
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed decarbonylative Suzuki–Miyaura cross-coupling of amides by carbon–nitrogen bond activation†
Tongliang
Zhou
b,
Chong-Lei
Ji
c,
Xin
Hong
 *c and
Michal
Szostak
*ab
*c and
Michal
Szostak
*ab
aCollege of Chemistry and Chemical Engineering, Key Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science and Technology, Xi'an 710021, China
bDepartment of Chemistry, Rutgers University, 73 Warren Street, Newark, NJ 07102, USA. E-mail: michal.szostak@rutgers.edu
cDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: hxchem@zju.edu.cn
First published on 3rd September 2019
Abstract
Palladium-catalyzed Suzuki–Miyaura cross-coupling or aryl halides is widely employed in the synthesis of many important molecules in synthetic chemistry, including pharmaceuticals, polymers and functional materials. Herein, we disclose the first palladium-catalyzed decarbonylative Suzuki–Miyaura cross-coupling of amides for the synthesis of biaryls through the selective activation of the N–C(O) bond of amides. This new method relies on the precise sequence engineering of the catalytic cycle, wherein decarbonylation occurs prior to the transmetallation step. The reaction is compatible with a wide range of boronic acids and amides, providing valuable biaryls in high yields (>60 examples). DFT studies support a mechanism involving oxidative addition, decarbonylation and transmetallation and provide insight into high N–C(O) bond activation selectivity. Most crucially, the reaction establishes the use of palladium catalysis in the biaryl Suzuki–Miyaura cross-coupling of the amide bond and should enable the design of a wide variety of cross-coupling methods in which palladium rivals the traditional biaryl synthesis from aryl halides and pseudohalides.
Introduction
Palladium-catalyzed cross-coupling reactions are fundamental methods for the construction of many important molecules in chemical synthesis and these reactions are widely used to expediate the synthesis of target motifs.1–3 In particular, due to broad generality, predictable functional group tolerance, high reaction selectivity and operational-simplicity, palladium-catalyzed cross-couplings have enabled rapid progress in the development of improved medicines, precise electronic devices, and multifunctional dyes that broadly benefit society.4–6While typical Suzuki–Miyaura cross-couplings employ aryl halides as electrophiles, unconventional C–X (X = O, N, S) cross-coupling partners have attracted significant attention because they enable an orthogonal selectivity paradigm.4–6 In this context, amides (X = CONR2) are particularly attractive as aryl electrophiles in the Suzuki–Miyaura cross-coupling because amides are among the most widespread functional groups in chemical science,7 including in the synthesis of pharmaceuticals, and play an essential role as linkages in peptides and proteins.8,9 While significant progress has been achieved in cross-coupling of amides enabled by selective metal insertion into the N–C(O) amide bond (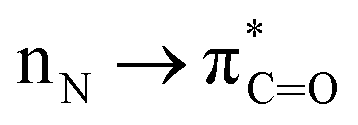 conjugation, 15–20 kcal mol−1 in planar amides),10–14 the palladium-catalyzed Suzuki–Miyaura biaryl cross-coupling of amides has been a major challenge (Fig. 1A).
conjugation, 15–20 kcal mol−1 in planar amides),10–14 the palladium-catalyzed Suzuki–Miyaura biaryl cross-coupling of amides has been a major challenge (Fig. 1A).
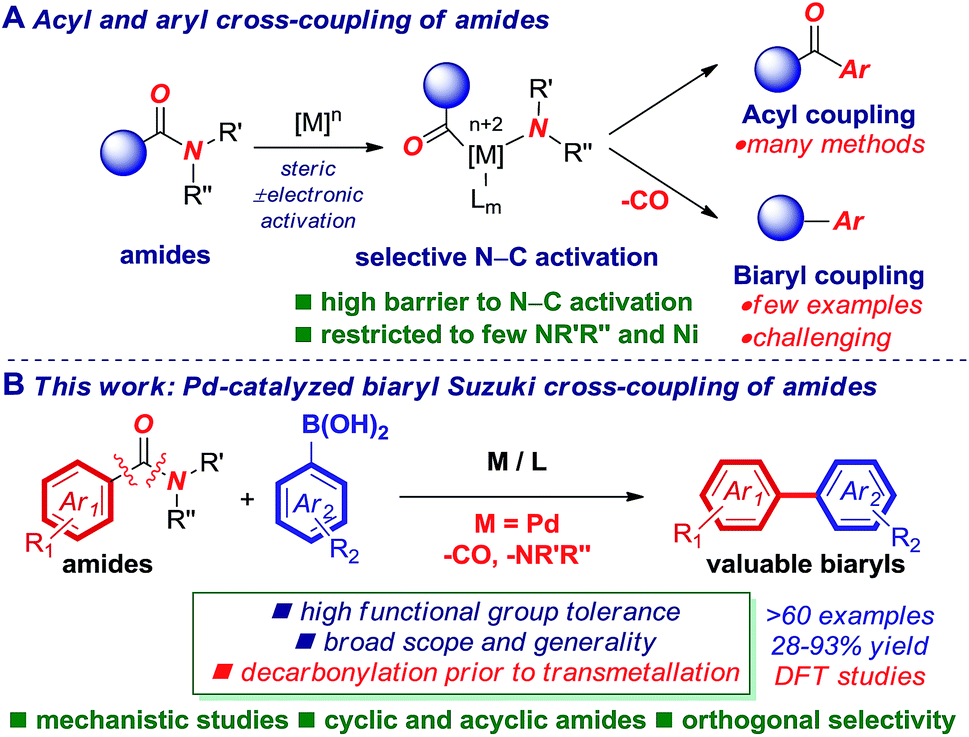 | ||
| Fig. 1 (A) Acyl- and decarbonylative cross-coupling of amides. (B) Pd-catalyzed decarbonylative biaryl cross-coupling of amides enabled by sequence engineering (this study). | ||
Herein, we disclose the first palladium-catalyzed decarbonylative Suzuki–Miyaura cross-coupling of amides for the synthesis of biaryls (Fig. 1B). This new method relies on the precise sequence engineering of the catalytic cycle, wherein decarbonylation occurs prior to the transmetallation step. The reaction proceeds through the selective activation of the N–C(O) bond of both cyclic and acyclic amides. DFT studies were conducted and support a mechanism involving oxidative addition, decarbonylation and transmetallation, and provide insight into the origin of the high N–C(O) bond activation selectivity. The reaction shows unprecedented generality and functional group tolerance in decarbonylative amide bond cross-coupling, providing valuable biaryls in high yields (>60 examples). Most crucially, the reaction establishes versatile palladium catalysis for the synthesis of high-value biaryls from amides, rivaling the substrate scope achieved with the traditional Suzuki cross-coupling of aryl halides.
Results and discussion
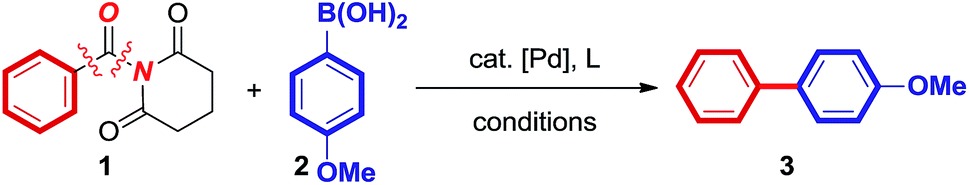
The decarbonylative biaryl Suzuki–Miyaura cross-coupling of amides is a challenging reaction, in which several elementary organometallic steps must occur in a well-engineered sequence.15–18 To date, very few examples of the biaryl Suzuki–Miyaura cross-coupling of amides have been reported; all of them limited to nickel-catalysis.15 The high reactivity of Ni has been ascribed to a facile CO migration, which permitted for transmetallation preceding decarbonylation.18 However, Ni-catalyzed biaryl Suzuki coupling of amides has been severely limited to specific substrate combinations and showed narrow functional group tolerance.In consideration of the tremendous utility of Pd-catalyzed cross-couplings in organic synthesis,19,20 we questioned whether general palladium catalysis might be applied for the decarbonylative biaryl Suzuki–Miyaura cross-coupling of amides by N–C(O) bond activation. Our investigation started with evaluation of the coupling of a challenging, electronically-neutral N-benzoyl glutarimide (1) with 4-methoxyphenyl boronic acid (2) in the presence of various Pd catalysts (Table 1); note that this combination is unsuccessful using Ni catalysis. After very extensive optimization (Table 1 and ESI†), we found that a catalytic system using Pd(dppf)Cl2 (5 mol%) in the presence of close to a stoichiometric amount of boronic acid (1.2 equiv.) and NaHCO3 (3.0 equiv.) in dioxane at 160 °C, delivered the desired biaryl product in 90% yield and >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 biaryl:ketone selectivity (entry 1). Interestingly, a comparable efficiency was observed using Pd(dppb)Cl2 (5 mol%) as the precatalyst (entry 2). The use of a weak base is crucial (entries 8–15). From the outset, we hypothesized that the use of a weak base would slow down transmetallation,21 permitting for decarbonylation preceding aryl transfer. Furthermore, the effect of boronic acid stoichiometry (entries 1–8) as well as the counterion (entries 19–20) is the key in determining the selectivity for the formation of biaryl, as expected from decarbonylation vs. transmetallation selectivity. It is worthwhile to note the impact of the reaction temperature on the selectivity (entries 21 and 22). The use of precatalysts22 (entries 1–3 vs. 19 and 20) simplifies the reaction set-up and facilitates CO de-insertion. It is further important to note that both bidentate and monodentate phosphane ligands are similarly effective (entries 1–7), consistent with the importance of decarbonylation prior to generating the aryl-Pd intermediate.17,18
:1 biaryl:ketone selectivity (entry 1). Interestingly, a comparable efficiency was observed using Pd(dppb)Cl2 (5 mol%) as the precatalyst (entry 2). The use of a weak base is crucial (entries 8–15). From the outset, we hypothesized that the use of a weak base would slow down transmetallation,21 permitting for decarbonylation preceding aryl transfer. Furthermore, the effect of boronic acid stoichiometry (entries 1–8) as well as the counterion (entries 19–20) is the key in determining the selectivity for the formation of biaryl, as expected from decarbonylation vs. transmetallation selectivity. It is worthwhile to note the impact of the reaction temperature on the selectivity (entries 21 and 22). The use of precatalysts22 (entries 1–3 vs. 19 and 20) simplifies the reaction set-up and facilitates CO de-insertion. It is further important to note that both bidentate and monodentate phosphane ligands are similarly effective (entries 1–7), consistent with the importance of decarbonylation prior to generating the aryl-Pd intermediate.17,18
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | 2 (equiv.) | Yield 3b (%) | Selectivityc |
|
a Conditions: amide (1.0 equiv.), Ar–B(OH)2, [Pd] (5 mol%), base (3 equiv.), dioxane (0.125 M), 160 °C, 12 h.
b GC/1H NMR yields.
c Refers to biaryl:ketone selectivity.
d [Pd] (3 mol%). Note that in entries 9, 10, 13 and 14, the ketone is formed in 92–98% yields.
e Toluene.
f DME.
g NMP.
h [Pd] (10 mol%), L (20 mol%). Entry 19: ketone formed in 71%.
i 140 °C.
j 120 °C. See ESI for full details.
|
|||||
| 1 | Pd(dppf)Cl2 | NaHCO3 | 1.2 | 90 | >10:1 |
| 2 | Pd(dppb)Cl2 | NaHCO3 | 1.2 | 88 | 90:10 |
| 3 | Pd(PCy3)2Cl2 | NaHCO3 | 1.2 | 83 | 84:16 |
| 4 | Pd(dppf)Cl2 | NaHCO3 | 2.0 | 82 | 86:14 |
| 5 | Pd(PCy3)2Cl2 | NaHCO3 | 2.0 | 70 | 72:26 |
| 6 | Pd(PPh3)2Cl2 | NaHCO3 | 2.0 | 67 | 69:31 |
| 7 | Pd(dcypf)Cl2 | NaHCO3 | 2.0 | 65 | 66:34 |
| 8 | Pd(PCy3)2Cl2 | NaHCO3 | 1.05 | 83 | 85:15 |
| 9d | Pd(PCy3)2Cl2 | K2CO3 | 2.0 | <10 | 6:94 |
| 10d | Pd(PCy3)2Cl2 | K3PO4 | 2.0 | <2 | <5:>95 |
| 11d | Pd(PCy3)2Cl2 | KHCO3 | 2.0 | 50 | 53:47 |
| 12d | Pd(PCy3)2Cl2 | Na2CO3 | 2.0 | 15 | 56:44 |
| 13d | Pd(PCy3)2Cl2 | KF | 2.0 | <2 | <5:>95 |
| 14d | Pd(PCy3)2Cl2 | KOAc | 2.0 | <2 | <5:>95 |
| 15d | Pd(PCy3)2Cl2 | — | 2.0 | <2 | nd |
| 16d,e | Pd(PCy3)2Cl2 | NaHCO3 | 2.0 | <2 | <5:>95 |
| 17d,f | Pd(PCy3)2Cl2 | NaHCO3 | 2.0 | 16 | 55:45 |
| 18d,g | Pd(PCy3)2Cl2 | NaHCO3 | 2.0 | 31 | 51:49 |
| 19h | Pd(OAc)2/PCy3 | NaHCO3 | 1.2 | <2 | <5:>95 |
| 20h | PdCl2/PCy3 | NaHCO3 | 1.2 | 63 | 79:21 |
| 21i | Pd(dppb)Cl2 | NaHCO3 | 1.2 | 78 | 80:20 |
| 22j | Pd(dppb)Cl2 | NaHCO3 | 1.2 | 47 | 53:47 |
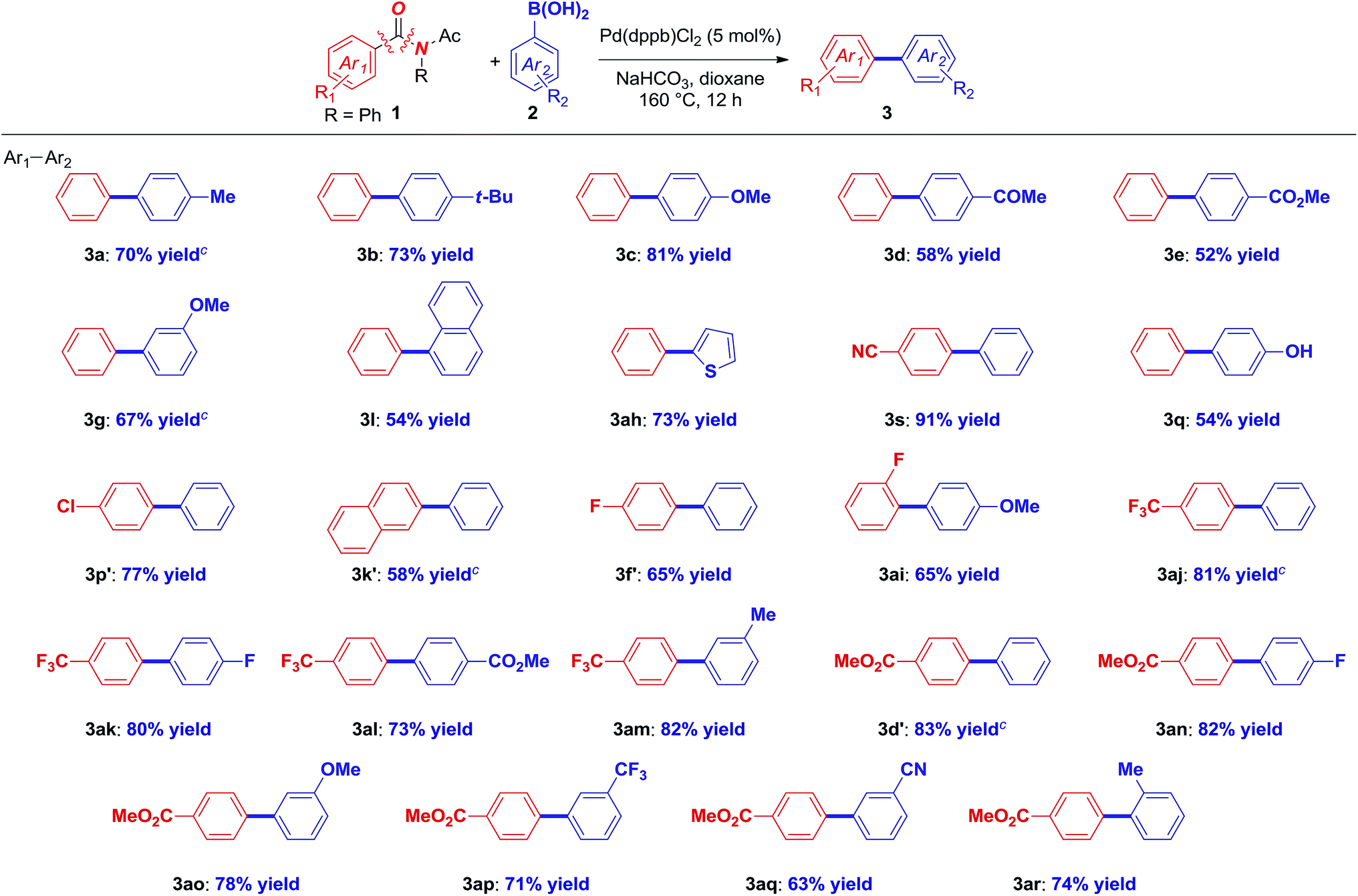
With optimized conditions in hand, the scope of this novel Suzuki–Miyaura biaryl cross-coupling of amides was next investigated (Table 2). The functional group tolerance and generality of this Pd-catalyzed method is remarkable. A broad range of electron neutral, electron-donating and electron-withdrawing boronic acids is compatible (3a–3f), all using the challenging electron-neutral, parent amide electrophile that is not suitable using Ni. Substitution at the meta-position with electronically-diverse boronic acids is well-tolerated (3g–3i). Sterically-hindered, polyaromatic, heterocyclic, including dioxolane, pyridine and thiophene boronic acids coupled with high levels of selectivity (3j–3o). Strikingly, the reaction is compatible with halides, including chlorides, as well as phenols, aldehydes, esters and nitriles (3p–3s), providing effective handles for further functionalization by established methods. In addition, electronically-diverse amides bearing representative electron-withdrawing, electron-donating and important fluorine-containing substituents are transferred with high selectivity across various boronic acids (3s–3ag), including such groups as prone to O–N cleavage isoxazolyl, aliphatic cyclopropyl, and sterically-hindered mesityl (3w–3y). The synthetic potential is highlighted in the cross-coupling of carboxyphenylboronic acid (3ag). Since amides can be ultimately derived from carboxylic acids, this preliminary result suggests the viability of iterative cross-coupling and is performed in the presence of an electrophilic nitrile handle, which serves as another orthogonal amide precursor. These results are for the first time comparable to the scope achieved using the classical Suzuki cross-coupling of halides and pseudohalides,1–6 and are unprecedented for any cross-coupling of amides to date.10–18

Remarkably, the optimized conditions are suitable for the cross-coupling of acyclic N-acetyl (N–Ac) amides (Table 3). A broad range of boronic acids and amides, including neutral, electron-rich and electron-withdrawing coupling partners, is compatible (3a–3ah). Notably, Pd-catalysis enables the coupling of an array of sensitive functional groups, such as halides, ethers, esters and nitriles (3c, 3e, 3s, 3q, 3p′) with high selectivity. Furthermore, the reaction delivers fluorinated biaryls of great importance in medicinal chemistry (3f′–3am) as well as biaryls bearing multiple electrophilic handles (3aq) as well as steric hindrance (3ar). It is noteworthy that the cleavage of the N-activating group, the main hurdle in decarbonylative cross-coupling of acyclic amides, is not observed under these mild conditions. This very rare use of acyclic amides in decarbonylative cross-coupling significantly expands the scope of biaryl Suzuki synthesis of amides and suggests a broad generality of this reactivity platform.
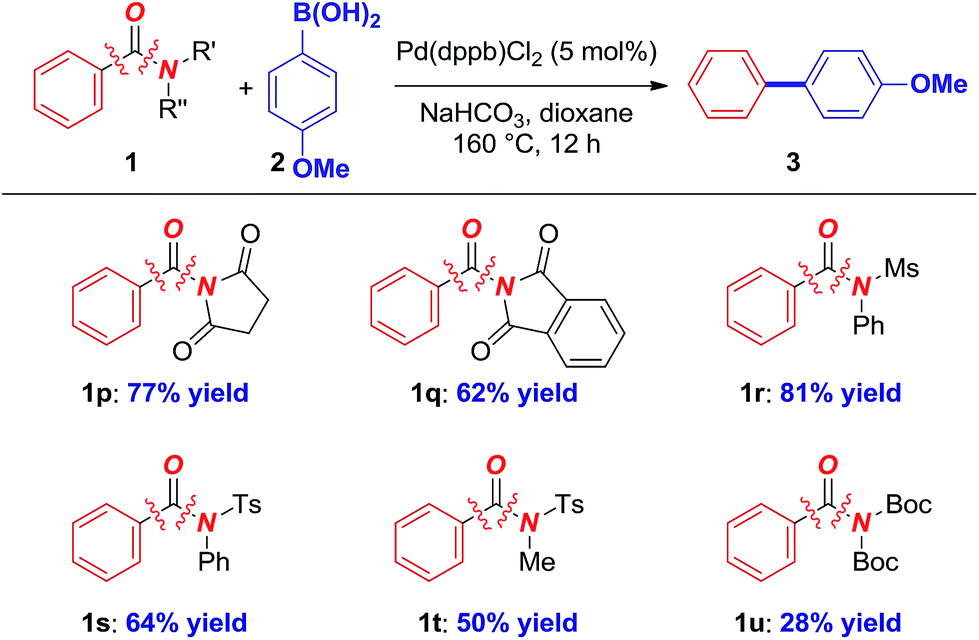
Furthermore, other amides, including N-succinimide (1p), N-phthalimide (1q), atom-economic N-Ms (1r) and acyclic N-Ts sulfonamides (1s–1t) are suitable substrates for the coupling (Table 4). Interestingly, even the highly challenging N-Boc2 amide (1u) that is prepared directly from 1° amide14c and prone to deactivation by a facile N-Boc cleavage afforded a promising yield in the coupling. These preliminary results bode well for the development of general biaryl syntheses from various amides, which is beyond the scope of Ni catalysis.
Several additional points should be noted: (1) at this stage, cross-coupling of alkenyl-amides proceeds in 30% unoptimized yield (1-cinnamoylpiperidine-2,6-dione). (2) Full selectivity for the cross-coupling of aryl bromides in the presence of amide electrophiles is observed. This allows to establish the following order of reactivity: Ar–Cl < Ar–C(O)–NR2 < Ar–Br. (3) Di-methyl and di-phenyl amides are recovered unchanged from the reaction, as expected from the amidic resonance (PhCONMe2, RE = 16.5 kcal mol−1; PhCONPh2, 12.7 kcal mol−1). (4) Ar–Bpin are not suitable substrates under the reaction conditions (<20% yield); pleasingly, MIDA boronates are competent nucleophiles (4-chlorophenyl MIDA boronate, 72% yield). (5) Although at this stage tetra-ortho-substituted biaryls are beyond the scope of the reaction, the cross-coupling of unactivated 1-benzoylpiperidine-2,6-dione with mesitylene-2-boronic acid proceeds in promising 48% yield. Further studies are in progress to develop improved conditions and new ligands for decarbonylative cross-coupling reactions of amides.
DFT studies were conducted to gain insight into the reaction mechanism (Fig. 2), using the experimental amide substrate 1 and mode ligand dmpe.11a,18,23 The substrate-coordinated complex 4 undergoes a facile C–N bond cleavage occurs viaTS6, leading to the acylpalladium intermediate 7. Subsequent decarbonylation occurs through TS8 to generate the arylpalladium species 9. 9 then undergoes the CO dissociation, and subsequent transmetallation viaTS12 generates the LPd(Ph)2 intermediate 14. In TS12, the base complexes with the boronic acid, which promotes the efficiency of the rate-determining transmetallation and lowers the overall reaction barrier. This is consistent with previous mechanistic studies,24 and corroborated the importance of weak base for the reaction success (Table 1).25 From 14, the aryl–aryl reductive elimination is quite efficient through TS15, leading to the product-coordinated complex 16. 16 eventually undergoes the product extrusion and regenerates the palladium(0) catalyst. Based on the DFT-computed free energy profile, the on-cycle resting state is the acylpalladium intermediate 7. The rate-determining step is the transmetallation viaTS12, which requires a overall barrier of 29.5 kcal mol−1 (7 to TS12).
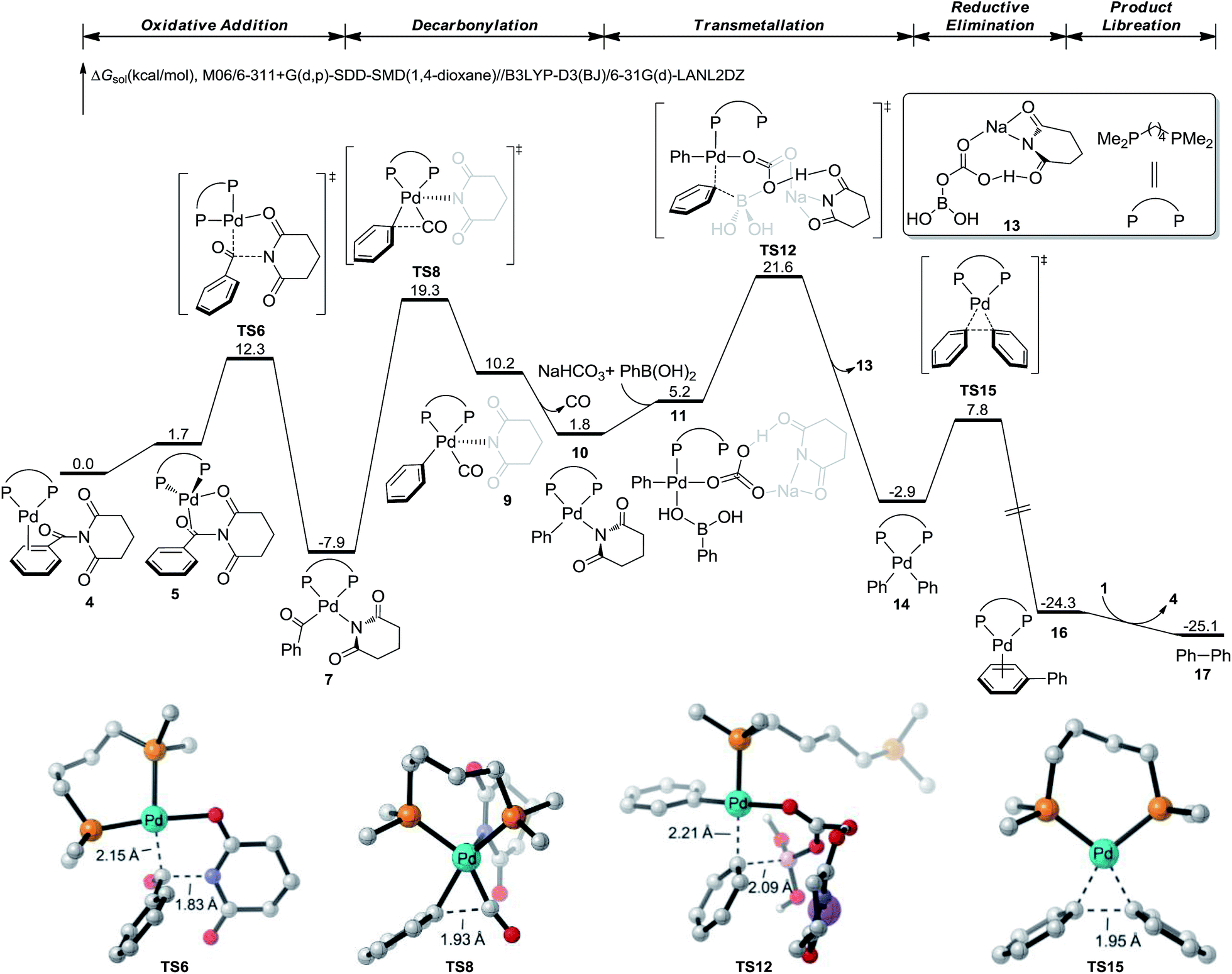 | ||
| Fig. 2 DFT-calculated reaction energy profile of Pd-catalyzed decarbonylative biaryl Suzuki–Miyaura cross-coupling of amides. See ESI† for computational details. | ||
We conducted additional competition studies to shed light on the mechanism (Fig. 3). (1) Intermolecular competition experiments revealed that electron-deficient boronic acids couple preferentially (4-Ac:4-MeO = 93:7, glutarimide; 4-Ac:4-MeO = 92:8, N–Ac), consistent with coordination of the leaving group to boron, while electron-poor amides are more reactive (4-CN:4-H >95:5, glutarimide; 4-CF3:4-H, 82:18, N–Ac), consistent with facility of metal insertion.11 (2) Furthermore, the reaction is not significantly affected by steric hindrance on either boronic acid (4-Me:2-Me = 54:46, glutarimide; 4-Me:2-Me = 47:53, N–Ac) or amide (4-Me:2-Me = 55:45, N–Ac), consistent with decarbonylation preceding transmetallation.21
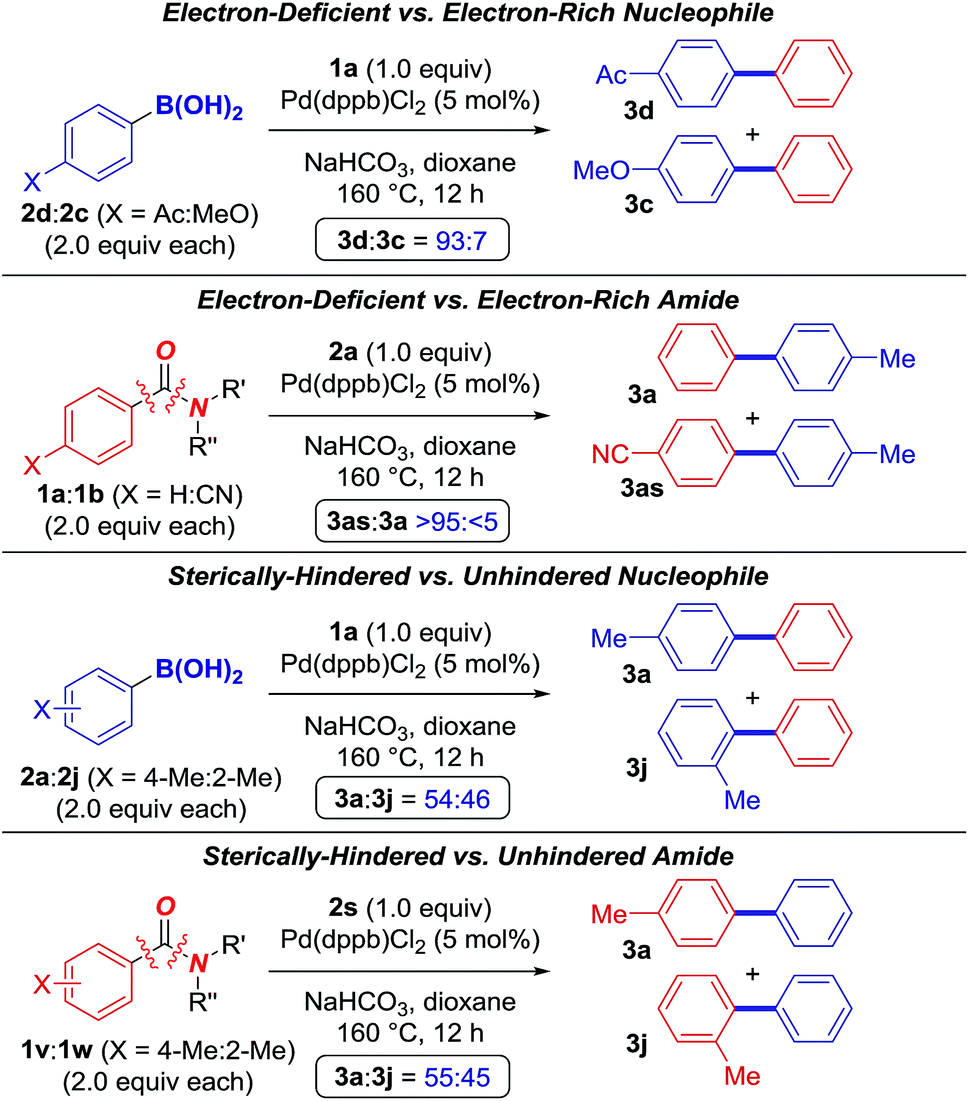 | ||
| Fig. 3 Intermolecular competition experiments. | ||
Conclusions
In conclusion, we have developed the first palladium-catalyzed decarbonylative Suzuki–Miyaura cross-coupling of amides for the synthesis of biaryls. A catalyst system derived from a Pd(II) precatalyst and a mild base enabled the direct route to biaryls from amides by selective carbon–nitrogen bond cleavage. This Pd-catalyzed reaction shows high generality and is compatible with a broad range of electronically-diverse cross-coupling partners. Furthermore, we demonstrated that a wide range of amides including both N-cyclic and N-acyclic are readily amendable to the cross-coupling. The key finding enabling the synthesis of biaryls was realization that decarbonylation must occur prior to the transmetallation step in palladium catalytic cycle. DFT studies demonstrated that mechanism involving decarbonylation prior to transmetalation is likely operative. Given the great importance of palladium-catalyzed Suzuki–Miyaura cross-couplings in chemical science, we believe that this reaction has a significant potential to enhance the utility of amides in cross-coupling reactions of general interest.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the NSF (CAREER CHE-1650766, M. S.), Rutgers University (M. S.), NSFC (21702182 and 21873081, X. H.), the Fundamental Research Funds for the Central Universities (2019QNA3009, X. H.) and Zhejiang University (X. H.) for generous financial support. The Bruker 500 MHz spectrometer used in this study was supported by the NSF-MRI grant (CHE-1229030). Calculations were performed on the high-performance computing system at the Department of Chemistry, Zhejiang University.Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) A. de Meijere, S. Bräse and M. Oestreich, Metal-Catalyzed Cross-Coupling Reactions and More, Wiley, 1st edn, 2014 CrossRef; (c) G. A. Molander, J. P. Wolfe and M. Larhed, Science of Synthesis: Cross-Coupling and Heck-Type Reactions, Thieme, 1st edn, 2013 Search PubMed; (d) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412 RSC.
- For excellent perspectives on the historical importance of cross-couplings, see: (a) X. F. Wu, P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9047 CrossRef CAS; (b) C. C. C. Johansson-Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062 CrossRef CAS.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722 CrossRef CAS.
- For leading reviews on cross-couplings in chemical industry, see: (a) C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027 CrossRef CAS; (b) M. Beller and H. U. Blaser, Organometallics as Catalysts in the Fine Chemicals Industry, Springer, 2012 CrossRef; (c) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed; (d) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senanayake, Adv. Synth. Catal., 2011, 353, 1825 CrossRef CAS; (e) M. L. Crawley and B. M. Trost, Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, Wiley, 2012 CrossRef; (f) A. Molnar, Palladium-Catalyzed Coupling Reactions: Practical Aspects and Future Developments, Wiley, 2013 CrossRef.
- T. J. Colacot, New Trends in Cross-Coupling, The Royal Society of Chemistry, 1st edn, 2015 Search PubMed.
- For recent pertinent reviews, see: (a) P. G. Gildner and T. J. Colacot, Organometallics, 2015, 34, 5497 CrossRef CAS; (b) L. C. Campeau and N. Hazari, Organometallics, 2019, 38, 3 CrossRef CAS; (c) I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137 CrossRef CAS.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry, Biochemistry and Materials Science, Wiley-VCH, 1st edn, 2003 Search PubMed.
- (a) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS; (b) A. B. Hughes, Amino Acids, Peptides and Proteins in Organic Chemistry, Wiley, 2011 CrossRef; (c) A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807 CrossRef CAS; (d) S. Ruider and N. Maulide, Angew. Chem., Int. Ed., 2015, 54, 13856 CrossRef CAS.
- Reactions involving amide bonds are among the most commonly employed in chemical industry: (a) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS; for a lead reference on using amides in polymer chemistry, see: (b) K. Marchildon, Macromol. React. Eng., 2011, 5, 22 CrossRef CAS; for an example of non-planar amides in biochemistry, see: (c) C. Lizak, S. Gerber, G. Michaud, M. Schubert, Y. Y. Fan, M. Bucher, T. Darbare, M. Aebi, J. L. Reymond and K. P. Locher, Nat. Commun., 2013, 4, 2627 CrossRef.
- Reviews on N–C amide cross-coupling: (a) S. Shi, S. P. Nolan and M. Szostak, Acc. Chem. Res., 2018, 51, 2589 CrossRef CAS PubMed; (b) G. Meng and M. Szostak, Eur. J. Org. Chem., 2018, 20–21, 2352 CrossRef; (c) J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413 CrossRef CAS; for reviews on acyl-metals, see: (d) L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef; (e) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef; for reviews on twisted amides, see: (f) M. Szostak and J. Aubé, Chem. Rev., 2013, 113, 5701 CrossRef CAS; (g) R. Szostak and M. Szostak, Molecules, 2019, 24, 274 CrossRef.
- For representative acyl coupling, see: (a) L. Hie, N. F. F. Nathel, T. K. Shah, E. L. Baker, X. Hong, Y. F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79 CrossRef CAS; (b) G. Meng and M. Szostak, Org. Lett., 2015, 17, 4364 CrossRef CAS; for a reductive coupling, see: (c) S. Ni, W. Zhang, H. Mei, J. Han and Y. Pan, Org. Lett., 2017, 19, 2536 CrossRef CAS; for a review, see: (d) J. Buchspies and M. Szostak, Catalysts, 2019, 9, 53 CrossRef , and references cited therein.
- For representative decarbonylative coupling, see: (a) G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2015, 54, 14518 CrossRef CAS; (b) C. Liu and M. Szostak, Angew. Chem., Int. Ed., 2017, 56, 12718 CrossRef CAS; (c) H. Yue, L. Guo, H. H. Liao, Y. Cai, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 4282 CrossRef CAS; (d) H. Yue, L. Guo, S. C. Lee, X. Liu and M. Rueping, Angew. Chem., Int. Ed., 2017, 56, 3972 CrossRef CAS.
- For additional representative studies, see: for a representative tandem reaction, see: (a) J. A. Walker, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228 CrossRef CAS; for a biomimetic esterification, see: (b) C. C. D. Wybon, C. Mensch, K. Hollanders, C. Gadals, W. A. Herrebout, S. Ballet and B. U. W. Maes, ACS Catal., 2018, 8, 203 CrossRef CAS; for a Cr-catalyzed N–C activation, see: (c) C. Chen, P. Liu, M. Luo and X. Zeng, ACS Catal., 2018, 8, 5864 CrossRef CAS; for a σ bond N–C activation, see: (d) Z. B. Zhang, C. L. Ji, C. Yang, J. Chen, X. Hong and J. B. Xia, Org. Lett., 2019, 21, 1226 CrossRef CAS.
- For studies on amide bond destabilization, see: (a) R. Szostak, S. Shi, G. Meng, R. Lalancette and M. Szostak, J. Org. Chem., 2016, 81, 8091 CrossRef CAS; (b) R. Szostak and M. Szostak, Org. Lett., 2018, 20, 1342 CrossRef CAS; (c) G. Meng, S. Shi, R. Lalancette, R. Szostak and M. Szostak, J. Am. Chem. Soc., 2018, 140, 727 CrossRef CAS PubMed; for a review, see: (d) K. B. Wiberg, Acc. Chem. Res., 1999, 32, 922 CrossRef CAS.
- (a) S. Shi, G. Meng and M. Szostak, Angew. Chem., Int. Ed., 2016, 55, 6959 CrossRef CAS; (b) C. Liu, G. Li, S. Shi, G. Meng, R. Szostak, R. Lalancette and M. Szostak, ACS Catal., 2018, 8, 9131 CrossRef CAS.
- For pertinent studies on the mechanism of amide bond distortion, see: (a) A. Greenberg and C. A. Venanzi, J. Am. Chem. Soc., 1993, 115, 6951 CrossRef CAS; (b) A. Greenberg, D. T. Moore and T. D. DuBois, J. Am. Chem. Soc., 1996, 118, 8658 CrossRef CAS; (c) R. Szostak, J. Aubé and M. Szostak, Chem. Commun., 2015, 51, 6395 RSC; (d) R. Szostak, J. Aubé and M. Szostak, J. Org. Chem., 2015, 80, 7905 CrossRef CAS; (e) C. Cox and T. Lectka, Acc. Chem. Res., 2000, 33, 849 CrossRef CAS.
- For a review on decarbonylative cross-coupling of amides, see: C. Liu and M. Szostak, Org. Biomol. Chem., 2018, 16, 7998 RSC.
- For a study on Ni catalysis in biaryl Suzuki–Miyaura cross-coupling of amides, see: C. L. Ji and X. Hong, J. Am. Chem. Soc., 2017, 139, 15522 CrossRef CAS PubMed.
- For representative reviews on biaryls, see: (a) J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS; (b) L. J. Gooßen, K. Gooßen and C. Stanciu, Angew. Chem., Int. Ed., 2009, 48, 3569 CrossRef; (c) C. E. I. Knappke and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2010, 49, 3568 CrossRef CAS.
- For representative studies on biaryl coupling, see: (a) L. J. Gooßen, G. Dong and L. M. Levy, Science, 2006, 313, 662 CrossRef; (b) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC; (c) K. Muto, J. Yamaguchi, D. G. Musaev and K. Itami, Nat. Commun., 2015, 6, 7508 CrossRef; for studies on decarbonylation of anhydrides, see: (d) J. B. Johnson and T. Rovis, Acc. Chem. Res., 2008, 41, 327 CrossRef CAS.
- D. V. Partyka, Chem. Rev., 2011, 111, 1529 CrossRef CAS.
- (a) N. Hazari, P. R. Melvin and M. M. Beromi, Nat. Rev. Chem., 2017, 1, 25 CrossRef CAS; (b) H. Li, C. C. C. Johansson-Seechurn and T. J. Colacot, ACS Catal., 2012, 2, 1147 CrossRef CAS.
- (a) L. Liu, P. Chen, Y. Sun, Y. Wu, S. Chen, J. Zhu and Y. F. Zhao, J. Org. Chem., 2016, 81, 11686 CrossRef CAS; (b) Z. Y. Xu, H. Z. Yu and Y. Fu, Chem.–Asian J., 2017, 12, 1765 CrossRef CAS.
- (a) M. Sumimoto, N. Iwane, T. Takahama and S. Sakaki, J. Am. Chem. Soc., 2004, 126, 10457 CrossRef CAS; (b) A. A. C. Braga, N. H. Morgon, G. Ujaque and F. Maseras, J. Am. Chem. Soc., 2005, 127, 9298 CrossRef CAS; (c) A. A. C. Braga, N. H. Morgon, G. Ujaque, A. Lledós and F. Maseras, J. Organomet. Chem., 2006, 691, 4459 CrossRef CAS.
- DFT-computed transmetallation barriers with and without base are included in the ESI (Fig. S1†)..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. See DOI: 10.1039/c9sc03169c |
| This journal is © The Royal Society of Chemistry 2019 |