
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An exceptionally stable octacobalt-cluster-based metal–organic framework for enhanced water oxidation catalysis†
Ning-Yu
Huang‡
a,
Jian-Qiang
Shen‡
ab,
Zi-Ming
Ye
a,
Wei-Xiong
Zhang
 a,
Pei-Qin
Liao
*a and
Xiao-Ming
Chen
a
a,
Pei-Qin
Liao
*a and
Xiao-Ming
Chen
a
aMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry, Sun Yat-Sen University, Guangzhou 510275, China. E-mail: liaopq3@mail.sysu.edu.cn
bDepartment of Chemical and Biomolecular Engineering, University of California, Los Angeles, CA 90095, USA
First published on 10th September 2019
Abstract
Extensive efforts have been devoted to developing efficient and durable catalysts for water oxidation. Herein, we report a highly stable metal–organic framework that shows high catalytic activity and durability for electrically driven (an overpotential of 430 mV at 10 mA cm−2 in neutral aqueous solution) and photodriven (a turnover frequency of 16 s−1 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles) water oxidation, representing the best catalyst for water oxidation reported to date. Computational simulation and isotope tracing experiments showed that the μ4-OH group of the {Co8(μ4-OH)6} unit participates in the water oxidation reaction to offer an oxygen vacancy site with near-optimal OH− adsorption energy.
000 cycles) water oxidation, representing the best catalyst for water oxidation reported to date. Computational simulation and isotope tracing experiments showed that the μ4-OH group of the {Co8(μ4-OH)6} unit participates in the water oxidation reaction to offer an oxygen vacancy site with near-optimal OH− adsorption energy.
Introduction
The replacement of fossil fuels with hydrogen generated by water splitting is a very attractive solution to the present energy problem. In order to develop a practical technology based on these elements, durable and efficient water oxidation catalysts need to be developed.1,2 However, the water oxidation process involves a four-electron process, leading to slow kinetics.3,4 For this reason, improving the efficiency of water oxidation catalysts is still a challenging task, despite the considerable achievements that have been made in recent years.5–7The active oxygen species (namely, the O˙ radical) has been accepted as the important intermediate for the formation of hydroperoxy (OOH) species and for the subsequent conversion to O2 molecules.8 Under alkaline conditions, the M–O˙ (M = metal) species is generated from the oxidation of the M–OH species, enhancing the adsorption energy of the reactant OH−, which might be beneficial to the formation of *OH for the oxygen evolution reaction (OER) intermediates. However, if OH− binds too strongly, it will occupy available surface sites and poison the reaction.9 Therefore, optimizing the OH− adsorption energy to a near-optimal value might be beneficial to accelerate the reaction kinetics.10 Obviously, regulating the coordination number of the oxygen atom of OH− could be the most effective strategy to optimize the adsorption energy (Scheme 1). For instance, Jin et al. incorporated gold clusters onto the CoSe2 catalyst to increase the coordination number of the OH− from 1 to 2, resulting in the enhancement of activity.11 Nevertheless, the binding affinity was still too weak, leading to a large amount of energy input required to produce *OH.
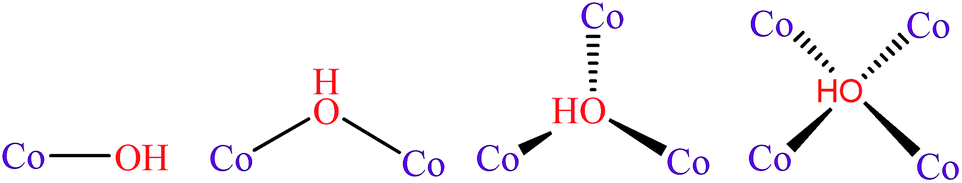 | ||
| Scheme 1 Proposed models of the reactant hydroxyl ion coordinated to the cobalt ions during the water oxidation reaction. | ||
Metal–organic frameworks (MOFs), as crystalline porous materials with high surface areas and outstanding structural designability, can combine the advantages of both homogeneous and heterogeneous catalysts. Recently, MOFs have emerged as potential catalysts for water oxidation,12,13 the hydrogen evolution reaction,14–17 carbon dioxide reduction,18–22etc.14,23–25 Nevertheless, as similar to traditional catalysts, the reported MOF catalysts also suffer from poor stability and low catalytic activity.26,27 Among the various types of MOFs, metal-azolate frameworks (MAFs) are famous for their extraordinary chemical stabilities.28 In addition, the high connectivity of the metal cluster could enhance the stability of the framework.29,30 Considering the relatively high activities and earth-abundance of cobalt ions, and that the multinuclear metal cluster might have a favourable OH− adsorption energy, a combination of the highly connected cobalt-hydroxide unit and the azolate bridging ligand is the best choice. Here, we report a highly stable, octacobalt cluster based MAF with both extraordinarily high activity and durability. We demonstrated that the metal site, capping four coplanar cobalt ions, indeed serves as a highly efficient active site for water oxidation.
