
Canalicular domain structure and function in matrix-free hepatic spheroids†
Vikas Raj
Sharma‡
ab,
Ananya
Shrivastava‡
 a,
Benoit
Gallet
c,
Elizaveta
Karepina
ab,
Peggy
Charbonnier
a,
Mireille
Chevallet
a,
Pierre-Henri
Jouneau
*b and
Aurélien
Deniaud
*a
a,
Benoit
Gallet
c,
Elizaveta
Karepina
ab,
Peggy
Charbonnier
a,
Mireille
Chevallet
a,
Pierre-Henri
Jouneau
*b and
Aurélien
Deniaud
*a
aUniv. Grenoble Alpes, CNRS, CEA, IRIG, Laboratoire de Chimie et Biologie des Métaux, 38000 Grenoble, France. E-mail: aurelien.deniaud@cea.fr
bUniv. Grenoble Alpes, CEA, IRIG, MEM, 38000 Grenoble, France. E-mail: pierre-henri.jouneau@cea.fr
cUniv. Grenoble Alpes, CEA, CNRS, IBS, 38000 Grenoble, France
First published on 13th November 2019
Abstract
Liver is pivotal in organism metabolism. This organ is receiving nutriments from the portal vein and then storing, metabolizing, distributing in the circulation or excreting excess and xenobiotics in bile. Liver architecture and hepatocyte polarization are crucial to achieve these functions. To study these mechanisms in details, relevant cell culture systems are required, which is not the case with standard 2D cell culture. Besides, primary hepatocytes rapidly de-differenciate making them inefficient in forming physiological system. Herein, we used an hepatoma-derived cell line to produce matrix-free hepatic spheroids and developed an integrated structural cell biology methodology by combining light sheet fluorescence microscopy and 3D electron microscopy to study their function and structure. Within these spheroids, hepatocytes polarize and organize to form bile canaliculi active for both organics and inorganics excretion. Besides, live imaging revealed the high dynamic of actin networks in basal membranes compared to their high stability in the apical pole that constitutes bile canaliculi. Finally, the first structure of active bile canaliculi was solved at nm resolution and showed the very high density of microvilli coming from all cells constituting the canaliculus. Therefore, this study is the first comprehensive and in-depth functional and structural study of bile canaliculi in a physiological-relevant context.
1. Introduction
Liver is central for metabolism, performing more than 500 functions ranging from carbohydrate storage and distribution to xenobiotic transformations and excretion. These are made possible thanks to a very specific architecture within lobules that constitute the functional unit in the liver. These hexagonal-like structures possess periportal regions at the angle of the hexagone composed of the triad, hepatic artery, portal vein and bile duct, and a central area where blood is distributed through the central vein. Between both regions, rows of hepatocytes maintain liver function by sustaining two countercurrent flow systems – the uptake, processing and secretion of sinusoidal blood components along with the synthesis and secretion of bile.1 Hepatocytes are able to do so because of their inherent structural and functional polarity, allowing them to form a crucial cell layer that separates sinusoidal blood from the canalicular bile,2 the so-called hepatocyte cords. The polarization of the hepatic epithelium has therefore to be very peculiar compared to most tissues that form typical columnar polarization.1,3 On the one hand, hepatocytes basal membranes are facing the space of Disse. They are in contact with the extracellular matrix and sinusoids that delivers blood arriving from portal vein. On the other hand, lateral membranes are interacting with neighboring hepatocytes and form the apical domains that are segregated by tight junctions. At this apical pole, canalicular domains are formed between two adjacent hepatocytes. These domains can merge together to form an extended canalicular network consisting of micron-sized diameter channels known as the bile canaliculus.1,3 These canalicular structure serves as sites of bile secretion for the hepatocytes that is collected in the bile duct at the periportal zone.3 Then, bile flows to the intestine where it has multiple functions: cancels out action of gastric acid pepsin; emulsifies lipids; takes part in formation of micelles; promotes secretion of enteric hormones (secretin and cholecystokinin); prevents adhesion of proteins and bacteria; enhances gastrointestinal motility; activates proteases including trypsin. Through the detergent-like properties of bile, cholestasis, the lack of bile flow, could cause hepatocellular damage such as extracellular accumulation of bile acids, fibrosis, and ultimately cirrhosis under chronic condition.4The canalicular network is crucial for liver function and requires this peculiar hepatocyte polarization that is functionally achieved by the targeting of specific proteins to the apical pole that can serve as markers of polarization. Several membrane proteins are thus constitutively or transiently targeted to the canalicular membranes to perform the transport of physiologic molecules or xenobiotics into the bile. These proteins belong to the large protein family of the ABC transporters and are generally classified as multidrug resistance proteins with the two sub-families, MDR and Mrp (for review5). These proteins can export various molecules without modification or after conjugation to glucuronide or glutathione. However, some of these transporters are involved in physiologic mechanism, such as the bile salt export pump (BSEP) that is required to export bile salt derivatives. Finally, some proteins are trafficking to canalicular membranes in response to stimuli. This is the case of ATP7B, a P-type ATPase that transports copper (Cu) (for review6). Liver has a central role in organismal Cu homeostasis with ATP7B being pivotal for this function. In basal conditions, ATP7B is located at the Golgi membranes and provides Cu for ceruloplasmin maturation that is then excreted into the blood where it constitutes around 90% of circulating Cu.7 Upon hepatic Cu concentration increases, ATP7B migrates to canalicular membranes to favor biliary and thereof fecal Cu excretion. This mechanism is critical since it constitutes the only way to avoid Cu accumulation in the organism.
In order to perform functional studies, different in vitro hepatic cultures have been developed such as sandwich cultures or spheroids.8 The latter has been widely developed in various ways because of its simplicity and its capacity to mimic drug metabolism and toxicity that is of fundamental interest in the field.8 Indeed, it has been shown that hepatocytes grown in 3D recover metabolic functions closer to those in the liver, while monolayer cultures lead to a decrease of functions such as albumin and urea production, and in liver specific gene expression or cytochrome P450 activity.9–14 Moreover, primary hepatocytes rapidly dedifferentiate outside of the liver context. Spheroids, also known as microtissues, are spherical 3D self-assembled clusters of cells with direct cell–cell contacts that can be formed using a variety of techniques with or without the use of scaffolds15,16 that can be obtained both with primary hepatocytes or hepatocarcinoma-derived cell lines. Spheroids possess significant similarity in the morphological and functional characteristics with liver tissue even when they are obtained using cell lines such as HepG2/C3A.17 Indeed, it has been described that both primary hepatocytes and HepG2/C3A grown in 3D demonstrate higher metabolic activity12–14,18,19 and could organize bile canaliculi. The functionality of biliary excretion in these spheroids have been proven using fluorescent dyes.12,19 However, analyses of canaliculi ultrastructure in these models were very limited because of the use of a single spheroid section from chemically fixed samples observed by electron microscopy. Therefore, round shaped areas with microvilli were visualized.14,20 Some of the studies presented clearly dilated bile canaliculi that could be due to sample preparation. In any case, none of these studies performed three-dimensional structural analysis that is essential to properly characterize the ultrastructure. Bile canaliculi in contrast with blood capillary does not have their own wall and are formed by cell membranes of adjacent hepatocytes. The smaller canaliculi run into bigger ducts and the overall biliary tract is a highly ramified tubular network for which the macroscopic structure has been solved recently together with the measure of bile flow.4 This pioneer study provided a very nice model of biliary system functioning in vivo. In the current work, we made use of light sheet fluorescence microscopy (LSFM, for review21) to characterize matrix-free spheroids in terms of polarization and bile canaliculi excretion functions. LSFM provides the unique capability to image the whole spheroid and make complete 3D reconstructions of the object of interest. It was possible to show that these spheroids are active for the excretion of any kind of species, organic or inorganic, validating the interest of the biological model for the study of metal homeostasis for instance. In addition, live LSFM imaging enabled to track actin networks dynamics in various conditions. Finally, the ultrastructure of the inside of the spheroid was obtained at few nanometer resolution thanks to Focused Ion Beam-Scanning Electron Microscopy (FIB-SEM, for review22). It was thus possible to obtain the first structure of a bile canaliculus that revealed the very high density of intertwined microvilli, which explains the features we could observe by fluorescence microscopy. Overall, this work provided a comprehensive view of this spheroid model and shows the potential of the combination of LSFM and FIB-SEM both in the fields of biomaterials development and characterization and in developmental and cellular biology.
2. Methods
2.1 Spheroid preparation
The cells used for the preparation of spheroids, HepG2/C3A were provided by the ATCC. HepG2/C3A cells were maintained in Eagle's Minimum Essential Medium supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin–streptomycin to prevent contamination at 37 °C and in the presence of 5% CO2. Spheroids were obtained in two steps. First, freshly trypsinized HepG2/C3A cells were seeded in 30 μL drops of 125 cells in complete media onto the flat surface of Petri dish lid that is then inverted above the dish filled with 5 mL of PBS and left for 72 hours at 37 °C to form spheroids. The obtained spheroids were then transferred to ultra low adherence plates (GravityTRAP™ ULA Plate InSphero). Media was renewed every 2-day. When indicated 6% matrigel was added in the first step, i.e. cell seeding in drops.2.2. Analysis of canalicular transporter functions and immunostaining
For canalicular excretion experiments, the spheroids were incubated with 5 μM 5-chloromethylfluoresceindiacetate (CMFDA) or 5 μM cholyl-lysyl-fluorescein (CLF) in growth medium for 30 min at 37 °C. For Cu-related experiments, spheroids were incubated overnight with 200 μM bathocuproinedisulfonic acid (BCS), 10 or 100 μM copper chloride solution (CuCl2) in growth medium. After the incubation, spheroids were washed in PBS twice and then fixed for 30 minutes with formalin (10%) or 3 minutes with methanol on ice. After fixing, spheroids were washed again in PBS.Fixed spheroids were permeabilized with 0.5% Triton X-100 in Tris-Buffered Saline with 0.05% Tween-20 in PBS for 60 minutes at room temperature and then blocked to prevent the nonspecific binding of the antibodies, with 0.1% Triton X-100/3% BSA with 0.05% Tween20 in PBS for 30 minutes at room temperature. Spheroids previously incubated with CMFDA or CLF, were incubated with Hoechst diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 and Phalloidin diluted 1:500 in antibody buffer (0.1% Triton X-100/1% BSA with 0.05% Tween20 in PBS) for 60 minutes at room temperature. For immunostaining, after fixation, the spheroids were incubated with antibodies against ATP7B (Abcam ab124973, 1:50) and/or Golgin-97 (Invitrogen A21270, 1:100) and/or Mrp2 (Abcam ab3373, 1:50) and/or ZO1 (Thermo Fisher 40-2200, 1:50) in antibody buffer for 60 minutes at room temperature. Spheroids were then incubated with secondary antibodies diluted 1:1000 (Alexa Fluor 488 goat anti-rabbit antibody and/or secondary Alexa Fluor 568 or 488 goat anti-mouse antibody, Life Technologies) in antibody buffer, with Hoechst diluted 1:1000 for 60 minutes at room temperature. For all the above-mentioned experimental conditions, the spheroids were washed in PBS in between subsequent steps. The spheroids were finally imaged using LSFM.
:1000 and Phalloidin diluted 1:500 in antibody buffer (0.1% Triton X-100/1% BSA with 0.05% Tween20 in PBS) for 60 minutes at room temperature. For immunostaining, after fixation, the spheroids were incubated with antibodies against ATP7B (Abcam ab124973, 1:50) and/or Golgin-97 (Invitrogen A21270, 1:100) and/or Mrp2 (Abcam ab3373, 1:50) and/or ZO1 (Thermo Fisher 40-2200, 1:50) in antibody buffer for 60 minutes at room temperature. Spheroids were then incubated with secondary antibodies diluted 1:1000 (Alexa Fluor 488 goat anti-rabbit antibody and/or secondary Alexa Fluor 568 or 488 goat anti-mouse antibody, Life Technologies) in antibody buffer, with Hoechst diluted 1:1000 for 60 minutes at room temperature. For all the above-mentioned experimental conditions, the spheroids were washed in PBS in between subsequent steps. The spheroids were finally imaged using LSFM.
For live imaging with SiR-actin (Tebu), spheroids were incubated for 1 hour with 1 μM SiR-actin and 10 μM verapamil to inhibit Mrp proteins and nuclei were stained with Hoechst. Then spheroids were rinsed in complete culture medium and prepared for acquisition.
2.3. Light sheet fluorescence microscopy, sample preparation and analysis
Prior to imaging, the spheroids were embedded in 1% (w/w) low melting point agarose (Roth). In order to do so, a 1% (w/w) low melting point agarose in PBS was made liquid by heating at 75 °C and then cooled down to 39 °C. Once it was equilibrated at 39 °C, 20 μL agarose was added to the spheroids. The molten agarose gel containing the spheroids was quickly aspirated in a glass capillary using a tight-fitting plunger and then allowed to gelify inside the capillary. Once the agarose forms a rigid cylinder, the glass capillary was placed in the sample holder for Lightsheet Z.1 microscope, adapted to hold the capillaries at precise translational and rotational positions. The sample holder with the capillary was then mounted in the chamber, pre-filled with PBS. The image acquisition was then carried out by pushing out the embedded spheroids out of the glass capillary to position them in front of the objectives. For live imaging, 0.75% agarose was made in complete cell culture medium and the chamber was maintained at 37 °C and filled with cell culture medium.ZEISS Lightsheet Z.1 LSFM was used to acquire multiview images of the spheroids at sub-cellular resolution. The spheroids were imaged with Lightsheet Z.1 detection optics 20×/1.0 (water immersion) with dual side illumination, where the sample was illuminated by the light sheet sequentially from the left and from the right, producing two images for each plane. The spheroids were illuminated with three different laser tracks with excitation wavelengths of 405 nm, 488 nm, and 561 nm, followed by image acquisition at detection wavelengths of 420–470 nm, 505–545 nm, and 575–615 nm, respectively. Z stack images were acquired from 4 different viewing angles (0°, 90°, 180°, 270°) and they were subsequently processed in ZEN 2014 for Lightsheet Z.1. The two images for each plane from dual side illumination were fused together. Multiview recordings were registered using Lightsheet Z.1 Multiview Processing and fused into one 3D dataset. The fused 3D dataset was imported to the modular software arivis Vision 4D to make 3D rendering, segmentation and/or videos.
