
Self-delivery nanoparticles of an amphiphilic irinotecan–enediyne conjugate for cancer combination chemotherapy†
Jing
Li
a,
Baojun
Li
a,
Lili
Sun
b,
Bing
Duan
b,
Shuai
Huang
a,
Yuan
Yuan
 b,
Yun
Ding
*a and
Aiguo
Hu
*a
b,
Yun
Ding
*a and
Aiguo
Hu
*a
aShanghai Key Laboratory of Advanced Polymeric Materials, School of Materials Science and Engineering, East China University of Science and Technology, Shanghai 200237, China. E-mail: yunding@ecust.edu.cn; hagmhsn@ecust.edu.cn
bThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, 200237, China
First published on 17th November 2018
Abstract
An amphiphilic small molecular drug self-delivery system was designed by linking a hydrophilic topoisomerase I inhibitor irinotecan (Ir) with a lipophilic cytotoxic enediyne (EDY) antibiotic through an ester bond. The maleimide-based EDY with a pendant carboxyl group was synthesized in four steps from commercially available reagents. The EDY compound possesses the ability to generate radical intermediates at physiological temperature as demonstrated by electron spin resonance analysis and further causes DNA-cleavage and tumor cell suppression. The self-delivery system prepared by the combination of two anticancer drugs, EDY and Ir, formed nanoparticles’ self-assembly with a size of around 60 nm in aqueous solution, enabling the drugs to accumulate in tumor tissues through the enhanced permeability and retention effect. With high drug loading capacity (100%), the Ir–EDY nanoparticles entered tumor cells through endocytosis and possessed strong synergistic effects, inducing tumor cell death through the cell apoptosis pathway efficiently.
Introduction
Cancer is a worldwide devastating disease which causes over 8 million deaths each year.1 According to the GLOBOCAN results, about 14.1 million new cancer cases occurred in 2012.2 To fight against this terrible disease, there has been tremendous progress over the past few decades in cancer therapy including surgical excision, immunotherapy, radiation therapy and chemotherapy.2,3 Among these therapeutics, chemotherapy relying on the high cytotoxicity of chemotherapeutic agents has become a predominant modality for cancer treatment due to its high efficiency compared with other treatments.4 Exploring new chemotherapeutic agents is therefore of great significance for the treatment and cure of cancer.Since the late 1980s, great research attention has been paid to a family of natural compounds called enediynes, which are known to be endowed with high anticancer activities. Commonly known natural enediyne antibiotics include over 20 kinds of compounds, such as calicheamicin,5 dynemicins,6 neocarzinostatin,7 esperamicin,8 uncialamycin,9 kedarcidin,10 and C-1027.11 In a physiological environment, these enediyne compounds undergo Bergman or Bergman-like cyclization to generate highly reactive diradical intermediates. The diradicals can induce DNA cleavage or crosslinking inside tumor cells through abstracting the H atoms from the DNA's sugar-phosphate backbone. This unique mode of action endows natural enediyne antibiotics with a higher cytotoxicity to tumor cells than traditional chemotherapeutic agents. For example, the natural enediyne calicheamicin is around 1000 times more effective than one commonly used chemotherapy drug, doxorubicin.12,13 Due to their promising anticancer and antitumor potential, the U.S. Food and Drug Administration (FDA) have approved two antibody drug conjugates, Mylotarg® and Besponsa®, with enediynes as the “warhead”.14,15 Despite the excellent feature of natural enediynes, the availability of these compounds either through extraction from microorganisms or from total synthesis is severely limited. Therefore, there have been great efforts devoted to the design and synthesis of simple artificial enediynes in order to study and enhance their biological properties.12,16
Conventional small molecular drugs often suffer from their inherent limitations such as poor bioavailability, nonspecific selectivity, rapid blood/renal clearance, low accumulation in tumors and severe multidrug resistance (MDR).17 To address these issues, the employment of assistant nanocarriers has been investigated for many years. Nano-vehicles such as self-assembled nanoarchitectures,18–21 liposomes,22 vesicles,23 protein nanoparticles,24,25 and inorganic nano-frameworks26,27 have been widely studied for drug delivery. Most of these nano-vehicles were designed to possess the enhanced permeability and retention (EPR) effect28 and demonstrated better therapeutic efficacy against tumors than small molecular drugs. Nevertheless, it is very complicated to fabricate active targeting nanocarriers, and their drug carrying capacities are not always satisfactory.29–31 Besides, most carriers have no therapeutic effect and the metabolites of the carriers may cause unexpected toxicity32 such as mitochondrial damage, cardiovascular effects, platelet aggregation, and inflammation. To overcome these problems, there have been tremendous research efforts devoted to the development of new self-delivery drug systems in recent years,33–35 which mainly involve active amphiphilic drug–drug conjugates self-assembled into a nanoscale structure to realize intracellular delivery by themselves. This mode of drug-delivery has excellent drug loading capacities (up to 100% for pure nanodrugs) and no carrier-induced toxicity and immunogenicity. For example, Yan et al. described an amphiphilic drug–drug conjugate based on two types of anticancer drugs including a hydrophilic irinotecan (Ir) and a hydrophobic chlorambucil (Cb) via an ester linkage and achieved remarkable anticancer activity both in vitro and in vivo.36
In our previous work, we found that when an acyclic enediyne compound was annulated with a maleimide group at the ene position, the Bergman cyclization of the enediyne was greatly facilated37 to generate reactive radicals at low temperature, possessing excellent DNA cleavage ability and high cytotoxicity towards tumor cell lines.38 Inspired by the advancement of drug self-delivery systems, herein, we designed and synthesized a small molecular anticancer drug irinotecan (Ir) and enediyne (EDY) conjugate. Ir is a hydrophilic derivate of camptothecin which can kill cancer cells via binding to DNA topoisomerase I, leading to transcription inhibition,39,40 while maleimide-based acyclic enediynes were designed with two alkyl chains as a hydrophobic molecule which can generate diradicals to abstract H atoms from the DNA's sugar-phosphate backbone and finally kill cancer cells. As shown in Fig. 1a, the hydrophilic Ir and the hydrophobic EDY were coupled through an ester bond to form amphiphilic Ir–EDY conjugates. When dispersed in water, they self-assembled into nanoparticles (Fig. 1b). During the transport in the blood vessel, the nanoparticles would accumulate in tumor tissues through the EPR effect (Fig. 1c). Once they enter the tumor cells by receptor-mediated endocytosis41,42 (Fig. 1d), the acidic microenvironment of lysosomes would hydrolyze the ester bond between Ir and EDY and release free Ir and EDY simultaneously. Both Ir and EDY can damage tumor cells through different ways and finally kill cancer cells.
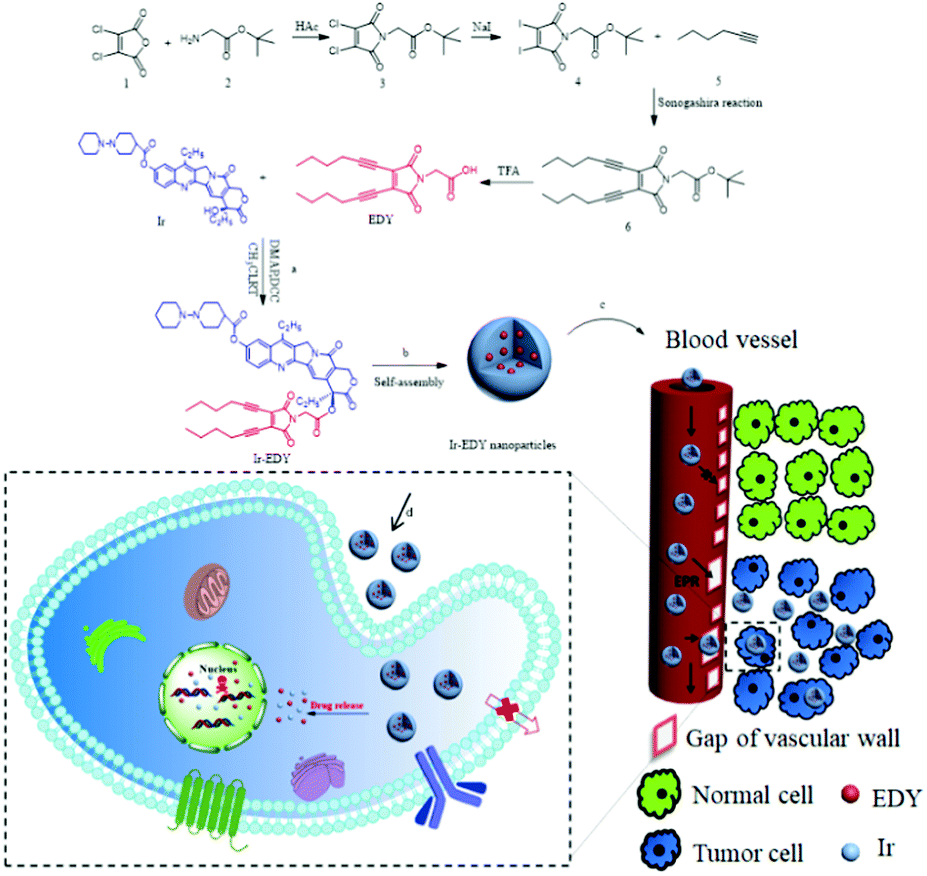 | ||
| Fig. 1 Schematic illustration of amphiphilic drug–drug conjugate (ADDC) from synthesis, self-assembly and self-delivery. (a) Synthesis of Ir–EDY ADDC through esterification in the DCC/DMAP-catalyzed system. (b) Ir–EDY ADDC self-assembles into nanoparticles in water. (c) Ir–EDY ADDC nanoparticles access tumor tissues by means of their leaky vasculatures (EPR effect). (d) Ir–EDY ADDC nanoparticles enter tumor cells by endocytosis. | ||
Experimental
Materials
Toluene and tetrahydrofuran (THF) were dried over calcium hydride and distilled before use. Other chemicals were of commercial grade and used as received. tert-Butyl 2-(3,4-diiodo-2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)acetate (4) was synthesized according to a literature procedure with minor modification.43 The detailed synthesis procedure for the precursors is presented in the ESI.†Synthesis of lipophilic enediyne (EDY)
Synthesis of Ir–EDY conjugate
Self-assembly of Ir–EDY nanoparticles
The Ir–EDY conjugate (30 mg) was dissolved in dimethylsulfoxide (DMSO, 3 mL) and stirred at room temperature for 5 min. Then the DMSO solution was added dropwise into deionized water (7 mL) under gentle stirring for 30 min. Subsequently, the solution was transferred into a dialysis bag (MWCO = 1000 g mol−1) and dialyzed against deionized water for 48 h, during which the water was renewed every 8 h. The final volume of the solution was adjusted to 30 mL by adding deionized water to reach a concentration of 1 mg mL−1 for further experiments.ESR study of EDY and corresponding PBN adduct
Electron spin resonance (ESR) spectroscopy studies were carried out with freshly prepared enediyne dissolved in methanol (40 mg mL−1) or enediyne (40 mg mL−1) with excess PBN dissolved in DMSO. The spectra were recorded on a Bruker EMX-8/2.7C ESR spectrometer.DNA cleavage experiments of EDY
Freshly prepared EDY were dissolved in DMSO (2 μL) at concentrations of 200 mM and 100 mM respectively. Each EDY solution was added to a solution of supercoiled plasmid PBR 322DNA (0.5 μg μL−1) in TE buffer (pH 7.6, 4 μL). Control samples consisted of a solution of supercoiled plasmid PBR 322DNA (0.5 μg μL−1) in TE buffer (pH 7.6, 4 μL) separately incubated with or without 2 μL DMSO. All the systems were incubated at 37 °C for 72 h. After incubation, each system (5 μL) was mixed with a 6× loading buffer (1 μL) and subjected to 0.8% agarose gel electrophoresis at 128 V (101 mA) for 30 min, stained by ethidium bromide and then the gel was photographed on a UV transilluminator (FR-200A) and analyzed by scanning densitometry.Critical aggregation concentration (CAC) measurements
A known amount of Nile red in CH2Cl2 was added to a series of vials and then the CH2Cl2 was evaporated. The amount was chosen to give a Nile red concentration of 1 × 10−6 M in the final solution. A measured amount of Ir–EDY nanoparticle solution was added into each vial and deionized water was added to the vials to make the concentrations of the nanoparticles ranging from 2 to 1024 μM. The vials were vibrated at room temperature overnight, and then the fluorescence emission intensity at a wavelength of 650 nm (excited at 485 nm) was measured. The critical aggregate concentration is obtained from the two linear regressions based on the fluorescence intensities against [Ir–EDY] where they intersect.Cell internalization
The cellular uptake behaviour was observed using HeLa cells (a human uterine cervix carcinoma) as a model cell line with confocal laser scanning microscopy (CLSM). In brief, HeLa cells were seeded in 6-well plates at 2.5 × 105 cells per well in 1 mL of complete Dulbecoo's modified Eagle's medium (DMEM) and incubated for 24 h, followed by removal of the culture medium and addition of EDY or Ir–EDY nanoparticle solution (0.5 mL DMEM medium) at the concentration of 50 μM. The control group was HeLa cells incubated without any additives. After incubation at 37 °C for 6 h or 24 h, the culture medium was removed, and the cells were washed with PBS two times. Subsequently, the cells were fixed with 2.5% glutaraldehyde at room temperature for 30 min, and the slides were rinsed with PBS three times. Finally, 100 μL 10 μg mL−1 propidium iodide (PI) solution and 0.5% Triton solution were added and the cells were cultured at 37 °C for another 15 min. The slides were rinsed with cold PBS three times. The resulting slides were mounted and observed using a LEICA TCS SP8 fluorescence microscope.Cell viability
Cell viability tests were investigated by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay.46 HeLa cells were cultured in DMEM containing 10% fetal bovine serum (FBS) at 37 °C under a humidified atmosphere containing 5% CO2. The cells were trypsinized until they reached 70% confluence in the tissue culture flasks with a buffered saline solution containing 0.25% trypsin and 0.03% EDTA. Cells were seeded into a 96-well plate at a density of 3 × 104 cells per well in 200 mL of the cell culture medium. After 24 h of incubation, the culture medium was removed and the cells were then exposed to 0.78, 1.57, 3.13, 6.25, 12.5, 25, 50 or 100 μM concentrations of EDY, Ir, mixture of Ir and EDY, or the Ir–EDY nanoparticles. The concentrated EDY or Ir was prepared by dissolving EDY or Ir in DMSO to reach 100 mM and then diluted using cell culture medium to 100 μM (the concentration of DMSO was 0.1%). The mixture ratio of EDY and Ir was 1/1 (v/v) at the same concentration, and the culture medium (with 0.1% DMSO) was used as a blank control. After incubation for another 48 h, 10 μL of sterile filtered MTT stock solution (5 mg mL−1) in PBS (pH 7.4) was added to each well and the cells were further incubated at 37 °C for 4 h to allow the yellow dye to transform into blue crystals. After removal of the supernatant MTT/medium, the purple MTT-formazan crystals were dissolved by adding 150 μL of DMSO to each well of the plate, and then the plate was shaken for 15 min. Finally, the optical density was measured using a microplate reader at a wavelength of 570 nm. The spectrophotometer was normalized using a culture medium without cells. Cell viability (%) related to control wells containing cell culture medium without HAPN suspension was calculated by [OD]test/[OD]control.Cell apoptosis assay
HeLa cells were seeded in 6-well plates at 5.0 × 105 cells per well in 2 mL of complete DMEM and cultured for 24 h. The cells were treated with Ir, EDY and Ir–EDY nanoparticles at the concentration of 25 μM for 24 h. HeLa cells without the treatment were used as a control. For quantitative measurements of apoptosis, the treated cells were harvested and washed twice with ice-cold PBS, and stained with FITC-A and PI according to the manufacturer's instructions. 2 × 104 events per sample were counted and analyzed by flow cytometry (BD FACSCalibur, USA).Results and discussion
Synthesis and characterization of EDY and Ir–EDY conjugate
The synthetic route to EDY and Ir–EDY is shown in Fig. 1. EDY was synthesized through a Sonogashira reaction between diiodomaleimide and hex-1-yne followed by the hydrolysis of tert-butyl ester in the presence of trifluoroacetic acid. The chemical structure of EDY was confirmed by 1H NMR (Fig. S3, ESI†), 13C NMR (Fig. S4, ESI†) and HR-MS spectroscopy. The molecular structure of EDY was carefully designed so that it could enter tumor cells efficiently. According to the Lipinski's Rule of Five (Ro5),47 a compound is more likely to exhibit poor absorption or permeation when two or more of the following physicochemical criteria are fulfilled: the molecular weight is greater than 500; the calculated octanol–water partition coefficient, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, is greater than 5; there are more than 5 hydrogen-bond donors or the number of hydrogen-bond acceptors (nitrogen or oxygen atoms) is greater than 10. The molecular weight of EDY is 315, below the criterion of 500; the calculated hydrophobicity (expressed via logP) of EDY is 2.48, lower than 5; the molecular formula of EDY is C18H21NO4, consisting of less than 10 hydrogen-bond acceptors. Therefore, EDY would possess the potential to enter the tumor cells, which was proved by a confocal laser scan microscopy (CLSM) study (see below, Fig. 6).
P, is greater than 5; there are more than 5 hydrogen-bond donors or the number of hydrogen-bond acceptors (nitrogen or oxygen atoms) is greater than 10. The molecular weight of EDY is 315, below the criterion of 500; the calculated hydrophobicity (expressed via logP) of EDY is 2.48, lower than 5; the molecular formula of EDY is C18H21NO4, consisting of less than 10 hydrogen-bond acceptors. Therefore, EDY would possess the potential to enter the tumor cells, which was proved by a confocal laser scan microscopy (CLSM) study (see below, Fig. 6).
The amphiphilic Ir–EDY conjugate was synthesized through esterification using DCC/DMAP as the catalyst. Compared with the 1H NMR spectrum (Fig. 2A) of Ir, the peak at 4.12 ppm (a) attributed to the hydroxyl proton disappears completely after esterification. The proton signals at 7.64 ppm (b) and 1.88 ppm (d) belong to the pyridone ring and the methylene (CH3CH2–, lactonic ring) of Ir respectively, shifted to 7.33 ppm (b′) and 2.17 ppm (d′) accordingly in the 1H NMR spectrum of the Ir–EDY conjugate. Compared with EDY, the two proton signals at 4.32 ppm (c) belonging to the methylene (CH2CO–) group was buried in the broad peak from 4.4–4.7 ppm (c′). All the signals attributed to the protons in Ir or EDY can be found in the 1H NMR spectrum of Ir–EDY (Fig. 2A). In addition, compared with the 13C NMR spectrum (Fig. 2B) of Ir, the peak at 174.0 ppm (a) attributed to the carbonyl group (lactonic ring) shifted to 166.4 ppm (a′), and compared with EDY, the peak at 166.8 ppm (b) related to –CON– on the maleimide ring didn’t change in the 13C NMR spectrum of the Ir–EDY (b′). Moreover, the signal at 172.1 ppm (c) belonging to –COOH shifted to 166.8 ppm (c′) which is ascribed to –OCOCH2– (carbonyl group) of the newly formed ester bond in Ir–EDY after esterification. The molecular weight of Ir–EDY was characterized by mass spectroscopy (Fig. S7, ESI†). Ir–EDY was also characterized by UV-vis (Fig. S8, ESI†) and fluorescence spectroscopy (Fig. S10, ESI†). From the UV-vis spectrum, the Ir–EDY conjugate exhibits two obvious absorption bands at around 288 nm and 360 nm. A 5 nm blue-shift of the Ir–EDY conjugate was observed compared to that of free Ir and EDY (365 nm). All experimental results confirmed that Ir–EDY has been successfully synthesized.
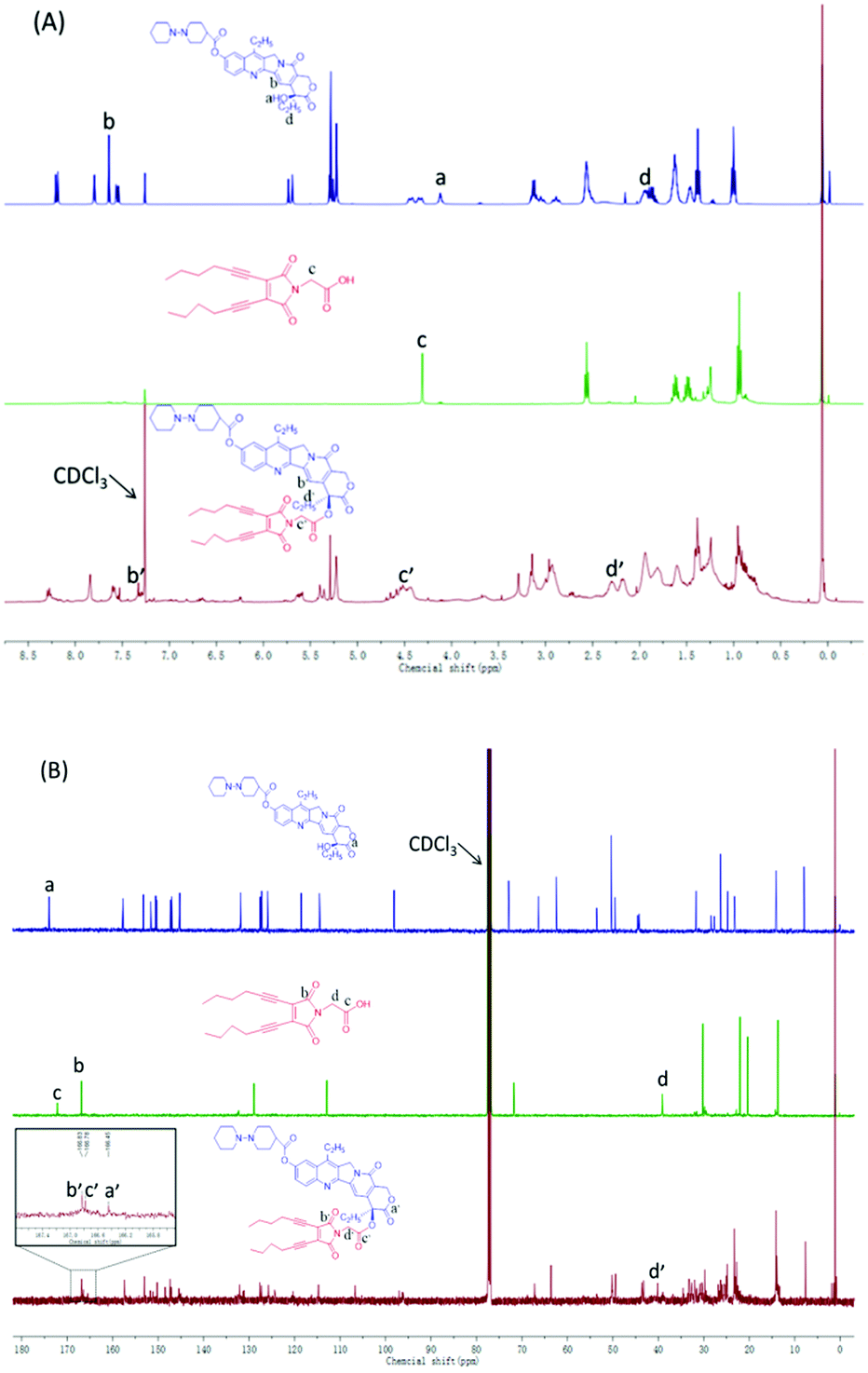 | ||
| Fig. 2 (A) 1H NMR and (B) 13C NMR spectra of Ir (top), EDY (middle) and Ir–EDY (bottom) in CDCl3. | ||
Preparation and characterization of Ir–EDY nanoparticles
Ir–EDY NPs were prepared by adding a DMSO solution of Ir–EDY conjugate dropwise into water, followed by removal of DMSO through dialysis and adjusting the concentration of the Ir–EDY NPs to 1 mg mL−1 as a stable and light yellow solution (Fig. 3C-inset). The size and morphology of Ir–EDY NPs were characterized using SEM, TEM, and DLS. The Ir–EDY NPs in water possess the typical Tyndall phenomenon (Fig. S11, ESI†). The average hydrodynamic diameter of Ir–EDY nanoparticles is about 60 nm with a narrow distribution as determined by DLS (Fig. 3C). Additionally, the DLS measurements were carried out at different time intervals (3, 6, 9, 12, 15, 18, 21 and 24 days) to evaluate the stability of the nanoparticles (Fig. S9, ESI†). The results proved that Ir–EDY NPs were stable enough and capable of long time storage at 4 °C in the dark. The SEM and TEM images in Fig. 3A and B indicate that Ir–EDY NPs are spherical with an average diameter of nearly 50 nm, which is smaller than the DLS results because of the shrinkage in the dry state during sample preparation. The inset of Fig. 3B presents a typical enlarged TEM image of one nanoparticle. Apparently, the Ir–EDY NPs consist of many small spherical building blocks. These small building blocks are smaller than 5 nm, indicating that the blocks were conventional micelles self-assembled to form the Ir–EDY conjugates. Then the secondary aggregation of these blocks generated the Ir–EDY nanoparticles. This mechanism is similar to the small micelle aggregate (SMA) of a multimicelle aggregate (MMA),48 which described that the dendritic multiarm copolymers first self-assemble into small micelles and then the small micelles further aggregate into large multimolecular micelles.36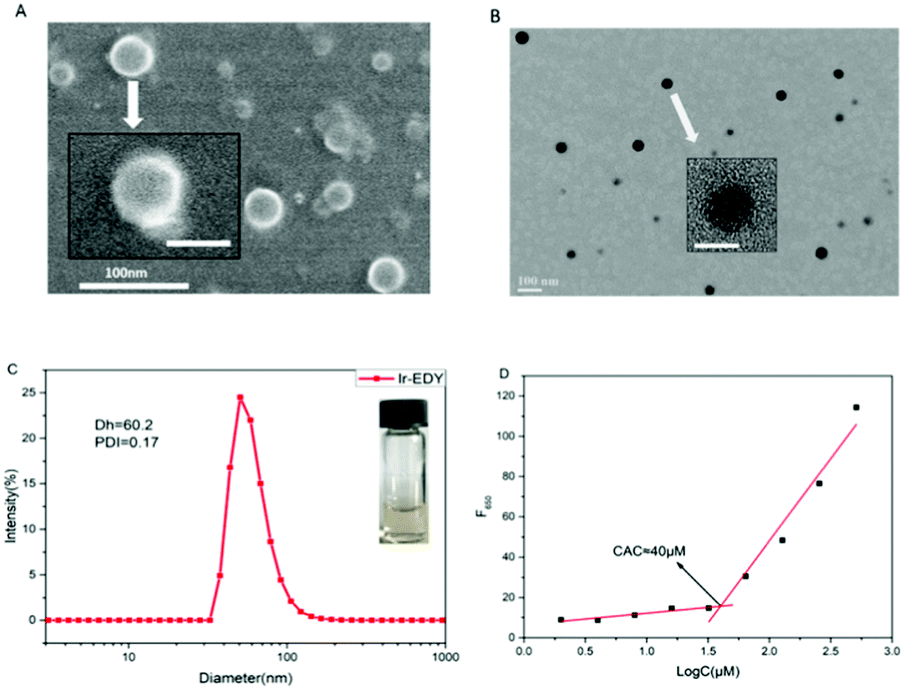 | ||
| Fig. 3 SEM (A) and TEM (B) images of Ir–EDY nanoparticles. Inset: The amplified image of one nanoparticle. Scale bars: 100 nm (outside), 50 nm (inset). (C) DLS curve of Ir–EDY nanoparticles. The polydispersity index (PDI) = 0.17, and the average size (Dh) = 60.2 nm. Inset: A digital photograph of Ir–EDY nanoparticles in water. (D) Relationship between the fluorescence intensity and Ir–EDY concentration in water. Emission intensity at 650 nm (F650) of Nile red as a function of concentrations of Ir–EDY nanoparticles in water. [Nile red] = 1.0 × 10−6 mol L−1, λex = 485 nm. | ||
The CAC measurements were performed to investigate the self-assembly behavior of Ir–EDY by using Nile red as a fluorescent probe.49 Nile red does not emit fluorescence in aqueous solution but its fluorescence emission would increase substantially in the hydrophobic environments of micelles.50–52Fig. 3D shows that the CAC value of the Ir–EDY NPs corresponding to the observed inflection point is about 40 μM.
Radical nature and DNA cleavage characteristics of EDY
It is well acknowledged that enediyne compounds undergo a Bergman or Bergman-like reaction to generate diradical intermediates, endowing them with DNA-cleavage ability. To verify the radical nature of this transformation, the freshly prepared EDY in methanol was subjected to ESR spectroscopy analysis. As shown in Fig. 4A, a clear signal appears at 3520 G, indicating the presence of carbon free radicals.37 The splitting of the peak suggests that there are at least two different kinds of radical species, either generated from asymmetric cyclization of EDY or partial coupling (or abstracting H-donor) of the symmetrical diradical intermediates. The unstable carbon radical can be trapped by radical-trapping agents and transform into a stable free radical for further analysis.53 Usuki et al. conducted a spin-trapping experiment on naturally occurring enediyne calicheamicin to confirm the generation and evolution of the radical species in ethanol solution.54 Inspired by this, the carbon radicals generated from EDY were trapped with phenyl tert-butyl nitrone (PBN) and subjected to ESR spectroscopy analysis. As shown in Fig. 4B, the ESR spectrum of the mixture of EDY and PBN exhibits five lines, indicating the diradical intermediates generated from EDY reacted with two equivalents of PBN, and formed closely interacting nitroxide diradicals,55 corroborating the formation of radical species from the EDY compound.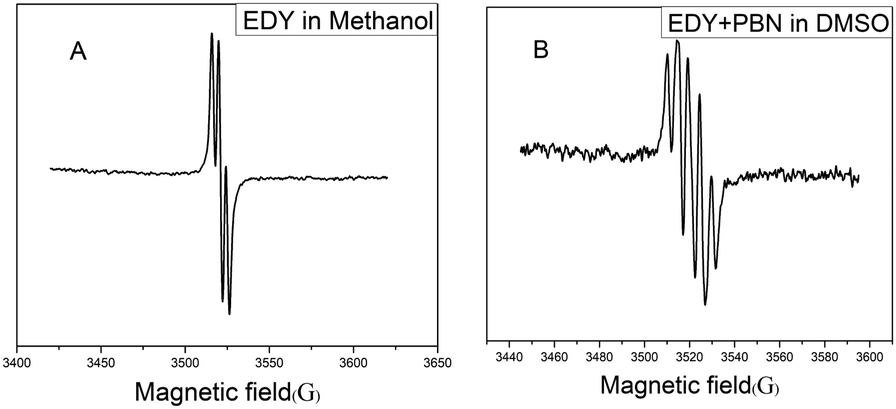 | ||
| Fig. 4 (A) ESR spectrum of EDY in methanol at 37 °C, (B) ESR spectrum of the PBN-adduct in DMSO at 37 °C. | ||
DNA cleavage experiments were further conducted to prove the ability of the diradical intermediates to abstract H atoms from the DNA's sugar-phosphate backbone and cause DNA cleavage from native supercoiled form (Form I) to circular relaxed form (Form II, single-strand cleavage).12 Briefly, the supercoiled plasmid PBR 322 DNA (isolated from E. coli) was incubated with EDY at 37 °C for 72 h and then subjected to agarose gel electrophoresis (Fig. 5). The supercoiled DNA is significantly converted to Form II in the presence of EDY (lane 3 and lane 4). In comparison, the control samples (free DNA or DNA with DMSO) were almost not affected. These results proved that the EDY compound is able to generate reactive radicals at physiological temperature to cause DNA cleavage. This characteristic enables EDY itself to act as a potential antitumor agent.
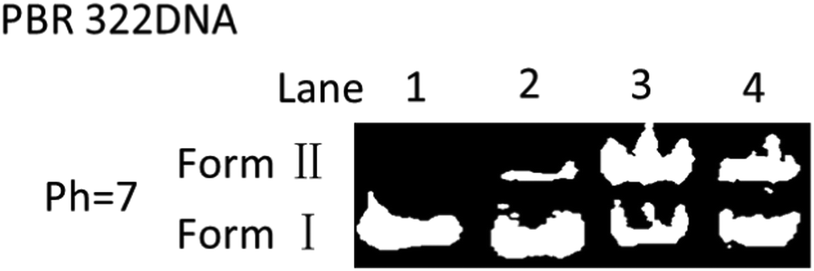 | ||
| Fig. 5 Cleavage of DNA by EDY at different concentrations for 72 h at 37 °C. Lane 1: PBR 322DNA (4 μL) in TE buffer (2 μL, pH 7.6); lane 2: PBR 322DNA (4 μL) + DMSO (2 μL); lane 3: PBR 322DNA (4 μL) + EDY (67 mM) in DMSO (2 μL); lane 4: PBR 322DNA (4 μL) + EDY (33 mM) in DMSO (2 μL). | ||
Cell internalization of EDY and Ir–EDY NPs
The fluorescence emission spectra of Ir, EDY and Ir–EDY NPs are shown in Fig. S10 (ESI†). Ir shows bright violet fluorescence in water (λmax: 420 nm) and EDY shows moderate blue fluorescence in methanol (λmax: 470 nm). After the formation of the conjugate, the fluorescence emission is dominated by the Ir part. The fluorescent property of these drugs can be used to directly study the cell internalization behavior through CLSM. In these experiments, HeLa cells were chosen as model cells. HeLa cells possess negligible fluorescence under the experimental conditions (Fig. 6a). The weak blue fluorescence in Fig. 6b shows that EDY have entered the nucleus region, which was stained by PI showing red fluorescence. These findings indicate that the EDY can enter the tumor cell nuclei efficiently through diffusion or permeation in a short time, proving the successful molecular design of EDY according to the Lipinski's Rule of Five (Ro5). As EDY was proved to be able to scissor DNA through the generation of radical species (see above), it is able to kill the tumor cells when it is internalized into the nuclei of tumor cells. Fig. 6c and d show the CLSM of HeLa cells incubated with Ir–EDY NPs for different time periods. After incubation for 6 h (Fig. 6c), the blue fluorescence of Ir–EDY NPs is mainly located in the cytoplasm, while the blue fluorescence becomes stronger and extends to the whole tumor cells (in both cytoplasm and nuclei) after 24 h incubation (Fig. 6d). These results demonstrate that Ir–EDY NPs are first internalized into the cells through endocytosis and enter the cytoplasm (Fig. 1d). Upon further incubation, the Ir–EDY NPs disassembled to release Ir and EDY, showing a stronger blue fluorescence and diffusion into the cell nucleus, where both Ir and EDY supress the copying of DNA through different mechanisms (Fig. 1-drug release), which laid a solid foundation for synergism as discussed below.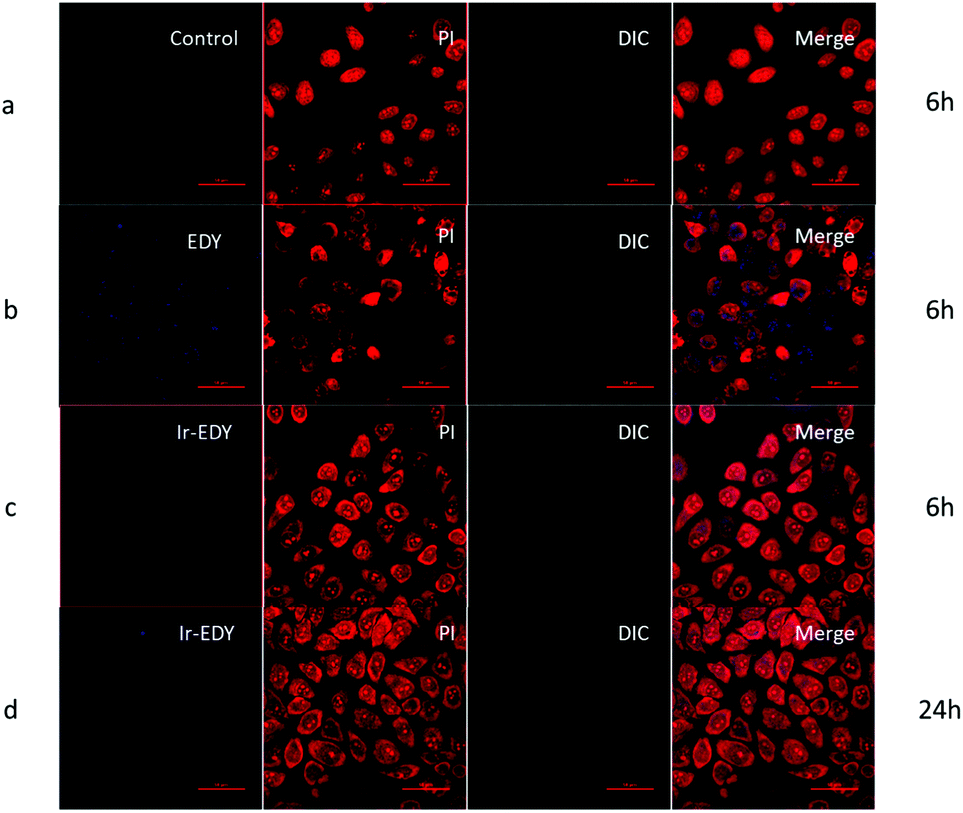 | ||
| Fig. 6 CLSM images of HeLa cells treated with EDY, or Ir–EDY for 6 h or 24 h. Control are HeLa cells incubated without any treatment to avoid the influence of the cells’ autofluorescence. Cell nuclei are stained with PI. The scale bar represents 50 μm. | ||
The cellular uptake of Ir–EDY NPs was further quantified using flow cytometric analysis. Fig. 7 shows that the fluorescence intensity of HeLa cells increases with the incubation time, which can be attributed to the continuous cellular uptake of Ir–EDY nanoparticles.
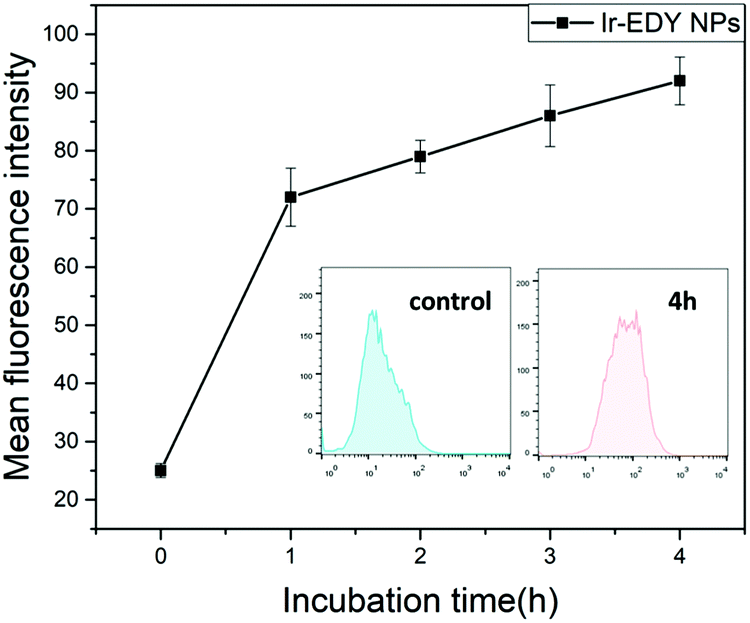 | ||
| Fig. 7 The relative geometrical mean fluorescence intensities of HeLa cells incubated with Ir–EDY nanoparticles for different time periods and then analyzed by flow cytometry. Inset: Representative flow cytometry histogram profiles of HeLa cells cultured with Ir–EDY nanoparticles for 4 h, the untreated cells are used as control. | ||
Cytotoxicity and synergism assay
The ability of small molecular drugs such as EDY, Ir, their mixture, and Ir–EDY NPs to inhibit the proliferation of tumor cells was evaluated by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay using HeLa cells as the model, and the cells treated with 0.1% DMSO were used as the control. From Fig. 8A, it can be seen that EDY inhibits the proliferation of HeLa cells efficiently after 48 h incubation. The IC50 concentration of EDY is comparable with that of Ir and many other clinically used antitumor agents such as doxorubicin and cisplatin,56,57 confirming the high cell inhibition ability of EDY. Ir–EDY NPs exhibit significant inhibition to the cell proliferation (IC50 = 12 μM) compared to that of other drug formulations (IC50, Ir = 15 μM, EDY = 19 μM, Ir + EDY mixture = 16 μM), the IC50 values calculated using Graphpad Prism also support this conclusion (Table S12, ESI†). It is interesting to find that the therapeutic efficacy of Ir–EDY is dependent on the drug concentration. When the concentration of Ir–EDY NPs is lower than CAC (40 μM), the cytotoxicity of Ir–EDY NPs is similar to or worse than Ir or Ir + EDY mixture, however when the concentration increases to around the CAC value, the cytotoxicity of the Ir–EDY NPs is better than other drug formulations. This phenomenon indicates that there might be a synergistic effects between Ir and EDY released from the Ir–EDY NPs.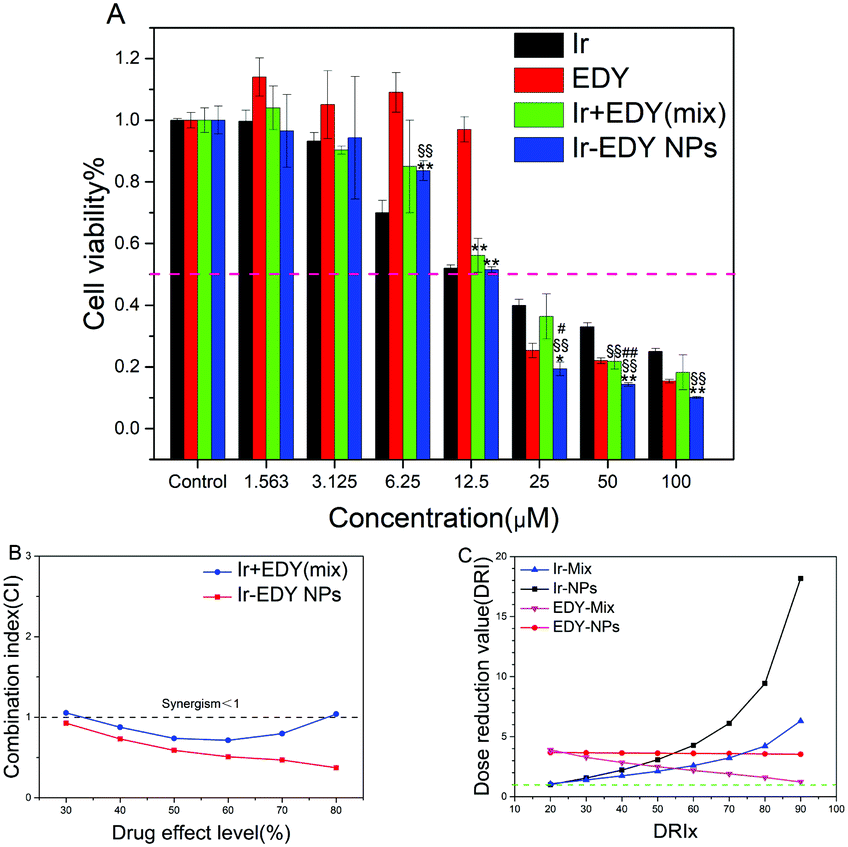 | ||
| Fig. 8 (A) In vitro cytotoxicity of EDY, Ir, Ir/EDY mixture, and Ir–EDY nanoparticles to HeLa cells determined by the MTT assay. The data are presented as average ± standard error (n = 3). A significant decrease in the cell viability compared with the Ir-treated cells is denoted by “§” (P < 0.05) and “§§” (P < 0.01), a significant decrease compared with EDY-treated cells is denoted by “*” (P < 0.05) and “**” (P < 0.01), and a significant decrease compared with Ir + EDY (Mix)-treated cells is denoted by “#” (P < 0.05) and “##” (P < 0.01). (B) Combination index (CI) plots for Ir/EDY mixture and Ir–EDY nanoparticles against HeLa cells. (C) The DRI values of Ir + EDY mixture and Ir–EDY nanoparticles. | ||
To investigate the synergistic effects proposed above, combination index (CI) values are calculated with computer simulation based on Chou-Talalay equation,58 CI = D1/Dm1 + D2/Dm2, where D1 and D2 are the concentrations of drug 1 and drug 2 that are used in combination to produce some specified effect (e.g., 50% inhibition of cell viability), while Dm1 and Dm2 are the doses of single drugs to achieve the same effect. CI values are typically plotted against the drug effect level (ICx values). The values of CI <1, =1, or >1 correspond to the effects of synergism, additivity, and antagonism respectively.59 As shown in Fig. 8B, the CI values of Ir–EDY NPs are less than 1 in a wide range of drug effect levels (30–80%), while the CI values of Ir + EDY mixture are near or above the line of CI = 1. In addition, the CI values of Ir–EDY NPs are much lower than those of the Ir + EDY mixture at each corresponding ICx value, proving that the synergistic effect of Ir–EDY NPs is much better than the simple mixture of Ir and EDY. The possible reason could be that the Ir–EDY NPs enter the cells through endocytosis, and the Ir and EDY released from the Ir–EDY NPs can keep the 1:1 molar ratio with a better synergistic effect, while the mixture of Ir and EDY can hardly reach this effect through diffusion.
Dose reduction value (DRI) is another key parameter to evaluate the synergistic effect.59 The DRI represents the dosage reduction of a single drug used alone to maintain its anticancer efficacy and is plotted against the drug effect level (DRIx values). DRI values are calculated by computer simulation software calcuSyn. As shown in Fig. 8C, the DRI values of both Ir + EDY mixture and Ir–EDY NPs are above the line of DRI = 1, meanwhile the DRI values of both Ir and EDY in Ir–EDY NPs are much higher than those in Ir + EDY mixture. All the findings indicate that the design of Ir–EDY NPs could largely reduce the dosage of free drugs while maintaining a more effective anticancer efficacy than other drug formulations.
Cell apoptosis assay study
Most small molecular chemotherapy drugs kill cancer cells by cell apoptosis.60 To confirm whether cell apoptosis is the main mechanism for Ir–EDY NPs to induce the death of cancer cells, we employed the FITC-Annexin V/PI apoptosis detection kit to qualify the cell apoptosis ratio. HeLa cells treated with Ir, EDY and Ir–EDY NPs at a concentration of 25 μM were applied, and the cells without any treatment were considered as the control. Flow cytometry graph shows that the ratio of apoptosis cells is 22.4% and 33.1% induced by Ir or EDY, the result increases to 63.6% when incubated with Ir–EDY nanoparticles, and the necrotic cells were not significant in all groups (Fig. 9). When compared with other formulations, the Ir–EDY nanoparticles promote a much higher apoptotic rate of HeLa cells with the same dose. The results demonstrate that Ir–EDY NPs can induce tumor cell death through cell apoptosis efficiently compared with other formulations.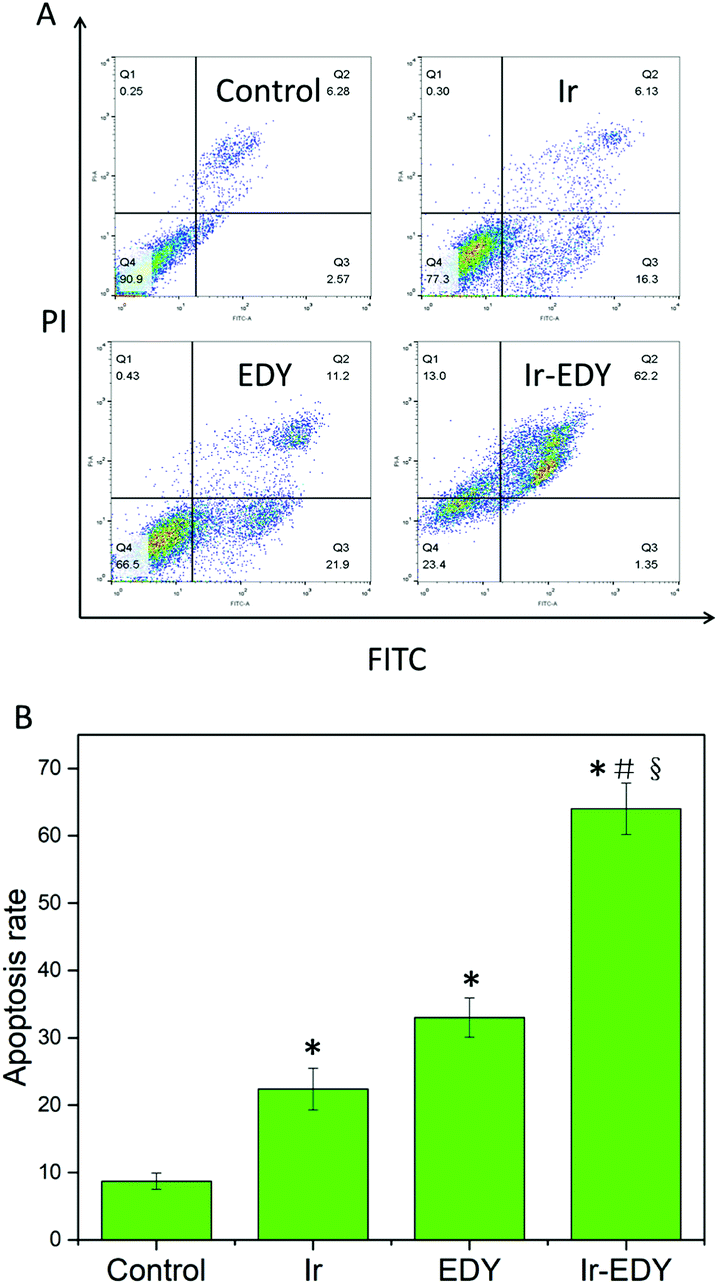 | ||
| Fig. 9 (A) Flow cytometry analysis for apoptosis of HeLa cells induced by Ir, EDY and Ir–EDY nanoparticles at the concentration of 25 μM for 24 h. Lower left, living cells; lower right, early apoptotic cells; upper right, late apoptotic cells; upper left, necrotic cells. Inserted numbers in the profiles indicate the percentage of the cells present in this area. (B) Ratio of apoptotic HeLa cells based on the results of flow cytometry measurements. Values represent mean ± SD (n = 3). A significant increase in the apoptosis rate compared with the control is denoted with “*” (P < 0.01), a significant increase compared with Ir-treated cells is denoted with “#” (P < 0.01), and a significant increase compared with EDY-treated cells is denoted with “§” (P < 0.01). | ||
Conclusions
In summary, we have synthesized a lipophilic enediyne, which undergoes Bergman cyclization or Bergman-like reaction to generate active diradicals leading to DNA cleavage. The EDY itself is able to enter the cell nuclei and exhibits cytotoxicity to tumor cells. Based on the lipophilicity of EDY, an amphiphilic drug couple Ir–EDY was prepared via a simple ester linkage for highly effective cancer combination chemotherapy. The amphiphilic Ir–EDY conjugate self-assembles into nanoparticles in water, and the Ir–EDY NPs can be easily self-delivered and accumulate in tumor tissues due to the EPR effect. After entering the tumor cell through endocytosis, the ester linkage in Ir–EDY NPs would break due to the acidic microenvironment and release the Ir and EDY respectively. The released free Ir and EDY further damage tumor cells synergistically through different mechanisms, and finally induce tumor cell death through the cell apoptosis pathway efficiently. The EDY based drug self-delivery nanoparticles are suggested to have potential application for chemotherapy in future cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (21474027, 21503078, 21871080), the Fundamental Research Funds for the Central Universities (22221818014), and Shanghai Leading Academic Discipline Project (B502) is greatly acknowledged. AH thanks the “Eastern Scholar Professorship” support from Shanghai Local Government. JL thanks Mr Yuequn Wu and Prof. Runhui Liu for their kindly help in cell experiments.Notes and references
- R. Siegel, J. Ma, Z. Zou and A. Jemal, Ca-Cancer J. Clin., 2014, 64, 9–29 CrossRef PubMed.
- L. A. Torre, F. Bray, R. L. Siegel, J. Ferlay, J. Lortet-Tieulent and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 87–108 CrossRef.
- V. P. Chauhan and R. K. Jain, Nat. Mater., 2013, 12, 958 CrossRef CAS.
- Y. Cao, R. A. DePinho, M. Ernst and K. Vousden, Nat. Rev. Cancer, 2011, 11, 749–754 CrossRef CAS.
- M. D. Lee, J. K. Manning, D. R. Williams, N. A. Kuck, R. T. Testa and D. B. Borders, J. Antibiot., 1989, 42, 1070–1087 CrossRef CAS.
- M. Konishi, H. Ohkuma, K. Matsumoto, K. Saitoh, T. Miyaki, T. Oki and H. Kawaguchi, J. Antibiot., 1991, 44, 1300–1305 CrossRef CAS.
- N. Ishida, K. Miyazaki, K. Kumagai and M. Rikimaru, J. Antibiot., Ser. A, 1965, 18, 68–76 CAS.
- J. Golik, J. Clardy, G. Dubay, G. Groenewold, H. Kawaguchi, M. Konishi, B. Krishnan, H. Ohkuma, K. Saitoh and T. W. Doyle, J. Am. Chem. Soc., 1987, 109, 3461–3462 CrossRef CAS.
- K. C. Nicolaou, J. S. Chen, H. J. Zhang and A. Montero, Angew. Chem., Int. Ed., 2008, 47, 185–189 CrossRef CAS.
- J. E. Leet, D. R. Schroeder, S. J. Hofstead, J. Golik, K. L. Colson, S. Huang, S. E. Klohr, T. W. Doyle and J. A. Matson, J. Am. Chem. Soc., 1992, 114, 7946–7948 CrossRef CAS.
- M. Hirama, K. Akiyama, T. Tanaka, T. Noda, K.-I. Iida, I. Sato, R. Hanaishi, S. Fukuda-Ishisaka, M. Ishiguro, T. Otani and J. E. Leet, J. Am. Chem. Soc., 2000, 122, 720–721 CrossRef CAS.
- M. Kar and A. Basak, Chem. Rev., 2007, 107, 2861–2890 CrossRef CAS PubMed.
- N. Zein, A. M. Sinha, W. J. McGahren and G. A. Ellestad, Science, 1988, 240, 1198–1201 CrossRef CAS.
- A. Thomas, B. A. Teicher and R. Hassan, Lancet Oncol., 2016, 17, e254–e262 CrossRef CAS.
- P. F. Bross, J. Beitz, G. Chen, X. H. Chen, E. Duffy, L. Kieffer, S. Roy, R. Sridhara, A. Rahman, G. Williams and R. Pazdur, Clin. Cancer Res., 2001, 7, 1490–1496 CAS.
- R. Romeo, S. V. Giofre and M. A. Chiacchio, Curr. Med. Chem., 2017, 24, 3433–3484 CrossRef CAS.
- K. M. Huttunen, H. Raunio and J. Rautio, Pharmacol. Rev., 2011, 63, 750–771 CrossRef CAS.
- L. Gu, A. Faig, D. Abdelhamid and K. Uhrich, Acc. Chem. Res., 2014, 47, 2867–2877 CrossRef CAS.
- Y. Li, D. Maciel, J. Rodrigues, X. Shi and H. Tomás, Chem. Rev., 2015, 115, 8564–8608 CrossRef CAS.
- K. Miyata, R. J. Christie and K. Kataoka, React. Funct. Polym., 2011, 71, 227–234 CrossRef CAS.
- H. Wei, R.-X. Zhuo and X.-Z. Zhang, Prog. Polym. Sci., 2013, 38, 503–535 CrossRef CAS.
- D. V. Volodkin, A. G. Skirtach and H. Möhwald, Angew. Chem., Int. Ed., 2009, 48, 1807–1809 CrossRef CAS.
- J. Song, J. Zhou and H. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS.
- A. MaHam, Z. Tang, H. Wu, J. Wang and Y. Lin, Small, 2009, 5, 1706–1721 CrossRef CAS.
- K. Fan, C. Cao, Y. Pan, D. Lu, D. Yang, J. Feng, L. Song, M. Liang and X. Yan, Nat. Nanotechnol., 2012, 7, 459 CrossRef CAS.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS.
- J. Zhang, Z.-F. Yuan, Y. Wang, W.-H. Chen, G.-F. Luo, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, J. Am. Chem. Soc., 2013, 135, 5068–5073 CrossRef CAS.
- H. Maeda, Adv. Drug Delivery Rev., 2015, 91, 3–6 CrossRef CAS.
- K. Cai, X. He, Z. Song, Q. Yin, Y. Zhang, F. M. Uckun, C. Jiang and J. Cheng, J. Am. Chem. Soc., 2015, 137, 3458–3461 CrossRef CAS.
- J. Zhang, S. Li, F.-F. An, J. Liu, S. Jin, J.-C. Zhang, P. C. Wang, X. Zhang, C.-S. Lee and X.-J. Liang, Nanoscale, 2015, 7, 13503–13510 RSC.
- D. Yu, P. Peng, S. S. Dharap, Y. Wang, M. Mehlig, P. Chandna, H. Zhao, D. Filpula, K. Yang, V. Borowski, G. Borchard, Z. Zhang and T. Minko, J. Controlled Release, 2005, 110, 90–102 CrossRef CAS.
- S. M. Moghimi, A. C. Hunter and J. C. Murray, FASEB J., 2005, 19, 311–330 CrossRef CAS.
- S.-Y. Qin, A.-Q. Zhang, S.-X. Cheng, L. Rong and X.-Z. Zhang, Biomaterials, 2017, 112, 234–247 CrossRef CAS.
- Y. Wang, P. Huang, M. Hu, W. Huang, X. Zhu and D. Yan, Bioconjugate Chem., 2016, 27, 2722–2733 CrossRef CAS PubMed.
- M. Hu, P. Huang, Y. Wang, Y. Su, L. Zhou, X. Zhu and D. Yan, Bioconjugate Chem., 2015, 26, 2497–2506 CrossRef CAS.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef CAS.
- S. Sun, C. Zhu, D. Song, F. Li and A. Hu, Polym. Chem., 2014, 5, 1241–1247 RSC.
- D. Song, S. Sun, Y. Tian, S. Huang, Y. Ding, Y. Yuan and A. Hu, J. Mater. Chem. B, 2015, 3, 3195–3200 RSC.
- I. Husain, J. L. Mohler, H. F. Seigler and J. M. Besterman, Cancer Res., 1994, 54, 539–546 CAS.
- Y. Pommier, Nat. Rev. Cancer, 2006, 6, 789 CrossRef CAS.
- H. J. Gao, W. D. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–9474 CrossRef CAS.
- L. A. Bareford and P. W. Swaan, Adv. Drug Delivery Rev., 2007, 59, 748–758 CrossRef CAS.
- M. Dubernet, V. Caubert, J. Guillard and M.-C. Viaud-Massuard, Tetrahedron, 2005, 61, 4585–4593 CrossRef CAS.
- A. Souffrin, C. Croix and M.-C. Viaud-Massuard, Eur. J. Org. Chem., 2012, 2499–2502 CrossRef CAS.
- S. Huang, J. Li, B. Li and A. Hu, J. East China Univ. Sci. Technol., 2017, 43, 1–7 CAS.
- Y. J. Lu, S. H. Yang, C. M. Chien, Y. H. Lin, X. W. Hu, Z. Z. Wu, M. J. Wu and S. R. Lin, Toxicol. In Vitro, 2007, 21, 90–98 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS.
- Y. Wang, B. Li, Y. Zhou, Z. Lu and D. Yan, Soft Matter, 2013, 9, 3293–3304 RSC.
- Y. Shen, E. Jin, B. Zhang, C. J. Murphy, M. Sui, J. Zhao, J. Wang, J. Tang, M. Fan, E. Van Kirk and W. J. Murdoch, J. Am. Chem. Soc., 2010, 132, 4259–4265 CrossRef CAS.
- T. G. W. Edwardson, K. M. M. Carneiro, C. K. McLaughlin, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2013, 5, 868 CrossRef CAS.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- C. Shi, D. Guo, K. Xiao, X. Wang, L. Wang and J. Luo, Nat. Commun., 2015, 6, 7449 CrossRef.
- N. Mifsud, V. Mellon, K. P. U. Perera, D. W. Smith and L. Echegoyen, J. Org. Chem., 2004, 69, 6124–6127 CrossRef CAS.
- T. Usuki, M. Kawai, K. Nakanishi and G. A. Ellestad, Chem. Commun., 2010, 46, 737–739 RSC.
- G. R. Luckhurst, Mol. Phys., 1966, 10, 543–550 CrossRef CAS.
- S. Dhar and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 22199–22204 CrossRef CAS PubMed.
- H. Y. He, J. N. Zhao, R. Jia, Y. L. Zhao, S. Y. Yang, L. T. Yu and L. Yang, Molecules, 2011, 16, 10685–10694 CrossRef.
- T.-C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS.
- T.-C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- M. O. Hengartner, Nature, 2000, 407, 770 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c8tb02367k |
| This journal is © The Royal Society of Chemistry 2019 |