Experimental section
Materials and methods
All reagents were commercially available and used without further purification. 1,4-Benzenedi(1H-1,2,3-triazole (H2bdt) was synthesized according to the method in the literature. Elemental analyses (EA) were conducted using an Elementar Vario EL analyzer. X-ray photoelectron spectroscopy (XPS) measurements were performed with a VG Scientific ESCALAB 250 instrument. Powder X-ray diffraction (PXRD) patterns were collected on a Bruker D8-Advance diffractometer with Cu Kα radiation and a LynxEye detector. Variable-temperature PXRD data were collected on a Rigaku SmartLab X-ray diffractometer (Cu-Kα, λ = 1.54056 Å). Thermogravimetric (TG) analyses were performed on a TA Q50 thermogravimetric analyzer under nitrogen gas at a heating rate of 10 °C min−1. Scanning electron microscope (SEM) images were obtained from an ultra-high-resolution electron microscope (FE-SEM, SU8010). Gas sorption isotherms were measured on a Micromeritics ASAP 2020M instrument. Before the sorption experiments, the as-synthesized samples were first solvent exchanged with MeOH, and then activated for 12 h at 150 °C under vacuum. N2 (99.999%) was used for all measurements. The temperature was controlled by a liquid nitrogen bath (77 K).Synthesis of [Co8(OH)4(H2O)2(bdt)6]·guest (denoted as MAF-48 or Co4-bdt)
A mixture of Co(OAc)2·4H2O (17.5 mg, 0.75 mmol), H2bdt (10.5 mg, 0.5 mmol), triethylamine (TEA, 0.2 mL), H2O (1.0 mL) and N,N-diethylacetoacetamide (DEF, 4.0 mL) was stirred for 30 minutes in air, transferred to a 100 mL vial and sealed with a screw cap, heated in an oven at 160 °C for 72 h, and then cooled to room temperature at a rate of 10 °C h−1, giving red cubic crystals. The resulting red microcrystalline powders were washed with EtOH three times and then immersed in 1 M KOH (yield 78%). EA calc. (%) for [Co8(OH)6(bdt)4(Hbdt)2]·12H2O·5MeOH (C65H88N36Co8O23): C, 35.28; H, 4.01; N, 22.79; found: C 35.44, H 4.02, N 22.91.Crystal structure determination
Diffraction data of Co4-bdt were collected on a Rigaku XtaLAB P300DS-detector diffractometer (Cu Kα). All structures were solved by direct methods and refined with the full-matrix least-squares technique on F2 by the SHELXTL-2014 software package. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed geometrically. The PLATON SQUEEZE treatment was applied, because all guest solvent molecules were extremely disordered and could not be modeled. Detailed structure determination parameters and crystallographic data are given in Table S1.†Results and discussion
The solvothermal reaction of Co(OAc)2 and H2bdt in N,N-diethylformamide (DEF) afforded red cubic crystals of [Co8(OH)6(bdt)4(Hbdt)2] (MAF-48, Co4-bdt). Single-crystal X-ray analysis revealed that Co4-bdt consists of an fcu network constructed of 12-connected Co8(μ4-OH)6(Rtrz)12 (Rtrz− = 1,2,3-triazolate group) clusters and 2-connected bdt2− ligands (Fig. 1 and Table S1†), and this is isostructural with [Ni8(OH)4(H2O)2(bdp)6] (Ni4-bdp, H2bdp = 4,4′-benzene-1,4-diylbis(1H-pyrazole)).31 At each face of {Co8(μ4-OH)6}, the hydroxyl anion links four coplanar CoII ions in a typical μ4 coordination mode to form a {Co4(μ4-OH)} unit, which is an underlying catalytic active site for the water oxidation reaction. X-ray photoelectron spectroscopy (XPS) of Co4-bdt showed that the metal ions are all divalent (Fig. S1†).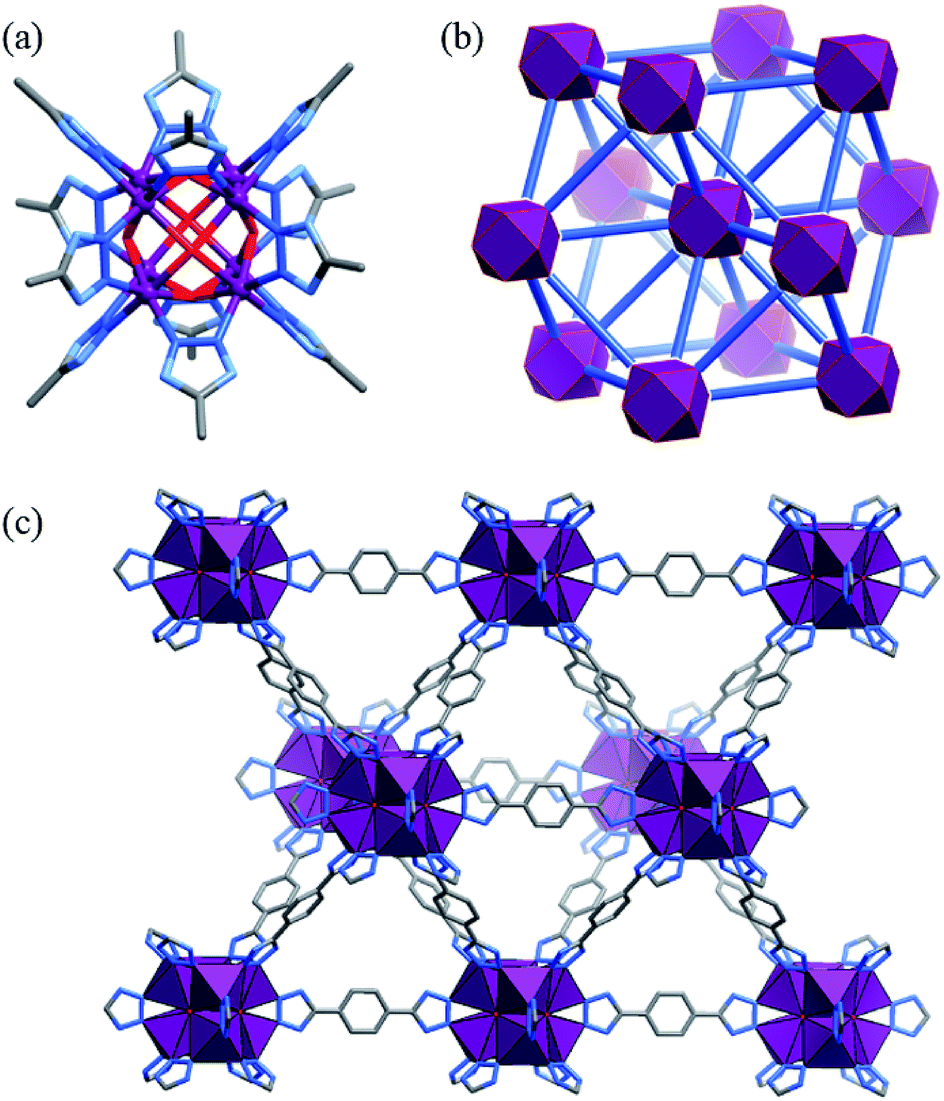 | ||
| Fig. 1 (a) The Co8(μ4-OH)6(Rtrz)12 cluster (hydrogen atoms are omitted for clarity), (b) the network topology (octanuclear clusters and bistriazolate ligands are simplified as violet polyhedra and blue sticks, respectively), and (c) 3D coordination framework (CoN3O3 units are shown as violet polyhedra) of Co4-bdt. | ||
The purity of Co4-bdt was preliminarily demonstrated by the scanning electron microscopy (SEM) images and transmission electron microscopy (TEM) images (Fig. S2†). Thermogravimetric (TG) and powder X-ray diffraction (PXRD) measurements of methanol-exchanged Co4-bdt indicated that its guest molecules can be removed above 100 °C, and the host framework can be stable up to 300 °C (Fig. S3 and S4†). Moreover, Co4-bdt can remain intact in 6 M KOH (Fig. S5†) for at least 3 days, representing the most alkali stable MOF.32,33 A N2 sorption isotherm was measured for Co4-bdt at 77 K (Fig. S6†), which shows typical type-I characteristics, with a saturation uptake of 594 cm3 (STP) g−1, corresponding to a pore volume of 0.928 cm3 g−1 (crystallographic value of 0.924 cm3 g−1). Furthermore, the Brunauer–Emmett–Teller (BET) and Langmuir surface areas of Co4-bdt were calculated to be 2266 and 2570 m2 g−1, respectively.
The photodriven water oxidation (PWO) experiment of Co4-bdt was performed under visible light in water with [Ru(bpy)3]SO4 (bpy = 2,2′-bipyridine) as the photosensitizer and Na2S2O8 as the sacrificial electron acceptor, and these are the typical reaction conditions used in the literature. A Clark-type oxygen electrode was used to monitor in situ the amount of evolved O2 dissolved in the solution (Fig. 2a and S7†). O2 rapidly formed at an initial turnover frequency (TOF) of 3.05 ± 0.03 s−1. Due to the consumption of the sacrificial electron acceptor, the production rates slowly decreased (Fig. S8†). Except for PSII, the performance of Co4-bdt is higher than that for all other known heterogeneous catalysts,26,27,34–39 and is comparable to the best homogeneous catalysts under the same conditions40–43 (Tables S2 and S3†). Since the process that limits catalytic turnover is the oxidative quenching of the Ru excited state,44–46 the photocatalytic water oxidation experiments using [Ru(bpy)3]3+ as the chemical oxidant without Na2S2O8 were carried out. It can be seen that the TOF values increased and that the catalysts work under pseudo first order conditions (Fig. S9†). Importantly, the actual TOF value of Co4-bdt is as high as 15.7 s−1 (Fig. S10†), which is higher than that of the best PWO catalyst (13 s−1).47–50
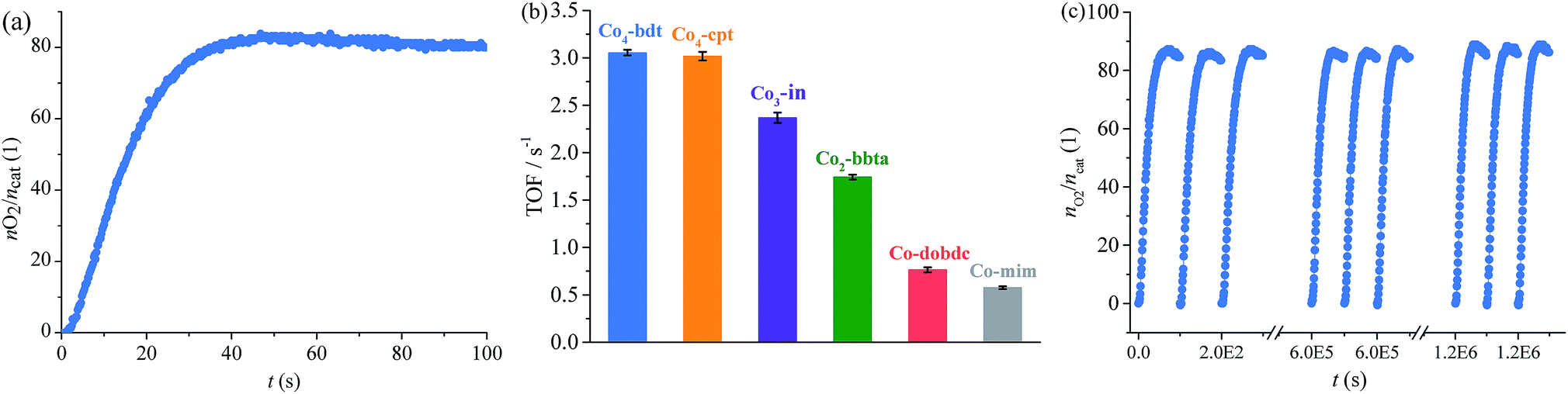 | ||
| Fig. 2 (a) Kinetics of O2 formation in the photocatalytic system using Co4-bdt as the catalyst. (b) Comparison of the TOF values for the photodriven water oxidation reactions. (c) O2 production profiles of the repeated photocatalytic water oxidation reactions using Co4-bdt as the catalyst. Reaction conditions: [Ru(bpy)3]SO4 (0.03 μmol), catalyst (0.5 nmol), borate buffer (pH = 9, 2 mL), Na2S2O8 (0.1 μmol), LED light (λ = 450 ± 5 nm), and 25 °C. | ||
Thanks to the excellent stability, the TOF of Co4-bdt remained 3.05 s−1 (Fig. 2c) after 12000 runs, indicating that the turnover number (TON) is larger than one million. Notably, the TON value of Co4-bdt is two orders higher than that of the best catalysts.51 To directly determine the TON of the Co4-bdt, time-dependent oxygen evolution experiments were carried out with chemical oxidant [Ru(bpy)3]3+. As shown in Fig. S11,†Co4-bdt gave a TON of 1.2 × 106, which is consistent with that obtained from the recycling experiment (Fig. 2c). After the reactions, the Co4-bdt catalyst was recovered from the reaction mixture and was found to be almost unchanged according to the N2 sorption isotherm measurements (Fig. S6†), the PXRD patterns and the SEM images (Fig. S12†). Furthermore, inductively coupled plasma-mass spectrometry showed that just 0.14% of the Co ions in Co4-bdt were leached into the reaction solution after the reaction, and the filtrate showed negligible catalytic activity (Fig. S7†), which confirmed the heterogeneous nature and the stability of the catalyst. The high stability of Co4-bdt might be ascribed to the high connectivity of the second building units (SBUs)29 and the more stable cobalt–N (nitrogen atom) coordination bonds.28,32
In order to study the mechanism, we selected five other cobalt-based MOFs with variable metal-hydroxide units and an isostructural MOF, [Ni8(OH)6(bdt)4(Hbdt)2] (Ni4-bdt), for comparison (Fig. S13–S15†): (i) [CoII8(μ4-OH)6(cpt)6] (Co4-cpt, Hcpt = 4-(4′-carboxyphenyl)-1,2,4-triazole) is made up of octanuclear cobalt-hydroxide {Co8(μ4-OH)6} clusters and cpt− ligands;52 (ii) [Co6(μ3-OH)2(in)4(HCOO)6] (Co3-in, Hin = isonicotinic acid) is made up of asymmetric triangular units of {Co3(μ3-OH)}, in− and oxalate ligands.53 Each hydroxyl anion is linked to three adjacent CoII ions in a typical μ3 coordination mode to build the {Co3(μ3-OH)} cluster; (iii) [Co2(μ-OH)2(bbta)] (MAF-X27-OH/Co2-bbta, H2bbta = 1H,5H-benzo-(1,2-d:4,5-d′)bistriazole) bears a pair of μ-OH− ligands at the cis-positions of its open metal site.32 Each hydroxyl anion links two adjacent CoII ions in a typical bidentate coordination mode to build a {Co2(μ-OH)} unit; (iv) [Co2(dobdc)] (Co-MOF-74/Co-dobdc, H4dobdc = 2,5-dihydroxyl-1,4-benzenedicarboxylic acid) is composed of square-pyramidal Co ions and dobdc4− ligands.54 The square-pyramidal Co ion can coordinate to a terminal water molecule or to a hydroxyl anion to form a distorted octahedral mode; (v) [Co(mim)2] (ZIF-67/Co-mim, Hmim = 2-methylimidazole) is constructed of tetrahedral Co ions and mim− ligands.55 In addition, the tetrahedral Co ion can coordinate to a terminal water molecule or to a hydroxyl anion to form a distorted trigonal-bipyramidal mode.
The PWO experiment was performed under the same conditions. As calculated from the initial O2 production rates (Fig. 2b and S16†), the turnover frequency (TOF) for O2 is as follows, Co4-bdt (3.05 ± 0.03 s−1) ≈ Co4-cpt (3.02 ± 0.05 s−1) > Co3-in (2.37 ± 0.05 s−1) > Co2-bbta (1.74 ± 0.03 s−1) > Ni4-bdt (1.21 ± 0.03 s−1) > Co-dobdc (0.77 ± 0.03 s−1) ≈ Co-mim (0.58 ± 0.01 s−1) (Tables S4 and S5†). Interestingly, for the cobalt ions, when the coordination number of the hydroxide ligand increases, the catalytic performance becomes better, and the catalytic performance of Co4-bdt is much higher than that of Ni4-bdt.
To demonstrate that the Co-manifold works with a four electron/four proton mechanism, the activity of Co4-bdt for water oxidation was studied by electrochemical characterization. Linear sweep voltammetry (LSV) was performed in water at pH = 7 (Fig. 3a, S17 and S18†). Assuming that the water oxidation reaction involves a four-electron process, the Faraday efficiency of Co4-bdt for water oxidation was measured to be virtually 100% (Fig. S19 and Table S6†). Importantly, Co4-bdt afforded a current density of 2.0 mA cm−2 at an overpotential of 352 mV, which is much lower than that for all other reported catalysts (Table S7†). The performance of Co4-bdt showed negligible changes after OER tests at 10 mA cm−2 for 24 h (Fig. 3b). Furthermore, PXRD patterns (Fig. S12†) and cyclic voltammetry curves (Fig. 3c) of Co4-bdt showed negligible changes after the electrochemical OER tests for 24 h. The electrocatalytic activity follows the order: Co4-bdt (352 mV) > Co4-cpt (355 mV) > Co3-in (385 mV) > Co2-bbta (489 mV) > Co-dobdc (544 mV) > Co-mim (638 mV) (Fig. S17a and Table S7†) and this is consistent with results observed from the photodriven water oxidation experiment. This phenomenon demonstrates the high catalytic activity of Co4-bdt in the water oxidation reaction.
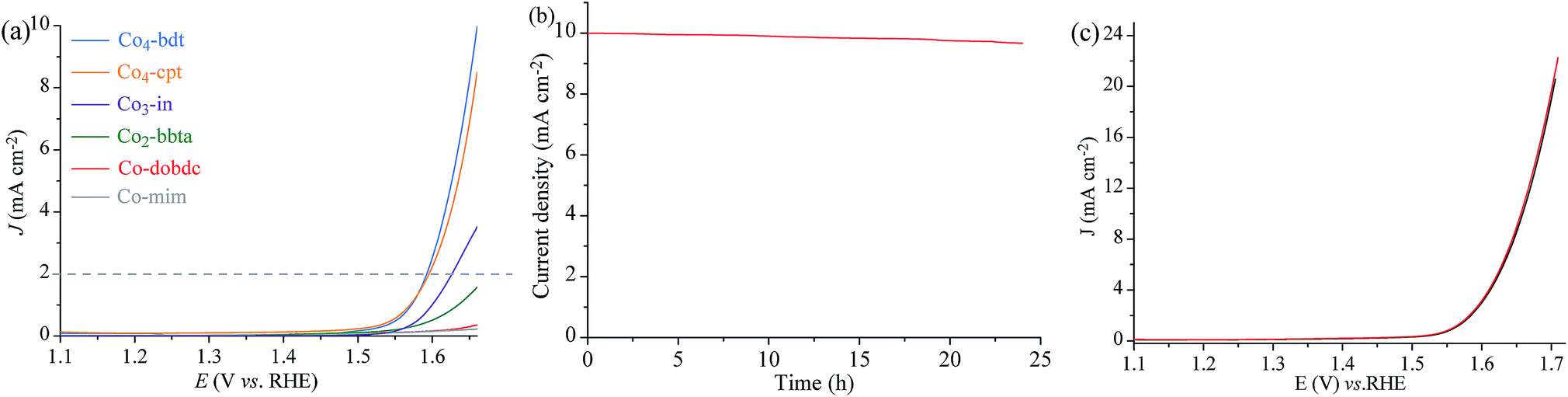 | ||
| Fig. 3 (a) LSV curves of Co4-bdt, Co4-cpt, Co3-in, Co2-bbta, Co-mim and Co-dobdc at pH = 7. (b) The chronopotentiometry curves of Co4-bdt at an overpotential corresponding to the current density of 10 mA cm−2 at pH = 7. (c) LSV curves of Co4-bdt before (black) and after (red) the electrochemical OER test at 10 mA cm−2 for 24 h at pH = 7. | ||
Isotope tracing experiments were carried out to investigate the role of μ4-OH− during the water oxidation reaction. The extent of 18O catalyst incorporation was calculated to be ca. 44.8 ± 1.1%, measured by the GC-MS analysis of the acidolyzed sample (Fig. S20 and Table S8†). During a representative water oxidation experiment with 18O labeled Co4-bdt as the photocatalyst, the photogenerated 18O16O (m/z = 34) could be clearly detected, while 18O16O was not detected in the water oxidation experiment with unlabeled Co4-bdt (Fig. S21, S22 and Table S9†). After the 18O-labeled Co4-bdt was immersed in pH = 9 aqueous solution H216O for 10 min, 18/16O2 intensity was the same as that of the fresh 18O-labeled sample (Fig. S23†). This demonstrates that the 16O/18O exchange behaviour between the water molecule and the cobalt-hydroxide {Co4(μ4-OH)} unit during the isotope tracing experiment can be neglected for the observed significant 18/16O2 intensity enhancement. This result indicates that the bridging OH− ligand does indeed participate in the reaction to offer an oxygen vacancy, which serves as the active site for water oxidation. This phenomenon was also observed for Co3-in and Co2-bbta (Fig. S21†). In other words, the site, capping four coplanar cobalt ions, indeed serves as the highly efficient active site for water oxidation. Such an active site for water oxidation is the first to be reported to date. It should be noted that not all oxygen atoms from the Co4 cluster participate in the reaction at the same time, and the O coordinated on the Co4 cluster will be replaced by the water molecule before the O2 formation. Therefore, although the O from the Co4 cluster indeed engages in O2 formation, the structure of Co4-bdt remains during and after the photodriven water oxidation reaction (Fig. 4a, S12 and S24†).
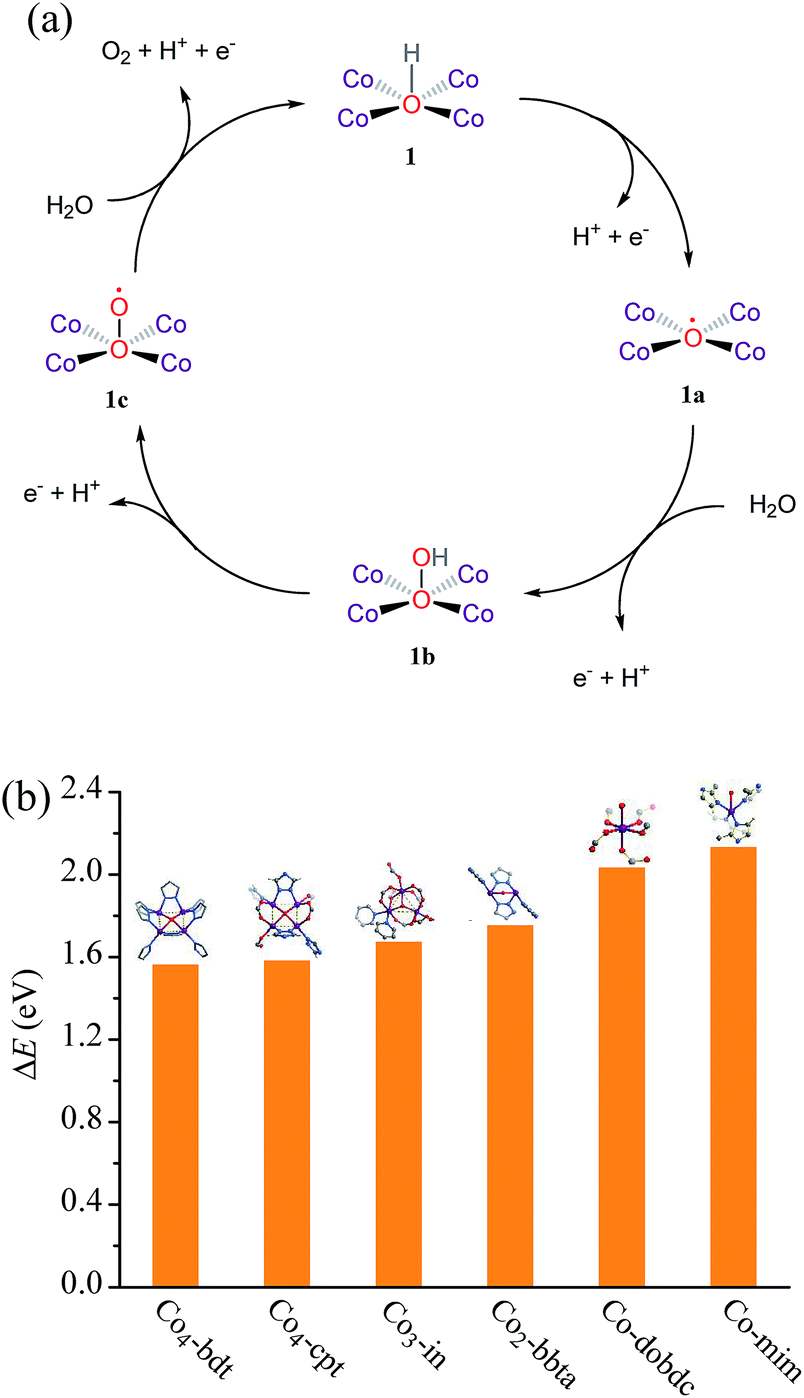 | ||
| Fig. 4 (a) A proposed reaction mechanism for using Co4-bdt as the catalyst for photodriven water oxidation. (1) The complex 1 is oxidatively activated by the photo-generated hole. (2) The nucleophilic attack of the water molecule forms the O–O bond. (3) The complex 1b is oxidized to complex 1c. (4) The complex 1c is further oxidized to evolve O2, accompanied by the regeneration of 1. (b) PDFT calculated adsorption energies of the reacting hydroxyl radical (ΔE = E(*OH) − E(*) − [E(H2O) − E(H)], * represents the catalyst). | ||
To further understand the relationship between the coordination number of the oxygen atom in OH− and the properties, we analyzed the adsorption energy of OH− adsorbed on the active site by periodic density functional theory (PDFT). The adsorption energy (ΔE) of OH− follows the order, Co-mim (2.13 eV) > Co-dobdc (2.03 eV) > Co2-bbta (1.75 eV) > Co3-in (1.67 eV) > Co4-cpt (1.58 eV) ≈ Co4-bdt (1.56 eV) > Ni4-bdt (1.19 eV) (Fig. 4b), and this implies that the OH− becomes more stable with an increase in the coordination number. The poor performance of Ni4-bdt is due to its too strongly OH− binding. By combining the PDFT simulation results and the isotope tracing experiments, it can be seen that the high catalytic performance of Co4-bdt might be ascribed to the fact that the OH− is appropriately stabilized by four coplanar cobalt ions.
Conclusions
In conclusion, through a combination of the highly connected cobalt-hydroxide unit and the azolate bridging ligand, we designed and synthesized cobalt azolate frameworks with high stability and excellent water oxidation performance. Since the oxygen atom is simultaneously coordinated by four coplanar cobalt ions, the reacting hydroxyl radical is appropriately stabilized during the water oxidation reaction, promoting the catalytic performance. These results should be insightful for understanding the structure–function relationship of water oxidation catalysts and for developing new MOF-based catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21701193, 21890380, and 21821003), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (2017BT01C161), the Guangdong Natural Science Funds for Distinguished Young Scholars (2018B030306009), and the Young Elite Scientists Sponsorship Program by CAST (2017QNRC001). Thank also go to Prof. Jie-Peng Zhang for his valuable suggestions.Notes and references
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115, 12974–13005 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed.
- X. Yang, Z. Chen, J. Xu, H. Tang, K. Chen and Y. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 15285–15293 CrossRef CAS PubMed.
- F. Zhang, A. Yamakata, K. Maeda, Y. Moriya, T. Takata, J. Kubota, K. Teshima, S. Oishi and K. Domen, J. Am. Chem. Soc., 2012, 134, 8348–8351 CrossRef CAS PubMed.
- Z.-J. Liu, X.-L. Wang, C. Qin, Z.-M. Zhang, Y.-G. Li, W.-L. Chen and E.-B. Wang, Coord. Chem. Rev., 2016, 313, 94–110 CrossRef CAS.
- X.-J. Su, M. Gao, L. Jiao, R.-Z. Liao, P. E. M. Siegbahn, J.-P. Cheng and M.-T. Zhang, Angew. Chem., Int. Ed., 2015, 54, 4909–4914 CrossRef CAS PubMed.
- H.-Y. Du, S.-C. Chen, X.-J. Su, L. Jiao and M.-T. Zhang, J. Am. Chem. Soc., 2018, 140, 1557–1565 CrossRef CAS PubMed.
- S. Peng, F. Gong, L. Li, D. Yu, D. Ji, T. Zhang, Z. Hu, Z. Zhang, S. Chou, Y. Du and S. Ramakrishna, J. Am. Chem. Soc., 2018, 140, 13644–13653 CrossRef CAS PubMed.
- X. Wang, H. Xiao, A. Li, Z. Li, S. Liu, Q. Zhang, Y. Gong, L. Zheng, Y. Zhu, C. Chen, D. Wang, Q. Peng, L. Gu, X. Han, J. Li and Y. Li, J. Am. Chem. Soc., 2018, 140, 15336–15341 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- S. Zhao, R. Jin, H. Abroshan, C. Zeng, H. Zhang, S. D. House, E. Gottlieb, H. J. Kim, J. C. Yang and R. Jin, J. Am. Chem. Soc., 2017, 139, 1077–1080 CrossRef CAS PubMed.
- J.-Q. Shen, P.-Q. Liao, D.-D. Zhou, C.-T. He, J.-X. Wu, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2017, 139, 1778–1781 CrossRef CAS PubMed.
- X.-L. Wang, L.-Z. Dong, M. Qiao, Y.-J. Tang, J. Liu, Y. Li, S.-L. Li, J.-X. Su and Y.-Q. Lan, Angew. Chem., Int. Ed., 2018, 57, 9660–9664 CrossRef CAS PubMed.
- W. Tu, Y. Xu, S. Yin and R. Xu, Adv. Mater., 2018, 30, 1707582 CrossRef PubMed.
- D. Li, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2018, 30, 1707377 CrossRef PubMed.
- G. Lan, Y.-Y. Zhu, S. S. Veroneau, Z. Xu, D. Micheroni and W. Lin, J. Am. Chem. Soc., 2018, 140, 5326–5329 CrossRef CAS PubMed.
- S. Pullen, H. Fei, A. Orthaber, S. M. Cohen and S. Ott, J. Am. Chem. Soc., 2013, 135, 16997–17003 CrossRef CAS PubMed.
- N. Kornienko, Y. Zhao, C. S. Kley, C. Zhu, D. Kim, S. Lin, C. J. Chang, O. M. Yaghi and P. Yang, J. Am. Chem. Soc., 2015, 137, 14129–14135 CrossRef CAS PubMed.
- Y. Wang, N.-Y. Huang, J.-Q. Shen, P.-Q. Liao, X.-M. Chen and J.-P. Zhang, J. Am. Chem. Soc., 2018, 140, 38–41 CrossRef CAS PubMed.
- Y. Lee, S. Kim, J. K. Kang and S. M. Cohen, Chem. Commun., 2015, 51, 5735–5738 RSC.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- P.-Q. Liao, J.-Q. Shen and J.-P. Zhang, Coord. Chem. Rev., 2018, 373, 22–48 CrossRef CAS.
- L. Jiao, Y. Wang, H.-L. Jiang and Q. Xu, Adv. Mater., 2018, 30, 1703663 CrossRef PubMed.
- H. Wang, Q.-L. Zhu, R. Zou and Q. Xu, Chem, 2017, 2, 52–80 CAS.
- L. Chi, Q. Xu, X. Liang, J. Wang and X. Su, Small, 2016, 12, 1351–1358 CrossRef CAS PubMed.
- Q. Xu, H. Li, F. Yue, L. Chi and J. Wang, New J. Chem., 2016, 40, 3032–3035 RSC.
- J.-P. Zhang, Y.-B. Zhang, J.-B. Lin and X.-M. Chen, Chem. Rev., 2012, 112, 1001–1033 CrossRef CAS PubMed.
- K. Wang, X.-L. Lv, D. Feng, J. Li, S. Chen, J. Sun, L. Song, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2016, 138, 914–919 CrossRef CAS PubMed.
- X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- N. M. Padial, E. Q. Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., 2013, 125, 8448–8452 CrossRef.
- X.-F. Lu, P.-Q. Liao, J.-W. Wang, J.-X. Wu, X.-W. Chen, C.-T. He, J.-P. Zhang, G.-R. Li and X.-M. Chen, J. Am. Chem. Soc., 2016, 138, 8336–8339 CrossRef CAS PubMed.
- H. J. Choi, M. Dinca, A. Dailly and J. R. Long, Energy Environ. Sci., 2010, 3, 117–123 RSC.
- D. Hong, Y. Yamada, T. Nagatomi, Y. Takai and S. Fukuzumi, J. Am. Chem. Soc., 2012, 134, 19572–19575 CrossRef CAS PubMed.
- F. A. Frame, T. K. Townsend, R. L. Chamousis, E. M. Sabio, T. Dittrich, N. D. Browning and F. E. Osterloh, J. Am. Chem. Soc., 2011, 133, 7264–7267 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- X. Du, Y. Ding, R. Xiang and X. Xiang, Phys. Chem. Chem. Phys., 2015, 17, 10648–10655 RSC.
- Y. Zhao, Y. Zhang, Y. Ding and M. Chen, Dalton Trans., 2015, 44, 15628–15635 RSC.
- Y. Yamada, K. Oyama, R. Gates and S. Fukuzumi, Angew. Chem., Int. Ed., 2015, 54, 5613–5617 CrossRef CAS PubMed.
- N. S. McCool, D. M. Robinson, J. E. Sheats and G. C. Dismukes, J. Am. Chem. Soc., 2011, 133, 11446–11449 CrossRef CAS PubMed.
- S. Berardi, G. La Ganga, M. Natali, I. Bazzan, F. Puntoriero, A. Sartorel, F. Scandola, S. Campagna and M. Bonchio, J. Am. Chem. Soc., 2012, 134, 11104–11107 CrossRef CAS PubMed.
- X.-B. Han, Z.-M. Zhang, T. Zhang, Y.-G. Li, W. Lin, W. You, Z.-M. Su and E.-B. Wang, J. Am. Chem. Soc., 2014, 136, 5359–5366 CrossRef CAS PubMed.
- X.-B. Han, Y.-G. Li, Z.-M. Zhang, H.-Q. Tan, Y. Lu and E.-B. Wang, J. Am. Chem. Soc., 2015, 137, 5486–5493 CrossRef CAS PubMed.
- H. Lv, J. Song, Y. V. Geletii, J. W. Vickers, J. M. Sumliner, D. G. Musaev, P. Kögerler, P. F. Zhuk, J. Bacsa, G. Zhu and C. L. Hill, J. Am. Chem. Soc., 2014, 136, 9268–9271 CrossRef CAS PubMed.
- M. Natali, M. Orlandi, S. Berardi, S. Campagna, M. Bonchio, A. Sartorel and F. Scandola, Inorg. Chem., 2012, 51, 7324–7331 CrossRef CAS PubMed.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- B. Das, B.-L. Lee, E. A. Karlsson, T. Åkermark, A. Shatskiy, S. Demeshko, R.-Z. Liao, T. M. Laine, M. Haukka, E. Zeglio, A. F. Abdel-Magied, P. E. M. Siegbahn, F. Meyer, M. D. Kärkäs, E. V. Johnston, E. Nordlander and B. Åkermark, Dalton Trans., 2016, 45, 13289–13293 RSC.
- A. F. Abdel-Magied, W. A. A. Arafa, T. M. Laine, A. Shatskiy, M. D. Kärkäs, B. Åkermark and E. V. Johnston, ChemCatChem, 2017, 9, 1583–1587 CrossRef CAS.
- J. W. Vickers, J. M. Sumliner, H. Lv, M. Morris, Y. V. Geletii and C. L. Hill, Phys. Chem. Chem. Phys., 2014, 16, 11942–11949 RSC.
- M. D. Kärkäs, Y.-Y. Li, P. E. M. Siegbahn, R.-Z. Liao and B. Åkermark, Inorg. Chem., 2018, 57, 10881–10895 CrossRef PubMed.
- Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 122, 9118–9121 CrossRef.
- R. J. Holmberg, M. Kay, I. Korobkov, E. Kadantsev, P. G. Boyd, T. Aharen, S. Desgreniers, T. K. Woo and M. Murugesu, Chem. Commun., 2014, 50, 5333–5335 RSC.
- Z. Li, L. Du, J. Zhou, L. Li, Y. Hu, Y. Qiao, M. Xie and Q. Zhao, New J. Chem., 2013, 37, 2473–2478 RSC.
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., 2005, 117, 6512–6516 CrossRef.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, crystal data and structure refinement results, thermogravimetric analyses, variable-temperature PXRD. CCDC 1881071. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc03224j |
| ‡ N.-Y. Huang and J.-Q. Shen contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |