
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed asymmetric intramolecular hydroarylation of allenes: access to functionalized benzocycles†
Dino
Berthold‡
,
Johannes
Klett‡
and
Bernhard
Breit
 *
*
Institut für Organische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, 79104 Freiburg im Breisgau, Germany. E-mail: bernhard.breit@chemie.uni-freiburg.de
First published on 10th September 2019
Abstract
A rhodium-catalyzed regio- and enantioselective cyclization of tethered allenylbenzenes is reported. Employing a RhI/Josiphos catalytic system a diverse set of 6-membered, α-chiral benzocycles were obtained, scaffolds found in many bioactive compounds. Moreover, a gram scale reaction as well as the application of suitable transformations are demonstrated.
Hetero- and carbocycles are ubiquitous in natural products, pharmaceuticals and functional molecules.1 In particular, benzocycles bearing an α-chiral carbon center are important structural motives in natural isolates ((−)-bruceol and (−)-Δ9-trans-tetrahydrocannabinol) and pharmaceuticals (levonantradol) (Fig. 1).2,3
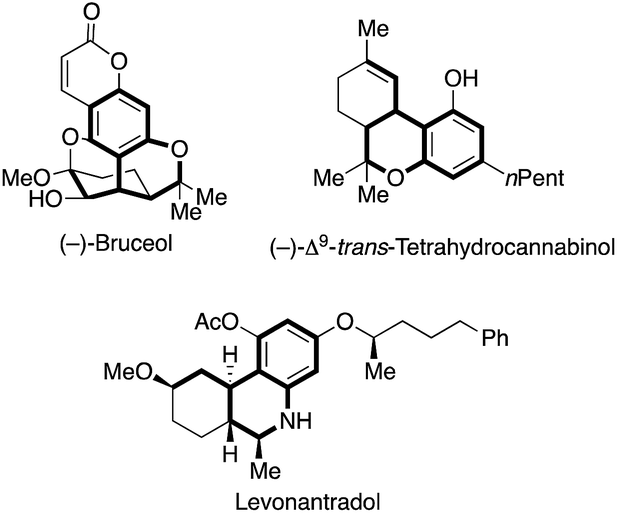 | ||
| Fig. 1 Natural products and bioactive compounds possessing an α-chiral benzocyclic scaffold. | ||
One general approach for the synthesis of α-chiral benzocycles is the intramolecular C-allylation of electron rich benzenes, in which these proceed as Friedel–Crafts-type alkylation reactions.4 In particular, the groups of Hamada and You contributed to the development of intramolecular, catalytic asymmetric dearomatization (CADA) reactions through Pd0- and IrI-catalyzed C-allylation of phenols and anilines.5 Furthermore, You and Carreira developed different asymmetric intramolecular allylic alkylations of phenols, anilines and general electron-rich arenes using IrI/phosphoramidite-based catalyst systems.6
