
A two-pronged anti-leukemic agent based on a hyaluronic acid–green tea catechin conjugate for inducing targeted cell death and terminal differentiation†
Kun
Liang
 a,
Ki Hyun
Bae
a,
Akiko
Nambu
b,
Bibek
Dutta
b,
Joo Eun
Chung
a,
Motomi
Osato
abcd and
Motoichi
Kurisawa
*a
a,
Ki Hyun
Bae
a,
Akiko
Nambu
b,
Bibek
Dutta
b,
Joo Eun
Chung
a,
Motomi
Osato
abcd and
Motoichi
Kurisawa
*a
aInstitute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669. E-mail: mkurisawa@ibn.a-star.edu.sg
bCancer Science Institute of Singapore, National University of Singapore, 14 Medical Drive, 117599 Singapore
cInternational Research Center for Medical Sciences, Kumamoto University, 2-2-1 Honjo, Chuo-ku, Kumamoto City, 860-0811 Japan
dDepartment of Paediatrics, Yong Loo Lin School of Medicine, National University of Singapore, 1E Kent Ridge Road, NUHS Tower Block, Level 12, 119228 Singapore
First published on 13th November 2019
Abstract
Acute myeloid leukemia (AML) is an aggressive malignancy that leads to a poor prognosis even with intensive chemotherapy. As the key feature of AML is the blockade of hematopoietic cell maturation, considerable attention has been paid to ‘differentiation therapy’ aimed at transforming AML cells into more mature, benign phenotypes using pharmacological agents. Here we report a hyaluronic acid–(−)-epigallocatechin-3-O-gallate (HA–EGCG) conjugate as a unique anti-leukemic agent, capable of selectively killing AML cells as well as promoting their terminal differentiation into monocytes and granulocytes. This ‘two-pronged’ effect of the HA–EGCG conjugate was demonstrated in two different AML cell lines (NB4 and HL60), but absent in a physical mixture (HA + EGCG), highlighting the importance of HA conjugation for targeting of EGCG moieties to AML cells. Moreover, administration of the HA–EGCG conjugate not only suppressed AML progression, but also prolonged survival in the HL60 xenograft mouse model. Our study suggests new opportunities for designing two-pronged anti-leukemic agents for more effective AML treatment.
Introduction
Acute myeloid leukemia (AML) is a malignant blood disorder with a poor prognosis. The 5-year survival rate of an adult with AML is only about 50% or lower.1 AML is generally characterized by the overproduction of myeloid blast cells that suffer from blockage in their differentiation pathways, which leads to the crowding out of normal blood cells and platelets. The stages in which the differentiation of AML blast cells is arrested define the subtypes of AML (AML-M0 to M7). Currently, the standard clinical treatment for AML is chemotherapy with or without hematopoietic stem cell transplantation involving administration of a combination of cytarabine and anthracycline, although the clinical outcomes have been marginal with a high risk of relapse.2In recent years, another therapeutic regimen has been implemented for acute promyelocytic leukemia (APL), the M3 subtype of AML. APL is typically characterized by the translocation of chromosomes 15 and 17: t(15;17)(q22;q21), resulting in the fusion of the retinoic acid receptor α gene (RARα) on chromosome 17 to the promyelocytic leukemia (PML) gene on chromosome 15, which subsequently causes differentiation blockage.3,4 Interestingly, it was discovered that these APL cells could be released from differentiation arrest using all-trans retinoic acid (ATRA), an analogue of vitamin A.5,6 This important finding led to the introduction of an alternative approach to APL treatment using ATRA to induce differentiation.7,8 The current combination regimen of ATRA-based differentiation therapy with arsenic trioxide-based chemotherapy has transformed AML-M3 to become the subtype with the best prognosis with a complete remission rate of over 90% and a dramatic elevation in the 5-year survival rate of up to 85%.8,9 However, the availability of such differentiation-inducing agents for AML to date has been limited and they have been found to elicit poor efficacy in other subtypes.10,11 Furthermore, studies citing ATRA resistance have also lately surfaced,12,13 highlighting the need to develop alternative treatment options for effective AML therapy.
Here we report a hyaluronic acid–(−)-epigallocatechin-3-O-gallate (HA–EGCG) conjugate for the treatment of AML via a two-pronged approach: targeted eradication and concurrent induction of terminal differentiation of myeloid blast cells. HA is a linear non-sulfated glycosaminoglycan frequently used in biomedical applications because of its non-immunogenicity, biodegradability and biocompatibility.14–16 More importantly, HA has been exploited as a targeting ligand for the cell surface receptor CD44,17,18 which is commonly overexpressed on leukemic blast cells in all AML subtypes.19 On the other hand, EGCG is a polyphenolic compound that constitutes the major component of green tea extract. Multiple studies have reported the beneficial biological effects of EGCG, including antioxidant, anti-inflammatory and anti-cancer properties.20–22 Particularly for AML treatment, EGCG has attracted significant interest because it exerts anti-proliferative and apoptosis-triggering activity against leukemic cells, while minimally affecting normal cells.23,24 Meanwhile, efforts have also been made recently to study the effect of EGCG in modulating AML cell differentiation. Britschgi et al. reported that EGCG boosted ATRA-induced neutrophil differentiation by increasing the expression of death-associated protein kinase 2 (DAPK2).25 Borutinskaitė et al. found that EGCG triggered the terminal maturation of AML cells via epigenetic modulations of genes involved in differentiation and cell cycle arrest.26 In addition, Moradzadeh et al. observed that EGCG enhanced APL cell differentiation via suppression of the PML-RARα gene.27 Despite these desirable features of EGCG, the lack of leukemia-targeting ability has limited its further application in AML therapy. We hypothesized that targeted eradication and differentiation induction of AML cells would be achieved by conjugating EGCG moieties to HA, which could serve as an AML-targeting ligand via HA-CD44 interactions (Fig. 1). To explore the potential of the HA–EGCG conjugate for AML therapy, we investigated its cytotoxicity, anti-clonogenic activity and differentiation-inducing effect on two different AML cell lines (NB4 and HL60), and further examined its anti-leukemic efficacy in the HL60 orthotopic mouse model.
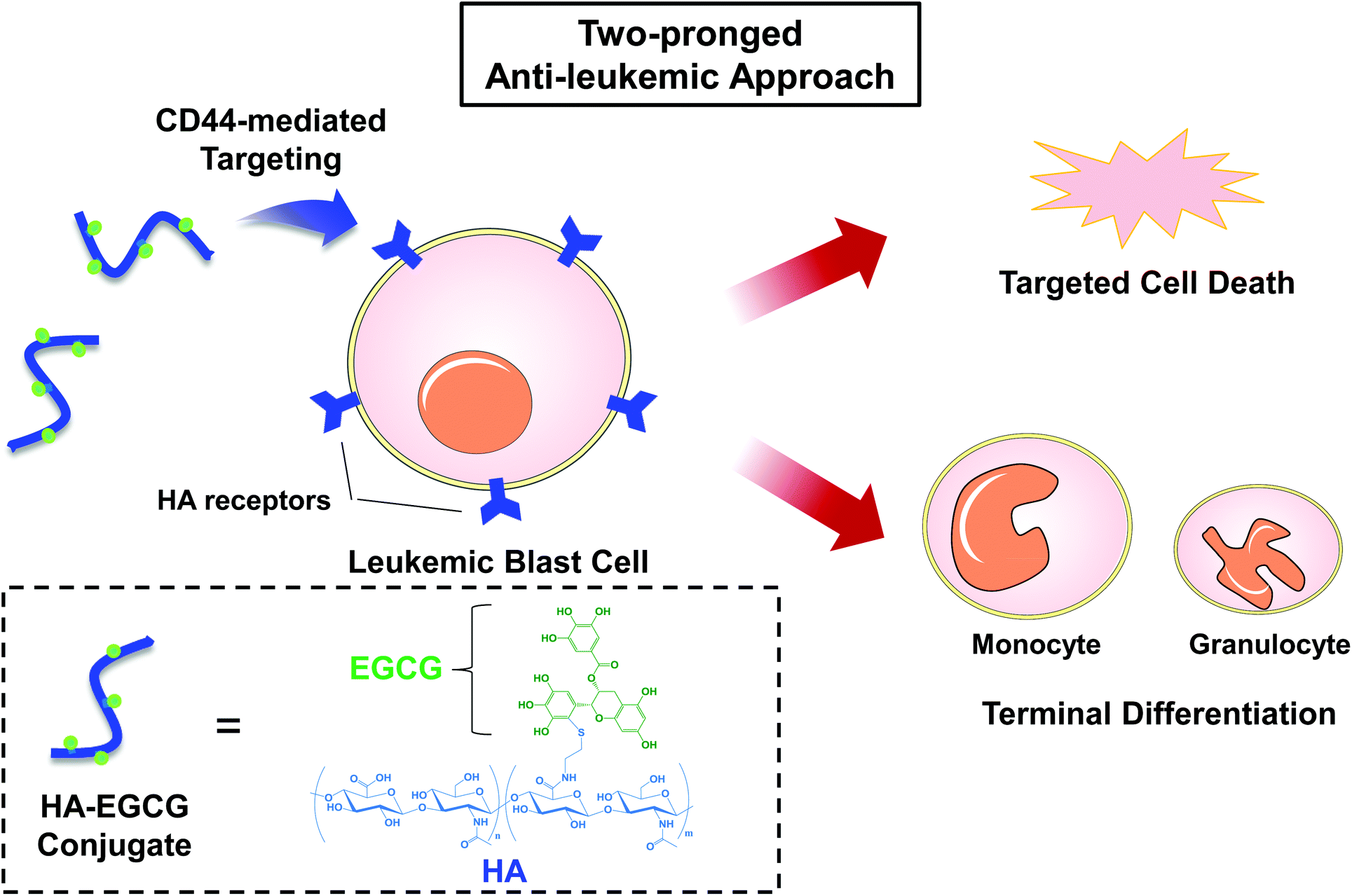 | ||
| Fig. 1 Proposed scheme of AML therapy using the HA–EGCG conjugate. Targeting of AML cells is achieved by binding of HA to CD44 receptors overexpressed on the cell surface. The HA–EGCG conjugate imparts an anti-leukemic effect by a combination of two effects – triggering targeted death of the leukemic blast cells and inducing their terminal differentiation into monocytes or granulocytes. | ||
Experimental
Materials
EGCG (minimum purity of 90%, TEAVIGO™) was obtained from DMS Nutritional Products Ltd (Basel, Switzerland). HA (average Mw = 90 kDa) was donated by JNC Corporation (Tokyo, Japan). Cystamine dihydrochloride was obtained from Merck Millipore Corporation (Darmstadt, Germany). 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were obtained from Tokyo Chemical Industry (Tokyo, Japan). ATRA was provided by Sigma-Aldrich (St Louis, MO, USA). Mouse anti-human antibody CD44 (Bu52), isotype control antibody and fluorescein isothiocyanate (FITC)-labelled secondary antibody were acquired from Bio-Rad Laboratories (Hercules, CA, USA). Fluorescently labelled mouse anti-human antibodies including FITC-conjugated CD11b (ICRF-44), APC/Cy7-conjugated CD14 (HCD14) and PE/Cy5-conjugated CD15 (SSEA-1) were obtained from Biolegend (San Diego, CA, USA).Synthesis of the HA–EGCG conjugate
The HA–EGCG conjugate was prepared by a two-step procedure established in our laboratory.28 In brief, firstly thiolated HA was prepared by introducing free thiol groups to the HA backbone. One gram of HA (2.5 mmol –COOH) was reacted with DMTMM (1.037 g, 3.75 mmol) and cystamine dihydrochloride (844.5 mg, 3.75 mmol) in 100 mL of phosphate buffer saline (PBS) (pH 7.4). After stirring for 24 h, 0.5 M TCEP solution (pH 7, 15 mL) was slowly added. The mixture was then stirred for another 2 h. The resulting solution was dialyzed (Mw cut-off: 3500 Da) using 0.1 M NaCl solution for 2 d, 25% ethanol for 1 d and deionized water for 2 d, successively, and then freeze-dried to produce thiolated HA. In the second step, thiolated HA (0.5 g) pre-dissolved in 70 mL of nitrogen-purged PBS (pH 7.4) was added dropwise to 30 mL of PBS (pH 7.4) which contained 1.55 g of EGCG. After adjustment of the pH value to 7.4, the solution was stirred for 3 h before adjusting the final pH value to 6. Dialysis of the solution (Mw cut-off: 3500 Da) was performed firstly against 25% ethanol for 1 d and then against deionized water for 2 d under nitrogen-purged conditions. Subsequently, the solution was freeze-dried to produce the HA–EGCG conjugate (∼95% yield). The degree of substitution (i.e. the average number of EGCG moieties per 100 disaccharide units of HA) was determined to be 6.6.Cell lines
The human AML cell lines HL60 and NB4, human embryonic kidney cell line HEK293 and primary human umbilical vein endothelial (HUVEC) cells were purchased from the American Type Culture Collection (Manassas, VA, USA). All AML cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 units per mL penicillin and 0.1 mg mL−1 streptomycin. The cells were maintained at a density of 2 × 105 to 1 × 106 cells per mL. HEK293 cells were cultured in DMEM medium supplemented with 10% FBS, while HUVEC cells were maintained in F-12K medium supplemented with 0.1 mg mL−1 heparin, 0.03–0.05 mg mL−1 endothelial cell growth supplement (ECGS) and 10% FBS. All cells were maintained in a humid incubator under 5% CO2 at 37 °C.Quantitative assessment of CD44 expression in cells
To elucidate the CD44 expression levels in various cells, 5 × 105 cells were suspended in PBS (pH 7.4) containing 0.2% (v/v) bovine serum albumin (BSA), and incubated with either anti-human CD44 antibody or isotype control antibody (2 μg mL−1) for 20 min at 4 °C. Subsequently, the cells were washed with cold PBS (pH 7.4) containing 0.2% (v/v) BSA and further incubated with FITC-tagged secondary antibody for another 20 min. The cells were then washed again and analyzed by flow cytometry using a fluorescence-activated cell sorter BD LSR II (BD Biosciences, San Jose, CA, USA).Evaluation of cellular binding of the HA–EGCG conjugate
For cellular binding analysis, the HA–EGCG conjugate was labelled with the fluorophore DyLight 488, according to the previous report.28 Briefly, 5 × 105 cells were seeded in 6-well plates followed by incubation with the DyLight 488-labelled HA–EGCG conjugate (500 μg mL−1) for 30 min at 25 °C. The cells were harvested and washed twice with cold PBS (pH 7.4) containing 0.2% (v/v) BSA, and then analyzed by flow cytometry using a fluorescence-activated cell sorter BD LSR II (BD Biosciences, San Jose, CA, USA).In vitro cell viability assay
All cells (HL60, NB4, HEK293 and HUVEC cells) were seeded at 1 × 104 cells per 100 mL per well in 96-well plates. All AML cells were incubated for 2 h, while HEK293 cells and HUVEC cells were allowed to attach overnight prior to drug treatments. Subsequently, the cells were treated with various concentrations of HA, EGCG, HA + EGCG mixture, and HA–EGCG conjugate for designated time-points. After drug treatments, the cell viability was evaluated using the CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega, Madison, WI, USA) following the manufacturer's instructions. The luminescence from each well was measured using a Tecan Infinite microplate reader (Tecan Group, Switzerland). The reported cell viability was derived based on the luminescence intensity, expressed as a percentage of the treated cells relative to untreated cells. All measurements were performed in triplicate.Colony forming assay
The colony forming assay was performed using HL60 and NB4 cells. Five hundred cells were seeded in 35 mm dishes in MethoCult H4434 Classic methylcellulose medium (StemCell Tec., Vancouver, BC, Canada) containing 1% antibiotic–antimycotic. The cells were subsequently treated with various concentrations of HA, EGCG, HA + EGCG mixture, and HA–EGCG conjugate. ATRA (1 μM) was also tested as a positive control. The number of colonies was determined using an inverted microscope after 14 days of culture. Colonies consisting of at least 40 cells were taken into consideration.Quantitative evaluation of the differentiation-inducing effect of the HA–EGCG conjugate on AML cells
AML cells were seeded at 1 × 105 cells per 1 mL per well in 24-well plates, and incubated for 2 h under 5% CO2 at 37 °C. The cells were then treated with the HA–EGCG conjugate at a concentration of 250 μg mL−1, together with equivalent concentrations of HA, EGCG, and HA + EGCG mixture. ATRA (1 μM) was included as a positive control. After three days of incubation, the cells were harvested and examined for signs of differentiation by assessing the cell-surface antigen expression. To label the cell surface antigens, the cells were suspended in PBS (pH 7.4) containing 0.2% (v/v) BSA and then incubated at 4 °C for 30 min with mouse anti-human FITC-conjugated CD11b, Cy7-conjugated CD14 and Cy5-conjugated CD15 antibodies (2 μg mL−1 each). The mouse IgG1 isotype antibody was used as a control. Afterwards, the cells were washed three times with PBS containing 0.2% (v/v) BSA and the extent of antibody binding was determined by flow cytometry using a fluorescence-activated cell sorter BD LSR II (BD Biosciences, San Jose, CA, USA). Each measurement included at least 1 × 104 cells and the analyses were considered informative when adequate numbers of events (>100) were collected in the enumeration gates. The percentages of stained cells were determined and compared to appropriate negative controls.Assessment of in vivo anti-leukemic efficacy of the HA–EGCG conjugate in an HL60 orthotopic mouse model
All animal procedures were performed in accordance with the National Advisory Committee for Laboratory Animal Research (NACLAR) guidelines and approved by the Singapore Biological Resource Centre's Institutional Animal Care and Use Committee. Anti-leukemic efficacy was evaluated using a previously developed AML orthotopic model based on non-obese diabetic (NOD)/LtSz-severe combined immunodeficiency (SCID) IL2Rγnull (NSG) mice.29,30 NSG male mice (6–8 weeks old) were irradiated with a sub-lethal dose of 2.5 Gy (60 cGy min−1) from a photon radiation source 24 h prior to the administration of 2 × 106 HL60 cells by intravenous injections via lateral tail veins. The mice were then treated with intravenous injections (200 μL) of either sterile PBS as the control or HA–EGCG solution (5 mg mL−1) 3 times a week for a total of 5 weeks. To obtain white blood cell counts, peripheral blood was collected by facial vein bleeding at designated time-points. Thirty microliters of blood were collected in heparin-coated tubes, and subsequently analyzed using a HEMAVET® 950FS hematology counter (Erba Diagnostic, Miami Lakes, FL, USA). At the end of the study, the mice were sacrificed and their spleens were collected and weighed. The body weight of the mice was measured during the experiment as an indicator of systemic toxicity. The mice were monitored bi-weekly for symptoms of disease (scruffy fur, tumor-like lumps, weakness and reduced mobility) and all mice showing any signs of distress were euthanized.Statistical analysis
Statistical analysis between two groups was conducted by unpaired Student's t-test, while statistical differences among multiple groups were examined using one-way ANOVA and Tukey post-hoc tests, where a P value smaller than 0.05 was regarded statistically significant. Survival analysis was performed using the Kaplan–Meier estimator and the log rank test was subsequently used to calculate the P value.Results and discussion
Cytotoxicity and anti-clonogenic activity of the HA–EGCG conjugate on AML cells
To date, the M3 subtype has been found to be the most amenable for differentiation therapy among all AML subtypes, since a majority of M3 cells carry a genetic mutation that generates the PML-RARα fusion protein, making them sensitive to ATRA.31,32 Henceforth, in this study, an AML-M3 cell line NB4 was selected in order to compare the efficacy of the HA–EGCG conjugate against an established differentiation agent such as ATRA. The efficacy of the HA–EGCG conjugate was also evaluated on another AML cell line, HL60 of the M2 subtype, which was arrested at an earlier stage of differentiation than M3.33 To exploit HA targeting to these AML cells, the expressions of CD44 in both NB4 and HL60 cells were first evaluated by flow cytometry. Elevated levels of expression of CD44 were observed in both cells (Fig. S1†) in accordance with the literature,19 indicating that these cells would be amenable for HA–EGCG therapy.To assess the leukemia-selective toxicity of the HA–EGCG conjugate, we used two normal cell types – human embryonic kidney cells (HEK293) and human umbilical vein endothelial cells (HUVEC) – for comparison. The HA–EGCG conjugate and EGCG were found to reduce the viabilities of all cell types in a concentration-dependent manner. Interestingly, the HA–EGCG conjugate demonstrated greater toxicity than EGCG in NB4 and HL60 cells after 72 h of treatment, whereas a reverse trend was observed in HEK293 and HUVEC cells (Fig. 2A). Furthermore, the HA–EGCG conjugate at a fixed concentration of 500 μg mL−1 dramatically reduced the cell viabilities of both AML cells by more than 98%. In contrast, 84.7 ± 1.2% of HEK293 cells and 73.3 ± 4.6% of HUVEC cells remained viable upon HA–EGCG treatment (Fig. 2B). Collectively, these results demonstrated the selectivity of HA–EGCG toxicity directed towards AML cells.
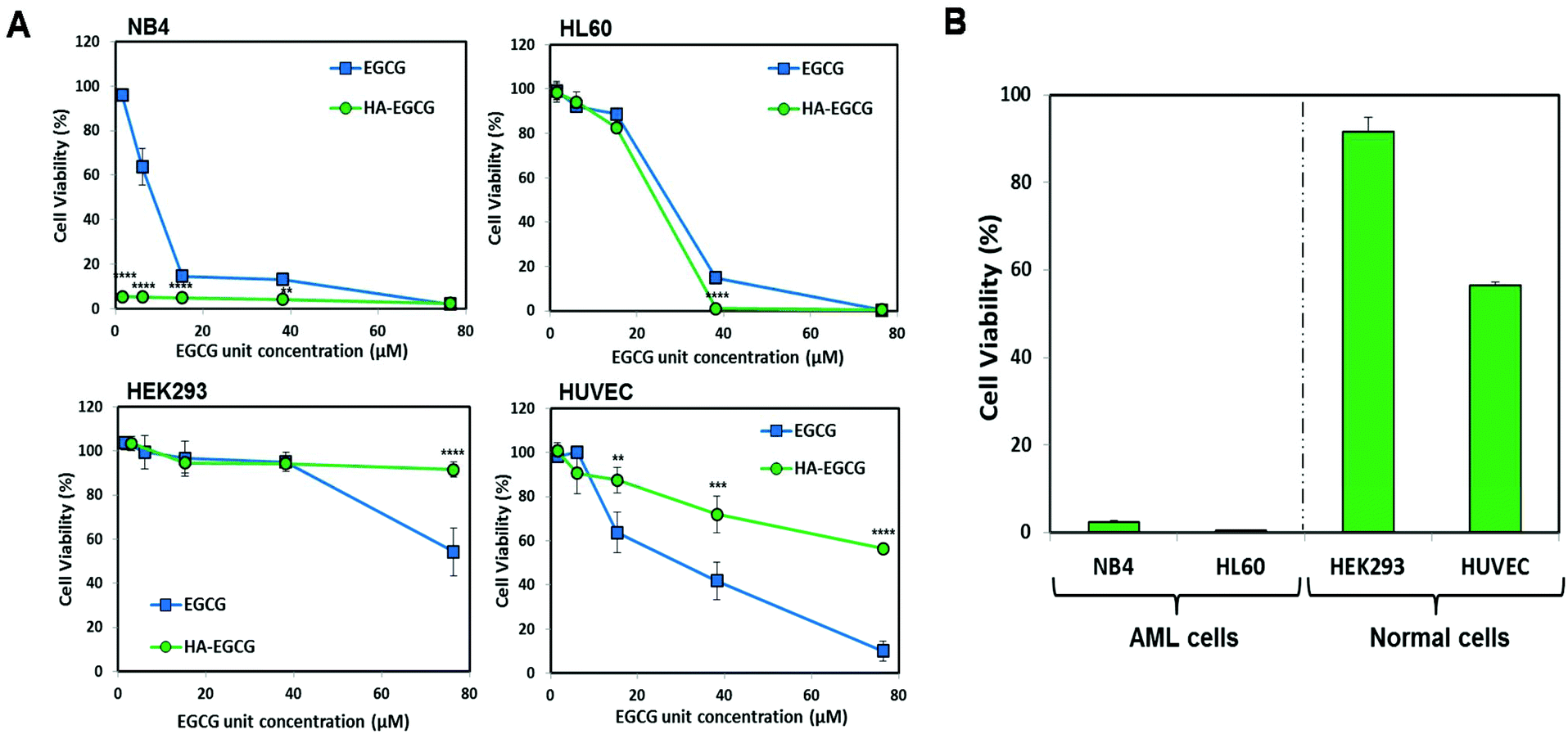 | ||
| Fig. 2 (A) The HA–EGCG conjugate was more toxic than EGCG alone at equivalent concentrations towards AML (NB4 and HL60) cells after 72 h of treatment, while the trend was reversed in normal (HEK293 and HUVEC) cells. (B) Viability of AML and normal cells after 72 h of treatment with the HA–EGCG conjugate at a EGCG unit concentration of 38 μM (HA–EGCG = 500 μg mL−1). Data shown are the means ± s.d.; n = 3. **P < 0.001; ***P < 0.001; ****P < 0.0001. | ||
To determine whether the conjugation between HA and EGCG was critical for the observed anti-leukemic activity of the HA–EGCG conjugate, we compared the cytotoxicity of the HA–EGCG conjugate with a physical mixture (HA + EGCG) and the individual constituents HA and EGCG at equivalent concentrations. The HA–EGCG conjugate exerted the greatest anti-leukemic activity among the four samples (Fig. 3A and B), leading to the decrease in the cell viabilities of NB4 and HL60 cells to 4.93 ± 0.38% and 0.87 ± 0.17% at 48 h, respectively (P < 0.001 versus all other groups). While EGCG alone and the HA + EGCG mixture exhibited similar moderate cytotoxicities, HA alone had little influence on cell viabilities, indicating that EGCG was mainly accountable for the observed cytotoxicity against AML cells. The enhanced cytotoxicity of the HA–EGCG conjugate relative to the HA + EGCG mixture suggests that coupling of HA to EGCG moieties improved their anti-leukemic activity possibly by targeting them towards AML cells via HA-CD44 interactions. To further clarify the CD44 targeting ability of the HA–EGCG conjugate, we investigated the binding of the DyLight 488-labelled HA–EGCG conjugate to cells expressing different levels of CD44 (Fig. S2†). Notably, the DyLight 488-labelled HA–EGCG conjugate exhibited a greater extent of binding to high CD44-expressing HL60 and NB4 cells as compared to HEK293 cells expressing very low levels of CD44 (Fig. S3 and Table S1†), suggesting that the preferential anti-leukemic activity of the HA–EGCG conjugate was likely due to its enhanced binding to AML cells via HA-CD44 interactions.
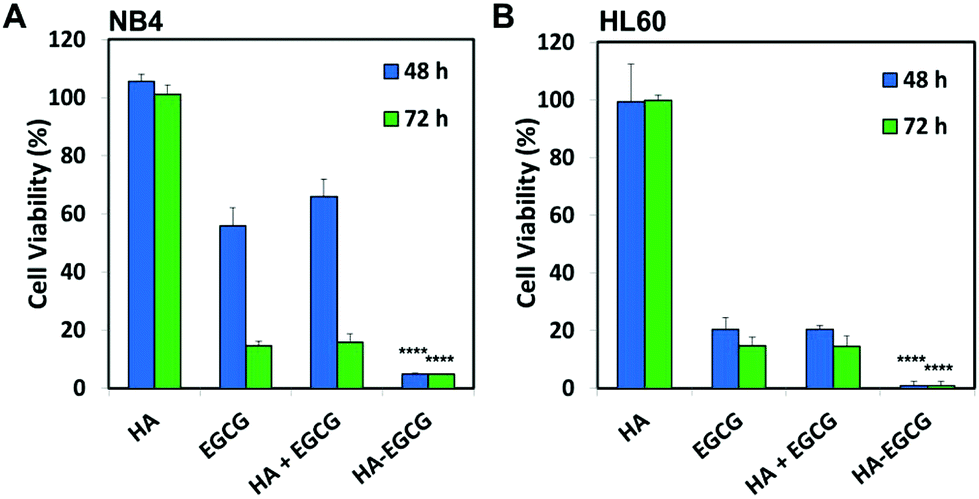 | ||
| Fig. 3 The HA–EGCG conjugate was more effective in killing both AML cell types (A) NB4 (40 μg mL−1) and (B) HL60 cells (10 μg mL−1) as compared to a physical mixture (HA + EGCG) and the individual constituents HA and EGCG at equivalent concentrations at both 48 and 72 h. Data shown are the means ± s.d.; n = 3. ****P < 0.0001 versus all other treatment groups. | ||
The effect of the HA–EGCG conjugate on clonogenic leukemic cells was further examined by the colony formation assay. These colony-forming leukemic cells belong to a subset of leukemic cells which possess high proliferative potential and self-renewal capacity, making them an important target for AML therapy.34,35 The HA–EGCG conjugate was the most effective at inhibiting colony formation in both NB4 and HL60 cells (Fig. 4A and B), which was in agreement with the result from the cytotoxicity assay. Although HA alone caused a slight reduction in the colony number, its effect was not as pronounced as that of the HA + EGCG mixture and EGCG alone, supporting the major role of EGCG in imparting toxicity. Overall, these findings indicated that the HA–EGCG conjugate was potent in killing AML cells and inhibiting their clonogenicity.
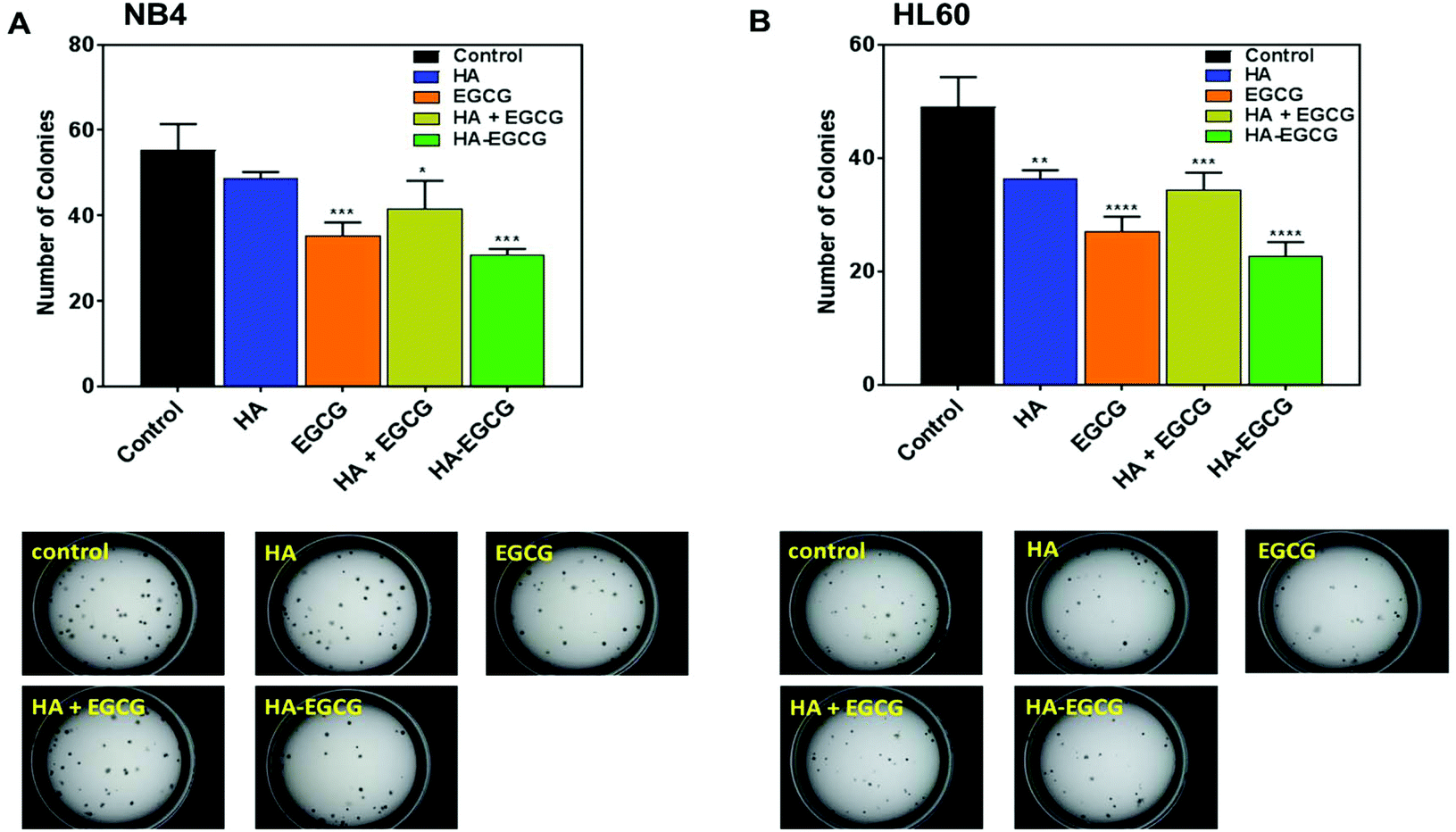 | ||
| Fig. 4 The HA–EGCG conjugate inhibited colony formation in (A) NB4 cells and (B) HL60 cells. Both AML cells were treated with the HA–EGCG conjugate (50 μg mL−1), HA + EGCG mixture, or HA and EGCG alone at equivalent concentrations. The microscopic images (bottom panel) depict the distribution of the colonies on day 14. Data shown are the means ± s.d.; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus control. | ||
Differentiation-inducing effect of the HA–EGCG conjugate on AML cells
To investigate the ability of the HA–EGCG conjugate to induce terminal differentiation in AML cells, we examined the expressions of three cell-surface antigens CD11b (common myeloid marker), CD14 (monocyte) and CD15 (granulocyte) following treatment with the HA–EGCG conjugate. As a positive control, the clinically approved differentiation-inducing agent ATRA was also used at its pharmacological concentration (1 μM).6,31 Flow cytometry analysis revealed that HA–EGCG treatment dramatically increased the population of NB4 cells expressing both CD14 and CD11b, indicating their maturation into monocyte-like cells (Fig. 5A). The proportion of such CD14+CD11b+ cells in the HA–EGCG group (35.6 ± 0.8%) was significantly higher than in all other treatment groups, including ATRA (Fig. 5B). While the HA + EGCG mixture and EGCG alone caused a moderate increase in the CD14+CD11b+ NB4 cell population, HA alone had only a marginal effect, suggesting that EGCG was mainly responsible for the induction of terminal differentiation. This finding was consistent with various previous reports citing EGCG-mediated differentiation of AML cells27,36 and augmentation of differentiation-inducing capability of ATRA.25 Similarly, a prominent population of NB4 cells expressing both CD15 and CD11b (Fig. 5C) appeared upon HA–EGCG treatment. The population of CD15+CD11b+ NB4 cells, indicative of granulocyte-like cells, was also significantly greater (71.5 ± 1.1%) in the HA–EGCG group than in all other groups (Fig. 5D). Impressively, the HA–EGCG conjugate was even more effective in inducing the terminal differentiation of NB4 cells than the clinically used agent ATRA, as indicated by 4- and 3-fold enhancement in the proportion of both CD14+CD11b+ (monocyte-like) and CD15+CD11b+ (granulocyte-like) cells respectively.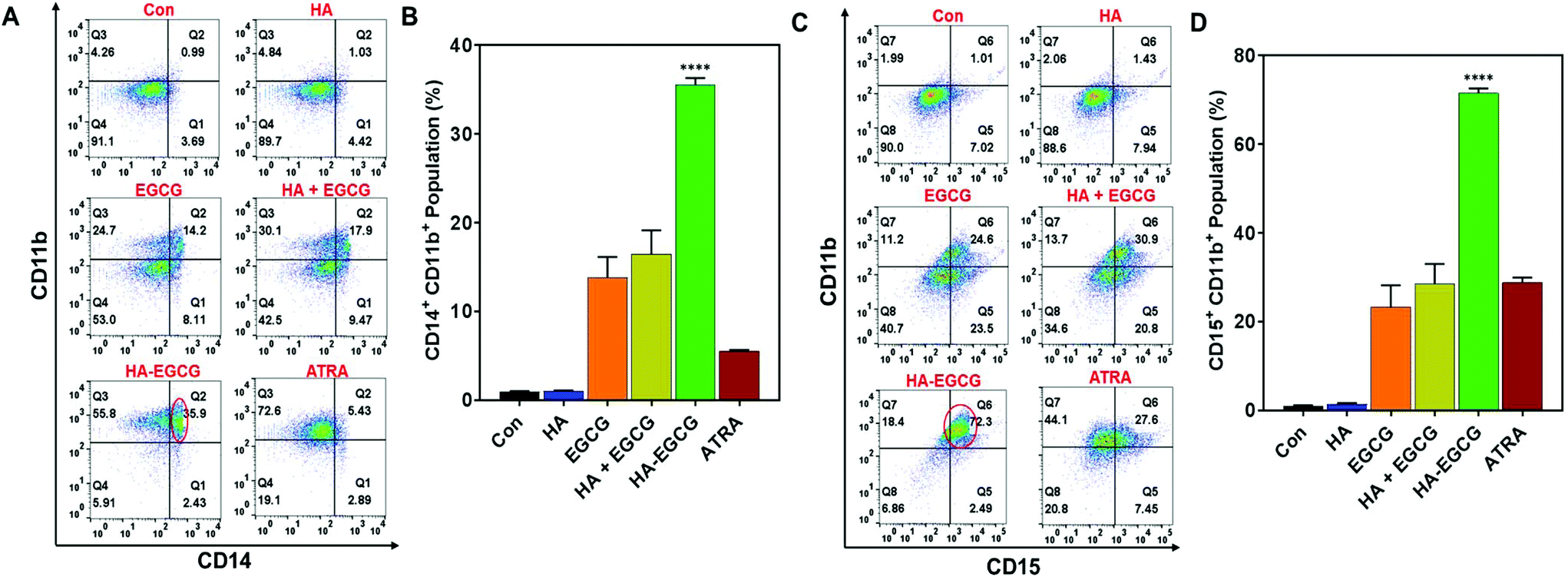 | ||
| Fig. 5 The HA–EGCG conjugate induced terminal differentiation of NB4 cells. (A) HA–EGCG treatment led to the emergence of a population of cells expressing both CD14 and CD11b (CD14+CD11b+), (B) which was significantly greater than in all other treatment groups. (C) HA–EGCG treatment also led to the emergence of a population of cells expressing both CD15 and CD11b (CD15+CD11b+), (D) which was significantly greater than in all other treatment groups. The populations in the flow cytometry analyses are indicated by red circles. Data shown are the means ± s.d.; n = 3. ****P < 0.0001 versus all other treatment groups. | ||
The differentiation-inducing effect of the HA–EGCG conjugate was also assessed in HL60 cells of the M2 subtype. We noted that the HA–EGCG conjugate induced terminal differentiation of HL60 cells into monocyte-like cells, as evident from the distinct emergence of CD14+CD11b+ HL60 cell population (8.29 ± 0.21%), which is more pronounced than all other groups, including ATRA (Fig. 6A and B). Moreover, HA–EGCG treatment led to a marked increase in granulocyte-like CD15+CD11b+ HL60 cells (5.35 ± 0.11%) when compared to all other groups (Fig. 6C and D). However, HL60 cells exhibited an attenuated response to HA–EGCG treatment as compared to NB4 cells probably due to the more “primitive” nature of the HL60 cells, which were obstructed at an earlier stage of maturation,33 making their differentiation more challenging. Collectively, these results revealed that the HA–EGCG conjugate has a superior capability to induce monocytic and granulocytic differentiation of AML cells than the HA + EGCG mixture and individual constituents HA and EGCG, as well as ATRA.
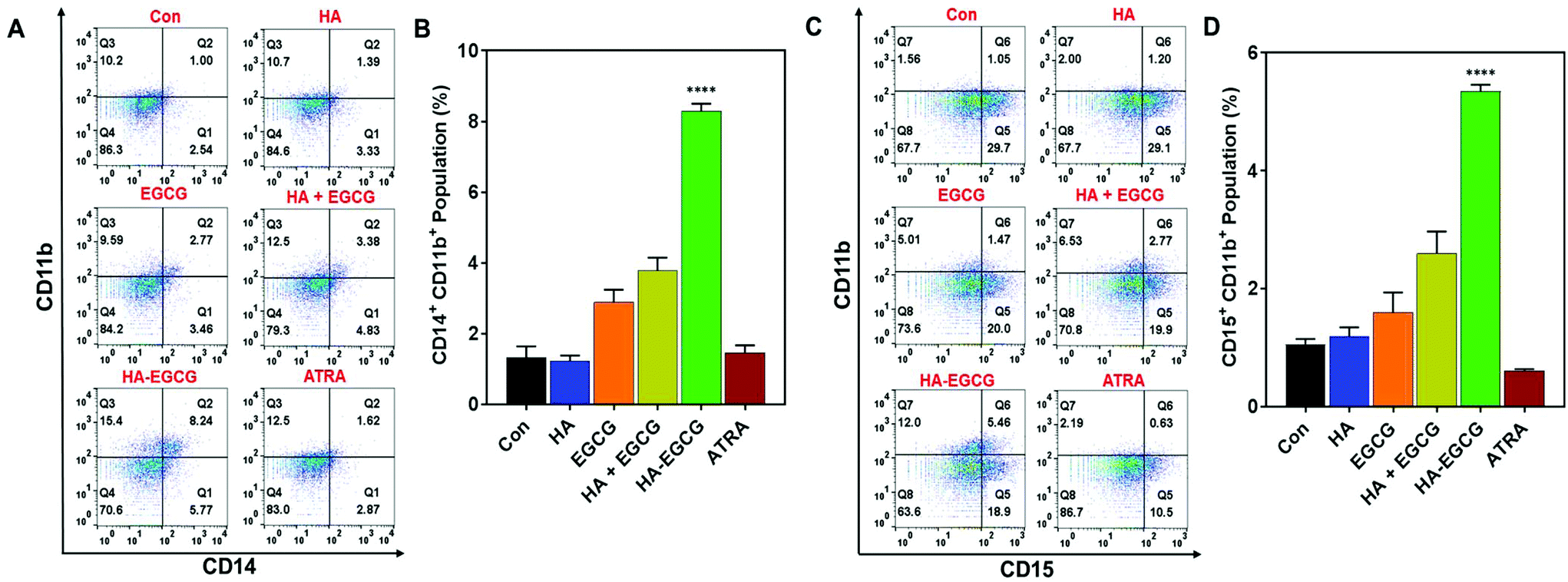 | ||
| Fig. 6 The HA–EGCG conjugate induced terminal differentiation of HL60 cells. (A) HA–EGCG treatment led to the emergence of a population of cells expressing both CD14 and CD11b (CD14+CD11b+), (B) which was significantly greater than in all other treatment groups. (C) HA–EGCG treatment also led to the emergence of a population of cells expressing both CD15 and CD11b (CD15+CD11b+), (D) which was significantly greater than in all other treatment groups. Data shown are the means ± s.d.; n = 3. ****P < 0.0001 versus all other treatment groups. | ||
In vivo anti-leukemic efficacy of the HA–EGCG conjugate in an AML orthotopic mouse model
As the HA–EGCG conjugate demonstrated distinctly superior AML cell eradication and differentiation-inducing effect compared to all other constituents, HA, EGCG, and HA + EGCG mixture, at equivalent concentrations in vitro, we proceeded to assess the anti-leukemic efficacy of the HA–EGCG conjugate in an orthotopic model of human AML in vivo.29,30 As illustrated in Fig. 7A, highly immunodeficient NOD-scid IL2Rγnull (NSG) male mice were sub-lethally irradiated (2.5 Gy) 24 h prior to inoculation of 2 × 106 HL60 cells on day 0; subsequently, the mice were randomly assigned to two groups which received either 50 mg kg−1 HA–EGCG conjugate or PBS via tail vein injections 3 times a week for a total of 5 weeks. PBS-treated mice showed a substantial increase in the white blood cell count from 2.6 ± 0.7 × 106 μL−1 to 8.7 ± 1.2 × 106 μL−1 in a period of 49 days, indicative of the aggressive proliferation of HL60 cells in the mice (Fig. 7B). Notably, HA–EGCG treatment effectively suppressed the progression of AML, as evident from the markedly lower white blood cell count (4.8 ± 0.7 × 106 μL−1). Our finding was well corroborated by a significant improvement in the survival rate of HA–EGCG treated mice (P < 0.01) (Fig. 7C) and the reduced weight of the spleen (Fig. 7D, 173 ± 53% compared to 317 ± 102% of PBS-treated mice), a common characteristic of decreased leukemic cell engraftment.29,37 Furthermore, minimal change in the body weight was observed during the course of treatment, suggesting that HA–EGCG treatment was not harmful to the mice (Fig. 7E). Taken together, these results demonstrated the efficacy of the HA–EGCG conjugate in the inhibition of AML progression in vivo.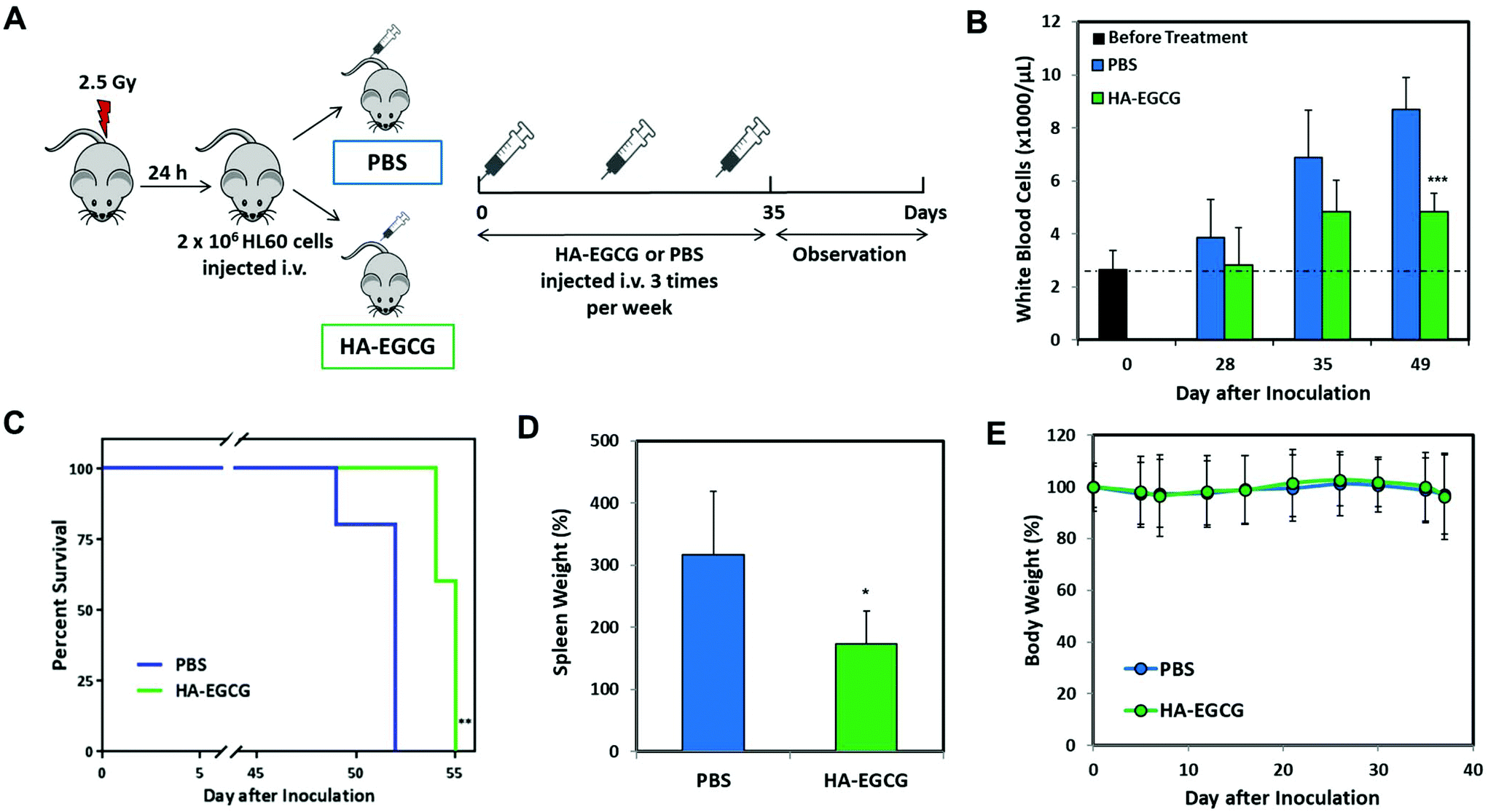 | ||
| Fig. 7 The HA–EGCG conjugate inhibited AML progression in vivo. (A) Schematic overview of the HA–EGCG efficacy evaluation in the HL60 orthotopic mouse model. (B) White blood cell count and human HL60 engrafted mice treated with i.v. injections of PBS or HA–EGCG conjugate (50 mg kg−1) at various days after i.v. inoculation of HL60 cells. (C) Kaplan–Meier survival analysis of mice treated with PBS or HA–EGCG conjugate. (D) Spleen weights of mice treated with PBS or HA–EGCG conjugate as compared to the spleen weight of normal, healthy mice. (E) Body weights of mice during the period of assessment. Data shown are the means ± s.d.; n = 5. *P < 0.05, **P < 0.01, ***P < 0.001 versus PBS-treated group. | ||
Conclusions
This study explored the potential of the HA–EGCG conjugate for AML therapy via a two-pronged strategy: targeted eradication and concurrent differentiation induction of myeloid blast cells. The in vitro cell viability assessment revealed that the AML cells were eliminated by HA–EGCG more preferentially than normal cells. The HA–EGCG conjugate also exhibited superior cytotoxicity, anti-clonogenic activity and differentiation-inducing effect on AML cells over the HA + EGCG mixture and EGCG alone. Moreover, the HA–EGCG conjugate demonstrated therapeutic efficacy and minimal toxicity in the HL60 orthotopic mouse model. The two-pronged anti-leukemic effect of the HA–EGCG conjugate may provide new opportunities for effective AML treatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).Notes and references
- B. Deschler and M. Lübbert, Cancer, 2006, 107, 2099–2107 CrossRef PubMed.
- M. S. Tallman, D. G. Gilliland and J. M. Rowe, Blood, 2005, 106, 1154–1163 CrossRef CAS PubMed.
- A. Kakizuka, W. H. Miller, K. Umesono, R. P. Warrell, S. R. Frankel, V. V. V. S. Murty, E. Dmitrovsky and R. M. Evans, Cell, 1991, 66, 663–674 CrossRef CAS.
- F. Grignani, M. Fagioli, M. Alcalay, L. Longo, P. P. Pandolfi, E. Donti, A. Biondi, F. Lo Coco, F. Grignani and P. G. Pelicci, Blood, 1994, 83, 10–25 CrossRef CAS.
- M. E. Huang, Y. C. Ye, S. R. Chen, J. R. Chai, J. X. Lu, L. Zhoa, L. J. Gu and Z. Y. Wang, Blood, 1988, 72, 567–572 CrossRef CAS PubMed.
- M. Lanotte, V. Martin-Thouvenin, S. Najman, P. Balerini, F. Valensi and R. Berger, Blood, 1991, 77, 1080–1086 CrossRef CAS.
- R. P. Warrell, S. R. Frankel, W. H. Miller, D. A. Scheinberg, L. M. Itri, W. N. Hittelman, R. Vyas, M. Andreeff, A. Tafuri, A. Jakubowski, J. Gabrilove, M. S. Gordon and E. Dmitrovsky, N. Engl. J. Med., 1991, 324, 1385–1393 CrossRef PubMed.
- Z. Wang and Z. Chen, Hematology, 2008, 111, 2505–2515 CAS.
- F. Lo-Coco, G. Avvisati, M. Vignetti, C. Thiede, S. M. Orlando, S. Iacobelli, F. Ferrara, P. Fazi, L. Cicconi, E. Di Bona, G. Specchia, S. Sica, M. Divona, A. Levis, W. Fiedler, E. Cerqui, M. Breccia, G. Fioritoni, H. R. Salih, M. Cazzola, L. Melillo, A. M. Carella, C. H. Brandts, E. Morra, M. von Lilienfeld-Toal, B. Hertenstein, M. Wattad, M. Lübbert, M. Hänel, N. Schmitz, H. Link, M. G. Kropp, A. Rambaldi, G. La Nasa, M. Luppi, F. Ciceri, O. Finizio, A. Venditti, F. Fabbiano, K. Döhner, M. Sauer, A. Ganser, S. Amadori, F. Mandelli, H. Döhner, G. Ehninger, R. F. Schlenk, U. Platzbecker and Gruppo Italiano Malattie Ematologiche dell'Adulto, German-Austrian Acute Myeloid Leukemia Study Group and Study Alliance Leukemia, N. Engl. J. Med., 2013, 369, 111–121 CrossRef CAS PubMed.
- D. Nowak, D. Stewart and H. P. Koeffler, Blood, 2009, 113, 3655–3665 CrossRef CAS PubMed.
- K. Petrie, A. Zelent and S. Waxman, Curr. Opin. Hematol., 2009, 16, 84–91 CrossRef CAS PubMed.
- A. Tomita, H. Kiyoi and T. Naoe, Int. J. Hematol., 2013, 97, 717–725 CrossRef CAS.
- B. C. Shaffer, J.-P. Gillet, C. Patel, M. R. Baer, S. E. Bates and M. M. Gottesman, Drug Resist. Updates, 2012, 15, 62–69 CrossRef CAS.
- H. Knopf-Marques, M. Pravda, L. Wolfova, V. Velebny, P. Schaaf, N. E. Vrana and P. Lavalle, Adv. Healthcare Mater., 2016, 5, 2841–2855 CrossRef CAS PubMed.
- H. Kim, H. Jeong, S. Han, S. Beack, B. W. Hwang, M. Shin, S. S. Oh and S. K. Hahn, Biomaterials, 2017, 123, 155–171 CrossRef CAS PubMed.
- N. V. Rao, H. Y. Yoon, H. S. Han, H. Ko, S. Son, M. Lee, H. Lee, D.-G. Jo, Y. M. Kang and J. H. Park, Expert Opin. Drug Delivery, 2016, 13, 239–252 CrossRef CAS PubMed.
- K. Liang, K. H. Bae, F. Lee, K. Xu, J. E. Chung, S. J. Gao and M. Kurisawa, J. Controlled Release, 2016, 226, 205–216 CrossRef CAS PubMed.
- Y. Zhu, J. Zhang, F. Meng, L. Cheng, J. Feijen and Z. Zhong, J. Mater. Chem. B, 2018, 6, 3040–3047 RSC.
- S. Ghaffari, G. J. Dougherty, A. C. Eaves and C. J. Eaves, Leukemia, 1996, 10, 1773–1781 CAS.
- C. S. Yang, X. Wang, G. Lu and S. C. Picinich, Nat. Rev. Cancer, 2009, 9, 429–439 CrossRef CAS PubMed.
- M. H. Pan and C. T. Ho, Chem. Soc. Rev., 2008, 37, 2558–2574 RSC.
- B. N. Singh, S. Shankar and R. K. Srivastava, Biochem. Pharmacol., 2011, 82, 1807–1821 CrossRef CAS PubMed.
- T. Nakazato, K. Ito, Y. Miyakawa, K. Kinjo, T. Yamada, N. Hozumi, Y. Ikeda and M. Kizaki, Haematologica, 2005, 90, 317–325 CAS.
- D. H. Han, J. H. Jeong and J. H. Kim, Anticancer Res., 2009, 29, 1417–1421 CAS.
- A. Britschgi, H.-U. Simon, A. Tobler, M. F. Fey and M. P. Tschan, Br. J. Haematol., 2010, 149, 55–64 CrossRef CAS PubMed.
- V. Borutinskaitė, A. Virkšaitė, G. Gudelytė and R. Navakauskienė, Leuk. Lymphoma, 2018, 59, 469–478 CrossRef PubMed.
- M. Moradzadeh, A. Roustazadeh, A. Tabarraei, S. Erfanian and A. Sahebkar, Phyther. Res., 2017, 1–9 Search PubMed.
- C. Liu, K. H. Bae, A. Yamashita, J. E. Chung and M. Kurisawa, Biomacromolecules, 2017, 18, 3143–3155 CrossRef CAS PubMed.
- A. Agliano, I. Martin-Padura, P. Mancuso, P. Marighetti, C. Rabascio, G. Pruneri, L. D. Shultz and F. Bertolini, Int. J. Cancer, 2008, 123, 2222–2227 CrossRef CAS PubMed.
- E. Saland, H. Boutzen, R. Castellano, L. Pouyet, E. Griessinger, C. Larrue, F. de Toni, S. Scotland, M. David, G. Danet-Desnoyers, F. Vergez, Y. Barreira, Y. Collette, C. Récher and J.-E. Sarry, Blood Cancer J., 2015, 5, e297 CrossRef CAS PubMed.
- C. Nervi, F. F. Ferrara, M. Fanelli, M. R. Rippo, B. Tomassini, P. F. Ferrucci, M. Ruthardt, V. Gelmetti, C. Gambacorti-Passerini, D. Diverio, F. Grignani, P. G. Pelicci and R. Testi, Blood, 1998, 92, 2244–2251 CAS.
- B. Mueller, T. Pabst and J. Fos, Blood, 2006, 107, 3330–3338 CrossRef CAS PubMed.
- D. S. Krause and R. A. Van Etten, Trends Mol. Med., 2007, 13, 470–481 CrossRef CAS PubMed.
- K. D. Sabbath, E. D. Ball and R. Larson, et al. , J. Clin. Invest., 1985, 75, 746 CrossRef CAS PubMed.
- D. Bonnet and J. E. Dick, Nat. Med., 1997, 3, 730–737 CrossRef CAS PubMed.
- H. L. Lung, W. K. Ip, C. K. Wong, N. K. Mak, Z. Y. Chen and K. N. Leung, Life Sci., 2002, 72, 257–268 CrossRef CAS.
- M. A. Papiez, J. Baran, K. Bukowska-Straková and W. Wiczkowski, Food Chem. Toxicol., 2010, 48, 3391–3397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01146c |
| This journal is © The Royal Society of Chemistry 2020 |