Hyaluronic acid/PEGylated amphiphilic nanoparticles for pursuit of selective intracellular doxorubicin release†
Received
8th September 2018
, Accepted 18th November 2018
First published on 20th November 2018
Abstract
Polyethylene glycol (PEG)-lyted cationic amphiphilic copolymers were employed as complexing agents with biocompatible anionic hyaluronic acid (HA) for the controlled release of doxorubicin (DOX). The overexpressed receptors to HA in a variety of cancerous cells enable preferential endocytosis of the HA-functionalized nanoparticles. Moreover, introduction of HA is supposed to diminish the unfavorable non-specific reactions in the biological milieu. Particularly, the drastic positive charge was validated post-endocytosis as a consequence of our strategic molecular design for the promotion of positive charges of cationic components. This deshielding effect of the anionic hyaluronic acid by endogenous hyaluronidase in endosomes and demotion of PEG at the endosome acid microenvironment consequently results in the structural rearrangement and favorable reaction of the resulting positive-charged structure with the intracellular species and structures, ultimately giving rise to liberation of the doxorubicin for the subsequent molecular pharmaceutic consequences. Simultaneously, the system containing quaternary ammonium salt and hydrophobic n-octyl acrylate (OA) possesses considerable antibacterial ability to alleviate anti-cancer drugs resistance. This delivery system is intended to overcome the intratumor bacteria-induced tumor resistance.
1. Introduction
The development of nanotechnology spurred the development of nanoparticle-based delivery strategies, termed as nanomedicine,1,2 to be capable of affording therapeutic efficacy to the targeted pathological site.3 Particularly, the chemotherapeutic drugs, once encapsulated into appropriate targeted delivery vehicles, are ideal to accomplish preferential transport to the targeted cells and facilely liberate the active chemotherapeutic drugs in the desirable subcellular compartment.4,5 To achieve this goal, in addition to targeted functionalities to the pathological cells,6,7 the reversible stabilities in the extracellular milieu and intracellular compartment are required with the goal of minimized pre-mature extracellular release of the encapsulated drugs and facilitated intracellular drug release.8 To fulfil this objective, a number of stimuli-responsive facilities were devised into the delivery nanoparticles to achieve accelerated drug release in response to endogenous stimuli (i.e., pH,9,10 glutathione,9 hypoxia,11 enzymes12 and other tumor markers) and exogenous stimuli (i.e., temperature,13 electromagnetic radiation and particle radiation).14–17 Nevertheless, most of the aforementioned strategies require sophisticated chemistry synthesis and exhaustive screening (or optimization) to attain adequate distinguishing capacity between the intracellular and extracellular milieus.
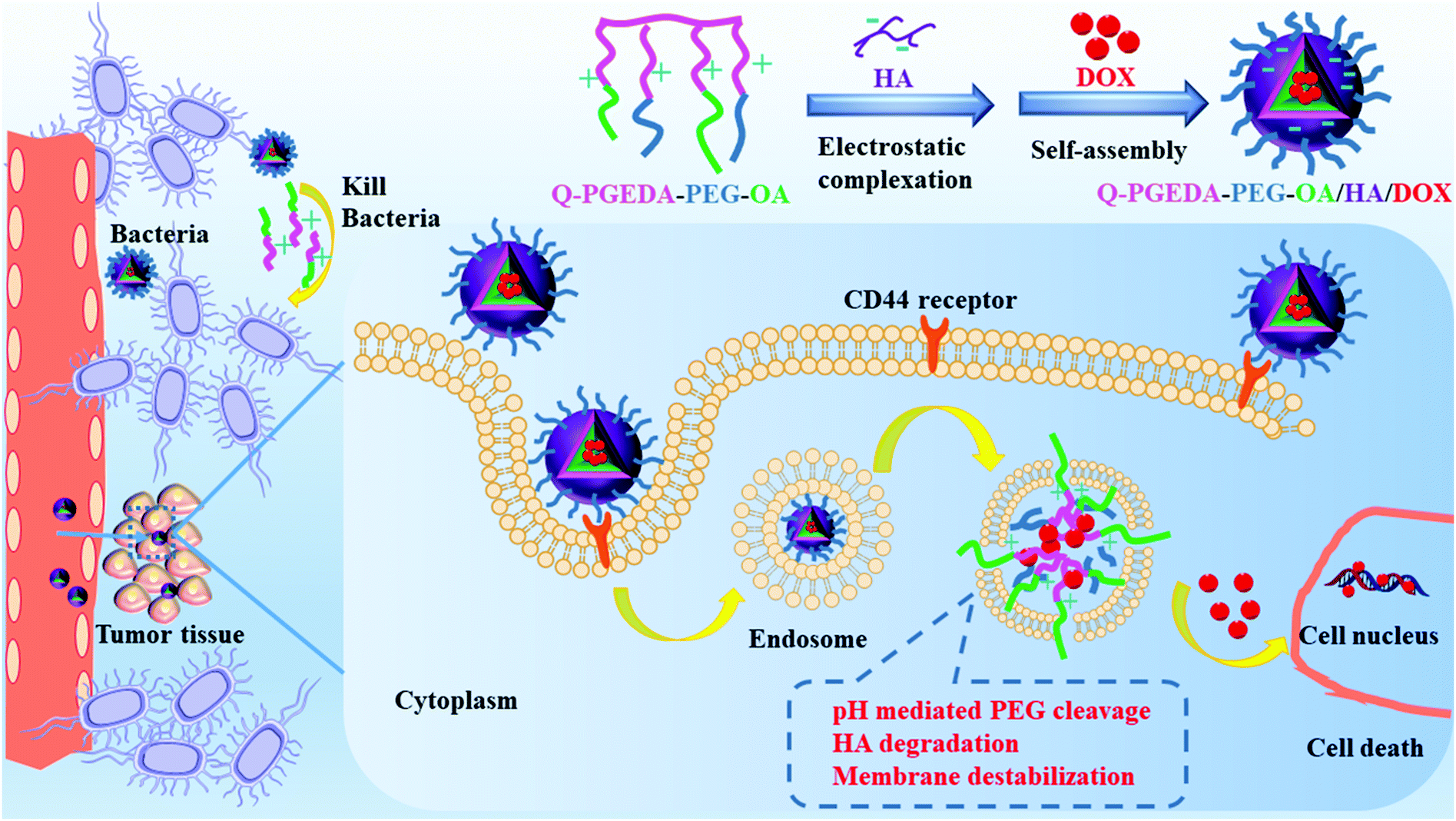
In nanoplatforms, polyethylene glycol (PEG)18 was used as the hydrophilic outer shell segment to endow the nanocarriers with in vivo longevity and specific capability of extravasation through the endothelium of inflammatory tissues via inducing steric repulsion of blood opsonins molecules, such as antibodies, that enhance phagocytosis.17 However, the neutral PEGylation will decrease the cell internalization and endosome escape potency. Among various PEG detachment strategies, introduction of a pH-responsive cleavable Schiff-base linkage between the positively charged polymer and PEG is a feasible route.19,20 In this study, we attempted to design a facile reversible electrostatic adsorption strategy in acquiring reversible stabilities between the intracellular and extracellular milieus, consequently leading to the liberation of the cytotoxic drugs selectively in the cell interior (Scheme 1). Of note, a cationic amphiphilic copolymer, consisting of PEGylated cationic quaternary amine groups21 and hydrophobic n-octyl acrylate segments, was employed to complex with hyaluronic acid (HA) as a drug delivery vehicle of DOX.
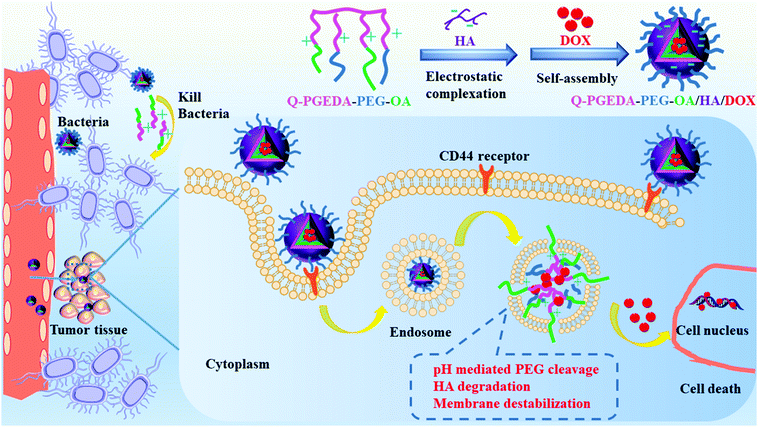 |
| | Scheme 1 Illustration of the formation and elevated antitumor and antimicrobial mechanism of Q-PGEDA-PEG-OA/HA/DOX nanocarriers. A PEGylated cationic amphiphilic copolymer was complexed with HA for controlled release of DOX via the electrostatic self-assembly. HA will bind with the CD44 receptor for increasing cellular internalization. Owing to the tumor-microenvironment and nanocarriers mediation, PEG was detached from the nanoparticles, accompanied with HA degradation and endosome membrane destabilization, which accelerated DOX release. Moreover, quaternary ammonium salt and hydrophobic OA of the polymer-killed bacteria in the tumor tissue to alleviate anti-cancer drugs resistance for preferable tumor inhibition. | |
Herein, anionic HA is intended to avoid the potential premature drug release due to the reaction of the positive-charged components and lipophilic components of the amphiphilic copolymer in the biological milieu.22 Note that diverse cancerous cell lines were reported to overexpress hyaluronic acid receptors.23 Hence, it is likely that preferential endocytosis of the HA-functionalized nanoparticles will occur.24 Furthermore, following cell endocytosis, the increase of positive charges was hypothesized to be present post-endocytosis considering our PEG detachment strategies, the rise in the number of positive charges of cationic components and the drop in the number of negative charges of anionic hyaluronic acid. Consequently, structural rearrangement to formulate a positive-charged structure would be expected for active reaction with the intracellular species and structures, ultimately accounting for liberation of the doxorubicin for the subsequent molecular pharmaceutic consequences.25 Hence, we believe the facile reversible adsorption strategy could serve as an important template to resolve non-specific premature chemotherapeutic drug release, which consequently could be developed for translation into a variety of lipophilic pharmaceutic substances.
There is growing evidence that microbes can influence the effectiveness of anti-cancer drugs. The nanoparticles are potentially useful to reduce cancer drugs resistance through bacterial inhibition. Bacterial inhibition is reported to enhance the antineoplastic activity of cancer drugs.26 Moreover, Yu et al.27 reported that Fusobacterium(F.) nucleatum was abundant in colorectal cancer tissues in patients with recurrence post-chemotherapy, accounting for promoted colorectal cancer resistance to chemotherapy. There is an urgent need to develop a new agent with both anticancer and antimicrobial activity to combat microbes in tumor tissue to alleviate cancer drugs resistance. We thus introduced quaternary ammonium salt and hydrophobic n-octyl acrylate (OA) into the polymer, which probably rendered the polymer with considerable antibacterial activity based on previous reports.28,29
2. Experimental section
2.1 Preparation of poly(ethylene glycol)ylated ethylenediamine-grafted polyglycerol methacrylate (PGEDA-PEG)
The synthesis of ethylenediamine-modified polyglycerol methacrylate (PGEDA) is reported in our previous published works30,31 (see ESI† for details). PGEDA-PEG was synthesized according to a similar method reported previously.20 In this procedure, the PGEDA (100 mg, 0.8 mmol) was suspended into 10 mL of pre-dried dimethyl sulfoxide (DMSO). Furthermore, the as-prepared methoxy poly(ethylene glycol) benzaldehyde (mPEG-CHO) (60 mg, 0.03 mol; its synthesis is briefly described in the ESI†) was slowly added into the above solution. Then, the mixture solution was stirred at 50 °C for 48 h. Subsequently, the reaction solution was transferred to dialysis (cut-off molecular weight 7 kDa), and dialyzed against absolute ethanol for 24 h, as well as against phosphate buffer solution (PBS) (pH = 7.4, 0.01 M) for 24 h. At last, the final copolymer product was obtained by freeze-drying (yield: 42.1%). 1H-NMR (400 MHz, DMSO) δ 7.91 (s, –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), 7.19 (s, –Ph), 6.64 (s, –Ph), 4.02 (m, –COO–C
N–), 7.19 (s, –Ph), 6.64 (s, –Ph), 4.02 (m, –COO–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–C(CH2)–OH), 3.62 (d, PEG peaks), 3.00 (m, NH2–C2–C2–NH–C2–), 1.92 (s, –C2–C(CH3)–), 0.93 (s, –CH2–C(C3)–).
2–C(CH2)–OH), 3.62 (d, PEG peaks), 3.00 (m, NH2–C2–C2–NH–C2–), 1.92 (s, –C2–C(CH3)–), 0.93 (s, –CH2–C(C3)–).
2.2 Preparation of n-octyl acrylate-grafted PGEDA-PEG quaternary ammonium salt (Q-PGEDA-PEG-OA)
First, PGEDA-PEG (50 mg) was dissolved in 20 mL of H2O/DMSO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture at room temperature, and then n-octyl acrylate (110 mg, 0.6 mmol) was added dropwise to the reaction solution and stirred for 48 h. Subsequently, the reaction solution was warmed up to 45 °C for another 24 h. Finally, the reaction was conducted overnight in the dark at the same temperature after the dropwise addition of the iodomethane (169 mg, 1.2 mmol). Consequently, the resulting product was precipitated in diethyl ether twice and dialyzed (cut-off molecular weight 7 kDa) against PBS (pH = 7.4, 0.01 M) for 24 h and 72 h, respectively. Finally, the product was lyophilized to obtain Q-PGEDA-PEG-OA (yields: 85%).
:1) mixture at room temperature, and then n-octyl acrylate (110 mg, 0.6 mmol) was added dropwise to the reaction solution and stirred for 48 h. Subsequently, the reaction solution was warmed up to 45 °C for another 24 h. Finally, the reaction was conducted overnight in the dark at the same temperature after the dropwise addition of the iodomethane (169 mg, 1.2 mmol). Consequently, the resulting product was precipitated in diethyl ether twice and dialyzed (cut-off molecular weight 7 kDa) against PBS (pH = 7.4, 0.01 M) for 24 h and 72 h, respectively. Finally, the product was lyophilized to obtain Q-PGEDA-PEG-OA (yields: 85%).
2.3 pH titration
Hyaluronic acid (40 mg) was first dissolved in deionized water (20 mL) and the pH value was adjusted to 12 with 1 M NaOH solution containing 150 mM NaCl. A pH meter (Mettler Toledo, Switzerland) with a microelectrode was used to measure the pH values. In this experiment, pH titration was carried out by adding small volumes (0.03 mL in increments) of 0.5 M HCl solution under stirring.32 In a similar manner, titration experiments of Q-PGEDA-PEG-OA (1 mg mL−1) were completed by adjusting the pH value to 2 with 0.5 M HCl solution initially. The pH change in the range of 2–12 was monitored as a function of total added volume of HCl or NaOH, respectively. Then, the relationship between pH and the protonation degree (α) of hyaluronic acid and Q-PGEDA-PEG-OA was calculated from the obtained titration curves, respectively.33
2.4 Statistics analysis
Significant differences in cell viability between any two groups were evaluated using a Student's t-test.
3. Results and discussion
3.1 Polymer synthesis and characterization
The amphiphilic copolymer of Q-PGEDA-PEG-OA was synthesized according to Fig. S1 (ESI†). The yielded Q-PGEDA-PEG-OA copolymer was characterized by 1H-NMR measurement. As shown in Fig. S2 (ESI†), concurrent peaks could be assigned to the chemical structure of Q-PGEDA-PEG-OA with chemical shifts at 8.41 ppm of (–HCN–) linkages and alkyl peaks of 1.25 ppm.
3.2 Particle size, zeta potential and morphology measurements of complexes
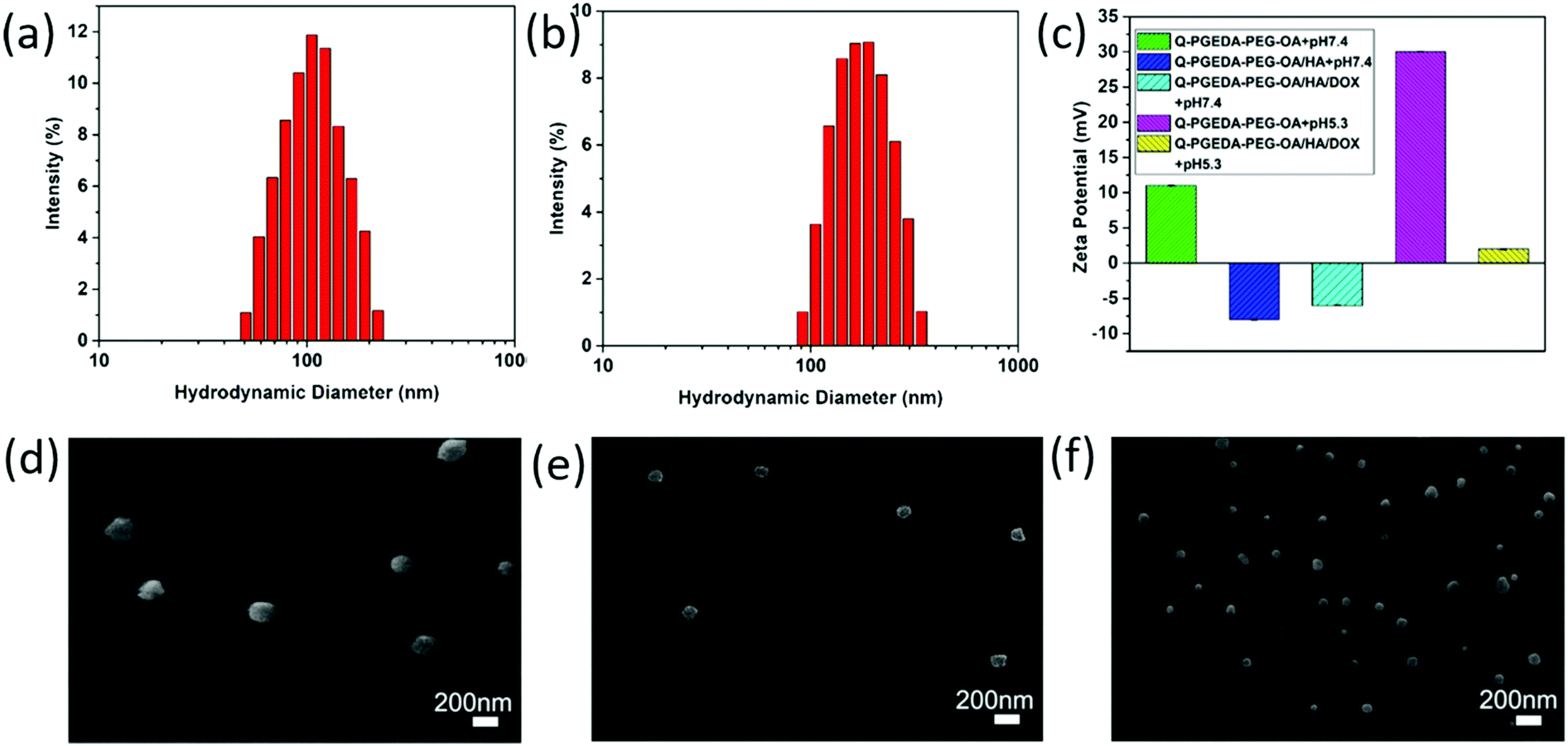
The zeta potential value of Q-PGEDA-PEG-OA is approximately +10 mV (Fig. 1c). This positive net charge is postulated to induce non-specific reactions in the biological milieu, e.g. diverse biological species and biological structures are identified with having a negative charge. For instance, the primary obstacles for nanomedicine, namely occurrence of protein adsorption, in particular opsonin adsorption, can induce reticuloendothelial system (RES) recognition and clearance.34,35 The relevant proteins are determined to be negatively charged with an intrinsic isoelectric point (pI) (albumin pI 4.9, fibrinogen pI 5.8, globulin pI 5.8). The fundamental factors that are capable of inducing RES capture (IGG pI 6.5–7.5, fibrinonectin pI 5.5–6.0, complementary factor C3b pI 5.7) were also verified to be negatively charged. Moreover, the cytoplasm membrane was determined to be uniformly negatively charged due to the presence of chondroitin sulfate,36 hyaluronic acid, heparan sulfate37 and so on. Therefore, we anticipate the reactions of the positive-charged nanoparticles in the biological milieu, and the structural disassembly to liberate the encapsulated drugs. To address this drawback, we attempted to introduce biocompatible anionic hyaluronic acid onto the surface of Q-PGEDA-PEG-OA. Note that hyaluronic acid is an anionic, nonsulfated glycosaminogly that can be distributed widely throughout connective, epithelial and neural tissues,38 which have been acknowledged as potential biocompatible materials in biomedical applications, e.g. to our best knowledge, hyaluronic acid has been validated as the only biocompatible organic material in biomedical implants, including dermal filler in cosmetic surgery. In this regard, the amphiphilic Q-PGEDA-PEG-OA copolymer was utilized to mix with a biocompatible hyaluronic acid in specific ratios. DOX was loaded into Q-PGEDA-PEG-OA/HA complexes via electrostatic interaction and intermolecular hydrophobic stacking. The average size of Q-PGEDA-PEG-OA was 153 nm with a high polydispersity index (PDI) of 1.0 (data not shown). Upon complexing with HA, the optimal size was obtained when the molar ratio of Q-PGEDA-PEG-OA and HA is 1:0.5 (Table S1, ESI†) with an average diameter of approximately 103 nm and polydispersity index (PDI) of 0.163. The final complexes (Q-PGEDA-PEG-OA/HA/DOX) possessed average diameters of approximately 201 nm and PDI of 0.182, suggesting the successful self-assembled formation (Fig. 1a and b). Potentially, the hydrophobic components of OA in Q-PGEDA-PEG-OA and the hydrophobic anthracycline residues of DOX could aggregate based on hydrophobic interactions into the internal compartment, in which anionic hyaluronic acid and cationic doxorubicin would be bound via electrostatic interactions, and the rest of the hydrophilic components including PEG and quaternary amine residues would be present as the surrounding external layer. Indeed, the zeta potential measurement confirmed our speculated architecture, wherein the positive net charge of Q-PGEDA-PEG-OA (approximately +10 mV) was reversed to negative charge on account of the addition of abundant anionic hyaluronic acid (Q-PGEDA-PEG-OA/HA, approximately −8 mV). The surface had a mild negative charge (approximately −4 mV) for DOX-loaded formulation (Fig. 1c). These results approved the facile attachment of hyaluronic acid and doxorubicin onto Q-PGEDA-PEG-OA. As shown in Fig. 1c, the zeta potential of Q-PGEDA-PEG-OA/HA/DOX was negative. While HA was the only negatively charged component of nanoparticles, which suggested that HA mainly existed on the surface of nanoparticle, which could supply the target effect of the nanoparticles. The self-assembled formation was transferred for scanning electron microscope (SEM) measurements. Consistent with our previous results, SEM images shows a spherical structure with an increased diameter of 175 ± 15 nm for Q-PGEDA-PEG-OA/HA/DOX (Fig. 1f) as compared to 100 ± 50 nm for Q-PGEDA-PEG-OA (Fig. 1d) and 96 ± 5 nm for Q-PGEDA-PEG-OA/HA (Fig. 1e), again verifying the self-assembled behaviors of hyaluronic acid and doxorubicin with the Q-PGEDA-PEG-OA. Specifically, the zeta potential of Q-PGEDA-PEG-OA was increased to +30 mV (Fig. 1c) upon addition of PBS (pH 5.3), which confirmed the pH-responsive detachment of mPEG-CHO.
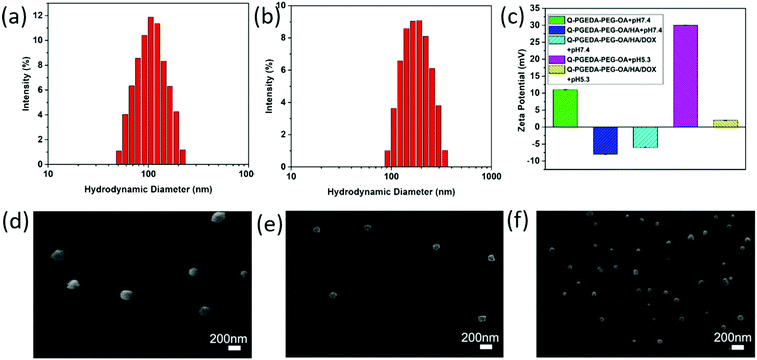 |
| | Fig. 1 Characterization of Q-PGEDA-PEG-OA, Q-PGEDA-PEG-OA/HA and Q-PGEDA-PEG-OA/HA/DOX. Particle size distributions of Q-PGEDA-PEG-OA/HA (a) and Q-PGEDA-PEG-OA/HA/DOX (b). (c) Zeta potentials of the copolymer and complexes at pH 7.4 or 5.3. SEM images of Q-PGEDA-PEG-OA (d), Q-PGEDA-PEG-OA/HA (e) and Q-PGEDA-PEG-OA/HA/DOX (f) nanoparticles. | |
3.3 Protonation degree (α) of Q-PGEDA-PEG-OA and HA
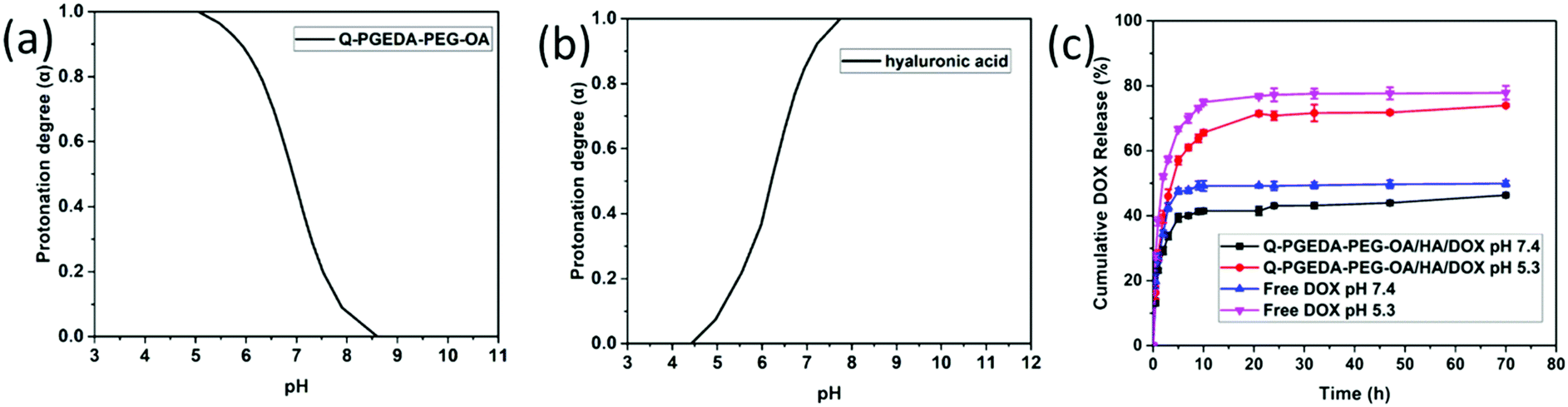
The electrostatically complexed components were designed with distinctive pH-responsive protonation behaviors. Namely, the numbers of positive charge in the polycationic components of Q-PGEDA-PEG-OA were designed to increase in response to the endosomal pH gradient (pH 5.3), while the numbers of negative charge in the polyanionic components of hyaluronic acid were designed to decrease in response to the endosomal pH gradient (pH 5.3). The subsequent potentiometric acid–base titration (titration curves not shown) confirmed our design strategies that the number of positive charges in Q-PGEDA-PEG-OA increased from approximately 20% at pH 7.4 to approximately 99% at pH 5.3. Moreover, the number of negative charges of hyaluronic acid decreased from approximately 92% at pH 7.4 to approximately 7.3% at pH 5.3 (Fig. 2a and b). Indeed, the zeta-potential measurement for the sample of Q-PGEDA-PEG-OA/HA/DOX demonstrated the rise in the zeta potential (+2 mV) upon incubation in acidic pH (5.3), in contrast to −4 mV at the physiological pH 7.4 (Fig. 1c). This growth in zeta-potential confirmed the promoted protonation of Q-PGEDA-PEG-OA. The yielded positive charge was conjectured to induce non-specific interactions in the intracellular species and cytoplasm structures, eventually accounting for the release of the loaded doxorubicin.
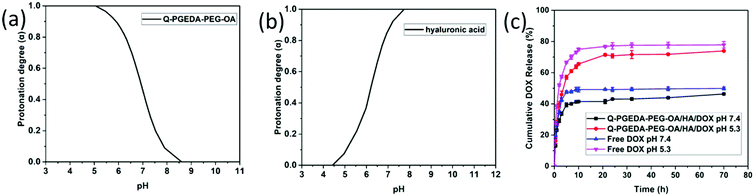 |
| | Fig. 2 Change in protonation degree (α) with pH (α/pH curve) of Q-PGEDA-PEG-OA (a) and hyaluronic acid (b). (c) Cumulative release profile (in %) of DOX in vitro from free DOX and Q-PGEDA-PEG-OA/HA/DOX nanoparticles in the PBS (pH 7.4 and 5.3). | |
3.4
In vitro drug release assay
The nanoparticles gave a high DOX loading content of 18.6%. Following cell internalization, the internalized nanoparticles were observed to experience acidic endosome entrapment; hence, it is imperative for the doxorubicin delivery vehicles to release the loaded DOX. Accelerated drug release was confirmed for the sample of Q-PGEDA-PEG-OA/HA/DOX at pH 5.3 as compared to that at pH 7.4. Particularly, approximately 75% of DOX was estimated to release from Q-PGEDA-PEG-OA/HA/DOX at 20 h post acidic incubation, as opposed to the limited release from Q-PGEDA-PEG-OA/HA/DOX in the physiological pH (merely 40% of doxorubicin released at 20 h) (Fig. 2c). A plausible reason for this accelerated drug release originates from the acidic incubation for a decrease in the interaction between the hyaluronic acid and DOX. Upon protonation of the carboxylic acid groups of hyaluronic acid, which prompted the detachment of DOX, doxorubicin payloads could be liberated from the self-assembly formation. Such a phenomenon was also observed by Du et al. when investigating the release of DOX from PAMA-DMMA nanogels with negatively charged carboxylate groups.39 The cumulative release property of free DOX was examined as the control. It indicated that free DOX was released at a faster rate compared with Q-PGEDA-PEG-OA/HA/DOX, showing that HA could prevent potential premature DOX release. The expedited liberation of doxorubicin could result in potent anti-tumor efficacy.
3.5 Targeting ability evaluation
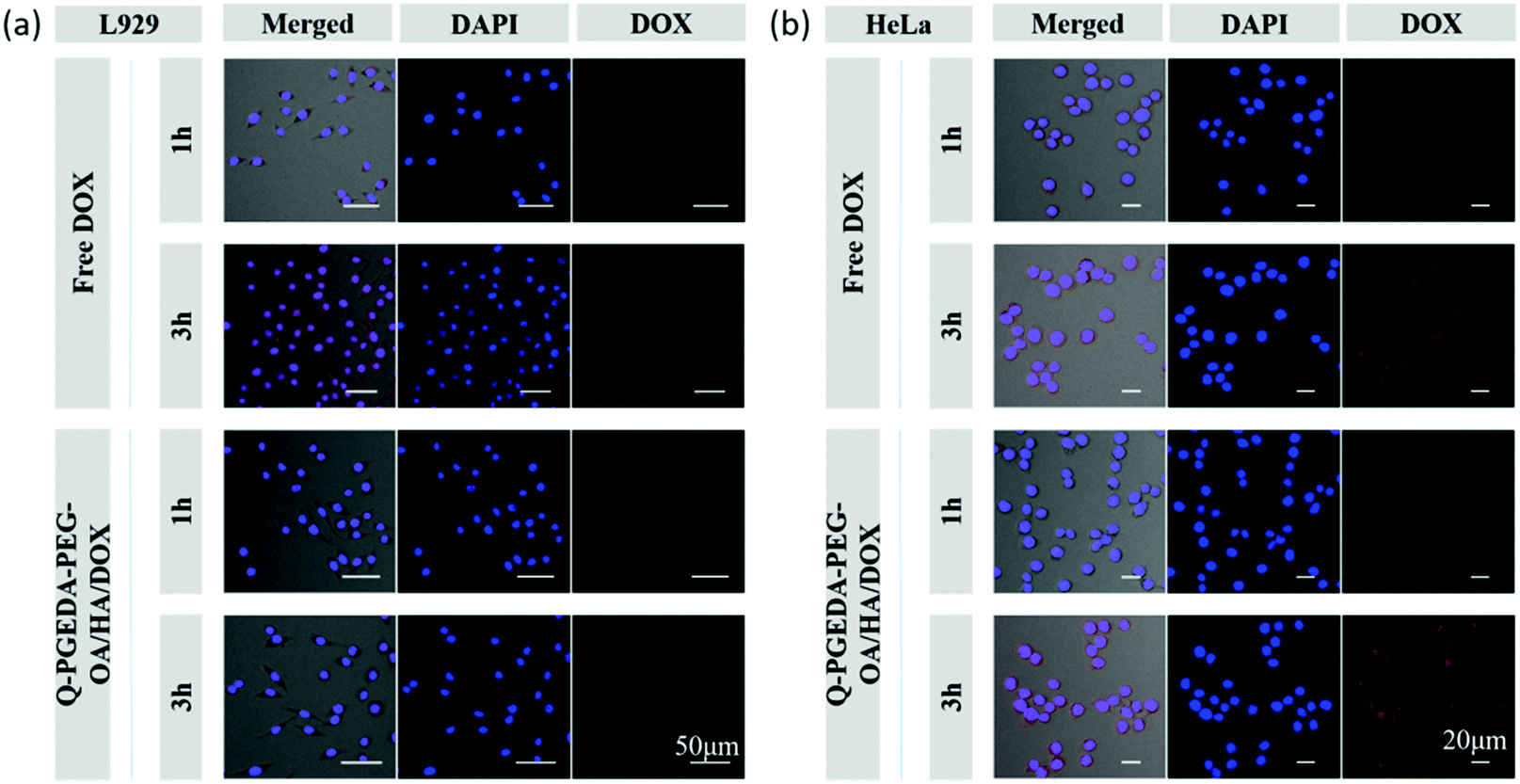
Of note, the mild negative charge of nanoparticles are particularly advantageous in the biological milieu in terms of reducing non-specific interactions and RES clearance, thereby possessing prolonged retention in the biological milieu. Moreover, hyaluronic acid is acknowledged to have favorable affinity to the cancerous cells given that a variety of hyaluronic acid receptors (e.g. CD44) have been identified to overexpress on the surface of diverse cancerous cells.40 Therefore, we would anticipate the preferential uptake of hyaluronic acid surface-functionalized nanoparticles into the cancerous cells. In this respect, we examined the cellular uptake activity of Q-PGEDA-PEG-OA/HA/DOX in cancerous cells (HeLa cells) and healthy cells (L929 cells) (Fig. 3). Quantitative analysis of cellular uptake of free DOX and Q-PGEDA-PEG-OA/HA/DOX was performed using flow cytometry (Fig. S4, ESI†). Remarkably higher cellular internalization of Q-PGEDA-PEG-OA/HA/DOX was observed for Hela cells relative to L929 cells. This result confirmed the functional role of hyaluronic acid for the pursuit of targeted transportation to the cancerous cells.
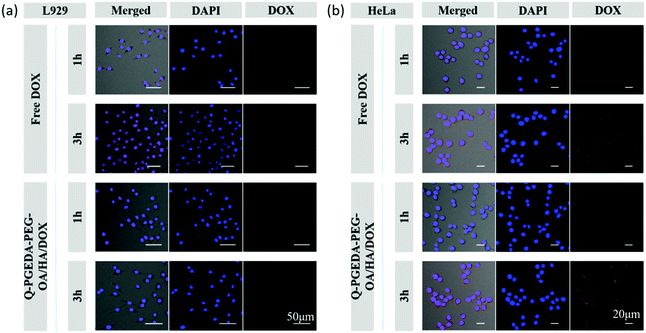 |
| | Fig. 3 Confocal laser scanning microscopy images of L929 (a) and HeLa (b) cells after incubation with free DOX and Q-PGEDA-PEG-OA/HA/DOX for 1 h and 3 h. | |
3.6 Biocompatibility and cell toxicity
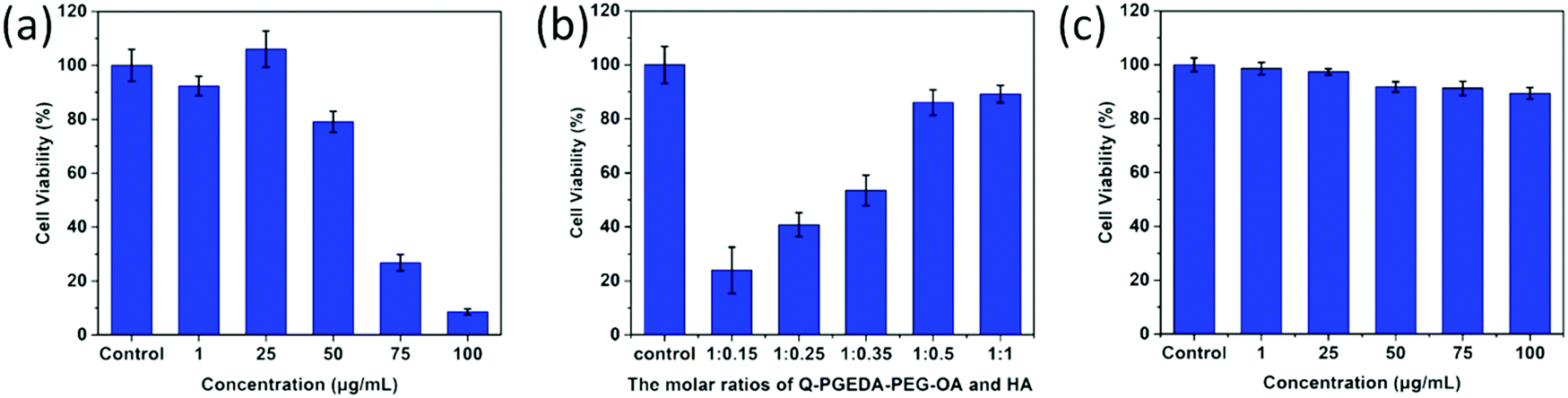
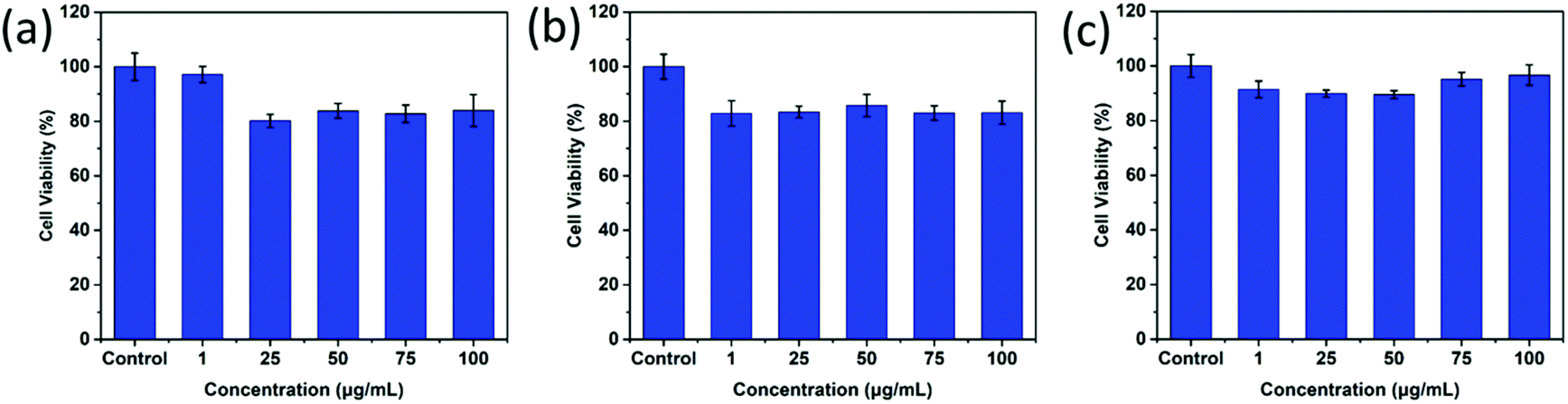
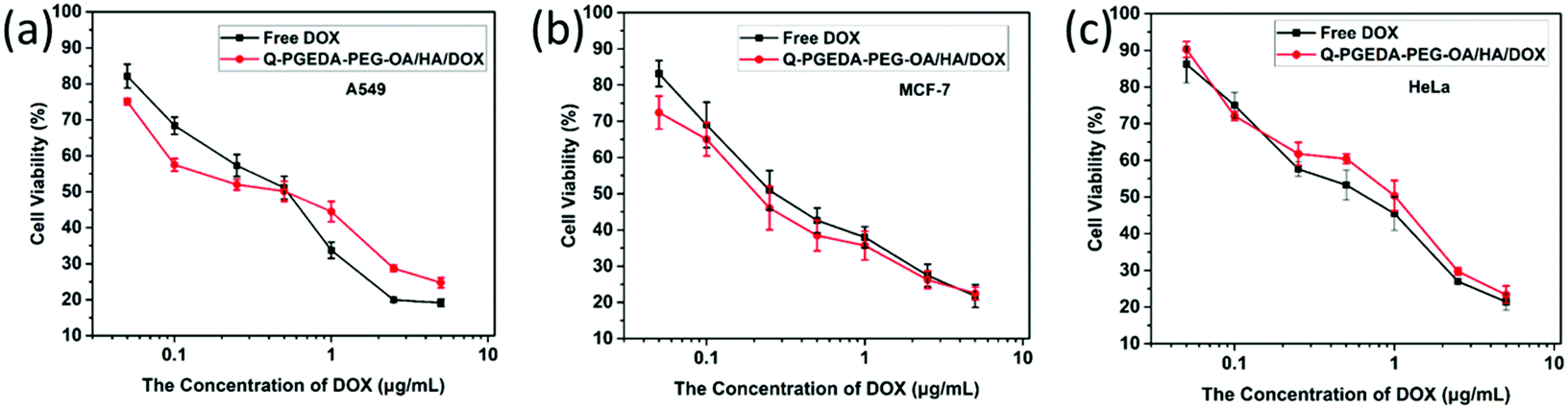
As desired drug delivery nanoparticles, biocompatibility plays a significant role in biomedical clinical applications. As shown in Fig. 4 and 5, blank micelles (Q-PGEDA-PEG-OA) exhibited incremental toxicity toward L929 cells at concentrations of 1 μg mL−1 to 100 μg mL−1 (Fig. 4a), which was due to the existence of increased positive charges of the quaternary ammonium salt at a high concentration. The cell viabilities of the Q-PGEDA-PEG-OA/HA micelles (75 μg mL−1) with increased HA coating ratio from 1:0.15 to 1:1 were carried out. As shown in Fig. 4b, the cytotoxicity of Q-PGEDA-PEG-OA/HA micelles was significantly reduced as the HA molar ratio increased, indicating the coating of HA significantly promoted the cellular compatibility of blank nanoparticles (Q-PGEDA-PEG-OA/HA). Based on the above experimental data, 1:0.5 was chosen as the optimal experimental condition for subsequent experiments. Furthermore, the healthy cells (Fig. 4c) and cancerous cells (Fig. 5) with Q-PGEDA-PEG-OA/HA (1:0.5) treatment achieved high cell viabilities. This prominent efficacy was attributed to the good biocompatibility of HA as well as the neutralized surface charge of the nanoparticles. To further evaluate the antitumor activity, Q-PGEDA-PEG-OA/HA/DOX complexes were incubated against different cancer cells at various DOX-concentrations using the MTT assay. Q-PGEDA-PEG-OA/HA/DOX revealed similar cytotoxicity as compared to free DOX (Fig. 6), which suggested encapsulated DOX still kept its excellent anticancer efficacy. The abundant cellular internalization as observed from confocal laser scanning microscopy (CLSM) (Fig. 3b) and accelerated intracellular release41 of DOX guarantee the effective anticancer activity. The half maximal inhibitory concentration (IC50) values of free DOX and Q-PGEDA-PEG-OA against different cancer cells were measured and the results are displayed in Table S2 (ESI†). The formulation with lower value of IC50 possessed the better antineoplastic activity of cancer drugs. The IC50 values are in accordance with antiproliferation effects against different cancer cells exhibited in Fig. 6.
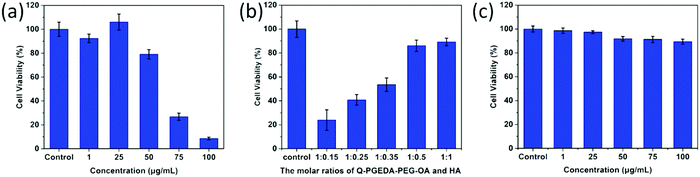 |
| | Fig. 4 Relative viabilities of L929 cells after incubation with Q-PGEDA-PEG-OA or Q-PGEDA-PEG-OA/HA: (a) at various Q-PGEDA-PEG-OA concentrations; (b) at various molar ratios of Q-PGEDA-PEG-OA (75 μg mL−1) and HA; (c) at similar molar ratio of Q-PGEDA-PEG-OA and HA (1:0.5, at various Q-PGEDA-PEG-OA concentrations). | |
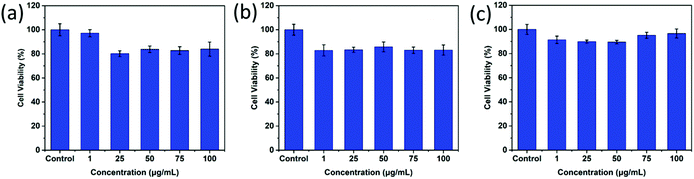 |
| | Fig. 5 Cytotoxicity assays of Q-PGEDA-PEG-OA/HA at various Q-PGEDA-PEG-OA concentrations (the molar ratio of Q-PGEDA-PEG-OA and HA was 1:0.5) against cancer A549 (a), MCF-7 (b) and HeLa cells (c). | |
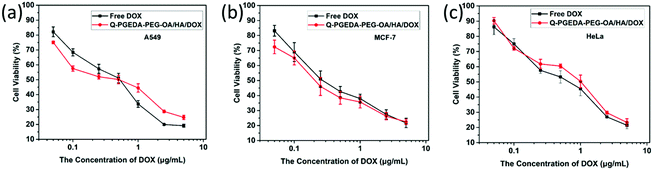 |
| | Fig. 6 Cellular viabilities of A549 (a), MCF-7 (b) and HeLa cells (c) treated with free DOX and Q-PGEDA-PEG-OA/HA/DOX at various DOX concentrations (the molar ratio of Q-PGEDA-PEG-OA and HA was 1:0.5). | |
3.7 Antimicrobial activity
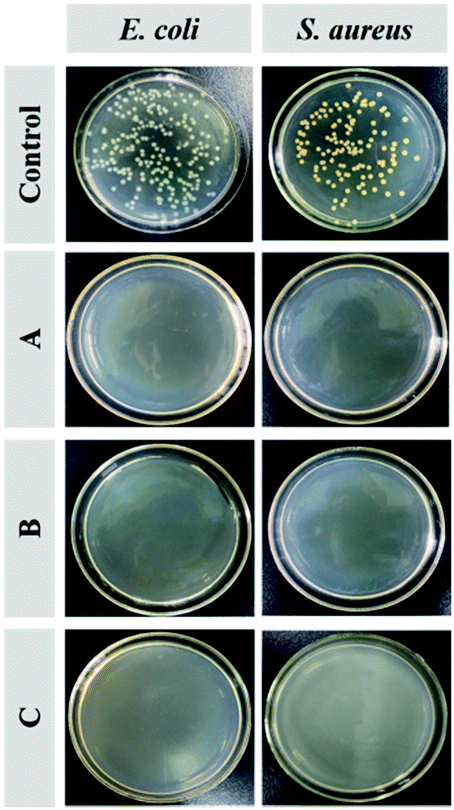
Fig. 7 shows the antimicrobial activity of the Q-PGEDA-EPG-OA and Q-PGEDA-EPG-OA/HA/DOX (at a final DOX concentration of 0.05 μg mL−1 or 5 μg mL−1) system to Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus). The quantitative assessment of the antimicrobial activity is displayed by the growth inhibition percentages (Fig. S5, ESI†). A remarkable E. coli and S. aureus inhibition effect of blank nanoparticles can be observed. Similarly, DOX-loaded nanoparticles with a DOX concentration of 5 μg mL−1 exhibited appreciable antibacterial activities (exceeding 99%) for treatment of both bacterial colonies, even when the concentration of DOX decreased to 0.05 μg mL−1. These results are attributed to the introduction of antimicrobial component OA,42,43 and the strong positive charge of quaternary ammonium salt exposed to the bacteria membrane, leading to bacterial membranolysis. Another noteworthy result was the cleavage of the hydrophobic segment of OA from Q-PGEDA-PEG-OA, as evidenced by the disappearance of the peak at 1.3 ppm of Q-PGEDA-PEG-OA in the presence of NaOH solution (0.015 M) via1H-NMR measurement (Fig. S3, ESI†). Appreciable cleavage was anticipated to resolve the chronic side effect concerns. Collectively, the nanoparticles could be successfully used to realize bacterial inhibition, which enhanced the antineoplastic activity of cancer drugs by means of reducing cancer drugs resistance.
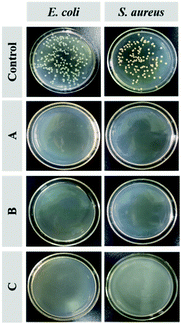 |
| | Fig. 7 Images of colony forming units (CFU) for E. coli and S. aureus treated with Q-PGEDA-PEG-OA (75 μg mL−1) (A), Q-PGEDA-PEG-OA/HA/DOX (5 μg mL−1) (B) and Q-PGEDA-PEG-OA/HA/DOX (0.05 μg mL−1) (C) on the Luria–Bertani (LB) agar plate. | |
4. Conclusions
To this end, we have proposed facile reversible HA/PEG-cationic amphiphilic nanoparticles for DOX delivery. The results demonstrated limited pre-mature drug release with PEG and HA coverage. The detachment of PEG and HA coverage was determined due to our strategic modulation of the charge stoichiometry for the cationic amphiphilic copolymer and HA in the subcellular endosome compartment, consequently accounting for facilitated drug release in the acidic endosome and eventually potent anti-tumor efficacy to the cancerous cells. The nanomedicine possessed both anticancer and antimicrobial agents to combat the microbes in tumor tissues to alleviate cancer drugs resistance. We believe the facile reversible adsorption strategy shed light on the development of intracellular-extracellular distinguishable nanomachines, thus opening doors for further development towards a broader range of biomedical applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors thank the National Natural Science Foundation of China (21674080), 131 talents program of Tianjin, Distinguished Professor of Tianjin, the Leading Talents Program of the Tianjin Educational Committee, Key program of Tianjin Municipal Natural Science Foundation (No. 18JCZDJC37700), Training Project of Innovation Team of Colleges and Universities in Tianjin (TD13-5020), and the Fundamental Research Funds for the Central Universities [No. DUT17RC(3)059] for financial support.
Notes and references
- M. Elsabahy, G. S. Heo, S. M. Lim, G. Sun and K. L. Wooley, Chem. Rev., 2015, 115, 10967–11011 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips and M. B. Carter, Nat. Mater., 2011, 10, 389–397 CrossRef CAS PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 1–19 CrossRef CAS PubMed.
- S. A. Amolegbe, Y. Hirano, J. O. Adebayo, O. G. Ademowo, E. A. Balogun, J. A. Obaleye, A. U. Krettli, C. Yu and S. Hayami, Sci. Rep., 2018, 8, 3078 CrossRef.
- L. Rajendran, H. J. Knölker and K. Simons, Nat. Rev. Drug Discovery, 2010, 9, 29–42 CrossRef CAS.
- Z. Feng, H. Wang, S. Wang, Q. Zhang, X. Zhang, A. A. Rodal and B. Xu, J. Am. Chem. Soc., 2018, 140, 9566–9573 CrossRef CAS.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and P. Dan, Nat. Commun., 2018, 9, 1–12 CrossRef CAS.
- Q. L. Li, S. H. Xu, H. Zhou, X. Wang, B. Dong, H. Gao, J. Tang and Y. W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664 CrossRef CAS.
- Y.-B. Long, W.-X. Gu, C. Pang, J. Ma and H. Gao, J. Mater. Chem. B, 2016, 4, 1480–1488 RSC.
- H. Tian, Z. Luo, L. Liu, M. Zheng, Z. Chen, A. Ma, R. Liang, Z. Han, C. Lu and L. Cai, Adv. Funct. Mater., 2017, 27, 1703197 CrossRef.
- X. Jia, Y. Zhang, Y. Zou, Y. Wang, D. Niu, Q. He, Z. Huang, W. Zhu, H. Tian, J. Shi and Y. Li, Adv. Mater., 2018, 1704490 CrossRef.
- X. Sun, H. Gao, G. Wu, Y. Wang, Y. Fan and J. Ma, Int. J. Pharm., 2011, 412, 52–58 CrossRef CAS.
- M. Elsabahy, S. Zhang, F. Zhang, Z. J. Deng, Y. H. Lim, H. Wang, P. Parsamian, P. T. Hammond and K. L. Wooley, Sci. Rep., 2013, 3, 3313 CrossRef.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk and M. Urban, Nat. Mater., 2010, 9, 101–111 CrossRef.
- Y. Jeong, Y. K. Jo, B. J. Kim, B. Yang, K. I. Joo and H. J. Cha, ACS Nano, 2018, 12, 8909–8919 CrossRef CAS.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS.
- T. Wang, Q. Chen, H. Lu, W. Li, Z. Li, J. Ma and H. Gao, Bioconjugate Chem., 2016, 27, 1949–1957 CrossRef CAS.
- W. Ye, Y. Zhao, H. Li, R. Na, F. Li, Q. Mei, M. Zhao and S. Zhou, Sci. Rep., 2015, 5, 14614 CrossRef CAS.
- Z. Jiang, Q. Chen, X. Yang, X. Chen, Z. Li, D. E. Liu, W. Li, Y. Lei and H. Gao, Bioconjugate Chem., 2017, 28, 2849–2858 CrossRef CAS.
- Q. Li, Y. Wu, H. Lu, X. Wu, S. Chen, N. Song, Y. W. Yang and H. Gao, ACS Appl. Mater. Interfaces, 2017, 9, 10180–10189 CrossRef CAS.
- S. Gerecht, J. A. Burdick, L. S. Ferreira, S. A. Townsend, R. Langer and G. Vunjak-Novakovic, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11298–11303 CrossRef CAS.
- M. Yu, S. Jambhrunkar, P. Thorn, J. Chen, W. Gu and C. Yu, Nanoscale, 2013, 5, 178–183 RSC.
- W. Miao, G. Shim, C. M. Kang, S. Lee, Y. S. Choe, H. G. Choi and Y. K. Oh, Biomaterials, 2013, 34, 9638–9647 CrossRef CAS.
- M. Li, W. Song, Z. Tang, S. Lv, L. Lin, H. Sun, Q. Li, Y. Yang, H. Hong and X. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 1781–1792 CrossRef CAS.
- L. T. Geller, M. Barzilyrokni, T. Danino, O. H. Jonas, N. Shental, D. Nejman, N. Gavert, Y. Zwang, Z. A. Cooper and K. Shee, Science, 2017, 357, 1156 CrossRef CAS.
- T. Yu, F. Guo, Y. Yu, T. Sun, D. Ma, J. Han, Y. Qian, I. Kryczek, D. Sun, N. Nagarsheth, Y. Chen, H. Chen, J. Hong, W. Zou and J. Y. Fang, Cell, 2017, 170, 548–563 CrossRef CAS.
- H. Yuan, Z. Liu, L. Liu, F. Lv, Y. Wang and S. Wang, Adv. Mater., 2014, 26, 4333–4338 CrossRef CAS PubMed.
- C. Krumm, S. Harmuth, M. Hijazi, B. Neugebauer, A. L. Kampmann, H. Geltenpoth, A. Sickmann and J. C. Tiller, Angew. Chem., Int. Ed. Engl., 2014, 53, 3830–3834 CrossRef CAS PubMed.
- M. C. Jones, H. Gao and J. C. Leroux, J. Controlled Release, 2008, 132, 208–215 CrossRef CAS.
- S. Chen, Q. Chen, Q. Li, J. An, P. Sun, J. Ma and H. Gao, Chem. Mater., 2018, 30, 1782–1790 CrossRef CAS.
- Y. Li, T. Zhao, C. Wang, Z. Lin, G. Huang, B. D. Sumer and J. Gao, Nat. Commun., 2016, 7, 13214 CrossRef.
- K. Miyata, M. Oba, M. Nakanishi, S. Fukushima, Y. Yamasaki, H. Koyama, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2008, 130, 16287–16294 CrossRef CAS.
- X. Guo, L. Wang, K. Duval, J. Fan, S. Zhou and Z. Chen, Adv. Mater., 2018, 30, 1705436 CrossRef.
- D. Oupicky, M. Ogris, K. A. Howard, P. R. Dash, K. Ulbrich and L. W. Seymour, Mol. Ther., 2002, 5, 463–472 CrossRef CAS.
- J. J. Young, C. C. Chen, Y. C. Chen, K. M. Cheng, H. J. Yen, Y. C. Huang and T. N. Tsai, Carbohydr. Polym., 2016, 137, 532–540 CrossRef CAS.
- M. Vanwildemeersch, J. Social Studies Res., 2006, 38, 111–113 Search PubMed.
- J. Li, Y. He, W. Sun, Y. Luo, H. Cai, Y. Pan, M. Shen, J. Xia and X. Shi, Biomaterials, 2014, 35, 3666–3677 CrossRef CAS.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed. Engl., 2010, 49, 3621–3626 CrossRef CAS.
- Y. P. Zhao, W. L. Ye, D. Z. Liu, H. Cui, Y. Cheng, M. Liu, B. L. Zhang, Q. B. Mei and S. Y. Zhou, Nanoscale, 2017, 9, 6264–6277 RSC.
- Z. Wang, X. Xue, Y. He, Z. Lu, B. Jia, H. Wu, Y. Yuan, Y. Huang, H. Wang, H. Lu, K. S. Lam, T.-Y. Lin and Y. Li, Adv. Funct. Mater., 2018, 1802159 CrossRef.
- J. Wu, S. Zhao, S. Xu, X. Pang, G. Cai and J. Wang, J. Mater. Chem. B, 2018, 6, 7462–7470 RSC.
- C. J. Waschinski, S. Barnert, A. Theobald, R. Schubert, F. Kleinschmidt, A. Hoffmann, K. Saalwächter and J. C. Tiller, Biomacromolecules, 2008, 9, 1764–1771 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8tb02370k |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Jinxia
An
a,
De-E
Liu
a,
Yongkang
Huang
a,
Rui
Yang
a,
Wei
Li
a,
Li
Chen
*a and
Hui
Gao
b,
Jinxia
An
a,
De-E
Liu
a,
Yongkang
Huang
a,
Rui
Yang
a,
Wei
Li
a,
Li
Chen
*a and
Hui
Gao