Despite their respective synthetic utility and their excellent selectivities, these methods represent allylic substitutions and thus violate the principle of atom economy by formation of stoichiometric amounts of a side product. In this context, the addition of C–H pronucleophiles to C–C multiple bonds is of great synthetic interest. Following this thought, Gagné and Hashmi developed an intramolecular, AuI-catalyzed addition of benzene derivatives to terminal and internal allenes for the preparation of racemic allylic benzocycles (Scheme 1).7a,b
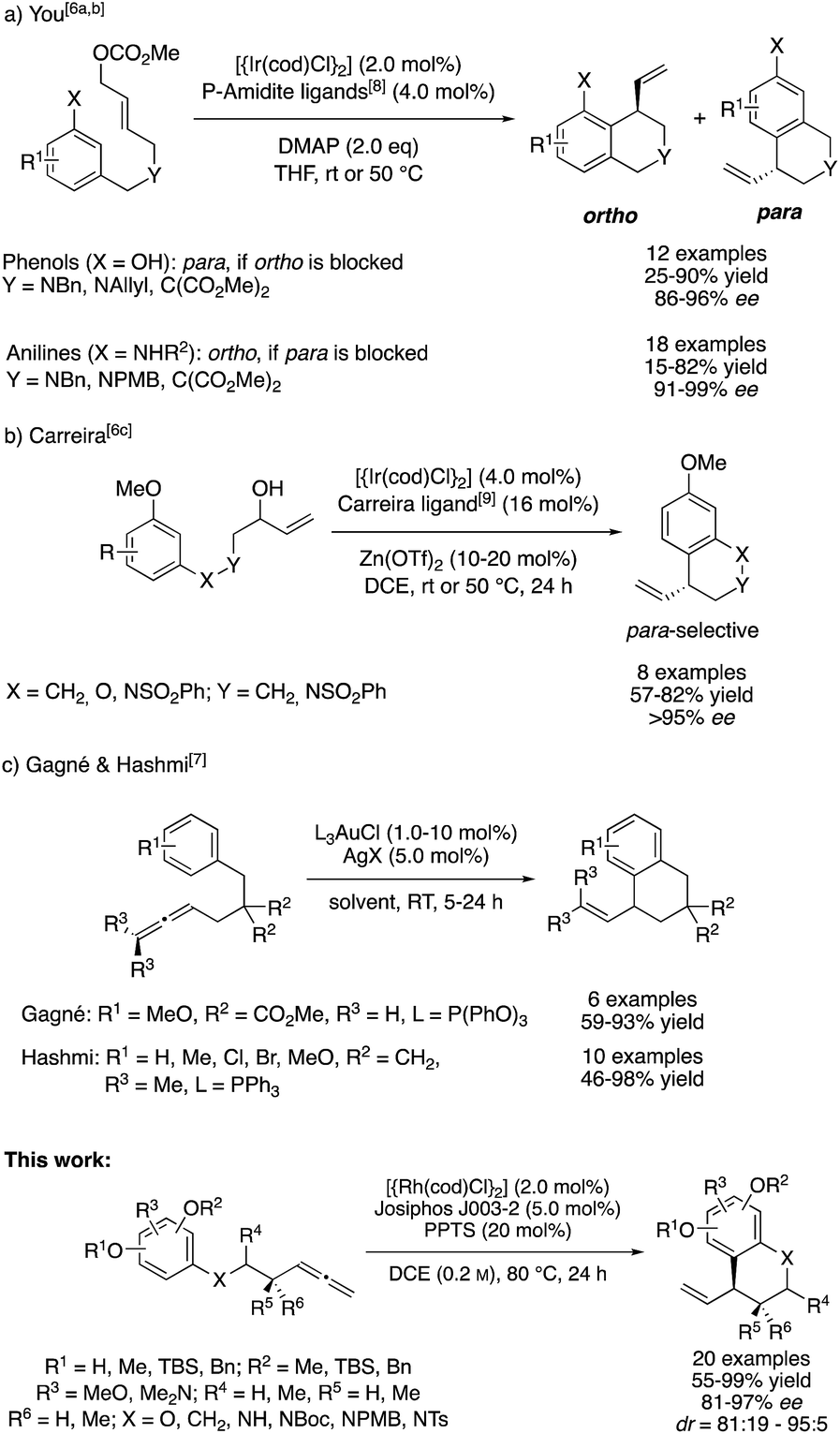 | ||
| Scheme 1 Strategies for the synthesis of allylic benzocycles. | ||
Throughout the last years, we reported on a series of RhI-catalyzed chemo-, regio- and enantioselective coupling reactions involving allenes10 and alkynes11 as allylic electrophile precursors with various pronucleophiles.12 Meanwhile, we were also able to expand this methodology toward asymmetric intramolecular transformations.13 However, the X–H pronucleophiles were always of an acidic character. On the basis of these previous studies, we wondered whether an allylic RhIII-complex, which is generated in the presence of a suitable Brønstedt-acid additive,14 can be trapped by a sufficiently nucleophilic reagent in a regio- and enantioselective fashion. In the recent past, Dong and our group confirmed this hypothesis by developing asymmetric additions of indoles to allenes and alkynes, respectively.15 In this context, we herein report a RhI-catalyzed, asymmetric addition of tethered allenylarenes providing the desired chiral benzocycles.
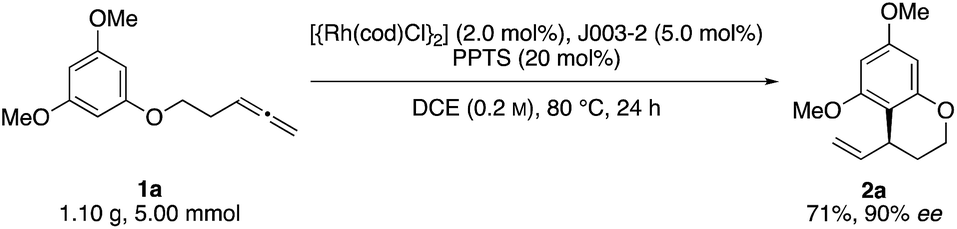
Initial reactivity assays were carried out using allenylbenzene 1a in the presence of [{Rh(cod)Cl}2] (2.0 mol%), dppf (L1) (5.0 mol%) and TFA (20 mol%) in DCE at 80 °C (Table 1, entry 1). To our delight we obtained the allylated benzocycle 2a in a high yield of 92%. Supporting our previously mentioned hypothesis, no conversion was observed in the absence of an acidic additive (entry 2). By screening numerous chiral bidentate ligands we were pleased to discover that Josiphos ligand J003-2 led to a formation of the desired product in good yield (70%) and promising enantioselectivity (54% ee) (entry 3). After evaluation of different acids and pyridinium salts, we observed that PPTS provided the desired product 2a in 73% yield and 90% ee (entry 4). In contrast, a similar yield with lower enantiomeric excess was obtained in the presence of free p-toluenesulfonic acid. These different results are possibly effects of different counter anions.16 As to be expected, similar results in terms of yield and enantioselectivity were obtained using the enantiomeric Josiphos ligand J003-1.17
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Additive | Solvent | Yieldb | eec |
| a Reactions were performed in 0.4 mmol scales. b Yield of isolated product. c The ee was determined by HPLC analysis using a chiral stationary phase. d dppf = 1,1′-bisdiphenylphosphinoferrocene. e J003-1 = enantiomer of Josiphos ligand J003-2. | |||||
| 1 | dppf (L1)d | TFA | DCE | 92% | rac |
| 2 | dppf (L1) | — | DCE | — | — |
| 3 | J003-2 | TFA | DCE | 70% | 54% |
| 4 | J003-2 | PPTS | DCE | 73% | 90% |
| 5 | J003-2 | p-TolSO3H | DCE | 80% | 66% |
| 6 | J003-1 | PPTS | DCE | 72% | −90% |
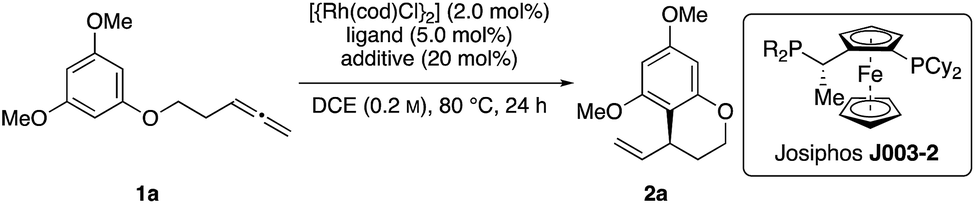
With these optimized conditions in hand, we sought to examine the reaction scope (Scheme 2). Towards this end, we subjected a variety of different benzenes to the indicated reaction conditions.18 We were pleased to see that several symmetrical allenylarenes were successfully converted into the corresponding products 2a–2h in good yields and high enantioselectivities. Aside from substrates that incorporate heteroatom tethers, benzocyclic product 2i was isolated in good yield and very good enantioselectivity, as well. Notably, even an iodide-substituted benzene – prone for enabling side reactions by facile oxidative addition of the rhodium catalyst – could be implemented in the catalysis. Furthermore, the double allylated product 2l could be synthesized after double ring closure in 64% yield, a dr of 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23 and 97% ee. Interestingly, the use of substrates 1m and 1n led to exclusive cyclization at the position para to the methoxy or amine substituent. This stands in contrast to observations by You, where product mixtures in dependence of the substituents were usually observed. Due to the presence of a TBSO-group in meta position, which is sterically more demanding than the hydroxy or methoxy group, the 6-membered ring was preferably closed in para-position for substrates 1o and 1p.19
:23 and 97% ee. Interestingly, the use of substrates 1m and 1n led to exclusive cyclization at the position para to the methoxy or amine substituent. This stands in contrast to observations by You, where product mixtures in dependence of the substituents were usually observed. Due to the presence of a TBSO-group in meta position, which is sterically more demanding than the hydroxy or methoxy group, the 6-membered ring was preferably closed in para-position for substrates 1o and 1p.19
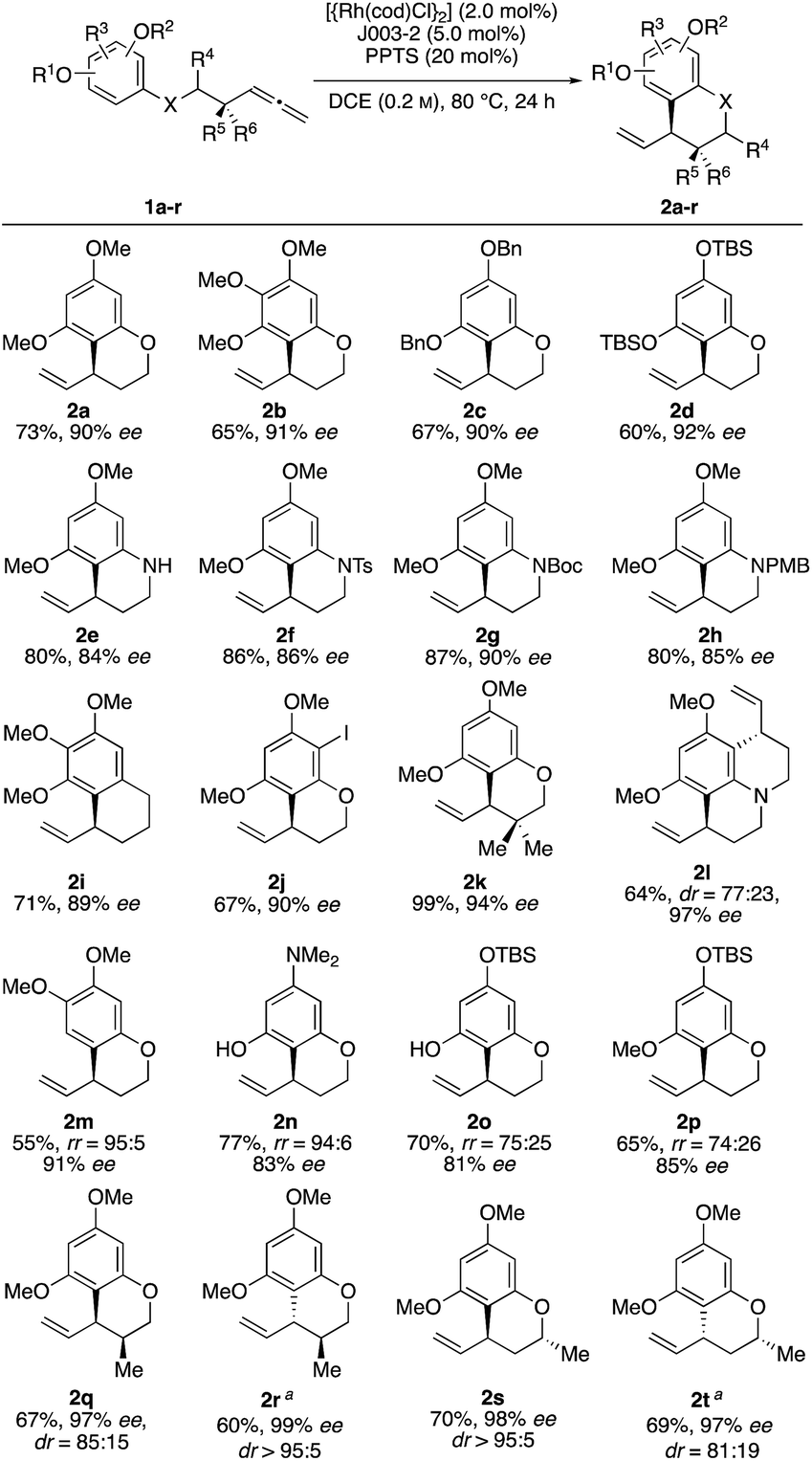 | ||
| Scheme 2 Scope of intramolecular addition of arenes to allenes. Reactions were performed in 0.4 mmol scales. aJ003-1 was used instead of J003-2. | ||
Finally, it was of interest to study catalyst control of diastereoselectivity employing substrates equipped with a stereogenic center either in α- or β-position relative to the allenyl substituent. Starting from the substrates 1qr and 1st, two diastereomeric products could be obtained using the two enantiomeric Josiphos ligands J003-1 and J003-2 respectively. As a result 2r and 2s were identified as the matched cases displaying very high diastereoselectivity. Conversely, 2q and 2t were the mismatched cases which were still formed in good diastereoselectivities. Based on the known absolute configuration of starting materials 1qr and 1st, the absolute configuration of the newly formed allyllic stereocenter could be determined by means of NOE NMR experiments (for details see ESI†).
Inspired by these synthetic possibilities, this new intramolecular allylation was applied to different functionalizations on the C–C double bond as well as the arene moiety. Preceding this, the synthetic utility of this methodology was highlighted by performing a gram-scale catalysis (Scheme 3).
 | ||
| Scheme 3 Gram-scale catalysis. | ||
To explore the synthetic utility of the obtained allylated benzocycles, we subjected 2a to various transformations. It was possible to functionalize the vinyl group in four different reactions while fully preserving the stereo center. 2a could be converted into the corresponding alcohol 3 in good yield by hydroboration. Hydroformylation of the terminal double bond, using our self-assembly ligand 6-diphenylphosphinopyridone (6-DPPon), furnished aldehyde 4 in good yield and excellent linear/branched selectivity (>95:5).20 Hoveyda–Grubbs II catalyzed cross metathesis of 2a with trans-stilbene provided the internal olefin 5 in excellent yield. Furthermore, the C–C double bond could be hydrogenated in very good yield. Finally, the nucleophilicity of the aromatic building block was to be used for regioselective Friedel–Crafts acylation or alkylation. For this purpose 2a was reacted with methyl vinyl ketone in the presence of catalytic BF3·Et2O yielding regioselectively the product 7 in 67% yield (Scheme 4).
 | ||
| Scheme 4 Various functionalizations of allylated benzocycle 2a. Reagents and conditions: (a) 9-BBN (1.05 eq.), THF (0.5 M), 0 °C – rt, 6 h; then H2O2 (30%, 3.00 eq.), NaOH (2.0 M, 3.00 eq.), 1 h, 91%, 90% ee; (b) [Rh(CO)2acac] (0.50 mol%), 6-DPPon (5.0 mol%), CO/H2 (1:1, 20 bar), toluene (0.13 M), 80 °C, 16 h, 70%, linear/branched >95:5, 89% ee; (c) Hoveyda–Grubbs II (10 mol%), trans-stilbene (10 eq.), DCE (0.2 M), 80 °C, 18 h, 95%, 91% ee; (d) Pd/C (10 wt%, 10 mol%), MeOH (0.1 M), rt, 18 h, 91%, 89% ee; (e) methyl vinyl ketone (1.3 eq.), BF3·Et2O (30 mol%), CH2Cl2 (0.2 M), 0 °C – rt, 18 h, 67%, 88% ee (6-DPPon = 6-diphenylphosphinopyridin-2-(1H)-one). | ||
On the basis of our previous studies of this catalytic system, we propose the following mechanism for this reaction (Scheme 5).15a,18 After initial preformation from [{Rh(cod)Cl}2] and Josiphos ligand J003-2, a rhodiumIII hydride complex A is generated upon reaction with PPTS,16 followed by hydrometallation of the allene moiety in 1a to give the allylrhodiumIII intermediate B. Subsequent nucleophilic attack of the arene releases arenium ion C, while simultaneously regenerating the RhI-catalyst. Rearomatization through elimination of HX then furnishes the benzocycle 2a.
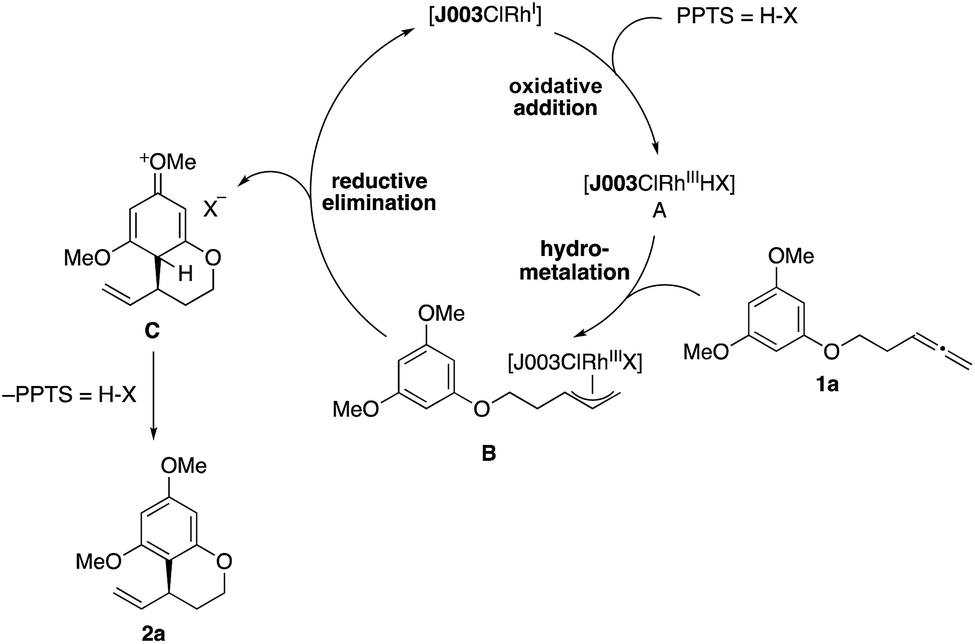 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In summary, we have accomplished an enantioselective, intramolecular addition of electron-rich benzenes to allenes in an atom-efficient manner by using a rhodium/Josiphos J003 based catalyst system. The benzocycles were synthesized in good yields and high stereoselectivities, thereby tolerating a broad range of benzenes and different substituted allenes. We also demonstrated possible opportunities for derivatization of either the allylic moiety or the benzene itself. Further investigations regarding the mechanism and the expansion of this method to alkynes as well as the application of this methodology in target oriented-synthesis are being pursued in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the DFG. We thank Dr Manfred Keller for NMR analysis.Notes and references
- For examples, see: (a) A. Kleeman, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances, Thieme, New York, 2001 Search PubMed; (b) D. J. Faulkner, Nat. Prod. Rep., 2002, 19, 1–48 CAS.
- For (–)-bruceol, see: (a) A. M. Duffield, P. R. Jefferies, E. N. Maslen and A. I. M. Rae, Tetrahedron, 1963, 19, 593–607 CrossRef CAS; (b) A. J. Day, J. H. Z. Lee, Q. D. Phan, H. C. Lam, A. Ametovski, C. J. Sumby, S. G. Bell and J. H. George, Angew. Chem., 2019, 131, 1441–1445 ( Angew. Chem., Int. Ed. , 2019 , 58 , 1427–1431 ) Search PubMed.
- For (–)-Δ9-trans-tetrahydrocannabinol and levonantradol, see: (a) M. A. Huestis, Chem. Biodiversity, 2007, 4, 1770–1804 CrossRef CAS PubMed; (b) P. J. Little, D. R. Compton, M. R. Johnson, L. S. Melvin and B. R. Martin, J. Pharmacol. Exp. Ther., 1988, 247, 1046–1051 CAS.
- For recent reviews concerning the IrI-catalyzed allylic substitution, see: (a) Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed; (b) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019 DOI:10.1021/acs.accounts.9b00209.
- For intramolecular, catalytic asymmetric dearomatization (CADA) reactions involving Friedel–Crafts-type allylic alkylation with phenols, naphthols and anilines, see: (a) T. Nemoto, Y. Ishigue, M. Yoshida, Y. Kohno, M. Kanematsu and Y. Hamada, Org. Lett., 2010, 12, 5020–5023 CrossRef CAS PubMed; (b) Y. Suzuki, T. Nemoto, K. Kakugawa, A. Hamajima and Y. Hamada, Org. Lett., 2012, 14, 2350–2353 CrossRef CAS PubMed; (c) M. Yoshida, T. Nemoto, Z. Zhao, Y. Ishige and Y. Hamada, Tetrahedron: Asymmetry, 2012, 23, 859–866 CrossRef CAS; (d) Q.-F. Wu, W.-B. Liu, C.-X. Zhuo, Z.-Q. Rong, K.-Y. Ye and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 4455–4458 CrossRef CAS PubMed; (e) Q. Cheng, Y. Wang and S.-L. You, Angew. Chem., Int. Ed., 2016, 55, 3496–3499 CrossRef CAS PubMed; (f) C.-X. Zhuo and S.-L. You, Angew. Chem., 2013, 125, 10240–10243 ( Angew. Chem., Int. Ed. , 2013 , 52 , 10056–10059 ) CrossRef; (g) H.-F. Tu, C. Zheng, R.-Q. Xu, X.-J. Liu and S.-L. You, Angew. Chem., 2017, 129, 3285–3289 ( Angew. Chem., Int. Ed. , 2017 , 56 , 3237–3241 ) CrossRef; (h) S.-B. Tang, H.-F. Tu, X. Zhang and S.-L. You, Org. Lett., 2019, 21, 6130–6134 CrossRef CAS PubMed.
- For intramolecular, IrI/phosphoramidite-ligand catalyzed allylic alkylation with phenols and anilines, see: (a) Q.-L. Xu, L.-X. Dai and S.-L. You, Org. Lett., 2012, 14, 2579–2581 CrossRef CAS PubMed; (b) Z.-K. Zhao, Q. Gu, X.-Y. Wu and S.-L. You, Chem.–Asian J., 2017, 12, 2680–2683 CrossRef CAS PubMed; (c) M. A. Schafroth, S. M. Rummelt, D. Sarlah and E. M. Carreira, Org. Lett., 2017, 19, 3235–3238 CrossRef CAS PubMed.
- (a) M. A. Tarselli and M. R. Gagné, J. Org. Chem., 2008, 73, 2439–2441 CrossRef CAS PubMed; (b) A. Fazeli, D. Pflästerer, M. Rudolph and A. S. K. Hashmi, J. Organomet. Chem., 2015, 795, 68–70 CrossRef CAS.
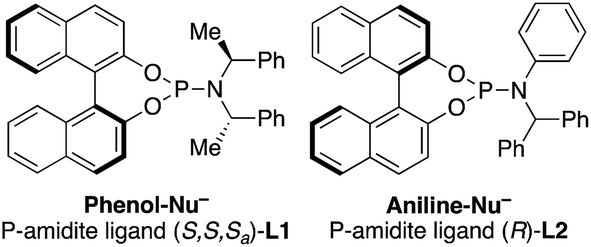
- The following ligands were used:
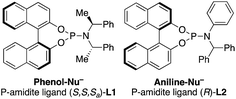 .
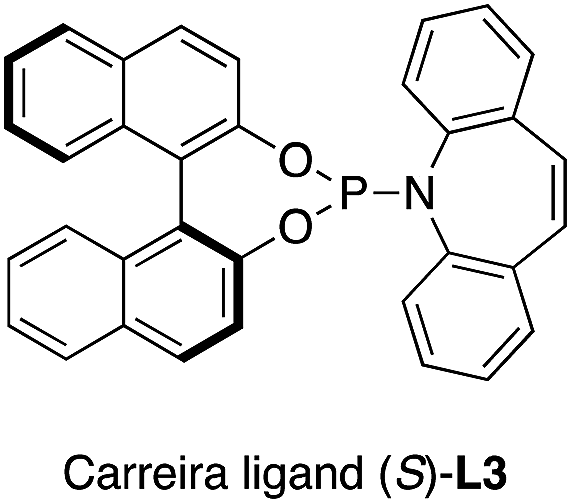
. - The following ligand was used:
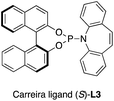 .
. - For a recent review, see: P. Koschker and B. Breit, Acc. Chem. Res., 2016, 49, 1524–1536 CrossRef CAS PubMed.
- For a recent review, see: A. M. Haydl, B. Breit, T. Liang and M. J. Krische, Angew. Chem., Int. Ed., 2017, 56, 11312–11325 ( Angew. Chem. , 2017 , 129 , 11466–11480 ) CrossRef CAS PubMed.
- For examples of asymmetric C–C bond formations: (a) T. M. Beck and B. Breit, Angew. Chem., Int. Ed., 2017, 56, 1903–1907 ( Angew. Chem. , 2017 , 129 , 1929–1933 ) CrossRef CAS PubMed; (b) C. P. Grugel and B. Breit, Org. Lett., 2018, 20, 1066–1069 CrossRef CAS PubMed; (c) C. P. Grugel and B. Breit, Chem.–Eur. J., 2018, 24, 15223–15226 CrossRef CAS PubMed.
- For recent examples on RhI-catalyzed cyclizations with allenes, see: (a) A. M. Haydl, D. Berthold, P. A. Spreider and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 5765–5769 ( Angew. Chem., Int. Ed. , 2016 , 128 , 5859–5863 ) CrossRef CAS PubMed; (b) S. Ganss and B. Breit, Angew. Chem., Int. Ed., 2016, 55, 9738–9742 ( Angew. Chem. , 2016 , 128 , 9890–9894 ) CrossRef CAS PubMed; (c) P. Steib and B. Breit, Angew. Chem., Int. Ed., 2018, 57, 6572–6576 ( Angew. Chem. , 2018 , 130 , 6682–6686 ) CrossRef CAS PubMed; (d) J. P. Schmidt and B. Breit, Chem. Sci., 2019, 10, 3074–3079 RSC; (e) D. Berthold, A. G. A. Geissler, S. Giofré and B. Breit, Angew. Chem., Int. Ed., 2019, 58, 9994–9997 ( Angew. Chem. , 2019 , 131 , 10099–10102 ) CrossRef CAS PubMed.
- J. Wolf and H. Werner, Organometallics, 1987, 6, 1164–1169 CrossRef CAS.
- (a) F. A. Cruz, Y. Zhu, Q. D. Tercenio, Z. Shen and V. M. Dong, J. Am. Chem. Soc., 2017, 139, 10641–10644 CrossRef CAS PubMed; (b) C. P. Grugel and B. Breit, Org. Lett., 2019, 21, 5798–5802 CrossRef CAS PubMed.
- Sulfonic acids are known for their ability to generate [Rh]–H species. For recent literature in this field, see: Y. Yang, E. G. Moschetta and R. M. Rioux, ChemCatChem, 2013, 5, 3005–3013 CrossRef CAS.
- For the screening of different ligands, additives, solvents and catalyst loadings see the ESI.†.
- Unfortunately, 7- and 8-membered benzocycles could not be obtained by this methodology. 1,1-Disubstituted and internal allenes were not feasible under the given conditions as well. For an overview of tested substrates, see ESI.†.
- It was necessary for the benzene moiety to incorporate at least three donating substituents for a successful outcome of the reaction. For an overview of tested substrates, see ESI.†.
- U. Gellrich, A. Meißner, A. Steffani, M. Kähny, H.-J. Drexler, D. Heller, D. A. Plattner and B. Breit, J. Am. Chem. Soc., 2014, 136, 1097–1104 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc03894a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |