50, 3116–3126 CrossRef CAS PubMed; (j) M. Kuritani, S. Tashiro and M. Shionoya, Inorg. Chem., 2012, 51, 1508–1515 CrossRef CAS PubMed; (k) M. Kuritani, S. Tashiro and M. Shionoya, Chem.–Asian J., 2013, 8, 1368–1371 CrossRef CAS PubMed; (l) R. Gramage-Doria, D. Armspach and D. Matt, Coord. Chem. Rev., 2013, 257, 776–816 CrossRef CAS; (m) C. Browne, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2014, 53, 10701–10705 CrossRef CAS PubMed; (n) T. Nakamura, Y. Kaneko, E. Nishibori and T. Nabeshima, Nat. Commun., 2017, 8, 129 CrossRef PubMed; (o) Y. Sakata, C. Murata and S. Akine, Nat. Commun., 2017, 8, 16005 CrossRef CAS PubMed.
- (a) L. S. Kaanumalle, C. L. D. Gibb, B. C. Gibb and V. Ramamurthy, J. Am. Chem. Soc., 2005, 127, 3674–3675 CrossRef CAS PubMed; (b) C. Peinador, E. Pía, V. Blanco, M. D. García and J. M. Quintela, Org. Lett., 2010, 12, 1380–1383 CrossRef CAS PubMed; (c) J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183–192 CrossRef CAS PubMed; (d) M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222–228 CrossRef PubMed; (e) P. Spenst and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 10165–10168 CrossRef CAS PubMed; (f) Y. Yamaki, T. Nakamura, S. Suzuki, M. Yamamura, M. Minoura and T. Nabeshima, Eur. J. Org. Chem., 2016, 1678–1683 CrossRef CAS; (g) G. Wu, C.-Y. Wang, T. Jiao, H. Zhu, F. Huang and H. Li, J. Am. Chem. Soc., 2018, 140, 5955–5961 CrossRef CAS PubMed.
- (a) A. Togni and R. L. Halterman, Metallocenes, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 1998 CrossRef; (b) D. Astruc, Modern Arene Chemistry, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2002 CrossRef; (c) D. Bandera, K. K. Baldridge, A. Linden, R. Dorta and J. S. Siegel, Angew. Chem., Int. Ed., 2011, 50, 865–867 CrossRef CAS PubMed; (d) T. Murahashi, K. Takase, M. Oka and S. Ogoshi, J. Am. Chem. Soc., 2011, 133, 14908–14911 CrossRef CAS PubMed; (e) M. Maruyama, M. König, D. M. Guldi, E. Nakamura and Y. Matsuo, Angew. Chem., Int. Ed., 2013, 52, 3015–3018 CrossRef CAS PubMed; (f) S. Lin, D. E. Herbert, A. Velian, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2013, 135, 15830–15840 CrossRef CAS PubMed.
- (a) E. A. H. Griffith and E. L. Amma, J. Am. Chem. Soc., 1974, 96, 5407–5413 CrossRef CAS; (b) J. L. Pierre, P. Baret, P. Chautemps and M. Armand, J. Am. Chem. Soc., 1981, 103, 2986–2988 CrossRef CAS; (c) H. V. R. Dias, Z. Wang and W. Jin, Inorg. Chem., 1997, 36, 6205–6215 CrossRef CAS; (d) M. Munakata, L. P. Wu, K. Sugimoto, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga, N. Maeno and M. Fujita, Inorg. Chem., 1999, 38, 5674–5680 CrossRef CAS; (e) M. Munakata, L. P. Wu and G. L. Ning, Coord. Chem. Rev., 2000, 198, 171–203 CrossRef CAS; (f) S. V. Lindeman, R. Rathore and J. K. Kochi, Inorg. Chem., 2000, 39, 5707–5716 CrossRef CAS PubMed; (g) M. Wen, M. Munakata, Y. Suenaga, T. Kuroda-Sowa and M. Maekawa, Inorg. Chim. Acta, 2002, 340, 8–14 CrossRef CAS; (h) M. Wen, M. Munakata, Y.-Z. Li, Y. Suenaga, T. Kuroda-Sowa, M. Maekawa and M. Anahata, Polyhedron, 2007, 26, 2455–2460 CrossRef CAS; (i) H. V. R. Dias and C. S. P. Gamage, Angew. Chem., Int. Ed., 2007, 46, 2192–2194 CrossRef CAS PubMed; (j) M. J. Fuchter, J. Schaefer, D. K. Judge, B. Wardzinski, M. Weimar and I. Krossing, Dalton Trans., 2012, 41, 8238–8241 RSC; (k) J. M. Maier, P. Li, J. Hwang, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2015, 137, 8014–8017 CrossRef CAS PubMed; (l) S. Liu and A. Y. Rogachev, ChemPhysChem, 2018, 19, 2579–2588 CrossRef CAS PubMed.
- M–π bonding is considered to be different from cation–π interaction, which is originated from electrostatic interaction between a positively charged cation and an electron-rich π-system. (a) J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303–1324 CrossRef CAS; (b) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- (a) K. Omoto, S. Tashiro, M. Kuritani and M. Shionoya, J. Am. Chem. Soc., 2014, 136, 17946–17949 CrossRef CAS PubMed; (b) W.-Y. Zhang, Y.-J. Lin, Y.-F. Han and G.-X. Jin, J. Am. Chem. Soc., 2016, 138, 10700–10707 CrossRef CAS PubMed; (c) M. Shimada, Y. Yamanoi, T. Ohto, S.-T. Pham, R. Yamada, H. Tada, K. Omoto, S. Tashiro, M. Shionoya, M. Hattori, K. Jimura, S. Hayashi, H. Koike, M. Iwamura, K. Nozaki and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 11214–11221 CrossRef CAS PubMed.
- The integral ratios of the 1H NMR signals of the included guest molecules encapsulated in [Ag2L1]2+ were evaluated to be smaller than those estimated from the structures of 1:1 complexes probably due to the fluxional dynamic movement of the included guest molecules within the nano-space, which caused broadening of the 1H NMR signals..
- The stabilities of the host–guest complexes were temperature dependent. The binding constant Ka estimated at 300 K seems to be smaller than that at 220 K, as revealed by the convergence of 1H NMR titration experiments..
- (a) A. Scarso, H. Onagi and J. Rebek Jr, J. Am. Chem. Soc., 2004, 126, 12728–12729 CrossRef CAS PubMed; (b) T. Matsuno, M. Fujita, K. Fukunaga, S. Sato and H. Isobe, Nat. Commun., 2018, 9, 3779 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. CCDC 1911739. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02619c |
| This journal is © The Royal Society of Chemistry 2019 |