Development of a cascade isothermal amplification approach for the sensitive detection of DNA methyltransferase†
Received
9th August 2018
, Accepted 27th November 2018
First published on 28th November 2018
Abstract
DNA methyltransferase (MTase) is an important epigenetic modification enzyme responsible for DNA methylation, and the dysregulation of DNA MTase activity is associated with various diseases in humans. Herein, we take advantage of the DNA lesion repair mechanism in vivo to develop a new fluorescence approach for the specific and sensitive detection of DNA methyltransferase (DNA MTase) on the basis of the DNA lesion repair-directed cascade isothermal amplification. Due to the high amplification efficiency of the uracil repair-mediated exponential isothermal amplification reaction (EXPAR), the efficient cleavage of endonuclease IV (Endo IV)-induced cyclic catalysis, and the low background signal caused by single uracil repair-mediated inhibition of nonspecific amplification, this approach exhibits high sensitivity with a detection limit of 0.014 U mL−1 for pure Dam MTase and 0.61 × 10−6 mg mL−1 for Dam MTase in E. coli cells and it can be further applied for the screening of DNA MTase inhibitors. More importantly, this approach can be applied to detect other DNA MTases by designing appropriate substrates, showing great potential in biomedical research and clinical diagnosis.
Introduction
DNA methylation is a chemical modification process catalyzed by DNA methyltransferase (DNA MTase) which can transfer an active methyl group from the methyl donor S-adenosylmethionine (SAM) to the cytosine or adenine residues of the target DNA sequence,1 and it plays pivotal roles in cell proliferation, senescence and gene transcription.2 The abnormal DNA methylation is related to several genetic disorders and has been considered as a predictive biomarker for various types of cancers such as colon,3 ovarian4 and prostate cancers.5 Notably, the aberrant DNA methylation patterns have close association with the abnormal DNA MTase activity,6 and DNA MTase has become a promising pharmacological target for anticancer therapies.7 Thus, the sensitive detection of DNA MTase activity and the screening of DNA MTase inhibitors are of great importance in clinical diagnosis and cancer therapeutics.
Traditional methods for DNA MTase assay include radioactive labeling of DNA substrates,8 high performance liquid chromatography (HPLC),9 and gel electrophoresis.10 However, they involve long assay procedures,8 use of expensive instruments9 and insecure isotope labeling,8,10 limiting their practical applications. Alternatively, some new approaches have been developed for the DNA MTase assay, including colorimetric,11 fluorescence,12 bioluminescence,13 electrochemical14 and electrogenerated chemiluminescence assays,15 but they are not sensitive enough to detect extremely low abundant DNA MTases. To improve the detection sensitivity, a variety of nucleic acid amplification approaches have been introduced for DNA MTase assay, including strand displacement amplification (SDA),16 rolling circle amplification (RCA)17 and exponential isothermal amplification reaction (EXPAR).18 The SDA-based colorimetric assay16 allows for the visual detection of DNA MTase, but it suffers from relatively poor sensitivity.16 The RCA-based chemiluminescence assay17 exhibits high sensitivity, but it involves multiple steps with a long assay time.19 The EXPAR-based fluorescence assay18 demonstrates high amplification efficiency and rapid amplification kinetics,20 but it inevitably suffers from a high background signal due to the nonspecific amplification.21 Therefore, the development of simple and sensitive methods for DNA MTase assay remains a great challenge.
Recently, DNA lesion repair in genomes has been attracting more and more attention.22 Uracil-DNA glycosylase (UDG) can specifically recognize and excise damaged uracil by catalyzing the cleavage of the N-glycosidic bond between the uracil and the DNA backbone, liberating the uracil base and simultaneously generating an abasic site (AP site).22 Endonuclease IV (Endo IV) is an endonuclease that may hydrolyze any intact AP site in double-stranded DNAs (dsDNAs).22 In this research, we take advantage of the enzyme-initiated DNA lesion repair mechanism22 to develop a new fluorescence approach for the sensitive detection of DNA MTase based on DNA lesion repair-directed cascade isothermal amplification. In contrast to the conventional nickases that cleave short recognition sequences, the combination of UDG and Endo IV can repair a single uracil to effectively inhibit the nonspecific amplification,23,24 leading to a lower background signal than the nickase-based amplification.16–18 The presence of Dam MTase enables the methylation of the hairpin substrate and the subsequent cleavage by DpnI to initiate the uracil repair-mediated EXPAR for generating the triggers. The resultant triggers can hybridize with the signal probes25,26 to actuate the Endo IV-induced cyclic cleavage of signal probes for liberating fluorescence signals. Due to the high amplification efficiency of the uracil repair-mediated EXPAR and the efficient cleavage of Endo IV-induced cyclic catalysis, this approach can detect DNA MTase activity with a detection limit of 0.014 U mL−1 and a large dynamic range of 4 orders of magnitude from 0.02 to 10 U mL−1. Moreover, it can be further applied for the quantification of intracellular DNA MTase activity and the screening of DNA MTase inhibitors.
Experimental
Chemicals and materials
All oligonucleotides (Table 1) were obtained from Sangon Biotechnology Co. Ltd (Shanghai, China). DNA adenine methyltransferase (Dam MTase), 10× Dam MTase reaction buffer (500 mM Trizma hydrochloride (Tris–HCl), 50 mM 2-mercaptoethanol (β-ME), and 100 mM ethylenediaminetetraacetic acid (EDTA), pH 7.5), S-adenosylmethionine (SAM), DpnI, 10× CutSmart buffer (500 mM potassium acetate (KAc), 200 mM tris–acetate, 100 mM magnesium acetate (Mg(Ac)2), and 1 mg mL−1 bovine serum albumin (BSA), pH 7.9), Bst DNA polymerase (large fragment), 10× ThermoPol reaction buffer (200 mM Tris–HCl, 100 mM potassium chloride (KCl), 100 mM ammonium sulfate (NH4)2SO4), (20 mM magnesium sulfate (MgSO4), and 1% Triton X-100, pH 8.8), uracil-DNA glycosylase (UDG), 10× UDG reaction buffer (20 mM Tris–HCl, 1 mM DTT, and 1 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0), endonuclease IV (Endo IV), 10× NEBuffer 3 (1000 mM sodium chloride (NaCl), 500 mM Trizma hydrochloride (Tris–HCl), 100 mM magnesium chloride (MgCl2), and 10 mM DL-dithiothreitol (DTT), pH 7.9), Haemophilus haemolyticus methyltransferase (Hhal MTase) and CpG methyltransferase (M.SssI MTase) were purchased from New England Biolabs (Ipswich, MA, USA). Deoxyadenosine triphosphate (dATP), deoxyguanosine triphosphate (dGTP), deoxycytidine triphosphate (dCTP) and deoxyuracil triphosphate (dUTP) were obtained from TaKaRa Biotechnology Co. Ltd (Dalian, China). SYBR Gold was purchased from Life Technologies (Carlsbad, CA, USA). Bovine serum albumin (BSA), 5-fluorouracil and other chemicals of analytical grade were obtained from Sigma-Aldrich Company (St. Louis, MO, USA).
Table 1 Sequences of the oligonucleotidesa
| Note |
Sequences (5′–3′) |
|
In the hairpin substrate, the Dam recognition site is marked by boldface. In the hairpin template, the binding sequence of the cleavage product of the hairpin substrate is shown in italic. In the signal probe, “X” indicates the abasic site mimic and the bases “T” in italic are those modified with FAM and TAMRA, respectively.
|
| Hairpin substrate |
ACT TAT CAG CTT AAG GAT CTT ATG TGC TGC TAG TCT AAG ATC CTT AAG CTG ATA AGT |
| Hairpin template |
TTC CCT CTC TCC TCG GTG CCC AGT GCT GCT TCT TAG ACT AGC AGC ACA TAA GA |
| EXPAR template |
TTC CCT CTC TCC TCG GTG CCC ATT CCC TCT CTC CTC GGT GCC C |
| Signal probe |
CCC T(FAM)CT CXC CT(TAMRA)C GGT GCC |
Polyacrylamide gel electrophoresis
The 15% nondenaturating polyacrylamide gel electrophoresis (PAGE) was performed to analyze the products of the cleavage reaction and uracil excision-activated EXPAR. The electrophoresis was carried out in 1× TBE buffer (9 mM Tris–HCl, 9 mM boric acid, and 0.2 mM EDTA, pH 7.9) at a constant voltage of 120 V for 50 min.
Detection of Dam MTase activity
The detection of Dam MTase activity involves four consecutive steps. First, all oligonucleotides were diluted with 1× Tris–EDTA buffer (10 mM Tris, 1 mM EDTA, pH 8.0) to prepare the stock solutions. The hairpin substrates and hairpin templates were diluted to 5 μM and 1 μM in a buffer (15 mM MgCl2, 10 mM Tris–HCl, pH 8.0), respectively, and then incubated at 95 °C for 5 min, followed by slowly cooling to room temperature to fold into the hairpin structures. The obtained hairpin substrates and hairpin templates were stored at 4 °C for further use. Second, the Dam MTase-directed cleavage reaction of the hairpin substrate was performed in 200 μL of reaction solution containing 0.5 μM hairpin substrate, 20 μL of 10× Dam buffer, 20 μL of 10× CutSmart buffer, 160 μM SAM, 10 U of DpnI and varying amounts of Dam MTase at 37 °C for 2 h, followed by inactivation at 80 °C for 20 min. Third, 4 μL of the cleavage products were added into 16 μL of the reaction solution containing 50 nM hairpin templates, 100 nM EXPAR templates, 350 nM signal probe, 2.8 U of Bst DNA polymerase, 1 U of UDG, 5 U of Endo IV, 2 μL of 10× ThermoPol buffer, 2 μL of 10× UDG buffer, 2 μL of 10× NEBuffer 3, and 100 μM dNTPs (dATP, dGTP, dCTP, and dUTP each). The DNA lesion repair-directed cascade isothermal amplification was performed at 37 °C for 110 min. Fourth, 20 μL of amplification products were diluted to 100 μL with ultrapure water. The fluorescence spectra were recorded using a RF-5301 spectrofluorophotometer at an excitation wavelength of 494 nm and the fluorescence intensity at 520 nm was recorded for data analysis.
Selectivity and inhibition assay
We used M.SssI MTase (10 U mL−1), HhaI MTase (10 U mL−1) and BSA (20 nM) as the interferences to investigate the selectivity of the proposed method. To study the effect of 5-fluorouracil on the activity of Dam MTase, varying concentrations of 5-fluorouracil were added to 200 μL of the reaction solution (0.5 μM hairpin substrate, 2 μL of 10× Dam buffer) and incubated at 37 °C for 15 min. Then 10 U mL−1 Dam MTase, 160 μM SAM, and 10 U of DpnI were added to the reaction solution and incubated at 37 °C for 2 h. The reaction was terminated by inactivation at 80 °C for 20 min. The Dam MTase activity was measured using the aforementioned procedure and its relative activity (RA) was calculated according to eqn (1):| | | RA = (Fi − Fo)/(Ft − Fo) | (1) |
where Fo is the fluorescence intensity in the absence of Dam MTase, Ft is the fluorescence intensity in the presence of Dam MTase, and Fi is the fluorescence intensity in the presence of both 5-fluorouracil and Dam MTase.
Bacterial cell culture and preparation of bacterial cell extracts
The colonies of E. coli cells (GW5100 and JM110) were incubated in 3 mL of liquid medium (5 g L−1 yeast extract, 10 g L−1 tryptone, 10 g L−1 NaCl) at 37 °C in a rotary shaker at 250 rpm for 12 h, respectively. Then 100 μL of cell suspensions from the E. coli cells were added into 3 mL of the medium and incubated at 37 °C for 2.5 h. At the exponential phase of growth, the E. coli cells were harvested and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm to obtain a cell pellet. After washing twice with pure water, the E. coli cells were lysed with RIPA lysis buffer and the concentration of the extracted protein was determined using the Bradford Protein Assay Kit. The cell lysate was freshly frozen and stored at −80 °C for further use.
000 rpm to obtain a cell pellet. After washing twice with pure water, the E. coli cells were lysed with RIPA lysis buffer and the concentration of the extracted protein was determined using the Bradford Protein Assay Kit. The cell lysate was freshly frozen and stored at −80 °C for further use.
Results and discussion
Principle of DNA MTase assay
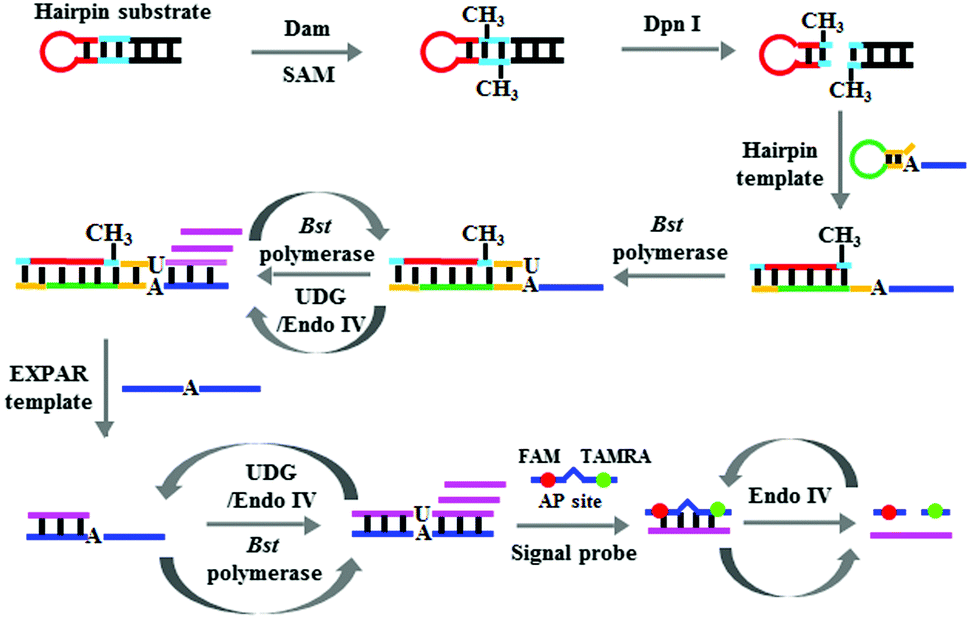
We used DNA adenine methylation (Dam) MTase as the model analyte27 and designed a hairpin probe and a signal probe for Dam MTase assay. The stem of the hairpin probe contains a palindromic 5′-G-A-T-C-3′ sequence which functions as the substrate of Dam MTase. The single-strand signal probe is designed with an AP site mimic that is flanked by nucleotides modified with a fluorophore (FAM) and a quencher (TAMRA), which is also a substrate of Endo IV.28 This assay involves four steps (Scheme 1): (1) Dam MTase-initiated DNA methylation, (2) DpnI-mediated cleavage of methylated DNA substrates,29 (3) uracil repair-mediated EXPAR, and (4) Endo IV-induced cyclic cleavage of signal probes for the liberation of fluorescence signals. The Dam MTase present can catalyze the methylation of adenine residue in the 5′-G-A-T-C-3′ sequence to generate the methylated product of 5′-G-mA-T-C-3′. Then the endonuclease DpnI will cleave the methylated substrate,29 leading to the release of a new hairpin structure containing a 13-nt loop and a 5-bp blunt terminus. Because the melting temperature of the new hairpin structure (31.5 °C) is much lower than the reaction temperature (37 °C), it is unstable and changed into a 23-nt ssDNA. To prevent the nonspecific amplification independent of the linear template in the absence of Dam MTase, we designed a template with a hairpin structure. The resultant 23-nt cleavage product may open the hairpin template and subsequently act as a primer to initiate the polymerization extension in the presence of the Bst DNA polymerase and four nucleotides (i.e., dATP, dGTP, dCTP and dUTP), generating a stable dsDNA duplex with a uracil (U) nucleotide incorporated. The uracil can be excised by UDG,30 creating an AP site which can be cleaved by Endo IV to generate a single nucleotide gap.31 This single nucleotide gap may act as a new replication site for the Bst DNA polymerase to induce the SDA reaction, producing abundant triggers (Scheme 1, pink color). With the addition of the EXPAR template (Scheme 1, blue color), the resultant trigger may hybridize with the EXPAR template and function as a primer to initiate a new polymerization extension, generating more dsDNA duplexes with one uracil (U). In the presence of UDG and Endo IV, the uracil lesion in the dsDNA duplex will be repaired through UDG-initiated base-excision repair, generating a new replication site for the initiation of new SDAs to produce a large number of triggers. Because the EXPAR template is strategically designed with two repeat sequences, the resultant triggers can hybridize with the free EXPAR templates to initiate cycles of polymerization and uracil-excision repair, eventually leading to an exponential amplification reaction and the generation of large amounts of triggers which can hybridize with the signal probes to form stable dsDNA duplexes. The signal probe in the dsDNA duplex may be cleaved by Endo IV, generating a distinct fluorescence signal and simultaneously releasing the trigger.24 The released triggers can hybridize with new signal probes to initiate cycles of hybridization, cleavage and release, eventually leading to an enhanced fluorescence signal.
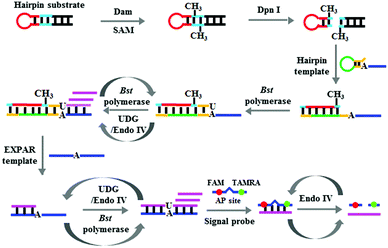 |
| | Scheme 1 Schematic illustration of the Dam MTase assay based on DNA lesion repair-directed cascade isothermal amplification. | |
Feasibility of the assay
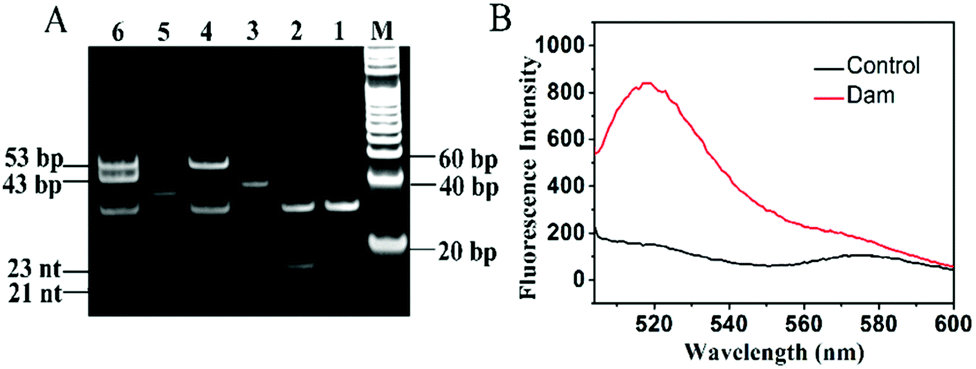
To demonstrate the feasibility of the proposed method, we analyzed the reaction products using 15% nondenaturating polyacrylamide gel electrophoresis (PAGE) with SYBR Gold as the fluorescence indicator. As shown in Fig. 1A, only one band of the hairpin substrate is observed in the absence of Dam MTase (Fig. 1A, lane 1), indicating no occurrence of the cleavage reaction in the presence of DpnI. In contrast, a new band of 23 nt is observed in the presence of both Dam MTase and DpnI (Fig. 1A, lane 2), suggesting that the hairpin substrate is methylated by Dam MTase and then cleaved by DpnI. With the addition of the hairpin template, Bst DNA polymerase, UDG and Endo IV (Fig. 1A, lane 3) into the reaction solution, the 23-nt cleavage products will hybridize with the hairpin templates to initiate the SDA reaction, producing long dsDNA duplexes and abundant triggers. As a result, the bands of the 53-bp dsDNA duplex and 21-nt trigger are observed (Fig. 1A, lane 4). When the EXPAR template (Fig. 1A, lane 5) is further added to the reaction solution, a new band of 43-bp dsDNA duplex is observed and the band of 21-nt trigger becomes brighter (Fig. 1A, lane 6) compared with that without the EXPAR template (Fig. 1A, lane 4), indicating that the presence of Dam MTase can direct the cleavage of the hairpin substrate in the presence of DpnI and subsequently initiate the uracil repair-mediated EXPAR. Moreover, we measured the fluorescence signal under different reaction conditions (Fig. 1B). In the presence of Dam MTase, the triggers generated from uracil repair-mediated EXPAR may hybridize with the signal probes, inducing cyclic cleavage of the signal probes for the generation of an enhanced fluorescence signal (Fig. 1B, red curve). In contrast, in the absence of Dam MTase, no trigger is generated, and consequently no signal probe is cleaved and no fluorescence enhancement is observed (Fig. 1B, black curve).
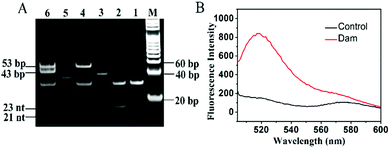 |
| | Fig. 1 (A) Nondenaturating PAGE analysis of the reaction products under different conditions. Lane M, DNA ladder marker; lane 1, hairpin substrate + DpnI; lane 2, hairpin substrate + Dam + DpnI; lane 3, hairpin template; lane 4, hairpin substrate + Dam + DpnI + hairpin template; lane 5, EXPAR template; lane 6, hairpin substrate + Dam + DpnI + hairpin template + EXPAR template. (B) Fluorescence monitoring of the amplification products in the presence (red curve) and absence of Dam MTase (control, black curve). The 4 U mL−1 Dam MTase, 0.5 μM hairpin substrate, 50 nM hairpin template and 100 nM EXPAR template were used in this experiment. | |
Improved sensitivity
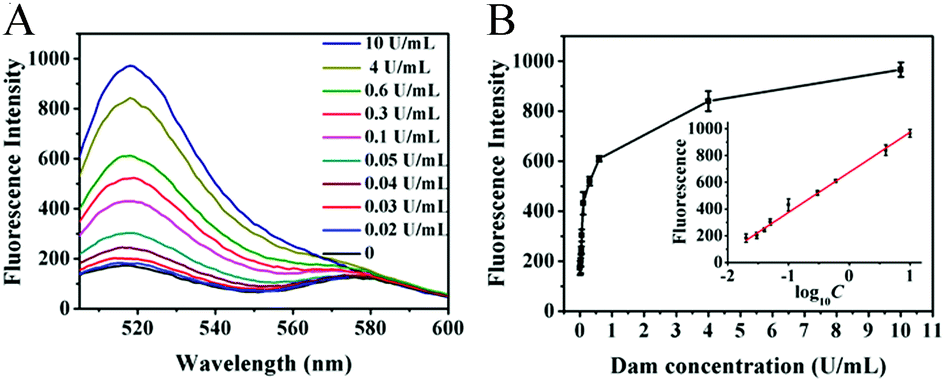
Under the optimized experimental conditions (ESI,† Fig. S1), we monitored the fluorescence signal in response to different concentrations of Dam MTase. Fig. 2A shows the variance in the fluorescence emission spectra in response to different concentrations of Dam MTase. In the logarithmic scale, the fluorescence intensity exhibits a linear correlation with the concentration of Dam MTase over a large range from 0.02 to 10 U mL−1 (Fig. 2B). The corresponding equation is F = 674.31 + 299.46log10C(R2 = 0.9959), where F is the fluorescence intensity at 520 nm and C is the Dam concentration (U mL−1). The detection limit is calculated to be 0.014 U mL−1 by evaluating the average response of the control group plus three times the standard deviation. The sensitivity of the proposed method is superior to those of the T7 exonuclease-assisted cyclic signal amplification-based assay (0.1 U mL−1),32 the DNAzyme-mediated amplification-based assay (0.2 U mL−1),33 and the nicking enzyme-assisted signal amplification-based assay (0.06 U mL−1).34 The improved sensitivity can be ascribed to: (1) the high amplification efficiency of the uracil repair-mediated EXPAR, (2) the efficient cleavage of Endo IV-induced cyclic catalysis, and (3) the low background signal caused by single uracil repair-mediated inhibition of nonspecific amplification.
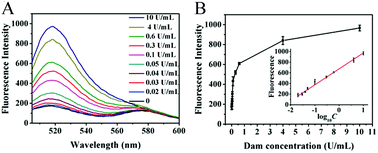 |
| | Fig. 2 (A) Measurement of the fluorescence emission spectra in response to different concentrations of Dam MTase. (B) Linear relationship between the fluorescence intensity and the logarithm of Dam MTase concentration. Error bars show the standard deviations of three independent experiments. | |
Detection specificity
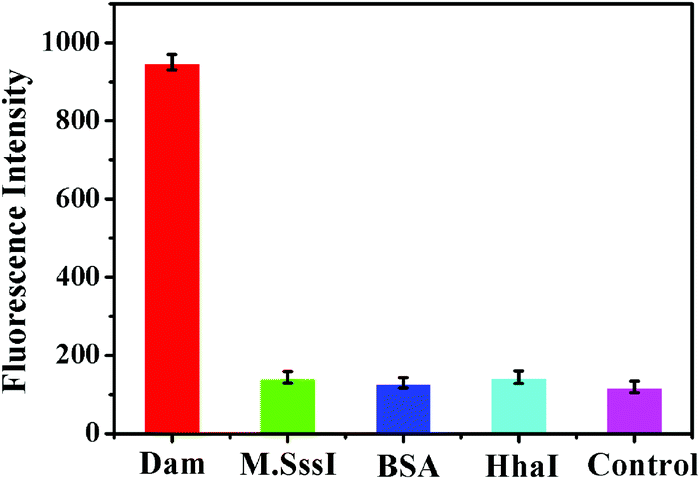
To evaluate the specificity of the proposed method, we employed HhaI MTase, M.SssI MTase and BSA as the nonspecific enzymes. M.SssI MTase and HhaI MTase are DNA methyltransferases that can specifically methylate the cytosine residues of 5′-C-G-3′ and 5′-G-C-G-C-3′ sequences in dsDNA, respectively, and BSA is an irrelevant protein. As shown in Fig. 3, the fluorescence intensity in response to Dam MTase (Fig. 3, red column) is much higher than those in response to M.SssI MTase (Fig. 3, green column), HhaI MTase (Fig. 3, cyan column), BSA (Fig. 3, blue column), and the control group (Fig. 3, purple column). This can be explained by the following factors: the cytosine residues in the 5′-G-A-T-C-3′ sequence can be methylated only by Dam MTase instead of M.SssI MTase, HhaI MTase and BSA. These results clearly demonstrate the good selectivity of the proposed method towards Dam MTase.
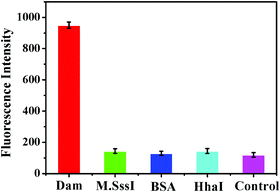 |
| | Fig. 3 Measurement of the fluorescence intensity in response to 10 U mL−1 Dam MTase (red column), 10 U mL−1 M.SssI MTase (green column), 20 nM BSA (blue column), 10 U mL−1 HhaI MTase (cyan column) and the control with only reaction buffer (purple column). Error bars represent the standard deviation from three independent experiments. | |
Inhibition assay
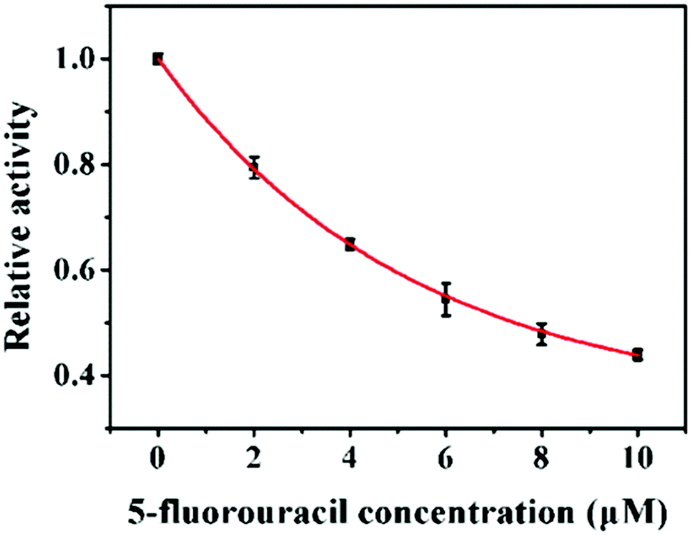
Aberrant DNA methylation is closely related to various cancers, and DNA MTase inhibitors can reverse the abnormal methylation by inhibiting the DNA MTase activity.35–38 Therefore, the screening of DNA MTase inhibitors is of great significance for cancer prevention and treatment. To verify the feasibility of the proposed method for DNA MTase inhibition assay, we used 5-fluorouracil as the model inhibitor. Previous reports demonstrated that 5-fluorouracil did not affect the activities of Endo IV and DpnI at a concentration of less than 10 μM39 and it exhibited no inhibition effect on the UDG activity in the millimolar concentration range.40 As shown in Fig. 4, after the incubation of 10 U mL−1 Dam MTase with different concentrations of 5-fluorouracil, the relative activity of DNA MTase decreases with the increasing concentration of 5-fluorouracil from 0 to 10 μM. The IC50 (i.e. the value of the inhibitor concentration that induces the decrease of enzyme activity by 50%) is calculated to be 7.44 μM, consistent with that obtained by T7 exonuclease-assisted cyclic signal amplification-based fluorescence assay (8.43 μM).32 This result indicates that the proposed method can be applied for the screening of Dam MTase inhibitors for drug discovery.
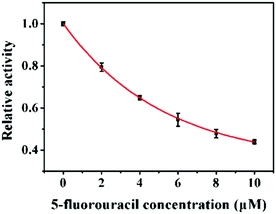 |
| | Fig. 4 Inhibition effect of 5-fluorouracil on the activity of Dam MTase. The concentration of Dam MTase is 10 U mL−1. Error bars show the standard deviations of three independent experiments. | |
Real sample analysis
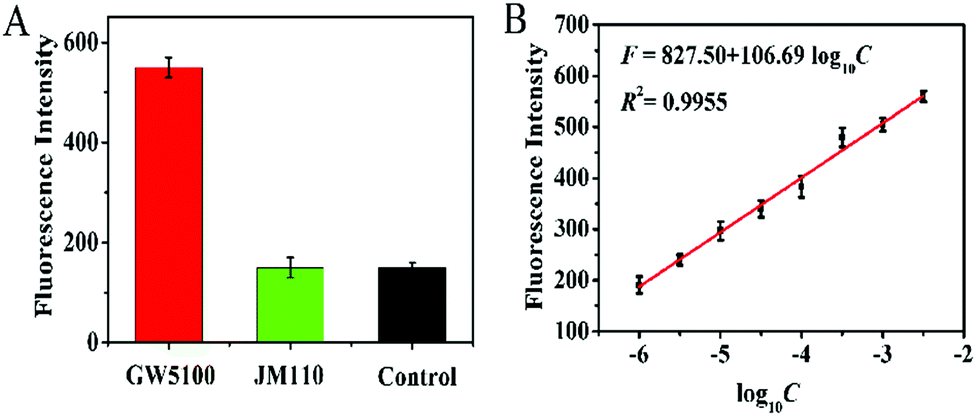
To investigate the feasibility of the proposed method for real sample analysis, we detected the Dam MTase activity in E. coli cells. We measured the Dam MTase activity in E. coli cells at the exponential growth stage because the level of Dam MTase in E. coli cells at the exponential growth stage is much higher than that in the stationary stage.41 As shown in Fig. 5A, a high fluorescence signal is detected in GW5100 (a Dam-positive E. coli), but no distinct fluorescence signal is observed in either JM110 (a Dam-negative E. coli) or the control group with only lysis buffer, suggesting that the obtained fluorescence signal is derived from Dam MTase instead of the interferences or the nonspecific amplification independent of Dam MTase. Notably, in the logarithmic scale, the fluorescence intensity exhibits a linear correlation with the concentration of total proteins extracted from GW5100 over a large range from 1.0 × 10−6 to 0.0032 mg mL−1 (Fig. 5B). The corresponding equation is F = 827.50 + 106.69log10C(R2 = 0.9955), where F is the fluorescence intensity and C is the concentration of total proteins (mg mL−1). The detection limit is determined to be 0.61 × 10−6 mg mL−1 based on the principle of 3 times the standard deviation over the signal of the negative control. These results demonstrate that the proposed method can be used to evaluate Dam MTase activity in complex biological samples.
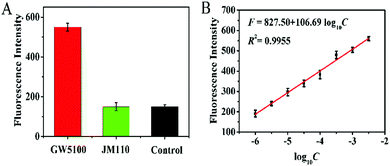 |
| | Fig. 5 (A) Measurement of fluorescence intensity in response to GW5100 (red column), JM110 (green column), and the control (black column). The total protein concentration extracted from GW5100 and JM110 is 0.0032 mg mL−1 and 0.0032 mg mL−1, respectively. (B) The fluorescence intensity shows a linear correlation with the logarithm of total protein concentration in the range from 1.0 × 10−6 to 0.0032 mg mL−1. Error bars represent the standard deviations of three independent experiments. | |
Conclusions
In conclusion, we take advantage of the base-excision repair mechanism in vivo to develop a new fluorescence approach for the specific and sensitive detection of DNA MTase using DNA lesion repair-directed cascade isothermal amplification. Due to the high amplification efficiency of uracil repair-mediated EXPAR, the efficient cleavage of Endo IV-induced cyclic catalysis, and the low background signal caused by single uracil repair-mediated inhibition of nonspecific amplification, this approach can provide a detection limit of 0.014 U mL−1 for pure Dam MTase and 0.61 × 10−6 mg mL−1 in E. coli cells. Moreover, this approach can be applied for the screening of DNA MTase inhibitors. In comparison with the reported Dam MTase assays,16–18,32–34 this approach has significant advantages of: (1) single-uracil repair instead of short recognition sequences cleaved by common nickases may efficiently prevent the nonspecific amplification, reducing the background signal, (2) the high efficiency of EXPAR and the efficient cleavage of Endo IV-induced cyclic catalysis endow this assay with high detection sensitivity, and (3) the amplification reaction is performed under homogeneous and isothermal conditions without the involvement of thermal cycling, washing and separation steps. Importantly, this approach can be further applied to detect other DNA MTases by simply designing appropriate substrates, showing great potential in biomedical research and clinical diagnosis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21405066, 21475053, 21325523, 21527811, 21735003, 21605098 and 21705097) and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China. We thank Prof. Bi-Feng Yuan at Wuhan University for providing the E. coli strains.
Notes and references
- X. Cheng and R. J. Roberts, Nucleic Acids Res., 2001, 29, 3784–3795 CrossRef CAS PubMed.
- W. Reik, W. Dean and J. Walter, Science, 2001, 293, 1089–1093 CrossRef CAS PubMed.
- J. P. J. Issa, P. M. Vertino, J. Wu, S. Sazawal, P. Celano, B. D. Nelkin, S. R. Hamilton and S. B. Baylin, J. Natl. Cancer Inst., 1993, 85, 1235–1240 CrossRef CAS PubMed.
- A. E. Bondurant, Z. Huang, R. S. Whitaker, L. R. Simel, A. Berchuck and S. K. Murphy, Gynecol. Oncol., 2011, 123, 581–587 CrossRef CAS PubMed.
- Y. Kobayashi, D. M. Absher, Z. G. Gulzar, S. R. Young, J. K. McKenney, D. M. Peehl, J. D. Brooks, R. M. Myers and G. Sherlock, Genome Res., 2011, 21, 1017–1027 CrossRef CAS PubMed.
- H. Zhang, M. Li, M. Fan, J. Gu, P. Wu and C. Cai, Chem. Commun., 2014, 50, 2932–2934 RSC.
- K. Mutze, R. Langer, F. Schumacher, K. Becker, K. Ott, A. Novotny, A. Hapfelmeier, H. Hofler and G. Keller, Eur. J. Cancer, 2011, 47, 1817–1825 CrossRef CAS PubMed.
- B. Y. Kim, O. S. Kwon, S. A. Joo, J. A. Park, K. Y. Heo, M. S. Kim and J. S. Ahn, Anal. Biochem., 2004, 326, 21–24 CrossRef CAS PubMed.
- S. Friso, S.-W. Choi, G. G. Dolnikowski and J. Selhub, Anal. Chem., 2002, 74, 4526–4531 CrossRef CAS PubMed.
- C. C. P. M. Bens, A. Voss and C. H. W. Klaassen, J. Clin. Microbiol., 2006, 44, 1875–1876 CrossRef CAS PubMed.
- T. Liu, J. Zhao, D. M. Zhang and G. X. Li, Anal. Chem., 2010, 82, 229–233 CrossRef CAS PubMed.
- T. Tian, H. Xiao, Y. Long, X. Zhang, S. Wang, X. Zhou, S. Liu and X. Zhou, Chem. Commun., 2012, 48, 10031–10033 RSC.
- C. Jiang, C. Y. Yan, C. Huang, J. H. Jiang and R. Q. Yu, Anal. Biochem., 2012, 423, 224–228 CrossRef CAS PubMed.
- N. B. Muren and J. K. Barton, J. Am. Chem. Soc., 2013, 135, 16632–16640 CrossRef CAS PubMed.
- Y. Li, X. Luo, Z. Yan, J. B. Zheng and H. L. Qi, Chem. Commun., 2013, 49, 3869–3871 RSC.
- Y. Zhao, F. Chen, M. Lin and C. Fan, Biosens. Bioelectron., 2014, 54, 565–570 CrossRef CAS PubMed.
- Y.-P. Zeng, J. Hu, Y. Long and C.-Y. Zhang, Anal. Chem., 2013, 85, 6143–6150 CrossRef CAS PubMed.
- Q. Xue, Y. Lv, S. Xu, Y. Zhang, L. Wang, R. Li, Q. Yue, H. Li, X. Gu, S. Zhang and J. Liu, Biosens. Bioelectron., 2015, 66, 547–553 CrossRef CAS PubMed.
- H. Y. Zhao, L. Wang and W. Jiang, Chem. Commun., 2016, 52, 2517–2520 RSC.
- F. Ma, Y. Yang and C.-Y. Zhang, Anal. Chem., 2014, 86, 6006–6011 CrossRef CAS PubMed.
- E. Tan, B. Erwin, S. Dames, T. Ferguson, M. Buechel, B. Irvine, K. Voelkerding and A. Niemz, Biochemistry, 2008, 47, 9987–9999 CrossRef CAS PubMed.
- L.-J. Wang, M. Ren, Q. Y. Zhang, B. Tang and C.-Y. Zhang, Anal. Chem., 2017, 89, 4488–4494 CrossRef CAS PubMed.
- L.-J. Wang, Z.-Y. Wang, Q. Y. Zhang, B. Tang and C.-Y. Zhang, Chem. Commun., 2017, 53, 3878–3881 RSC.
- D.-M. Zhou, W.-F. Du, Q. Xi, J. Ge and J.-H. Jiang, Anal. Chem., 2014, 86, 6763–6767 CrossRef CAS PubMed.
- S. Tyagi, S. A. Marras and F. R. Kramer, Nat. Biotechnol., 2000, 18, 1191–1196 CrossRef CAS PubMed.
- S. A. Marras, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30, e122 CrossRef PubMed.
- Y. Li, X. R. Zou, F. Ma, B. Tang and C.-Y. Zhang, Methods Appl. Fluoresc., 2017, 5, 012002 CrossRef PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, 1115–1121 CrossRef CAS PubMed.
- G. E. Geier and P. Modrich, J. Biol. Chem., 1979, 254, 1408–1413 CAS.
- Y. Xiang and Y. Lu, Anal. Chem., 2012, 84, 9981–9987 CrossRef CAS PubMed.
- J. D. Levin, A. W. Johnson and B. Demple, J. Biol. Chem., 1988, 263, 8066–8071 CAS.
- Y. F. Ma, L. N. Chen, L. L. Zhang, S. Q. Liao and J. J. Zhao, Analyst, 2015, 140, 4076–4082 RSC.
- X.-H. Zhao, L. Gong, X.-B. Zhang, B. Yang, T. Fu, R. Hu, W. H. Tan and R. Q. Yu, Anal. Chem., 2013, 85, 3614–3620 CrossRef CAS PubMed.
- Y. Zhao, F. Chen, Y. Wu, Y. Dong and C. Fan, Biosens. Bioelectron., 2013, 42, 56–61 CrossRef CAS PubMed.
- S. Y. Chen, H. Ma, W. Li, Z. Nie and S. Z. Yao, Anal. Chim. Acta, 2017, 970, 57–63 CrossRef CAS PubMed.
- Q. Wang, M. Pan, J. Wei, X. Q. Liu and F. A. Wang, ACS Sens., 2017, 2, 932–939 CrossRef CAS PubMed.
- J. Bao, X. T. Geng, C. J. Hou, Y. N. Zhao, D. Q. Huo, Y. Wang, Z. N. Wang, Y. Zeng, M. Yang and H. B. Fa, J. Electroanal. Chem., 2018, 814, 144–152 CrossRef CAS.
- Y. Chen, H. C. Yi, Y. Xiang and R. Yuan, Anal. Chim. Acta, 2018, 1001, 18–23 CrossRef CAS PubMed.
- W. Li, Z. Liu, H. Lin, Z. Nie, J. Chen, X. Xu and S. Yao, Anal. Chem., 2010, 82, 1935–1941 CrossRef CAS PubMed.
- Y. Zhang, C.-C. Li, B. Tang and C.-Y. Zhang, Anal. Chem., 2017, 89, 12408–12415 CrossRef CAS PubMed.
- X.-W. Xing, F. Tang, J. Wu, J.-M. Chu, Y.-Q. Feng, X. Zhou and B.-F. Yuan, Anal.
Chem., 2014, 86, 11269–11274 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02096e |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a and
Chun-yang
Zhang
a and
Chun-yang
Zhang