
High-performance organic light-emitting diodes with low-efficiency roll-off using bulky tetradentate [Pt(O^N^C^N)] emitters†
Mao
Mao‡
a,
Jiahuan
Peng‡
bc,
Tsz-Lung
Lam
 a,
Wai-Hung
Ang
a,
Huiyang
Li
b,
Gang
Cheng
*ad and
Chi-Ming
Che
*ad
a,
Wai-Hung
Ang
a,
Huiyang
Li
b,
Gang
Cheng
*ad and
Chi-Ming
Che
*ad
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong SAR, China. E-mail: ggcheng@hku.hk; cmche@hku.hk
bGuangdong Aglaia Optoelectronic Materials Co., Ltd, No. 8 of Xinhui Road, Foshan, 528300, China
cSchool of Material Science and Engineering, South China University of Technology, Guangzhou, 510640, China
dHKU Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong, 518053, China
First published on 11th April 2019
Abstract
High-performance organic light-emitting diodes (OLEDs) were realized by utilizing three robust platinum(II) complexes bearing tetradentate O^N^C^N ligands with a bridging tertiary amine (tetra-Pt-N), a biphenyl group with a spiro linkage (tetra-Pt-S) or a sterically encumbered 2,6-dimethyl-4-tert-butylphenyl moiety (tetra-Pt-M). By using a double-host emissive layer (EML) structure and a 10 nm-thick interlayer between the EML and the electron-transport layer (ETL), a maximum power efficiency of over 100 lm W−1, a low turn-on voltage of less than 2.5 V and low efficiency roll-off were achieved. A high external quantum efficiency (EQE) of 19.4% and 20.3% was maintained at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2 in OLEDs with tetra-Pt-N and tetra-Pt-M, respectively. Such high efficiency at high luminance could be attributed to the effective suppression of triplet–triplet annihilation (TTA) in our optimized device structure.
000 cd m−2 in OLEDs with tetra-Pt-N and tetra-Pt-M, respectively. Such high efficiency at high luminance could be attributed to the effective suppression of triplet–triplet annihilation (TTA) in our optimized device structure.
1. Introduction
Since the first efficient organic electroluminescent device was reported in 19871 and organic electro phosphorescent devices with metal complexes were demonstrated in 1998,2,3 tremendous research and development activities have been devoted to making organic light-emitting diodes (OLEDs) for a new generation of lighting sources and display devices because of their privileged features: energy saving, area lighting, lightweight, ultra-thin and solid state.4–9 Currently, OLEDs are extensively used in almost every kind of display from small-area display units in electronic products such as mobile phones to large-area televisions. In these applications, OLEDs usually work at low luminance in the range between 100 and 400 cd m−2.10 Nonetheless, for general illumination purposes, OLEDs are required to work at much higher luminance, i.e., up to 5000 cd m−2, and even 10000 cd m−2. Therefore, the realization of OLEDs with high efficiency at luminance levels up to 10000 cd m−2 remains a challenge that has to be overcome to utilize this new kind of light-emitting device for illumination purposes.11 Utilizing phosphorescent metal complexes as emitting dopants is an effective strategy to achieve OLEDs with high efficiency and high luminance due to the potential 100% internal quantum efficiencies originating from harvesting both singlet and triplet excitons.2 Bottom-emitting phosphorescent OLEDs (PHOLEDs) with an external quantum efficiency (EQE) over 30% and/or a power efficiency (PE) over 100 lm W−1 without any optical out-coupling technique have been reported in the literature.12–20 Nevertheless, the efficiency of PHOLEDs tends to decrease with luminance, which is referred to as efficiency roll-off.10 Indeed, the efficiency roll-off for most PHOLEDs is quite pronounced at high luminance values above 1000 cd m−2, which is mainly attributed to triplet–triplet annihilation (TTA) due to the long triplet state lifetimes of phosphorescent emitters and/or the imbalance of charges in the emissive layer (EML).10 Compared to fluorescent emitters with emission lifetimes of ∼10 ns,21 phosphorescent metal complexes display much longer excited state lifetimes of 0.5–50 μs for Pt(II) and Ir(III) complexes,2,22–24 and even longer for Au(III) and Pd(II) complexes.25–27 Broadening the recombination zone has been proven to be an effective means to suppress TTA via reducing the exciton densities in the EML of OLEDs. In this regard, several device structures have been proposed in the literature, such as (i) a double-EML structure composed of hole-transporting and electron-transporting EMLs,18 (ii) a single EML with uniform or gradient mixed hole- and electron-transport host materials,13,14,28,29 and (iii) a single EML with a bipolar host material capable of equally transporting both holes and electrons.30 In addition to the optimization of the EML, the charge mobility and energy levels of both the hole-transport layer (HTL) and the electron-transport layer (ETL) have been intensively studied to balance the charge carriers in the EML and to lower the driving voltage of the device.31–33 Kim and co-workers designed an efficient co-host single EML structure to fabricate OLEDs with [Ir(ppy)2tmd] (ppy = 2-phenylpyridine; tmd = 2,2,6,6-tetramethylheptane-3,5-diketonate).13,14 In these devices, TCTA (4,4′,4′′-tris(N-carbazolyl)-triphenylamine) and B3PYMPM (bis-4,6-(3,5-di-3-pyridylphenyl)-2-methylpyrimidine) were mixed to give an exciplex-forming double-host; B3PYMPM was also used as the ETL to eliminate the electron-injection barrier from the ETL to the EML and to lower the driving voltage. An EQEmax of 32.3% and PEmax of 142.5 lm W−1 were achieved with this device.13,14
Recently, the device performance of PHOLEDs based on Pt(II) complexes has been significantly improved.34–39 Chi and co-workers40 utilized planar [Pt(fppz)2] (fppz = 3-(trifluoromethyl)-5-(2-pyridyl)-pyrazolate) and its highly oriented emitting dipoles to boost the EQE of non-doped Pt-OLEDs to over 38%, which is the highest reported efficiency for bottom-emitting OLEDs without an optical enhancement technique. Nevertheless, because the emission of such non-doped OLEDs originated from the aggregation states of Pt(II) complexes, which have a large red-shift from that of the parental Pt(II) complexes, only low-energy (yellow to near infrared) emission could be realized.40–42 In addition, in terms of fabrication cost, using pristine expensive metal complexes as EMLs is quite expensive and poses a challenge for practical applications. Thus, the traditional host–guest structure EML could be more appealing for Pt-OLEDs targeted for practical applications. Examples of high-performance Pt-OLEDs with EQEs of over 20% have been reported; however, most of them still suffer from pronounced efficiency roll-off and relatively low PE at high luminance beyond 1000 cd m−2.6,24,35–37,39,40,43,44 As examples, although high EQEmax values of 24.8% and 25.7% were achieved in a blue device with PtON7 and a white device with Pt7O7,36–38 their efficiency roll-off was pronounced and resulted in EQE values of less than 15% and PE values of 30 lm W−1 for both devices at 1000 cd nm−2. Thus, a suitable device structure design to lower the efficiency roll-off of Pt-OLEDs is of great importance.
In this work, we designed a device structure with a modified double-host EML using three [Pt(O^N^C^N)] emitters with high photoluminescence quantum yields (PLQYs) and relatively short emission lifetimes. By systemically optimizing the individual layers of this device, a low turn-on voltage of 2.5 V, a high EQEmax of 24.5%, and a PEmax of 104.3 lm W−1 were realized in the device with tetra-Pt-M. An EQEmax of more than 20% and a PEmax of more than 100 lm W−1 were similarly achieved in the devices with tetra-Pt-N and tetra-Pt-S, respectively. Moreover, for the device with tetra-Pt-M, a high EQE of 20.3% was maintained at a high luminance of 10000 cd m−2, attributed to the effectively suppressed TTA via the broadening recombination zone in this device structure.
2. Results and discussion
2.1. Chemical structure and photophysical properties of tetra-Pt-N, tetra-Pt-S and tetra-Pt-M
As depicted in Chart 1, three tetradentate [Pt(O^N^C^N)] complexes, tetra-Pt-N, tetra-Pt-S and tetra-Pt-M, were studied as emitting dopants. The complex tetra-Pt-N has been reported,34 while tetra-Pt-S and tetra-Pt-M are newly developed. The syntheses of tetra-Pt-S and tetra-Pt-M are detailed in the ESI.† Our early report showed that [Pt(O^N^C^N)] complexes are prone to intermolecular Pt–Pt and/or ligand π–π interactions in the excited states, which may give rise to aggregate species formation with low-energy, broad-band emission and/or emission self-quenching, thereby leading to dopant concentration-dependent EL spectra and efficiency roll-off, which are both undesirable for a monochromic device.26,35 To address this issue, all Pt emitters described in this work feature sterically hindered motifs in the O^N^C^N framework in addition to the 3,5-di-tert-butylphenyl moiety on the interior pyridyl ring. Complexes tetra-Pt-N and tetra-Pt-S have a tertiary arylamine and a biphenyl spiro linkage, respectively, that are orthogonal to the molecular plane, while the ligand scaffold of tetra-Pt-M is appended by a bulky 2,6-dimethyl-4-tert-butylphenyl moiety on the exterior pyridyl ring.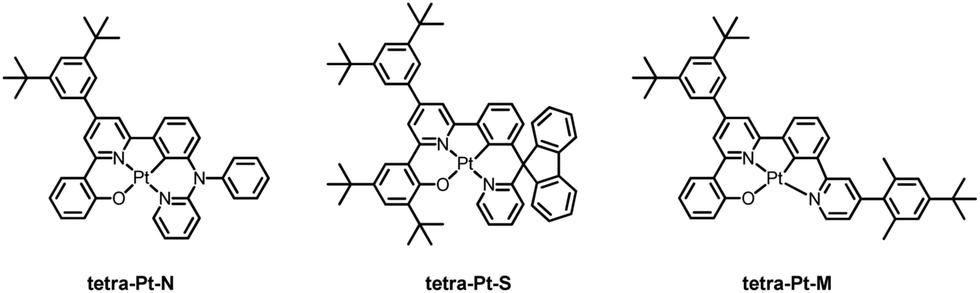 | ||
| Chart 1 Chemical structures of Pt(II) complexes tetra-Pt-N, tetra-Pt-S and tetra-Pt-M studied in this work. | ||
All Pt(II) complexes have high thermal stability with degradation temperatures ranging from 398 to 411 °C in thermogravimetric analysis (Fig. S1, ESI†). The highest occupied molecular orbital (HOMO)/lowest unoccupied molecular orbital (LUMO) levels of tetra-Pt-S and tetra-Pt-M, which were estimated from the onset of the redox waves in cyclic voltammograms (Fig. S1, ESI†), are −5.09 eV/−2.64 eV and −5.26 eV/−2.76 eV, respectively (Table S1, ESI†). The photophysical data of tetra-Pt-S and tetra-Pt-M are summarized in Table 1. Fig. 1a depicts the absorption spectra of tetra-Pt-S and tetra-Pt-M and their absorption profiles resemble those of previously reported [Pt(O^N^C^N)] congeners.34,35 In CH2Cl2 solutions, the two complexes exhibit intense absorption below ca. 320 nm with an absorptivity over 4 × 104 dm3 mol−1 cm−1, which originates from the intra ligand (IL) 1π–π* transitions of the O^N^C^N ligands. The moderately intense absorption beyond ca. 400 nm (ε = (0.4–1.3) × 104 dm3 mol−1 cm−1) can be attributed to the admixture of singlet metal-to-ligand charge-transfer (1MLCT) 1[5dπ (Pt) → π* (O^N^C^N)] and singlet intraligand charge-transfer (1ILCT) 1[π (phenolate) → π* (N^C^N)] transitions.34,45,46 These absorptions are slightly sensitive to the solvent polarity and shifted by ca. 5–15 nm in different solvents (Table S2, Fig. S2a and b, ESI†). Upon photoexcitation, tetra-Pt-S and tetra-Pt-M display strong yellowish green (λmax = 551/530 nm) and green emission (λmax = 515/510 nm) in CH2Cl2 solutions/PMMA films with emission lifetimes of 3.3/4.6 μs and 3.7/1.9 μs and emission quantum yields of up to 0.75 and 0.90, respectively (Fig. 1b). Both complexes display structureless emission bands that indicate the emissive excited states have mixed 3MLCT/3ILCT parentage as previously reported.34,45,46 Accordingly, the emission λmax span with a moderate range of 24 nm for tetra-Pt-S and 12 nm for tetra-Pt-M in various solvents (Table S2, Fig. S2c and d, ESI†). The red-shift of the emission of tetra-Pt-S relative to that of tetra-Pt-M is attributed to the two electron-donating tert-butyl groups on the phenolate moiety that effectively destabilize the HOMO (−5.09 eV for tetra-Pt-S and −5.26 eV for tetra-Pt-M). Notably, the relatively low self-quenching rate constants of tetra-Pt-S and tetra-Pt-M of approximately 107 mol−1 dm3 s−1 demonstrate the effectiveness of the bulky peripheral substituents and orthogonal 3D molecular configuration in suppressing the excited state intermolecular interactions.
| Complex | UV-vis absorption λabsa [nm] (ε × 104 [mol−1 dm3 cm−1]) | Emission | |||
|---|---|---|---|---|---|
| λ max [nm]; τ [μs] in solution | λ max [nm]; τ [μs] in film | Φ em in solution; Φemd in film | k q [mol−1 dm3 s−1] | ||
| a Determined in degassed CH2Cl2 at a concentration of 2 × 10−5 mol dm−3. b Determined as 5 wt% of a PMMA film. c Emission quantum yield was estimated with BPEA (9,10-bis(phenylethynyl)anthracene) in benzene as a standard with Φem = 0.85. d Determined with the Hamamatsu QY absolute PL quantum yield measurement system. e Self-quenching constant. | |||||
| tetra-Pt-S | 267 (5.4), 279 (5.5), 334 (1.8), 358 (1.6), 398 (0.7), 453 (sh, 0.4) | 551; 3.3 | 530; 4.6 | 0.17c; 0.75 | 2.2 × 107 |
| tetra-Pt-M | 284 (5.6), 301 (sh, 4.2), 374 (2.2), 403 (1.3), 428 (sh, 1.1) | 515; 3.7 | 510; 1.9 | 0.90d; 0.62 | 7.5 × 107 |
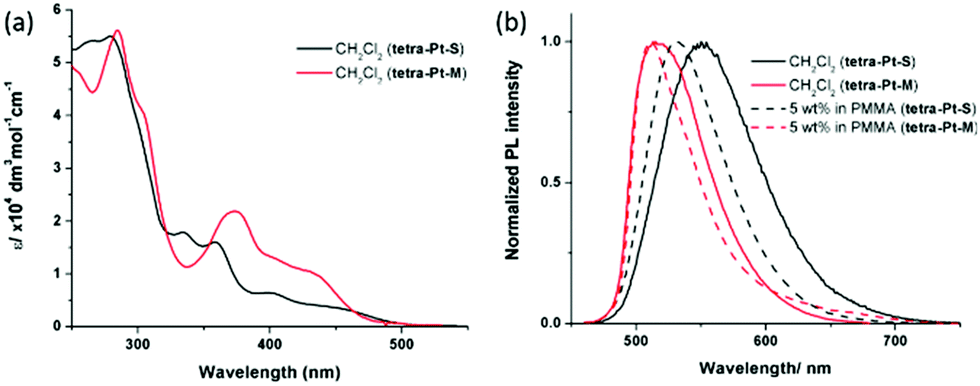 | ||
| Fig. 1 (a) UV-vis absorption spectra and (b) PL spectra of tetra-Pt-S and tetra-Pt-M in CH2Cl2 and PMMA at room temperature. | ||
2.2. Electroluminescent properties of [Pt(O^N^C^N)] complexes
As depicted in Fig. 2, the device structure was indium tin oxide ITO/MoO3 (2 nm)/TAPC (40 nm)/TCTA (10 nm)/TCTA:B3PYMPM:x wt% Pt(II) emitter (15 nm)/ETL (50 nm)/LiF (1.2 nm)/Al (100 nm). In these devices, TAPC (1,1-bis-(4-bis(4-methyl-phenyl)-amino-phenyl)-cyclohexane) was used as the HTL, TCTA was used as the electron-blocking layer, and B3PYMPM was used as the hole-blocking layer (Chart 2). A 1:1 molar ratio of TCTA and B3PYMPM was used as the double-host. Tetra-Pt-N, tetra-Pt-S or tetra-Pt-M was used as the emitter in the EML. For the ETL, B3PYMPM, TMPPPYTZ (2,4,6-tris(3-(3-(pyridin-3-yl)phenyl)phenyl)-1,3,5-triazine) or TMPYPB (1,3,5-tri(m-pyrid-3-yl-phenyl)) was used in the different devices to optimize the device performance. Fig. 2 depicts the proposed energy level diagrams of the organic materials used. The stepwise HOMO levels of TAPC (−5.6 eV) and TCTA (−5.83 eV) are beneficial for hole injection and transport; similarly, the LUMO levels of TMPYPB (−2.73 eV), TMPPPYTZ (−3.09 eV) and B3PYMPM (−3.2 eV) facilitate the injection and transport of electrons.13,14,34 In the EML, since the triplet energy (Et) of B3PYMPM:TCTA is 2.5 eV,14 being higher than those of tetra-Pt-N (2.23 eV), tetra-Pt-S (2.31 eV) and tetra-Pt-M (2.49 eV) (see Table S1, ESI†), triplet excitons of these Pt-emitters are effectively confined in the co-host of B3PYMPM:TCTA.
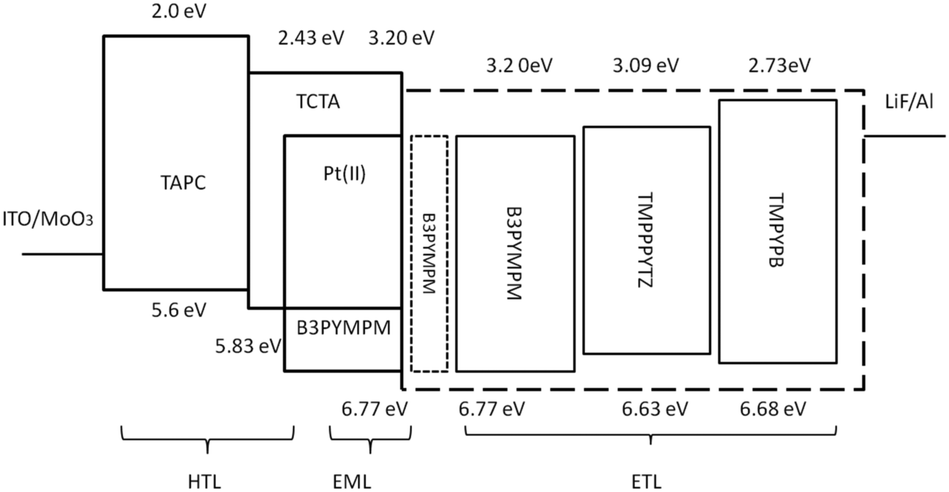 | ||
| Fig. 2 The device structure and energy diagram of the OLEDs. | ||
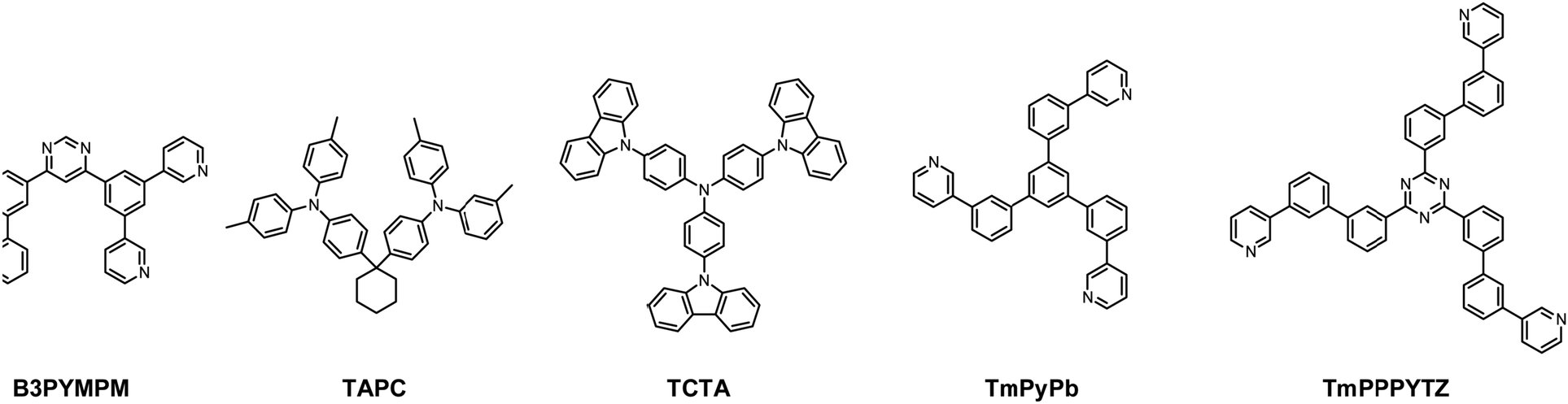 | ||
| Chart 2 The chemical structures of the host materials, hole-transport materials and electron-transport materials used in this work. | ||
First, single layers of B3PYMPM (device 1), TMPYPB (device 2) and TMPPPYTZ (device 3) as well as combined layers of B3PYMPM/TMPYPB (device 4) and B3PYMPM/TMPPPYTZ (device 5) were respectively used as the ETL in devices based on tetra-Pt-N with a fixed concentration of 6 wt% to investigate the influence of the ETL material on the Pt-OLEDs. As depicted in Fig. S4a (ESI†), the EL spectra of all devices closely resemble the PL spectrum of tetra-Pt-N in solution,34 indicating that the emission of these devices originates from tetra-Pt-N. As listed in Table 2, device 5 showed the highest maximum current efficiency (CEmax) of 72.3 cd A−1 and EQEmax of 20.6% among the tetra-Pt-N devices. Particularly, at high luminances of 1000 and 5000 cd m−2, the efficiency of device 5 was also superior to the other devices, due to the fact that the interlayer of B3PYMPM (10 nm) can effectively confine the excitons within the EML and the higher electron mobility of TMPPPYTZ (10−4–10−3 cm2 V−1 s−1)32 than those of B3PYMPM (∼10−5 cm2 V−1 s−1)47 and TMPYPB (10−4 cm2 V−1 s−1)48 in device 5. Without the interlayer of B3PYMPM, the efficiency of device 3 is relatively lower due to the shallow HOMO of TMPPPYTZ (−6.63 eV), which is not deep enough to confine the excitons within the EML. The turn-on voltages of devices 2 and 4, on the other hand, are higher, being 2.8 and 3.0 V, respectively, caused by the higher LUMO level (−2.73 eV) of TMPYPB, which increase the electron-injecting barrier from the cathode. For the same reason, the efficiencies of devices 2 and 4 are relatively lower due to the imbalanced charge density in the EML.
| Device no. | Interlayer/ETL (thickness) | V on (V) | CEb (cd A−1) | EQEc (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Max. | 1000 cd m−2 | 5000 cd m−2 | Max. | 1000 cd m−2 | 5000 cd m−2 | |||
| a Turn-on voltage at 1 cd m−2. b Current density. c External quantum efficiency. | ||||||||
| 1 | B3PYMPM (50 nm) | 2.4 | 70.9 | 64.0 | 51.3 | 18.8 | 18.3 | 13.6 |
| 2 | TMPYPB (50 nm) | 2.8 | 36.7 | 35.0 | 33.0 | 11.0 | 10.5 | 9.7 |
| 3 | TMPPPYTZ (50 nm) | 2.4 | 48.0 | 43.0 | 42.6 | 15.7 | 13.8 | 12.3 |
| 4 | B3PYMPM (10 nm)/TMPYPB (40 nm) | 3.0 | 36.0 | 32 | 23.9 | 10.2 | 9.05 | 6.8 |
| 5 | B3PYMPM (10 nm)/TMPPPYTZ (40 nm) | 2.4 | 72.3 | 66.1 | 53.2 | 20.6 | 19.0 | 15.3 |
To further improve the performance of device 5, devices with doping concentrations of tetra-Pt-N ranging from 6 to 16 wt% were fabricated and characterized. As depicted in Fig. S5a (ESI†), with increasing dopant concentration, slight red-shifts of the EL spectra appear, suggesting the presence of weak intermolecular interactions of tetra-Pt-N.34 As shown in Fig. S5b (ESI†), the EQE of the device increases with the doping concentration up to 12 wt%, and the EQEmax and CEmax were 21.0% and 73.2 cd A−1, respectively, in the device with 12 wt% tetra-Pt-N. Meanwhile, the efficiency roll-off of this device was largely suppressed, especially at a high luminance of 1000–5000 cd m−2 (EQE1000 = 20.2%, EQE5000 = 18.6%, see Table S3, ESI†).
To optimize the thickness of the EML, OLEDs with 12 wt% tetra-Pt-N and EML thicknesses from 10 to 20 nm were fabricated and characterized. As shown in Fig. S5c (ESI†) and Table 3, the device with an 18 nm thick EML (device 6) showed the highest EQEmax of 23.2% and a PEmax of 102.8 lm W−1. At high luminances of 1000, 5000 and 10000 cd m−2, device 6 maintained an EQE1000 of 22.8% (PE1000 = 82.9 lm W−1), an EQE5000 of 21.0% (PE5000 = 66.3 lm W−1) and an EQE10000 of 19.4% (PE10000 = 50.9 lm W−1). In addition, the high efficiency at high luminance also results in high luminance at high driving current; a maximum luminance (Lmax) of 116630 cd m−2 was achieved with optimized device 6, which is remarkably higher than that of the best reported OLED with tetra-Pt-N (Lmax = 51200 cd m−2, TCTA as the single host).34 We attribute this result to the broadened recombination zone in the double-host EML. Based on the optimized device structure of the OLED with tetra-Pt-N, we applied tetra-Pt-S (device 7) and tetra-Pt-M (device 8) in the same device structure as the emitting dopants. As depicted in Table 3 and Fig. 3, a low turn-on voltage of 2.5 V, a PEmax of 100.2 lm W−1 and an EQEmax of 21.3% were successfully achieved in optimized device 7, however, its efficiency roll-off was relatively pronounced, which was attributed to the relatively long emission lifetime of 4.6 μs of tetra-Pt-S in the thin film. Similarly, a PEmax of 104.3 lm W−1 and an EQEmax of 24.5% were achieved in optimized device 8. With a short emission lifetime of 1.9 μs for tetra-Pt-M, the efficiency roll-off was notably suppressed in device 8. The PE and EQE values only slightly decreased to 85.6 lm W−1 and 23.1%, respectively, at a luminance of 1000 cd m−2 (Table 3). Even at a very high luminance of 10000 cd m−2, an EQE10000 of 20.3% and a PE10000 of 52.0 lm W−1 were observed. To the best of our knowledge, these values are comparable to those of the best Ir(III)-OLEDs13–15,49 and Pt(II)-OLEDs36,37 at a high luminance of 1000–10000 cd m−2 without an out-coupling enhancement. The good OLED performance achieved in devices 7 and 8 indicates that our modified co-host device structure is a good general design for efficient [Pt(O^N^C^N)] complexes.
| Device no. | V on (V) | L max (cd m−2) | CEmaxc (cd A−1) | PEd (lm W−1) | EQEe (%) | Roll-offf (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max. | 1000 cd m−2 | 5000 cd m−2 | 10000 cd m−2 |
Max. | 1000 cd m−2 | 5000 cd m−2 | 10000 cd m−2 |
1000 cd m−2 | 5000 cd m−2 | 10000 cd m−2 |
||||
| a Turn-on voltage at 1 cd m−2. b Maximum luminance. c Maximum current efficiency. d Power efficiency. e External quantum efficiency. f (EQEmax − EQE)/EQEmax. | ||||||||||||||
| 6 | 2.4 | 116630 |
87.2 | 102.8 | 82.9 | 66.3 | 50.9 | 23.2 | 22.8 | 21.0 | 19.4 | 1.8 | 9.5 | 16.4 |
| 7 | 2.5 | 48340 |
82.9 | 100.2 | 62.9 | 33.6 | 20.0 | 21.3 | 19.2 | 15.7 | 11.2 | 9.8 | 26.2 | 47.4 |
| 8 | 2.5 | 82580 |
86.4 | 104.3 | 85.6 | 64.0 | 52.0 | 24.5 | 23.1 | 21.6 | 20.3 | 5.7 | 11.8 | 17.1 |
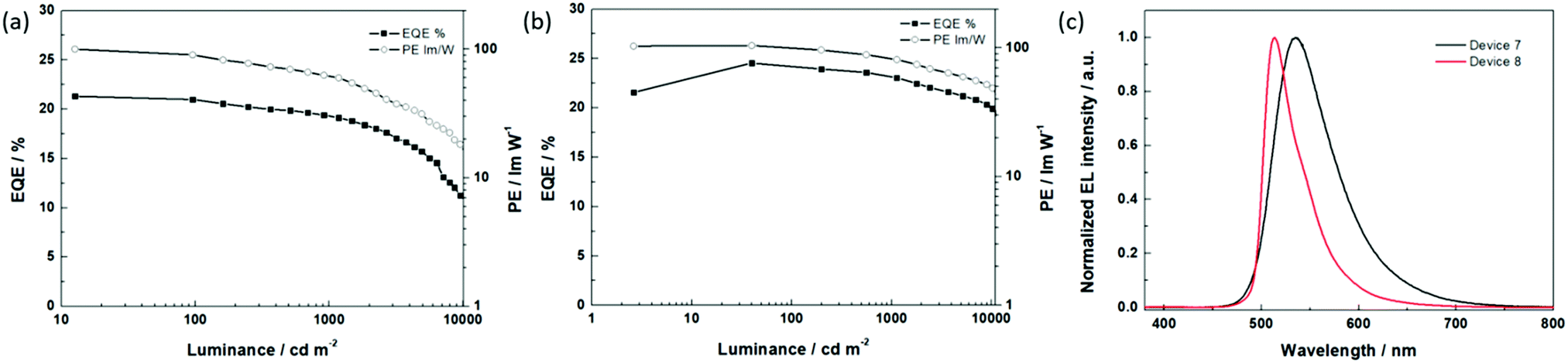 | ||
| Fig. 3 The EL characteristics of device 7 and device 8; (a) the EQE–PE–luminance characteristics of device 7; (b) the EQE–PE–luminance characteristics of device 8; and (c) the EL spectra of device 7 and device 8 at a luminance of 1000 cd m−2. | ||
The EQE value is mainly determined by the PLQY and out-coupling efficiency for properly designed phosphorescent OLEDs.23 The out-coupling efficiency is strongly influenced by the horizontal transition dipole moment of the EML and could be over 40% when the horizontal dipole ratio (Θ) of the EML is approaching unity.14 In our case, for the optimized OLEDs based on tetra-Pt-N, tetra-Pt-S and tetra-Pt-M, the out-coupling efficiencies were respectively 24.7, 23.9, and 25.9% as estimated from the EQE and PLQY values listed in Table 3 and Table S3 (ESI†).23 These out-coupling values are higher than the typical value of 20% for the devices with Lambert's emission distribution, attributed to the horizontally aligned co-host TCTA:B3PYMPM (Θ > 0.67) used.14,50
Preliminary studies on the operational stability of device 7 together with the ones with a single host of TCTA or B3PYMPM were undertaken. As depicted in Fig. S7 (ESI†), with an initial luminance of about 4000 cd m−2, the lifetime (LT50) of the device with the co-host EML (device 7) was longer than those of the devices with TCTA and B3PYMPM hosts by 12 and 5 times, respectively. The improved device lifetime could be the result of the broadened exciton recombine zone in the co-host EML of device 7. As shown in Fig. S8c and d (ESI†), an ultrathin (0.5 nm) layer of tetra-Pt-S was placed in different positions of the EML of OLEDs with a co-host or single host to detect the exciton distribution in these two kinds of devices.51–53 As depicted in Fig. S8a (ESI†), all EL spectra similarly originate from the emission of tetra-Pt-S without obvious emission from the host materials irrespective of the location of the ultra-thin layer of tetra-Pt-S in the EML, suggesting that the excitons are evenly distributed over the entire EML. As depicted in Fig. S8b (ESI†), pure tetra-Pt-S emission can only be observed when the ultra-thin layer of tetra-Pt-S is located at the EML/ETL interface. The emission of the TCTA host quickly increases when the ultra-thin layer moves to the EML/HTL interface, indicating that the density of excitons is the highest at the EML/ETL interface and decreases with the distance to this interface due to the hole-transporting property of TCTA.
3. Conclusions
We have optimized phosphorescent OLEDs based on three emissive [Pt(O^N^C^N)] complexes: tetra-Pt-N, tetra-Pt-S and tetra-Pt-M. By utilizing double-host EMLs and inserting a 10 nm-thick interlayer of B3PYMPM between the EML and the ETL of TMPPPYTZ, a high PEmax of 104.3 lm W−1 and an EQEmax of 24.5% were achieved in the device with tetra-Pt-M. Furthermore, the optimized OLEDs with tetra-Pt-N and tetra-Pt-M could achieve low-efficiency roll-off at a luminance of 10000 cd m−2, maintaining high EQEs of ≈20%. The diminished efficiency roll-off was attributed to the short emissive lifetimes of tetra-Pt-N and tetra-Pt-M as well as our modified double-host device structure with a broadened recombination zone in the EML, both of which could lower the exction density in the EML, thereby effectively suppressing TTA at high luminances. The excellent EL performance of these Pt(II)-OLEDs described in this work at practical luminances of 1000–10000 cd m−2 demonstrated that [Pt(O^N^C^N)] complexes are good candidates for high-brightness OLED applications, such as high-brightness OLED displays and lighting.
4. Experimental section
4.1. Photophysical measurements
All solvents for photophysical measurements were of HPLC grade. The thin films were fabricated by drop-casting from a chlorobenzene solution containing the Pt(II) complex and PMMA. The solvent was evaporated at room temperature and translucent films were obtained. Absorption spectra were measured on a Hewlett-Packard 8453 diode array spectrophotometer. Steady-state excitation and emission spectra were obtained on a SPEX Fluorolog-3 spectrophotometer and/or a Hamamatsu C11347 Quantaurus-QY Absolute PL quantum yield measurement system. Emission lifetime measurements were performed with a Quanta Ray DCR-3 pulsed Nd:YAG laser system (pulse output 355 nm, 8 ns).4.2. Device fabrication and characterization
Glass slides with a pre-patterned ITO electrode were employed as substrates for OLEDs and were cleaned in an ultrasonic bath with Decon 90 detergent and deionized water, rinsed with deionized water, cleaned in sequential ultrasonic baths of deionized water, acetone, and isopropanol, and subsequently dried in an oven for 1 h. OLEDs were fabricated in a Kurt J. Lesker SPECTROS vacuum deposition system with a base pressure of 10–8 mbar. In the vacuum chamber, organic materials were thermally deposited in sequence at a rate of 0.02–0.1 nm s−1. The doping process in the emitting layer was realized by co-deposition technology. Afterwards, LiF (1.2 nm) and Al (150 nm) were thermally deposited at rates of 0.012 and 0.2 nm s−1, respectively. Film thicknesses were determined in situ by calibrated oscillating quartz-crystal sensors. The luminance–current density–voltage characteristics (L–J–V), and EL spectra were simultaneously measured with a programmable Keithley model 2400 sourcemeter measurement unit and a Photoresearch PR-655 Spectrascan spectrorediometer. All devices were characterized at room temperature without encapsulation. The external quantum efficiency and power efficiency were calculated by assuming a Lambertian distribution. LiF was purchased from Sigma-Aldrich and MoO3, TAPC, TCTA, TMPYPB, TMPPPYTZ and B3PYMPM were purchased from Luminescence Technology Corp. All of these materials were used as received.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work is financially supported by the Innovation and Technology Fund (ITS/224/17FP) and Basic Research Program of Shenzhen (JCYJ20160229123546997, JCYJ20160530184056496, and JCYJ20170818141858021).Notes and references
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913–915 CrossRef CAS
.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS
- Y. Ma, H. Zhang, J. Shen and C. Che, Synth. Met., 1998, 94, 245–248 CrossRef CAS
- G. Cheng, G. K.-M. So, W.-P. To, Y. Chen, C.-C. Kwok, C. Ma, X. Guan, X. Chang, W.-M. Kwok and C.-M. Che, Chem. Sci., 2015, 6, 4623–4635 RSC
- T. Chiba, Y.-J. Pu and J. Kido, Adv. Mater., 2015, 27, 4681–4687 CrossRef CAS PubMed
- M. Cocchi, J. Kalinowski, D. Virgili, V. Fattori, S. Develay and J. A. G. Williams, Appl. Phys. Lett., 2007, 90, 163508 CrossRef
- J. Kalinowski, M. Cocchi, D. Virgili, V. Fattori and J. A. G. Williams, Adv. Mater., 2007, 19, 4000–4005 CrossRef CAS
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234–238 CrossRef CAS PubMed
- G. Zhou, Q. Wang, C.-L. Ho, W.-Y. Wong, D. Ma and L. Wang, Chem. Commun., 2009, 3574–3576 RSC
- C. Murawski, K. Leo and M. C. Gather, Adv. Mater., 2013, 25, 6801–6827 CrossRef CAS PubMed
- S. Reineke, M. Thomschke, B. Lüssem and K. Leo, Rev. Mod. Phys., 2013, 85, 1245 CrossRef CAS
- D. Zhang, J. Qiao, D. Zhang and L. Duan, Adv. Mater., 2017, 29, 1702847 CrossRef PubMed
- K.-H. Kim, C.-K. Moon, J.-H. Lee, S.-Y. Kim and J.-J. Kim, Adv. Mater., 2014, 26, 3844–3847 CrossRef CAS PubMed
- S.-Y. Kim, W.-I. Jeong, C. Mayr, Y.-S. Park, K.-H. Kim, J.-H. Lee, C.-K. Moon, W. Brütting and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 3896–3900 CrossRef CAS
- C.-Y. Kuei, W.-L. Tsai, B. Tong, M. Jiao, W.-K. Lee, Y. Chi, C.-C. Wu, S.-H. Liu, G.-H. Lee and P.-T. Chou, Adv. Mater., 2016, 28, 2795–2800 CrossRef CAS PubMed
- S. Lee, H. Shin and J.-J. Kim, Adv. Mater., 2014, 26, 5864–5868 CrossRef CAS PubMed
- H. Shin, S. Lee, K.-H. Kim, C.-K. Moon, S.-J. Yoo, J.-H. Lee and J.-J. Kim, Adv. Mater., 2014, 26, 4730–4734 CrossRef CAS PubMed
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS PubMed
- J. Lee, T.-H. Han, M.-H. Park, D. Y. Jung, J. Seo, H.-K. Seo, H. Cho, E. Kim, J. Chung, S.-Y. Choi, T.-S. Kim, T.-W. Lee and S. Yoo, Nat. Commun., 2016, 7, 11791 CrossRef CAS PubMed
- S.-H. Jeong, S.-H. Woo, T.-H. Han, M.-H. Park, H. Cho, Y.-H. Kim, H. Cho, H. Kim, S. Yoo and T.-W. Lee, NPG Asia Mater., 2017, 9, e411 CrossRef CAS
- S. R. Forrest, IEEE J Sel. Top. Quant., 2000, 6, 1072–1083 CAS
- C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky, M. E. Thompson and R. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622–1624 CrossRef CAS
- C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048–5051 CrossRef CAS
- K. Li, G. Cheng, C. Ma, X. Guan, W.-M. Kwok, Y. Chen, W. Lu and C.-M. Che, Chem. Sci., 2013, 4, 2630–2644 RSC
- M.-C. Tang, D. P.-K. Tsang, M. M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed
- G. Cheng, K. T. Chan, W.-P. To and C.-M. Che, Adv. Mater., 2014, 26, 2540–2546 CrossRef CAS PubMed
- P.-K. Chow, G. Cheng, G. S. M. Tong, C. Ma, W.-M. Kwok, W.-H. Ang, C. Y.-S. Chung, C. Yang, F. Wang and C.-M. Che, Chem. Sci., 2016, 7, 6083–6098 RSC
- S. Lee and C. W. Tang, J. Vac. Sci. Technol., B, 2011, 29, 062401 CrossRef
- S. W. Liu, X. W. Sun and H. V. Demir, AIP Adv., 2012, 2, 012192 CrossRef
- D. Zhang, L. Duan, Y. Li, H. Li, Z. Bin, D. Zhang, J. Qiao, G. Dong, L. Wang and Y. Qiu, Adv. Funct. Mater., 2014, 24, 3551–3561 CrossRef CAS
- S.-J. Su, Y. Takahashi, T. Chiba, T. Takeda and J. Kido, Adv. Funct. Mater., 2009, 19, 1260–1267 CrossRef CAS
- S.-J. Su, H. Sasabe, Y.-J. Pu, K. Nakayama and J. Kido, Adv. Mater., 2010, 22, 3311–3316 CrossRef CAS PubMed
- H. Ye, D. Chen, M. Liu, S.-J. Su, Y.-F. Wang, C.-C. Lo, A. Lien and J. Kido, Adv. Funct. Mater., 2014, 24, 3268–3275 CrossRef CAS
- G. Cheng, S. C. F. Kui, W.-H. Ang, M.-Y. Ko, P.-K. Chow, C.-L. Kwong, C.-C. Kwok, C. Ma, X. Guan, K.-H. Low, S.-J. Su and C.-M. Che, Chem. Sci., 2014, 5, 4819–4830 RSC
- P.-K. Chow, G. Cheng, G. S. M. Tong, W.-P. To, W.-L. Kwong, K.-H. Low, C.-C. Kwok, C. Ma and C.-M. Che, Angew. Chem., Int. Ed., 2015, 54, 2084–2089 CrossRef CAS PubMed
- T. Fleetham, J. Ecton, Z. Wang, N. Bakken and J. Li, Adv. Mater., 2013, 25, 2573–2576 CrossRef CAS PubMed
- T. Fleetham, G. Li, L. Wen and J. Li, Adv. Mater., 2014, 26, 7116–7121 CrossRef CAS PubMed
- G. Li, T. Fleetham and J. Li, Adv. Mater., 2014, 26, 2931–2936 CrossRef CAS PubMed
- J. Soellner, M. Tenne, G. Wagenblast and T. Strassner, Chem. – Eur. J., 2016, 22, 9914–9918 CrossRef CAS PubMed
- K. H. Kim, J.-L. Liao, S. W. Lee, B. Sim, C.-K. Moon, G.-H. Lee, H. J. Kim, Y. Chi and J.-J. Kim, Adv. Mater., 2016, 28, 2526–2532 CrossRef CAS PubMed
- K. T. Ly, R.-W. Chen Cheng, H.-W. Lin, Y.-J. Shiau, S.-H. Liu, P.-T. Chou, C.-S. Tsao, Y.-C. Huang and Y. Chi, Nat. Photonics, 2017, 11, 63–69 CrossRef
- Q. Wang, I. W. H. Oswald, X. Yang, G. Zhou, H. Jia, Q. Qiao, Y. Chen, J. Hoshikawa-Halbert and B. E. Gnade, Adv. Mater., 2014, 26, 8107–8113 CrossRef CAS PubMed
- V. N. Kozhevnikov, B. Donnio, B. Heinrich, J. A. G. Williams and D. W. Bruce, J. Mater. Chem. C, 2015, 3, 10177–10187 RSC
- M. Bachmann, D. Suter, O. Blacque and K. Venkatesan, Inorg. Chem., 2016, 55, 4733–4745 CrossRef CAS PubMed
- S. C. F. Kui, P. K. Chow, G. S. M. Tong, S.-L. Lai, G. Cheng, C.-C. Kwok, K.-H. Low, M. Y. Ko and C.-M. Che, Chem. – Eur. J., 2013, 19, 69–73 CrossRef CAS
- S. C. Kui, P. K. Chow, G. Cheng, C.-C. Kwok, C. L. Kwong, K.-H. Low and C.-M. Che, Chem. Commun., 2013, 49, 1497–1499 RSC
- H. Sasabe, D. Tanaka, D. Yokoyama, T. Chiba, Y.-J. Pu, K. Nakayama, M. Yokoyama and J. Kido, Adv. Funct. Mater., 2011, 21, 336–342 CrossRef CAS
- S.-J. Su, T. Chiba, T. Takeda and J. Kido, Adv. Mater., 2008, 20, 2125–2130 CrossRef CAS
- Y.-H. Zhou, Q.-L. Xu, H.-B. Han, Y. Zhao, Y.-X. Zheng, L. Zhou, J.-L. Zuo and H. Zhang, Adv. Opt. Mater., 2016, 4, 1726–1731 CrossRef CAS
- Y.-S. Park, S. Lee, K.-H. Kim, S.-Y. Kim, J.-H. Lee and J.-J. Kim, Adv. Funct. Mater., 2013, 23, 4914–4920 CrossRef CAS
- L. Zhu, Y. Zhao, H. Zhang, J. Chen and D. Ma, J. Appl. Phys., 2014, 115, 244512 CrossRef
- K. Udagawa, H. Sasabe, F. Igarashi and J. Kido, Adv. Opt. Mater., 2016, 4, 86–90 CrossRef CAS
- C. Cai, S.-J. Su, T. Chiba, H. Sasabe, Y.-J. Pu, K. Nakayama and J. Kido, Org. Electron., 2011, 12, 843–850 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, thermal gravimetric analysis, electrochemical and photophysical data of tetra-Pt-N, tetra-Pt-S and tetra-Pt-M and device data of OLEDs with tetra-Pt-N. See DOI: 10.1039/c9tc00682f |
| ‡ M. Mao and J. Peng contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |