
Selenium-containing polyurethane with elevated catalytic stability for sustained nitric oxide release†
Baoliu
Qu
abc,
Liguang
Yuan
abc,
Jinge
Li
ab,
Jie
Wang
ab,
Hongying
Lv
*ab and
Xiaoniu
Yang
 *ab
*ab
aState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Str. 5625, Changchun 130022, P. R. China. E-mail: xnyang@ciac.ac.cn; hongyinglv@ciac.ac.cn
bPolymer Composites Engineering Laboratory, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Renmin Str. 5625, Changchun 130022, P. R. China
cUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 24th November 2018
Abstract
Stable and controllable nitric oxide (NO) release at the physiological level from biomedical materials remains a challenge for NO-based therapy. NO-generating polymers have great potential to achieve this goal because they can catalytically decompose endogenous S-nitrosothiols (RSNOs) into NO. However, the current catalytic surfaces based on such polymers often suffer from loss of catalytic sites, which can influence the stability of NO release in their long-term application. In this work, we proposed a novel strategy to enhance the catalytic stability of NO-catalytic materials by incorporating catalytic sites into the polymer backbone. Selenium-containing polyurethane (PU-Se) was synthesized by using the catalyst 2,2′-diselenodiethanol (SeDO) as the chain extender. A series of PU/PU-Se blend films were prepared to investigate the effect of PU-Se content on the catalytic properties. The blend films exhibited excellent catalytic activity, and also showed outstanding catalytic stability in comparison with PU coated by diselenide/dopamine (PU-PDA-Se). Among these blend films, PU-Se-10 exhibited a stable NO release rate of 5.05 × 10−10 mol cm−2 min−1 after exposure to PBS buffer for 30 days. Moreover, the PU/PU-Se films exhibited decreased platelet activation/adhesion, low hemolysis ratio, excellent biocompatibility, and similar mechanical properties to PU. It is expected that the newly designed PU-Se has great potential in generating stable NO release at the physiological level for the long-term application of blood-contacting medical devices.
1. Introduction
Thrombosis is a common problem in the application of blood-contacting medical devices such as vascular grafts, catheters, and so on.1,2 The interface between blood and material is critical for platelet adhesion. Many strategies have been proposed to improve the hemo-compatibility of materials by manipulating the surface properties.3,4 NO has been demonstrated to be a natural inhibitor of platelet adhesion and activation, and healthy endothelial cells exhibit a NO flux of 0.5–4.0 × 10−10 mol cm−2 min−1.5–7 Thus, NO release from the polymer surface exhibits great potential in reducing thrombus due to its unique advantage of mimicking natural endothelium, which is the truly thrombo-resistant surface. A variety of NO-releasing polymers have been developed by doping or covalently bonding the NO donor to the polymer for applications in blood-contacting medical devices.8–10 Although these NO-releasing polymers have exhibited reduced platelet adhesion, challenges such as instability of NO donors, finite donor reservoir, leaching of unbound NO donors, and instability of NO release rate greatly limit their clinical applications.9,11–14Recently, NO-generating polymers have been widely explored due to their capability of generating NO by catalytically decomposing endogenous RSNOs at the interface between the polymer and blood. Commonly, these polymers are obtained by embedding the catalyst into the polymer matrix or immobilizing catalytic sites on the substrate surfaces.15–18 One type of the catalysts is the organoselenium compounds, which has been shown to effectively decompose RSNOs into NO through the glutathione peroxidase (GPx)-like catalytic reaction.19 Many Se-catalytic surfaces have been constructed by coating and grafting organoselenium compounds onto material surfaces,17,20,21 and it has been demonstrated that these surfaces significantly inhibited platelet adhesion because of the continuously generated NO. However, the Se-catalytic surfaces are often encountered with leaking or loss of catalytic sites, which can influence the stability of NO release and the following anti-platelet behavior in their long-term application.1 Meanwhile, the release rate of NO could greatly affect the performance of NO-generating polymers and the surrounding tissues, where the low rate may cause the failure of anti-thrombosis while the high rate may be harmful to the blood vessels.22 Therefore, developing NO-generating polymers with stable NO release at the physiological level is fairly important for the long-term application of blood-contacting medical devices.
PUs have been widely investigated in many biomedical devices due to their excellent mechanical properties, molecule designability, and good biocompatibility.23,24 However, platelet adhesion and activation on the PU surfaces can still be found when they are in contact with blood for a long term. Herein, we propose to introduce organoselenium catalytic sites (Se–Se covalent bond) into PU backbones in order to obtain PU-Se, which was blended with PU to achieve the stable NO-catalytic substrate (Scheme 1). SeDO was synthesized to be an organoselenium catalyst and incorporated into PU in the form of the chain extender, while BDO was employed as the chain extender to synthesize the common PU. Blend films with different ratios of PU and PU-Se were prepared by using a drop-coating method to adjust the catalytic site content. NO-catalytic effect and release rates of NO from these films were investigated at different soaking times in order to attain the stable NO release rate at the physiological level over a long-term period (30 days). Further, the anti-platelet effects and the mechanical properties of the blend films were also examined.
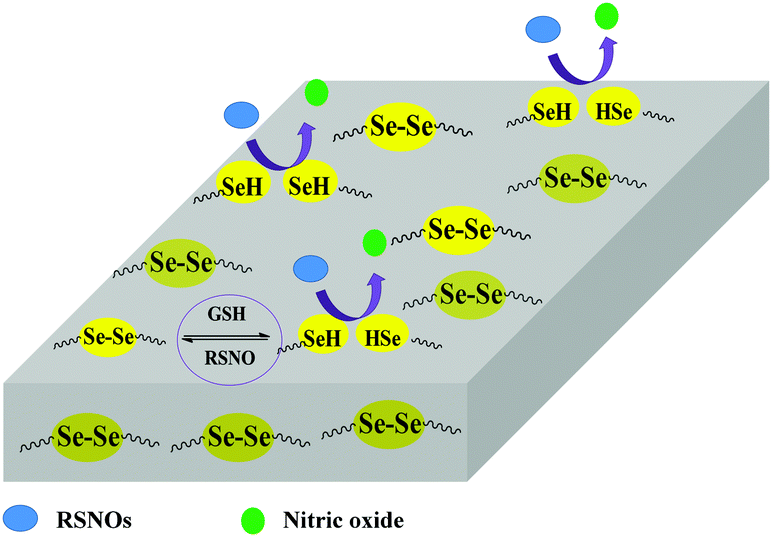 | ||
| Scheme 1 Schematic illustration of catalytic release of NO from PU/PU-Se blend films. | ||
2. Experimental
2.1 Materials
Selenium (Se), sodium borohydride (NaBH4), 2-bromoethanol, stannous octoate (Sn(Oct)2), hexamethylene diisocyanate (HDI), BDO and hexafluoroisopropanol (HFIP) were purchased from Energy Chemical Company. Dopamine hydrochloride (DH), glutathione (reduced, GSH), sodium nitrite (NaNO2), N-(1-naphthyl)ethylenediamine dihydrochloride (NED) and sulfanilic acid (SA) were purchased from Sigma-Aldrich. Poly(caprolactone)diol (PCL-diol, molecular weight (Mw) = 2000 g mol−1) was purchased from Jilian Scientific Ltd. Ethyl acetate (EtOAc), petroleum ether (PE), N,N-dimethylformamide (DMF), and diethyl ether (Et2O) were purchased from Beijing Chemical Works. All reagents except DMF were used as received without further purification, and DMF was dehydrated with sodium. S-Nitrosoglutathione (GSNO) was synthesized from GSH and NaNO2 according to the literature.25 The Griess agents were self-made from NED and SA. Mouse embryonic fibroblast (NIH-3T3) was kindly provided by Liquan Yang's laboratory of Jilin University.2.2 Synthesis of SeDO
SeDO was synthesized according to the reported method.26 Briefly, Se (3.0 g) was suspended in 25 mL degassed water under a nitrogen atmosphere, and then NaBH4 (3.0 g) dissolved in degassed water (25 mL) was gradually added into the above Se suspension. After the mixture turned clear, Se (3.0 g) was further added and the mixture was stirred at 90 °C for 30 min. Then, 2-bromoethanol (10.0 g) in water (25 mL) was added to the above mixture at 40 °C and stirred overnight. Finally, the solution was filtered and extracted with EtOAc (3 × 100 mL), which was evaporated under vacuum. The crude product was purified by column chromatography with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of EtOAc and PE as the eluent and orange viscous oil was obtained with a yield of 54.7%. 1H NMR (400 MHz, CDCl3) δ (ppm): 3.93 (m, 4H), 3.11 (m, 4H), 2.13 (s, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 61.82, 32.68; 77Se NMR (400 MHz, CDCl3) δ (ppm): 246.49 (s, 2Se) (Fig. S1, ESI†).
:1 mixture of EtOAc and PE as the eluent and orange viscous oil was obtained with a yield of 54.7%. 1H NMR (400 MHz, CDCl3) δ (ppm): 3.93 (m, 4H), 3.11 (m, 4H), 2.13 (s, 2H); 13C NMR (100 MHz, CDCl3) δ (ppm): 61.82, 32.68; 77Se NMR (400 MHz, CDCl3) δ (ppm): 246.49 (s, 2Se) (Fig. S1, ESI†).
2.3 Synthesis of PU and PU-Se
Both PU and PU-Se were synthesized via a two-step solvent polymerization: the PCL-diol (16.0 g) was dissolved in 200 mL anhydrous DMF at 70 °C in a flask, and was then degassed by argon for 25 min. Then, three droplets of Sn(Oct)2 and HDI (4.0 g) were added into the flask successively. After stirring at 70 °C for 3 h, SeDO (4.0 g) dissolved in 10 mL anhydrous DMF was added dropwise into the solution and the reaction was further carried out for 12 h. Finally, the orange solid of PU-Se (22.5 g) was obtained after precipitation in 1000 mL Et2O and drying in a vacuum. PU was also synthesized according to the same procedure with BDO as the extender.2.4 Film preparation
4 mL polymer solutions were prepared at a concentration of 10 wt% by dissolving the PU/PU-Se blend in HFIP with mass ratios of 100/0, 95/5, 90/10, 80/20 and 50/50. The blend films were prepared by drop coating the polymer solution (100 μL) onto a glass substrate (1 cm × 1 cm). After the solvent evaporation at room temperature for 2 h, the films were further annealed at 120 °C for 1 h to remove the residual solvent. For convenience, the blend films were named PU-Se-x, where x is the number of weight percentage of PU-Se.The PU-PDA-Se film was fabricated according to the literature.1 First, freshly prepared PU was immersed into 2 mg mL−1 dopamine solution (10 mM Tris buffer, pH 8.5) at 37 °C for 12 h in an open culture dish. Then, the poly-dopamine grafted PU was sonicated for 15 min in water to remove the weakly bonded dopamine, and then it was transferred into a solution of 1 mM selenocystamine (10 mM Tris buffer, pH 8.5) at 37 °C for another 12 h. After being sonicated to remove unbounded selenocystamine, PU-PDA-Se was finally obtained.
2.5 Polymer and film characterization
X-ray photoelectron spectroscopy (XPS) analysis was performed on a VG ESCALAB MK II X-ray photoelectron spectrometer using an MgKα source (hν = 1253.6 eV). The pressure in the chamber of the analysing lumen was 8 × 10−8 Pa, and the grazing angle for all samples was 30°. The binding energy scale was referenced by setting the C1s peak at 284.7 eV. Scanning electron microscopy (SEM) and energy dispersive X-ray spectrometry (EDS) were performed on an FEI XL-30 at an accelerating voltage of 10 kV, and the working distance is 10 mm. Gel permeation chromatography (GPC) was performed on a GC-2010 Plus system using DMF as the eluent at 80 °C and with polystyrene as the standard.2.6 Catalytic release of NO and Se leaking tests
The NO release catalyzed by the films was tested using a Griess assay method. Briefly, the film was peeled off from the glass substrate, and then immersed in 2 ml PBS (pH 7.4, consisting of 50 μM GSNO, 50 μM GSH, and 100 μM EDTA) at 37 °C. At an interval of every 20 minutes, 50 μL solution was added to a 96-well microtiter plate, and then 50 μL of Griess agents A and B was added successively. After reacting for 10 min in the dark, the absorbance at 540 nm was measured using a microplate reader.The catalytic stability of the blend films was measured on a month-long timescale. Briefly, the films were soaked in 50 μM GSH/PBS buffer at 37 °C, and taken out at the 1st, 3rd, 8th, 16th and 30th day respectively. After rinsing three times with PBS buffer carefully, the catalytic activity of the films was measured by Greiss reagents.
The Se leaching test was done by immersing one piece of PU/PU-Se film into 10 mL PBS buffer (consisting of 50 μM GSNO, 50 μM GSH, and 100 μM EDTA) at 37 °C in the dark for 24 h. Each day, the soaking solution was replaced with an equal volume of fresh PBS buffer. The soaking solutions of day 1, 3, 7, 11 and 16 were collected, and the Se concentration of these solutions was measured by inductively coupled plasma-optical emission spectroscopy (ICP-OES).
2.7 Hemocompatibility test
where ODtest, ODneg, and ODpos are the absorbance values of the test sample, negative control (saline), and positive control (water), respectively.
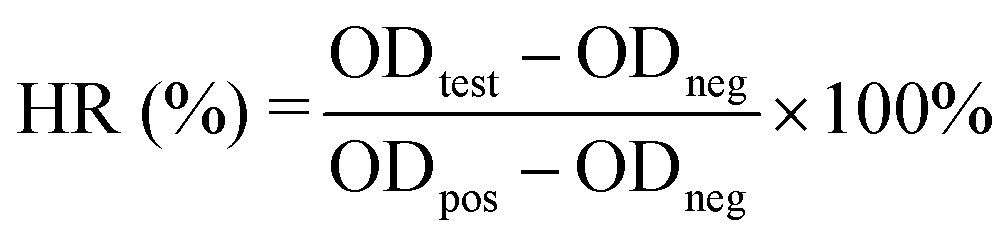
2.8 In vitro cytotoxicity
The in vitro cytotoxicity of the films was tested according to the ISO 10993-5 elution method, as the following: the sterilized films were immersed into Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 1% L-glutamine at 37 °C for 24 h to prepare the test extracts. NIH-3T3 cells were seeded in a 96-well plate at 10000 cells per well, and then incubated in a humidified atmosphere of 5% CO2 at 37 °C for 24 h. Then the normal DMEM was replaced by 100 μL test extract and incubated at 37 °C for 24 h. After that, the cell viability was quantitatively analyzed by an MTT assay. Positive and negative controls were performed by culturing the cells with normal DMEM and 0.64% phenol-contained DMEM, respectively.
2.9 Mechanical test
Uniaxial tensile tests were carried out using an electromechanical testing system (Instron 5982) at room temperature with a 10 kN load cell and a crosshead speed of 50 mm min−1. The dumbbell-shaped strips (length × width × thickness = 25 mm × 2 mm × 0.3 mm) were cut from the films and then pre-conditioned with a relative humidity of 55%, at 25 °C for at least 48 h. For each film, 5 samples were tested.3. Results and discussion
3.1 Synthesis and characterization
Firstly, the organoselenium catalyst, SeDO, was synthesized according to the previous literature, and its structure was demonstrated by 1H NMR, 13C NMR and 77Se NMR results (Fig. S1, ESI†). Then SeDO was used as the chain extender in the PU polymerization to form PU-Se, and the common PU was also synthesized with BDO as the chain extender. The synthetic protocol for them is displayed in Fig. 1A, and both of them were characterized by 1H NMR spectroscopy (Fig. 1B). Compared to PU, the new peaks at 4.18 ppm (–NH–COO–CH2–CH2–Se–) and 3.11 ppm (–NH–COO–CH2–CH2–Se–) were observed for PU-Se, indicating that SeDO was successfully incorporated into the PU backbone. The number average molecular weight (Mn) of PU was 27000 g mol−1 with a PDI of 2.30, and the Mn value of PU-Se was 24000 g mol−1 with a PDI of 2.76.
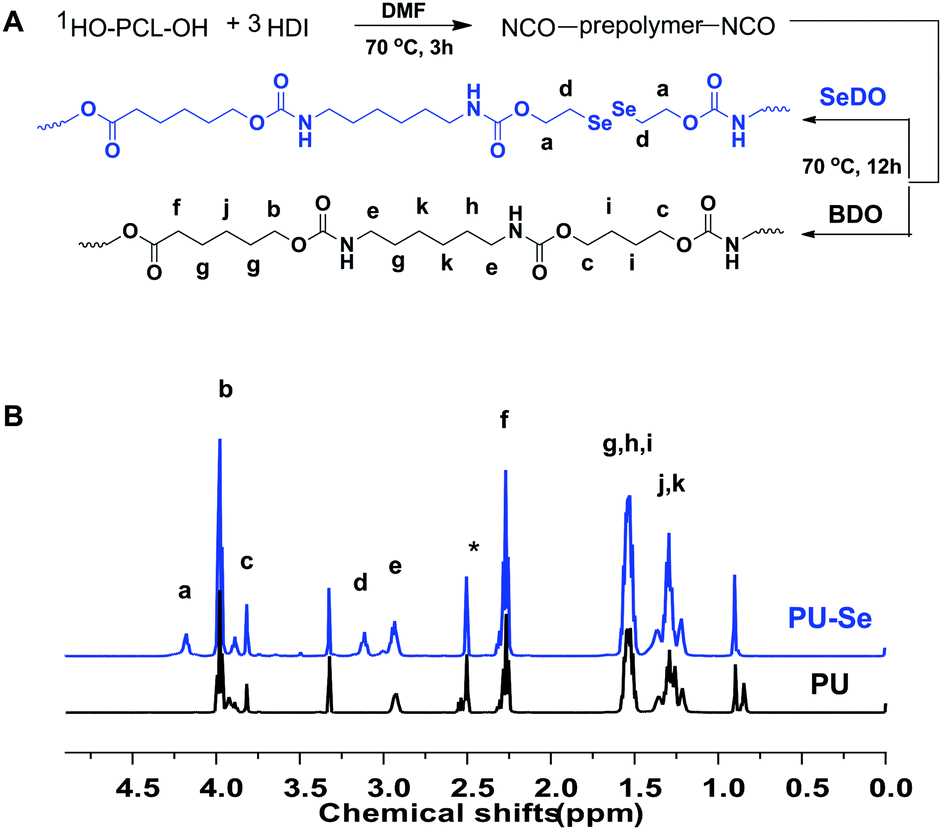 | ||
| Fig. 1 (A) Synthetic routes to PU and PU-Se. (B) 1H NMR spectra of PU and PU-Se. | ||
Polymer films were prepared by using a drop-coating method to measure their catalytic properties. The catalytic NO release rate could be influenced by the content of the catalytic sites on the film surface. In this work, the content of Se–Se bonds on the films was adjusted by manipulating the weight ratio of PU to PU-Se, and the Se content of the film surfaces was characterized by the XPS and EDS measurements. As shown in Fig. 2A, the peaks located at 54.9 eV and 55.7 eV contributed by Se 3d were observed, and the peak area was gradually enhanced with the decrease of the PU/PU-Se ratio. This indicates that the Se content on the film surfaces was gradually increased with the increase of PU-Se proportion. This result agreed well with the EDS results, as shown in Fig. 2B and Fig. S2 (ESI†). The surface morphology of the films was characterized by SEM (Fig. 2C–E), which shows that the surface becomes rough with the increase of PU-Se content. This may be attributed to the metal-like properties of Se, which has poor compatibility with polymers and thus results in the aggregation of Se–Se bonds on the film surface.
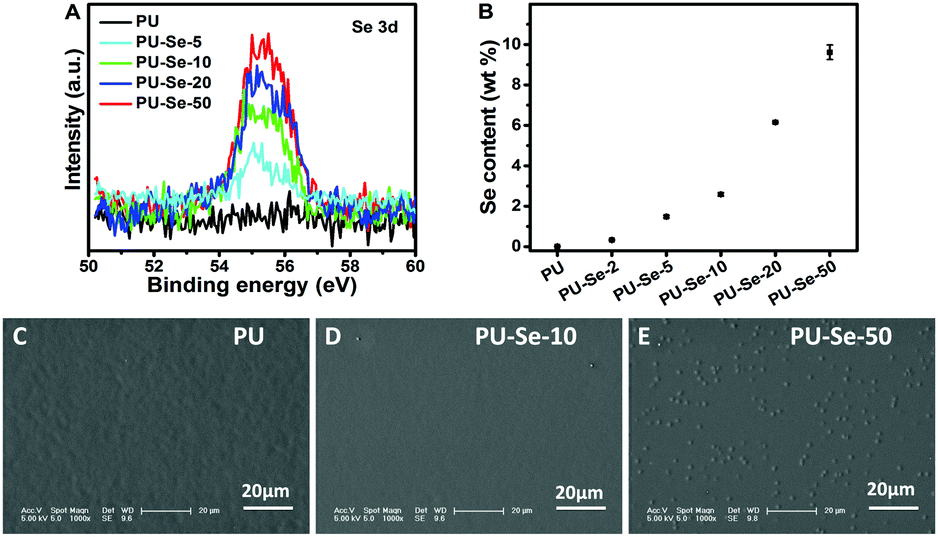 | ||
| Fig. 2 (A) XPS spectra of different films. (B) Surface Se content of the films measured by EDS. Surface morphology of (C) PU, (D) PU-Se-10 and (E) PU-Se-50. | ||
3.2 Catalytic activity and stability of films
It has been reported that diselenides have the capability of catalyzing RSNO into NO.1,17 The catalytic activity of SeDO was performed before examining the films, and GSNO was used as the donor molecule. It could be found that NO was generated from GSNO quickly when SeDO was added into pH 7.4 PBS solution (50 μM GSNO, 50 μM GSH and 100 μM EDTA), which indicates that SeDO has excellent NO-catalytic ability (Fig. S3, ESI†). Therefore, SeDO could be used as the chain extender to construct NO-generating PU. For the PU/PU-Se blend films, the in vitro catalytic curves were measured and the average NO release rate was calculated according to the curves. As shown in Fig. 3A and B, PU-Se-0.5 exhibited obvious NO generation with an average release rate of 6.7 ± 0.2 × 10−10 mol cm−2 min−1, while PU did not show catalytic activity. As the PU-Se content increases to 2%, 5%, 10% and 20%, the NO-releasing rate was much higher than that of PU-Se-0.5 but similar to each other, which indicates that the average NO release rate reaches the maximum (8.0 × 10−10 mol cm−2 min−1). This might be attributed to the change in the rate-determining factor which was the GSNO rate diffusing into the film/solution interface when the PU-Se content was more than 2 wt%, while it became the amount of catalytic sites when the PU-Se content is less than 2 wt%.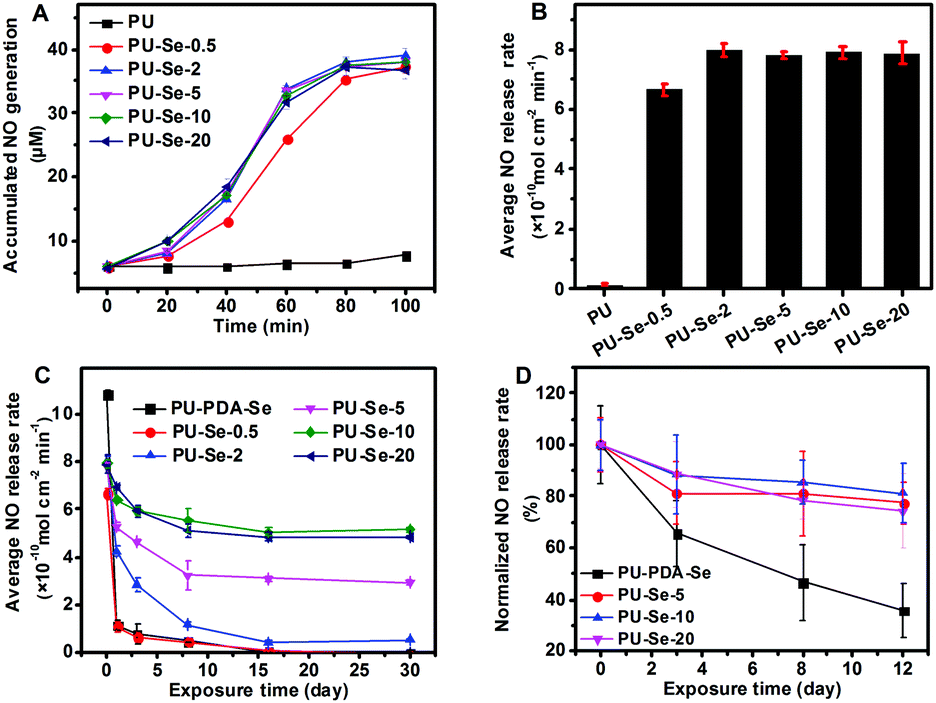 | ||
| Fig. 3 (A) Catalytic NO release curves of the PU and PU/PU-Se blend films. (B) The calculated average catalytic NO release rate of the films. (C) NO release rate of the films upon exposure to PBS at 37 °C for different times. (D) Normalized NO release rate of the films upon exposure to PRP at 0 °C for different times (mean ± SD, n = 4). | ||
Continuous and sufficient NO release from a catalytic surface is necessary to achieve its biological function in vivo. However, due to the weak bond energy of Se–Se (172 kJ mol−1),27 the current diselenide catalytic coatings were confronted with the challenges of loss of catalytic sites upon exposure to reduce agent-contained media, which severely impaired the catalytic stability of the coatings.1 In this work, the PU/PU-Se films were continuously exposed to GSH/PBS buffer at 37 °C, and the catalytic activities of the films were measured at intervals of 1 d, 3 d, 8 d, 16 d and 30 d, respectively, to investigate their catalytic stability. Meanwhile, the traditional diselenide–dopamine coating was prepared on the PU substrate (PU-PDA-Se) as the control set. As shown in Fig. 3C, the NO release rate of PU-PDA-Se and PU-Se-0.5 significantly decreased after 1 day of exposure, and then decreased slowly over time until it not being detected at the 16th day. For the blend films, a significant decrease in NO release rate was also found within a period of 11 days and then the NO release rates entered into a stable stage after 16 days of exposure and remained at 0.4 ± 0.06, 3.1 ± 0.09, 5.1 ± 0.18, and 4.8 ± 0.17 × 10−10 mol cm−2 min−1 for PU-Se-2, PU-Se-5, PU-Se-10, and PU-Se-20, respectively. This decline in the NO release rate could be attributed to the loss of catalytic sites, which was confirmed by the detection of Se in the extracts of the films (Fig. S4, ESI†). It is interesting that Se leaking from the Se-contained PU films also slowed down with the exposure time, and was stabilized after exposing in GSH/PBS buffer for 16 days, which is consistent with the catalytic stability result. From the above result, we infer that the weak Se–Se bond was broken by the reducing agent, and low molecular Se-contained segments could be extracted from the surface while high molecular segments were retained on the film, which resulted in the loss of catalytic sites until it reached stability after 16 days of exposure. Compared to PU-PDA-Se, PU/PU-Se films showed remarkable catalytic stability due to the incorporation of Se–Se bonds into the backbone of PU. The normalized NO release rate was shown in Fig. S5 (ESI†); among the blend films, PU-Se-10 showed the best catalytic stability with 61.22% of the initial NO release rate retained after exposure to PBS buffer for 30 days.
There is almost no difference in catalytic performance when the PU-Se content in the film was higher than 10 wt%, which is the critical point for the rate-determining factor changing from the amount of catalyst to kinetic diffusion of GSNO. Based on the result of catalytic stability, the PU-Se content of 2 to 10 wt% has great potential to give a continuous and stable NO release rate at the physiological level (0.5–4.0 × 10−10 mol cm−2 min−1). The catalytic stability of the films exposed to platelet-rich plasma was also measured, and the PU/PU-Se films showed good catalytic stability after 12 days of exposure, while PU-PDA-Se displayed a declined catalytic ability (Fig. 3D). Compared to soaking in PBS buffer, immersing into PRP exhibited a promoted catalytic performance for the same soaking periods. This may be attributed to the adsorption of certain selenoproteins (such as GPx) and platelets (surface bonding incorporates Cu+/Cu2+-containing enzymes or disulfide isomerase) from PRP, which could catalyze the decomposition of RSNOs.6,28,29
3.3 Blood compatibility of the films
The effect of catalytically generated NO by PU/PU-Se blend films on platelet adhesion was studied by SEM. As shown in Fig. 4A–E, adhesion of platelets was obviously inhibited on the PU/PU-Se blend films in comparison with that on the PU film. Meanwhile, the shape of the platelets on the blend films was round while the platelets on PU exhibited a spreading shape with pseudopodia, which implies the activation of the platelets.30 This indicates that the incorporation of the Se–Se bond into PU could enhance the inhibition of platelet adhesion and activation on the PU/PU-Se films.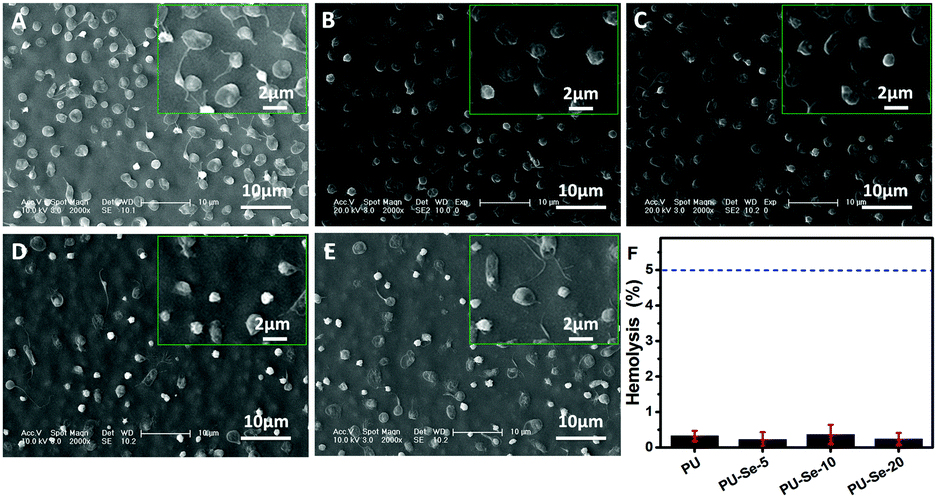 | ||
| Fig. 4 SEM images of platelet adhesion on (A) PU, (B) PU-Se-2, (C) PU-Se-5, (D) PU-Se-10 and (E) PU-Se-20 films. The insets show the platelet morphology under higher magnification. (F) Hemolysis test results of the films, where the hemolysis values of the positive control (water) and negative control (saline) were 100% and 0, respectively. | ||
Hemolysis refers to the process where the red blood cells rupture and their content releases into blood plasma. The released cytoplasm can aggravate the aggregation of platelets, which accelerates the formation of thrombus.3 Thus, hemolysis is another serious problem associated with the blood-contacting medical devices, and it has been reported that up to 5% hemolysis is permissible for biomaterials.31 Herein, the hemolytic activity of PU/PU-Se blend films was evaluated, and the results are shown in Fig. 4F. It can be observed that the hemolysis degree is less than 0.5% and has no statistical significance for all samples, which indicates that introducing organoselenium catalytic sites into PU backbones would not result in hemolysis, and the PU/PU-Se blend has potential to be used as the blood-contacting material.
3.4 Cytotoxicity
It has been reported that aliphatic Se species are toxic in some degree due to their capability of producing superoxide radicals.32,33 Thus, the cytotoxicity of the extracts of PU/PU-Se films was evaluated by an MTT assay. As shown in Fig. 5, PU and PU-Se-2 showed excellent cytocompatibility with the viability of NIH-3T3 cells around 100%, while the cell viability of the blend films was decreased slightly with the increase of PU-Se proportion, which may be attributed to oxidative damage of superoxide radicals produced from diselenide bonds. It should be noted the cell viability of the PU/PU-Se film was higher than 96% when the PU-Se content was below 10 wt%, indicating good cytocompatibility at a lower PU-Se content.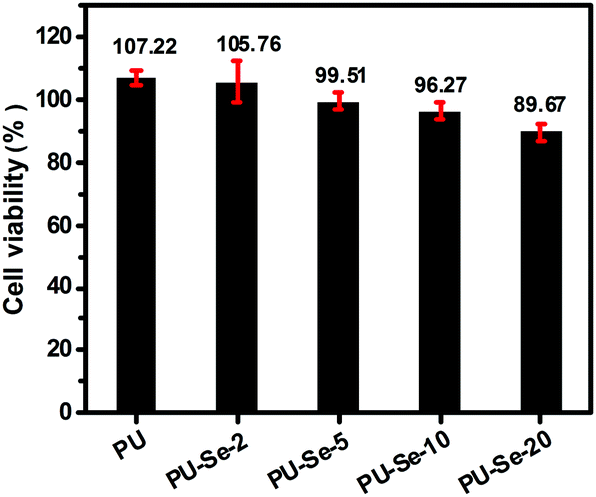 | ||
| Fig. 5 NIH-3T3 cell viability cultured by the 1 day extracts of the films, the negative control and positive control were 100% and 0% cell viability, respectively. (mean ± SD, n = 6). | ||
3.5 Mechanical property
The tensile behavior of the films was determined at a draft rate of 50 mm min−1 at room temperature. The representative stress–strain curves are shown in Fig. 6, and the calculated mechanical parameters are summarized in Table 1. Compared to PU, the tensile strength and initial modulus of PU/PU-Se gradually decreased with the increase of PU-Se content, while the fracture strain was maintained at around 1200%. This decrease in the mechanical properties of PU/PU-Se may be attributed to the weak Se–Se bonds in PU-Se, which might be adverse to the aggregation of the hard segments and the formation of intermolecular hydrogen bonding. However, when the PU-Se content was below 10 wt%, the decrease in tensile strength and initial modulus was minimal and could be negligible. For example, the tensile strength was slightly decreased from 41.6 MPa for PU to 39.8 MPa for PU-Se-10. In combination with the enhanced catalytic stability and excellent biocompatibility, PU-Se-10 is the optimized blend film and has great potential in long-term blood-contacting applications.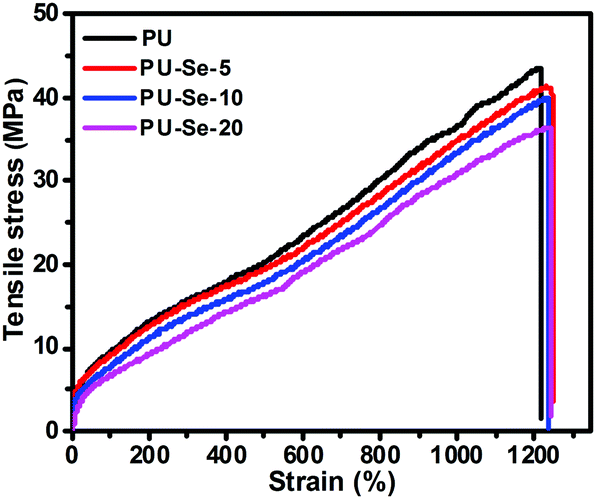 | ||
| Fig. 6 Tensile stress–strain curves of PU and PU/PU-Se blend films. | ||
| Sample | Initial modulus (MPa) | Tensile strength (MPa) | Fracture strain (%) |
|---|---|---|---|
| PU | 35.6 ± 2.5 | 41.6 ± 2.6 | 1225 ± 84 |
| PU-Se-5 | 38.7 ± 8.3 | 40.7 ± 3.8 | 1237 ± 35 |
| PU-Se-10 | 34.6 ± 3.9 | 39.8 ± 1.6 | 1152 ± 56 |
| PU-Se-20 | 23.7 ± 5.0 | 35.5 ± 1.2 | 1104 ± 28 |
4. Conclusion
In this work, a novel NO-generating strategy was proposed by designing and synthesizing PU-Se to achieve stable and controllable NO release at the physiological level. PU-Se was successfully synthesized by using the catalyst SeDO as the chain extender, and a series of PU/PU-Se blend films were prepared to investigate the effect of PU-Se content on the catalytic properties. These blend films exhibited excellent catalytic activities, and the highest NO release rate (8.0 × 10−10 mol cm−2 min−1) was obtained when the PU-Se content was above 2 wt%. Compared to PU-PDA-Se, the blend films also showed outstanding catalytic stability. Among them, PU-Se-10 showed the best catalytic stability, which retained 61.22% of the initial NO release rate after exposure to PBS buffer for 30 days. It is interesting that the blend films with 2–10 wt% PU-Se content could generate the stable NO release rate at the physiological level for 30 days. What's more, the PU/PU-Se blend films also demonstrated enhanced anti-platelet effect, low hemolysis ratio, excellent biocompatibility, and similar mechanical properties to PU. To the best of our knowledge, it is the first time to enhance the catalytic stability of NO-catalytic materials by incorporating catalytic sites into the polymer backbone. Using this method, stable and controllable NO release at the physiological level was achieved. We hope this kind of polymer will have great potential in fabricating biomedical devices for long-term blood-contacting applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21803069).References
- W. Cai, J. Wu, C. Xi, J. A. Ashe and M. E. Meyerhoff, Biomaterials, 2011, 32, 7774–7784 CrossRef CAS.
- J. An, S. Chen, J. Gao, X. Zhang, Y. Wang, Y. Li, S. Mikhalovsky, D. Kong and S. Wang, Sci. China: Life Sci., 2015, 58, 765–772 CrossRef CAS PubMed.
- H. Jiang, X. B. Wang, C. Y. Li, J. S. Li, F. J. Xu, C. Mao, W. T. Yang and J. Shen, Langmuir, 2011, 27, 11575–11581 CrossRef CAS PubMed.
- Y. Ji, Y. Wei, X. Liu, J. Wang, K. Ren and J. Ji, J. Biomed. Mater. Res., Part A, 2012, 100A, 1387–1397 CrossRef CAS.
- J. H. Li, S. S. Wang, D. B. Zhang, X. X. Ni and Q. Q. Zhang, Chin. J. Polym. Sci., 2016, 34, 805–819 CrossRef CAS.
- S. Goldstein, J. Lind and G. Merenyi, Chem. Rev., 2005, 105, 2457–2470 CrossRef CAS PubMed.
- W. V. Mark, K. Lih and C. L. James, Am. J. Physiol.: Heart Circ. Physiol., 1998, 274, 2163–2176 CrossRef.
- M. VanWagner, J. Rhadigan, M. Lancina, A. Lebovsky, G. Romanowicz, H. Holmes, M. A. Brunette, K. L. Snyder, M. Bostwick, B. P. Lee, M. C. Frost and R. M. Rajachar, ACS Appl. Mater. Interfaces, 2013, 5, 8430–8439 CrossRef CAS PubMed.
- A. Lutzke, J. B. Tapia, M. J. Neufeld and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2017, 9, 2104–2113 CrossRef CAS.
- G. E. Gierke, M. Nielsen and M. C. Frost, Sci. Technol. Adv. Mater., 2011, 12, 055007 CrossRef.
- A. R. Ketchum, M. P. Kappler, J. Wu, C. Xi and M. E. Meyerhoff, J. Mater. Chem. B, 2016, 4, 422–430 RSC.
- Y. Wo, Z. Li, E. J. Brisbois, A. Colletta, J. Wu, T. C. Major, C. Xi, R. H. Bartlett, A. J. Matzger and M. E. Meyerhoff, ACS Appl. Mater. Interfaces, 2015, 7, 22218–22227 CrossRef CAS.
- E. J. Brisbois, R. P. Davis, A. M. Jones, T. C. Major, R. H. Bartlett, M. E. Meyerhoff and H. Handa, J. Mater. Chem. B, 2015, 3, 1639–1645 RSC.
- E. J. Brisbois, T. C. Major, M. J. Goudie, M. E. Meyerhoff, R. H. Bartlett and H. Handa, Acta Biomater., 2016, 44, 304–312 CrossRef CAS PubMed.
- B. K. Oh and M. E. Meyerhoff, Biomaterials, 2004, 25, 283–293 CrossRef CAS PubMed.
- S. Hwang and M. E. Meyerhoff, Biomaterials, 2008, 29, 2443–2452 CrossRef CAS PubMed.
- W. Cha and M. E. Meyerhoff, Biomaterials, 2007, 28, 19–27 CrossRef CAS.
- P. T. Kao, I. J. Lee, I. Liau and C. S. Yeh, Chem. Sci., 2017, 8, 291–297 RSC.
- J. Chaudiere, O. Courtin and J. Leclaire, Arch. Biochem. Biophys., 1992, 296, 328–336 CrossRef CAS.
- Z. Yang, Y. Yang, K. Xiong, X. Li, P. Qi, Q. Tu, F. Jing, Y. Weng, J. Wang and N. Huang, Biomaterials, 2015, 63, 80–92 CrossRef CAS.
- Y. Wang, S. Chen, Y. Pan, J. Gao, D. Tang, D. Kong and S. Wang, J. Mater. Chem. B, 2015, 3, 9212–9222 RSC.
- J. MacMicking, Q. W. Xie and C. Nathan, Annu. Rev. Immunol., 1997, 15, 323–350 CrossRef CAS.
- J. B. Hibbs, R. R. Taintor and Z. Vavrin, Biochem. Biophys. Res. Commun., 1988, 157, 87–94 CrossRef CAS.
- J. W. Boretos and W. S. Pierce, J. Biomed. Mater. Res., 1968, 2, 121–130 CrossRef CAS PubMed.
- J. S. Stamler and J. Loscalzo, Anal. Chem., 1992, 64, 779–785 CrossRef CAS.
- K. Troels, S. Elisabeth, H. Ulla and B. Ole, Bioconjugate Chem., 1990, 1, 296–304 CrossRef.
- S. Ji, J. Xia and H. Xu, ACS Macro Lett., 2016, 5, 78–82 CrossRef CAS.
- A. C. John, Y. K. Sungmee, T. Diane, C. K. Murali, P. Roberto, B. M. James, V. Yoram, W. N. Raymond, C. Danae, M. M. Allen, B. G. Matthew and A. W. David, Anal. Biochem., 1996, 238, 150–158 CrossRef.
- M. P. Gordge, J. S. Hothersall and G. H. Neild, Br. J. Pharmacol., 1996, 119, 533–538 CrossRef CAS.
- S. L. Goodman, T. G. Grasel, S. L. Cooper and R. M. Albrecht, J. Biomed. Mater. Res., 1989, 23, 105–123 CrossRef CAS.
- D. Shim, D. S. Wechsler and T. R. Lloyd, Catheter. Cardiovasc. Interv., 1996, 39, 287–290 CrossRef CAS.
- J. J. Chen, L. M. Boylan and C. K. Wu, BioFactors, 2007, 31, 55–66 CrossRef CAS.
- P. Pacher, J. S. Beckman and L. Liaudet, Physiol. Rev., 2007, 87, 315–424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02264j |
| This journal is © The Royal Society of Chemistry 2019 |