In case of live imaging, one angle Z-stacks were recorded using excitation wavelengths of 405 and 638 nm and detection wavelengths of 420–470 nm and long pass 660 nm, respectively.
2.4. Sample preparation for FIB-SEM
For high pressure freezing 5 to 10 spheroids were dispersed in 2 μL of culture media supplemented with 20% FBS and loaded on the 200 μm side of a type A 3 mm gold platelet (Leica Microsystems), covered with the flat side of a type B 3 mm aluminum platelet (Leica Microsystems), and were vitrified by high-pressure freezing using an HPM100 system (Leica Microsystems). Next, the samples were freeze substituted at −90 °C for 80 h in acetone supplemented with 1% OsO4 and warmed up slowly (1 °C h−1) to −60 °C in an automated freeze substitution device (AFS2; Leica Microsystems). After 8 to 12 h, the temperature was raised (1 °C h−1) to −30 °C, and the samples were kept at this temperature for another 8 to 12 h before a step for 1 h at 0 °C, cooled down to −30 °C and then rinsed four times in pure acetone. The samples were then infiltrated with gradually increasing concentrations of EPON resin in acetone (1:2, 1:1, 2:1 [vol/vol] and pure) for 2 to 3 h while raising the temperature to 20 °C. Pure EPON resin was added at room temperature. After polymerization 24 h at 60 °C, 80 nm sections were obtained using an ultra-microtome UC7 (Leica Microsystems) and an Ultra 35° diamond knife (DiATOME) and were collected on formvar-carbon-coated 100 mesh copper or nickel grids. The thin sections were post-stained for 10 min with 2% aqueous uranyl acetate, rinsed and incubated for 5 min with lead citrate. Digital images were obtained using a Tecnai G2 Spirit BioTwin microscope (FEI) operating at 120 kV with an Orius SC1000 CCD camera (Gatan) in order to check the morphology of the cells in the spheroids and to select the area of interest for FIB-SEM imaging.
2.5. FIB-SEM acquisition
FIB-SEM experiments were performed using a Zeiss crossbeam 550 microscope. In a first step, the sample embedded in the resin was coated by a thin layer of Pt (few nm) to avoid charge accumulation. The surface of the sample to be analyzed was then protected by the deposition of 2 μm thick Pt or C layer, called protecting layer or pad on a surface of typically 20 × 30 μm. A large hole was then excavated with a Ga+ beam of very high current, 30 nA approximately. The surface of this hole was then trenched slice by slice with the Ga+ beam (typically with a current of 300 or 700 pA at a beam acceleration of 30 kV). Each new surface was imaged with the electron beam. This was done at a low accelerating voltage (1.5 kV) and at a very low current (1 nA approximately) and using the in-lens backscattered detector (EsB detector, with a bias of 600 V). Acquisitions have been done at a resolution of 10 nm to obtain a voxel size of 10 × 10 × 10 nm3.2.6. FIB-SEM analysis
Images were acquired as a series of 2-dimensional TIFF files. In order to compile a 3D tiff file format, the images were first registered in Fiji.23 During registration, the central image from the stack was taken as a reference and list of registered image slices was done with reference to central image. Translation technique was used to register. The stack of registered image slices was again checked in orthogonal view for alignment. If the misalignment persists, further processing was done using python scripts. The angle of misalignment was calculated using python and then set that angle to zero.After alignment, the stack of images was denoised and inverted (initial images were acquired in dark field regime). Median filter served as a nice tool in preserving the boundaries while removing noise. It selects the median value of the neighboring entries. The sharpness of the stack was further improved for clear bifurcation of boundaries. Finally, the black portion at the extremities of the stack created while registering was cropped out.
Segmentation of different objects (nuclei, vesicles, vacuoles, mitochondria, etc.) was done with Ilastik software.24 Mesh modification and cleaning of mesh was done in MeshLab25 and visualization was carried out in Paraview.26
3. Results and discussion
3.1. Matrix-free hepatic spheroids – hepatocyte polarization and biliary excretion
Various hepatic spheroids have been described in the last years with an interest in studying, in vitro, liver metabolic functions and thus determining the putative toxicity of drugs under development.9–11 In some systems, bile canaliculi-like structures have been observed thanks to CFMDA excretion assay, which requires a fully functional hepatic system to metabolize CMFDA first and then excrete the glutathione conjugate in bile canaliculi via Mrp2. It has been shown that the hepatocarcinoma-derived cell line, HepG2/C3A and not its parental cell line HepG2, can be used to make spheroids with active bile canaliculi and with liver functions recovered after 18 days.13,14,17 Besides, matrix-free hepatic spheroids can be obtained by placing few cells in a hanging drop. This process generates an initial cluster of cells leading to a spheroid without any geometrical constraint and with cells that did not attach to plastic support avoiding the generation of GPa forces that do not reflect growth conditions of hepatocytes within a liver that is in the kPa range.27 Therefore, it avoids the appearance of strong actin fibers at the site of cell adherence to the support as usually observed in other set-up, such as liver-on-chip (Fig. S1†). We chose to generate matrix-free spheroids and compared the HepG2/C3A with the HepG2 cell line. On the one hand, HepG2 cells did not polarize significantly since actin surrounding the cells is intense and evenly distributed (Fig. S2†). Only some focal point of very high intensity with Mrp2 co-localized can be observed. On the other hand, HepG2/C3A cells showed low intensity actin staining surrounding the cells plus high intensity actin staining delimiting an inter-cellular space (Fig. 1A). The latter possessed ovoid or elongated shape highlighting the possibility to grow row of cells as observed in the center of the spheroid in Fig. 1A. The polarization of our cellular system was further highlighted by immunostaining Mrp2 within the spheroids (Fig. 1B and C) that showed a very nice co-localization with the strong actin signal. Two regions with hepatocytes very nicely ordered can be observed (Fig. 1B, white arrows) with the apical pole of these rows of cells clearly delineated and evidenced by the Mrp2 localization that is not found in the other membranes of these cells, which are therefore basal membranes. Besides, small hot spots of colocalized Mrp2 and actin (Fig. 1B, grey arrows) were observed. These should correspond to initiation sites of apical poles. Based on Z-stack and multiview acquisitions, this spheroid was reconstructed in 3D and the Video S1† enables to look at the elongated bile canaliculus co-stained for actin (red) and Mrp2 (green). In this system, cells have to produce their own extracellular matrix to be able to grow in 3D with intercellular contacts and proper polarization. However, we evaluated the impact of added extracellular matrix in the form of matrigel that is not very well defined but universally used. This biomaterial did not change neither the polarization nor the shape of bile canaliculi within HepG2/C3A spheroids (Fig. S3†). Therefore, the following of the study was performed with HepG2/C3a spheroids grown without added extracellular matrix to have more physiologic and better controlled system.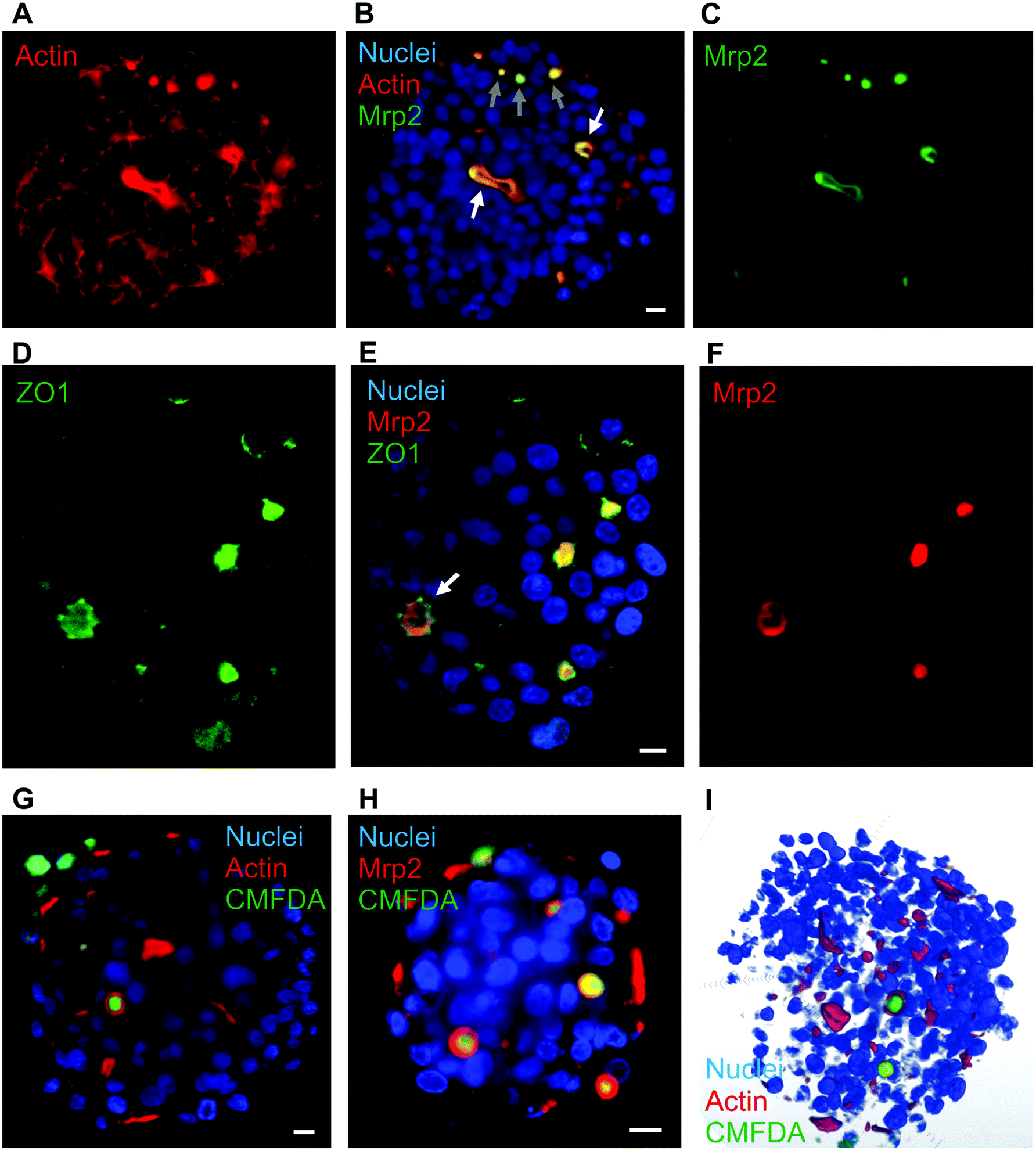 | ||
| Fig. 1 Hepatocyte polarization in matrix-free spheroids and biliary excretion. (A–C) Single plan LSFM of a matrix-free hepatic spheroid stained for actin (red), Hoechst (blue) and Mrp2 (green). The full volume is presented in Video S1.† White arrows show elongated bile canaliculi and grey arrows initiation sites. (D–F) Single plan LSFM of a matrix-free hepatic spheroid stained for ZO1 (green), Hoechst (blue) and Mrp2 (red). (G) Single plan LSFM of a matrix-free hepatic spheroid stained for actin (red), Hoechst (blue) and CMFDA (green). A full volume is shown in Video S2.† (H) Single plan LSFM of a matrix-free hepatic spheroid stained for Mrp2 (red), Hoechst (blue) and CMFDA (green). (I) 3D reconstructed spheroid stained for actin (red), Hoechst (blue) and CMFDA (green). Scale bars correspond to 10 μm. | ||
The polarization was further assessed by immunostaining ZO1, a protein involved in tight junctions. As expected, ZO1 was mainly localized at bile canaliculi structures co-localized with Mrp2 (Fig. 1D–F and Fig. S4A–F†). Interestingly, ZO1 is better defined than Mrp2 and surrounding it, confirming the role of ZO1 in sealing bile canaliculi at tight junctions. Besides, ZO1 can also be observed at low intensity surrounding cell membranes (Fig. S4G†) similar to what was observed with actin. Therefore, our spheroid model shows the hallmarks of hepatic polarization.
CMFDA excretion assay led to the green staining of a majority of this intercellular structures showing that they are bile canaliculi-like structure with active Mrp2-dependent transport (Fig. 1G and Fig. S5A†). The red stained structures that were not stained green are probably connected to the outside of the spheroids and CMFDA is excreted outside bile canaliculi. Oppositely to other groups we did not observe active bile canaliculi-like structures using the HepG2 cell line18 validating our choice for HepG2/C3A that is a robust cell line with functionality in spheroids close to primary hepatocytes. As observed with actin staining, intercellular structures were nicely stained for Mrp2 with the green CMFDA dye inside (Fig. 1H and Fig. S5B†). Altogether, this complementary imaging analysis revealed the presence of active bile-canaliculi like structure proving an appropriate polarization of HepG2/C3A cells in our matrix-free spheroid model. Besides, imaging was performed using LSFM that possesses the capability to acquire fluorescence images in thick sample (sub-mm) and the multi view acquisition mode enables to reconstruct a whole biological sample of 100–200 μm diameter spheroid at subcellular resolution (hundreds of nm resolution). Three-dimensional reconstruction of the whole spheroid can thus be obtained (Fig. 1I and Video S2†) and segmentation of subcellular or intercellular structures can be done such as bile canaliculi-like structures containing excreted CMFDA (Video S3†). LSFM is therefore the appropriate choice for spheroid analysis compared to confocal microscopy.
In vivo, bile canaliculi also possess physiologic excretion functions such as bile salt that are required for digestion in the intestine. These molecules are excreted by the BSEP protein that is located at the canalicular membrane. Spheroids exposed to cholyl-lysin-fluorescein, a fluorescent analogue of bile salt, accumulated within actin and Mrp2 stained intercellular structures (Fig. 2A and B) similarly to CMFDA staining. Therefore, within the spheroids, active Mrp2 and BSEP were present in the membranes of bile canaliculi. Interestingly, within these spheroids elongated bile canaliculi containing excreted CLF can also be found and reconstructed in 3D, green fluorescence wrapped in red fluorescence (Fig. 2C). This structure again highlights features of rows of hepatocytes polarized within spheroids.
 | ||
| Fig. 2 Bile salt excretion. (A) Single plan LSFM of matrix-free hepatic spheroids stained for actin (red), Hoechst (blue) and CLF (green). (B) Single plan LSFM of a matrix-free hepatic spheroid stained for Mrp2 (red), Hoechst (blue) and CLF (green). (C) Three different views of 3D reconstructed spheroid stained for actin (red), Hoechst (blue) and CLF (green) with a specific focus on a bile canaliculus filled with CLF. Scale bars correspond to 10 μm. | ||
3.2. Copper homeostasis in spheroids
Liver is also central for metal homeostasis, in particular Cu and iron, both having strong interplays together. The proper study of Cu homeostasis in liver-mimicking model requires fully polarized hepatocytes able to excrete matured ceruloplasmin on one side and excess Cu on the other side. The switch between these two pathways is the protein ATP7B that traffics between Golgi apparatus and bile canaliculi via lysosomal vesicles.28 In basal conditions, ATP7B was localized both at the Golgi apparatus, as shown by the co-localization with the Golgin-97, and in cytosolic vesicles most probably lysosomes based on the literature (Fig. 3A and Fig. S6A†). It is interesting to observe the inner part of the spheroids, clusters of hepatocytes polarized with diffuse ATP7B staining oriented towards what should be a bile canaliculus (Fig. S6A,† white arrow). This is consistent with the co-staining of actin and Mrp2 that showed elongated bile canaliculi structure (Fig. 1A–C and Video S1†). However, no canalicular localization was observed most probably because of the low frequency and transiency of these events (Fig. 3B and Fig. S6B†). These data showed that ATP7B are not stacked at the Golgi apparatus in basal conditions. This is consistent with a highly dynamic process that is crucial to maintain low Cu level within the cell. ATP7B can be driven towards a predominantly Golgi location using the Cu chelator BCS (Fig. 3C and Fig. S6C†). On the other side, upon overnight exposure to Cu, a co-localization between ATP7B and Mrp2 was visualized at structures typical of bile canaliculi (Fig. 3D, E and Fig. S7A, B and S8†), while actin and Mrp2 are not affected by the exposure to Cu (Fig. S7C†). In 10 μM Cu, part of ATP7B was still found in cytosolic vesicles, while 100 μM Cu pushed ATP7B to a full canalicular location. These data highlight the role of the pool of vesicular ATP7B for the trafficking of this protein and controlling the balance between Golgi and canalicular localization depending on Cu concentration. Besides, these results prove that Cu can diffuse through the whole spheroid and activates liver physiological pathways as in vivo. Therefore, matrix-free spheroid is an appropriate model to study metal homeostasis and is not hindered by a limited diffusion of metal ions.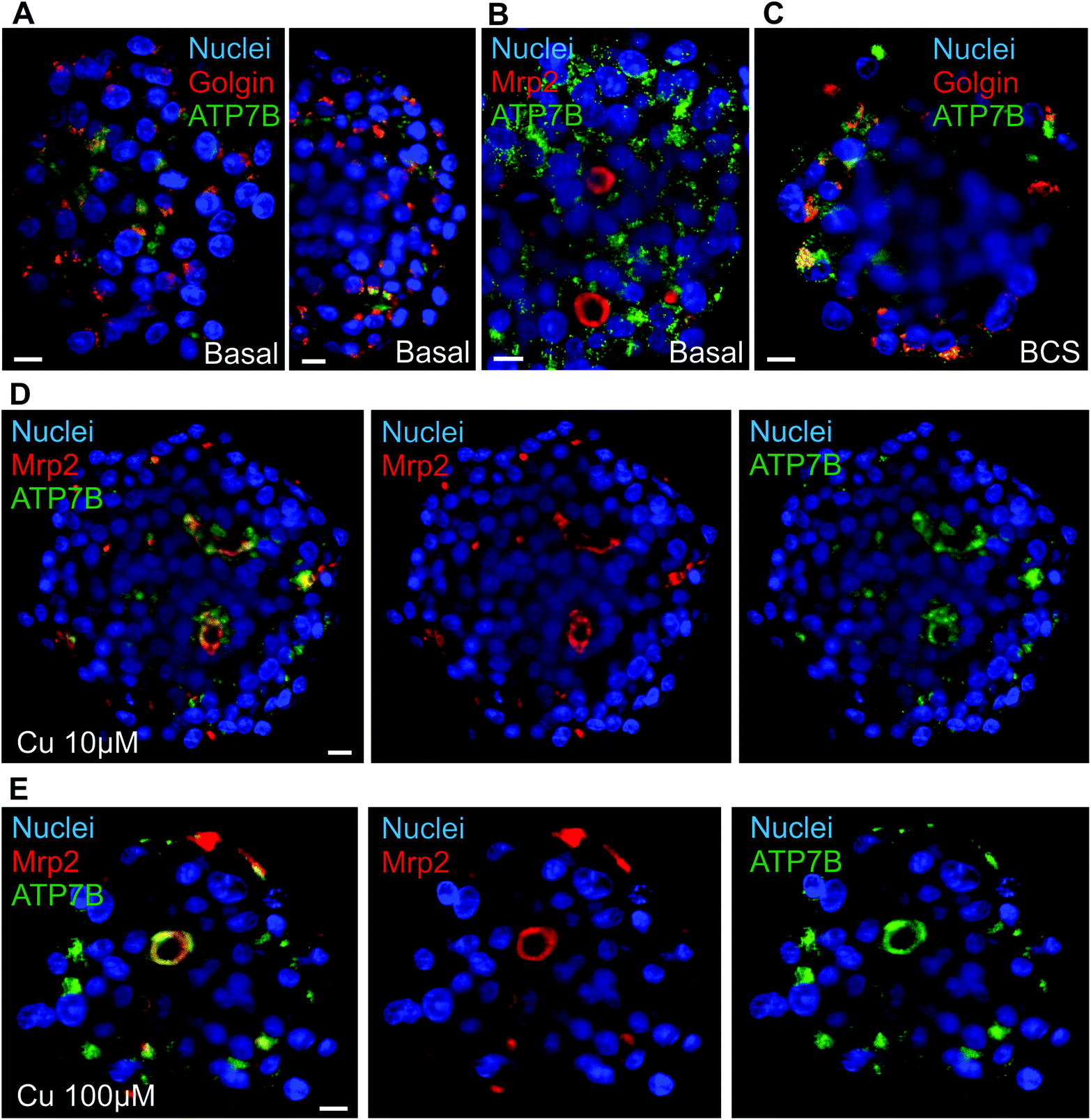 | ||
| Fig. 3 ATP7B trafficking within spheroids. (A) Single plan LSFM of matrix-free hepatic spheroids in basal conditions stained for Golgin-97 (red), Hoechst (blue) and ATP7B (green). (B) Single plan LSFM of a matrix-free hepatic spheroid in basal conditions stained for Mrp2 (red), Hoechst (blue) and ATP7B (green). (C) Single plan LSFM of a matrix-free hepatic spheroid in Cu-deprived conditions (200 μM BCS) stained for Golgin-97 (red), Hoechst (blue) and ATP7B (green). (D) Single plan LSFM of a matrix-free hepatic spheroid exposed to 10 μM Cu stained for Mrp2 (red), Hoechst (blue) and ATP7B (green). Same plan is presented with different combination of colors for a better visualization of the co-localization between ATP7B and Mrp2. (E) Single plan LSFM of a matrix-free hepatic spheroid exposed to 100 μM Cu stained for Mrp2 (red), Hoechst (blue) and ATP7B (green). Same plan is presented with different combination of colors for a better visualization of the co-localization between ATP7B and Mrp2. Scale bars correspond to 10 μm. | ||
3.3. Dynamics of actin networks in spheroids
Another advantage of LSFM is the possibility to perform live cell imaging that enables to follow biological processes in spheroids or small animal models. We showed that actin staining is an appropriate marker of bile canaliculi. Therefore, we used SiR-actin, which allows live imaging of F-actin,29 to investigate the dynamics of actin cytoskeleton in hepatic spheroids. On short time scale, 2 minutes with one image taken every 10 to 15 seconds, actin fibers were very stable both in basal conditions (Videos S4 and S5†) and in presence of Cu at 100 μM (Video S6†). However, longer time scales (60 to 120 minutes acquisition with 1 acquisition per 2-minute) enabled to observe actin network re-organization. In basal conditions, spheroids were relatively stable in size (Fig. 4 and Videos S7 and S8†) and actin is highly dynamic upon cell division (Fig. 4A, white arrows and Video S7†), a process that can be nicely revealed with this approach. On the contrary, bile canaliculi structure were very stable and only a decrease in fluorescence intensity was observed (Fig. 4B, white arrows and Video S8†). This decrease can either be due to photobleaching on such a long period of acquisition or to remobilization of part of the actin proteins. Interestingly, the comparison of the events caught with these two videos show that actin networks are highly dynamic in basal membranes, while very stable at apical sites. In presence of chenodeoxycholic acid (CDCA) (Fig. S9 and Video S9†), a bile salt that activates a pathway to export them into bile canaliculi or of Cu at 100 μM (Fig. 5, Fig. S10, and Videos S10 and S11†), the internal dynamic within the spheroid seems faster and it is accompanied with an expansion of the internal volume in some of the acquisitions. We assume that this is due to a stimulation of the hepatocytes by these compounds. In addition, similarly to basal conditions, cell division can be observed as in Fig. S10,† white arrows. In presence of Cu, we also managed to track an elongated bile canaliculi structure (Fig. 5, white arrow) that remains relatively stable as in basal conditions. Overall, live actin imaging within spheroids revealed the stability of the bile canaliculi once formed as opposed to highly dynamic actin networks visualized upon cell division.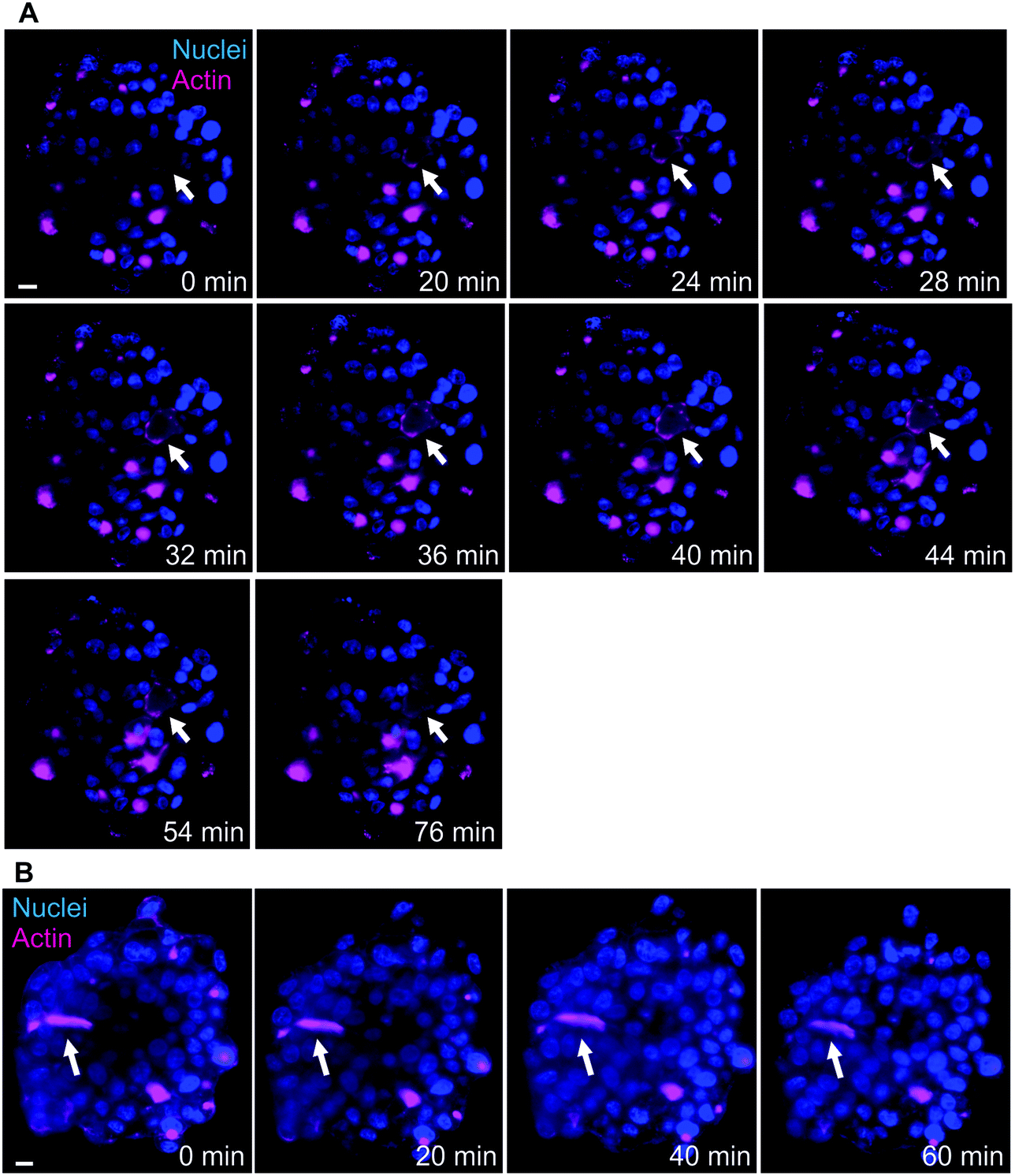 | ||
| Fig. 4 Actin network dynamics in basal conditions. (A) Live LSFM imaging of SiR-actin (pink) and Hoechst (blue). Selected time-points of a single plan LSFM within a spheroid in basal conditions. The full video corresponds to 80 minutes with one acquisition every 2 minutes (Video S7†). White arrows highlight the location of a site of actin re-organization upon cell division process. (B) Live LSFM imaging of SiR-actin (pink) and Hoechst (blue). Selected time-points of a single plan LSFM within a spheroid in basal conditions. The full video corresponds to 78 minutes with one acquisition every 2 minutes (Video S8†). White arrows highlight the location of a bile canaliculus. Scale bars correspond to 10 μm. | ||
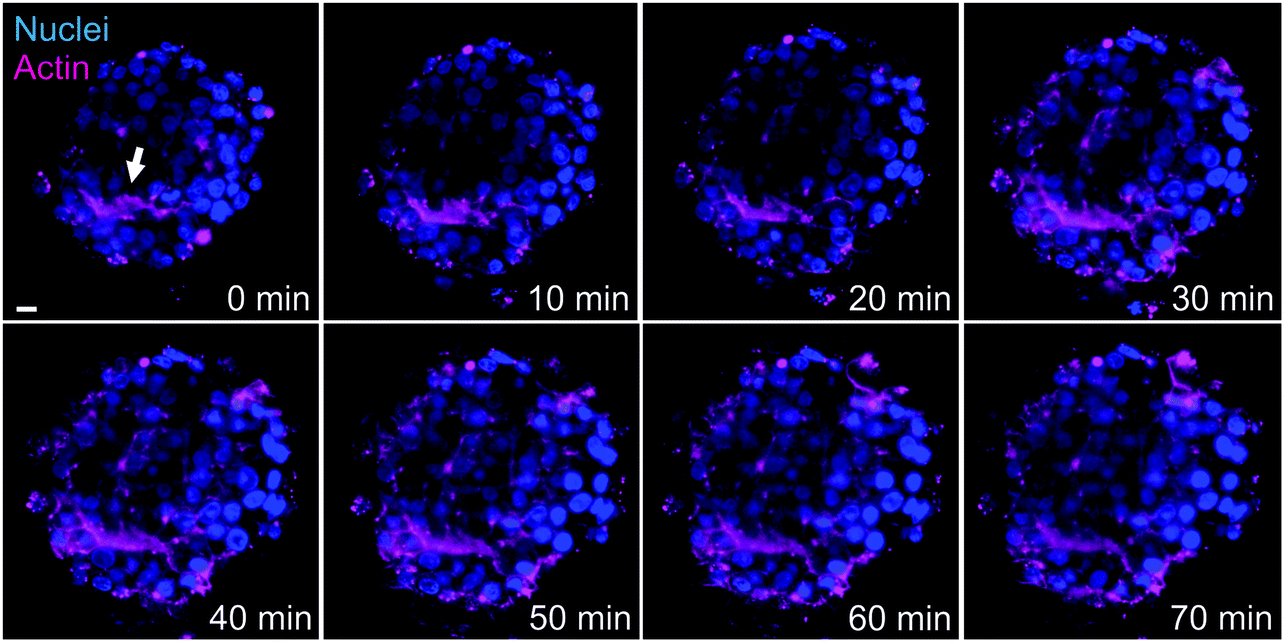 | ||
| Fig. 5 Actin network dynamics in Cu conditions. Live LSFM imaging of SiR-actin (pink) and Hoechst (blue). Selected time-points of a single plan LSFM within a spheroid exposed to 100 μM Cu. The full video corresponds to 80 minutes with one acquisition every 2 minutes (Video S10†). White arrow highlights the location of a bile canaliculus. The scale bar corresponds to 10 μm. | ||
The approach is therefore very powerful and could further be used to understand determinants involved in bile canaliculi formation and growth at the apical membrane versus actin surrounding basal membranes. Besides, the use of LSFM proved the interest and the validity of matrix-free spheroids and enabled to highlight key functional mechanisms of bile canaliculi excretion of both organic and inorganic compounds. However, the fine details of the ultrastructure of bile canaliculi are still missing.
3.4. Bile canaliculi structure within spheroids
Up to now, the ultrastructure of bile canaliculi was mainly analyzed based on ultrathin section of hepatocyte culture or of liver following chemical fixation and resin embedding. A single study proposed a low resolution 3D reconstruction of liver biliary tract using serial block face electron microscopy with a resolution of 40 nm between EM plan sections.4 This work provided a great overview of liver biliary functions coupled to low resolution ultrastructural data. To obtain a more detailed view of bile canaliculi ultrastructure, spheroid samples were analyzed by FIB-SEM, the only method able to provide a resolution down to 2 nm in the three space dimensions and applicable for biological samples embedded into plastic resin (for review22). Initial trials have been made using chemically fixed spheroids and bile canaliculi regions were easily identified as intercellular areas with membrane budding inside, the microvilli (Fig. S11A,† yellow arrow), and sealed by tight junctions that could not really be seen but that corresponds to the juxtaposition of two plasma membranes (Fig. S11A,† white arrows). However, typical artefacts of chemical fixation were observed, such as membrane ripening. High pressure freezing of the spheroids followed by resin embedding was also tested. Ultrastructure preservation was drastically improved as shown by nice membrane curvatures (Fig. 6A and Video S12†). Besides, several features looks shrunken following chemical fixation such as bile canaliculi lumen and microvilli. It is therefore clear that high-pressure freezing is required for the analysis of native and high-resolution ultrastructure of liver-mimicking model and especially for bile canaliculi structure reconstruction. Visual inspection of 2D electron micrographs showed the presence of nicely preserved bile canaliculi formed between several hepatocytes with typical tight junctions and microvilli (Fig. 6A and B, white and yellow arrows, respectively). Mitochondria were also nicely preserved with the inner and outer membranes visible (Fig. 6A and C). More interestingly, endoplasmic reticulum surrounding mitochondria were observed with parallel membrane curvatures for the two organelles highlighting inter-organelle organized areas oppositely to the images from chemically fixed sample (Fig. 6Cvs. Fig. S11B†). Indeed, it is now known that transient physical interactions between these two organelles exists (Fig. 6C, white arrows)30 and mediates specific signaling involving calcium transfer, for instance.31 The possibility to visualize such intracellular details is of paramount importance for the study of cellular signaling pathways in a liver-like model. However, this is just an example of the potential of the method developed in this study but all organelles, nuclei, vacuoles, etc., are nicely preserved and can be reconstructed in 3D. Based on the stack of 2D electron micrographs, we reconstructed an entire volume within a spheroid and segmented the different organelles (Fig. 7A and Video S13†): nuclei from the different surrounding hepatocytes (pink), mitochondrial network (green) and the intercellular space corresponding to bile canaliculi (grey). The final segmentation of the mitochondrial network is very good and shows a quite dense network with high connectivity (Fig. 7B). | ||
| Fig. 6 Ultrastructures within a high-pressure frozen spheroid. Electron micrographs extracted from FIB-SEM stack of images of a matrix-free spheroid. (A) Full field images of the area acquired in 3D extracted from the stack shown in Video S12.† (B) Zoom in on a bile canaliculus region (yellow arrow) defined as an intercellular space with microvilli and delimited by tight junctions (electron dense areas (black), white arrows). (C) Images showing various mitochondria, sometimes clearly interconnected. Endoplasmic reticulum is often found closely surrounding mitochondria and even some contact sites between the two organelles can be observed (white arrows). | ||
 | ||
| Fig. 7 3D reconstruction and segmentation of the inside of a high-pressure frozen spheroid. (A) Using the stack of images shown in Video S12† the inside of the spheroid was reconstructed and segmented in the different organelles and represented with nuclei (pink), mitochondria (green), vesicles (blue) and bile canaliculi (grey). The reconstruction and segmentation is shown in Video S13.† (B) 3D volume of the extracted mitochondrial segment that shows the connectivity of the mitochondrial network. (C) Electron micrograph of a single plan showing a bile canaliculus region. (D) 3D volume of the bile canaliculus segment that shows the density of intertwined microvilli and the high packing of the surrounding basal membranes. (E) Segmentation of the plasma membranes of the different hepatocytes surrounding the bile canaliculus shown in (D). 3D visualization with each cell membrane with a different color and showing sequentially only 2 or 3 plasma membranes at a time. | ||
As shown previously, the bile canaliculi are fully functional in these matrix-free spheroids, it was therefore particularly pertinent to perform an in-depth analysis of their structure considering them as an appropriate model for liver bile canaliculi. Intercellular areas with microvilli and closed by tight junctions were easily found (Fig. 7C). These regions are full of vesicle-like structures in 2D electron micrographs. From the 3D reconstruction of the FIB-SEM dataset, it was possible to segment the volume corresponding to a bile canaliculus (Fig. 7D) enabling to observe that all vesicle-like structures correspond to section view of microvilli. They are highly intertwined and all of them are attached to a plasma membrane meaning that exocytosis mechanisms are not observed in bile canaliculi. This high density enables to provide a very large membrane surface area in which Mrp2, BSEP, ATP7B, and other membrane transporters are inserted in order to favor excretion mechanisms. This also explains the high intensity and low resolution fluorescence signal obtained for Mrp2, ATP7B and actin staining since they are present all along these microvilli. Indeed, actin fibers enables the building of the microvilli and together with myosin their contraction favors bile secretion.32 Besides, microvilli with homogenous diameters were determined of about 0.3 μm. To get a better vision of the contribution of the different cells forming the canaliculus, this volume was again segmented to separate the different cells. The different plasma membranes are represented with different colors (Fig. 7E and Video S14†). Each cell surrounding the bile canaliculus is showing the same features with protrusions, i.e. the microvilli, and very flat membranes outside of canaliculus area tightly packed together with membranes of adjacent cells. This analysis is therefore consistent with functional data providing the ultrastructural basis of this polarized hepatic system. This is the first time the structure of a bile canaliculus is resolved at such a resolution in a liver-like model. Therefore, the use of FIB-SEM enabled to make a major progress in the structural characterization of hepatocytes organization within spheroids, which opens the way for deeper analysis of cellular events in this system.
4. Conclusions
In this study, we used LSFM and FIB-SEM imaging approaches to analyze the structure and function of matrix-free hepatic spheroids. Both methods are appropriate for the study of 3D cell culture model since their large size impairs the use of more conventional cell biology imaging methods such as confocal microscopy and electron microscopy of cell sections. LSFM enabled to follow any kind of canalicular excretion events including inorganics, which is very interesting regarding the central role of liver in metal homeostasis. Besides, LSFM showed that matrix-free spheroid is a very good model of liver biliary excretion since it recapitulates its functions and presents a great polarization as shown by the colocalization of Mrp2 and actin at the apical membranes, while the other cell membranes does not contain Mrp2 at all. An observation that is similar to what has been described recently in single liver cell polarization model.32 LSFM has also been used for live cell imaging that revealed the dynamics of actin networks in apical versus basal membranes where actin networks are highly stable and very dynamic, respectively. Furthermore, FIB-SEM analysis performed on highly preserved spheroid sample enabled to reconstruct at an unprecedented resolution a bile canaliculus, which showed the very high density of intertwined microvilli, which can be correlated with immunofluorescence images of canalicular proteins. The future would ideally be to implement LSFM and FIB-SEM in a correlative way to be able to reconstruct at nanometer resolution a specific region of interest. The current work thus paves the way for deep study of any kind of biomaterials in order to characterize them in more details both structurally and functionally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the CEA DRF-Impulsion grant FIB-Bio. This work used the platforms of the Grenoble Instruct-ERIC Center (ISBG: UMS 3518 CNRS-CEA-UGA-EMBL) with support from FRISBI (ANR-10-INSB-05-02) and GRAL (ANR-10-LABX-49-01) within the Grenoble Partnership for Structural Biology (PSB). The EM facility is headed by Guy Schoehn and supported by the Rhône-Alpes Region, the Fondation Recherche Medicale (FRM), the fonds FEDER and the GIS-Infrastrutures en Biologie Sante et Agronomie (IBISA). This research is part of the LabEx SERENADE (grant ANR-11-LABX-0064) and the LabEx ARCANE and CBH-EUR-GS (grant ANR-17-EURE-0003).Notes and references
-
A. Treyer and A. Müsch, in Comprehensive Physiology, ed. R. Terjung, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013 Search PubMed
.
- P. Gissen and I. M. Arias, J. Hepatol., 2015, 63, 1023–1037 CrossRef PubMed
- C. L. Slim, S. C. D. van IJzendoorn, F. Lázaro-Diéguez and A. Müsch, Bioarchitecture, 2014, 4, 47–52 CrossRef PubMed
- K. Meyer, O. Ostrenko, G. Bourantas, H. Morales-Navarrete, N. Porat-Shliom, F. Segovia-Miranda, H. Nonaka, A. Ghaemi, J.-M. Verbavatz, L. Brusch, I. Sbalzarini, Y. Kalaidzidis, R. Weigert and M. Zerial, Cell Syst., 2017, 4, 277–290 CrossRef CAS PubMed
- N. Okamura, T. Sakaeda and K. Okumura, Pers. Med., 2004, 1, 85–104 CrossRef CAS PubMed
- C. Hartwig, S. A. Zlatic, M. Wallin, A. Vrailas-Mortimer, C. J. Fahrni and V. Faundez, Curr. Opin. Cell Biol., 2019, 59, 24–33 CrossRef CAS
- N. E. Hellman and J. D. Gitlin, Annu. Rev. Nutr., 2002, 22, 439–458 CrossRef CAS PubMed
- S. R. Khetani, D. R. Berger, K. R. Ballinger, M. D. Davidson, C. Lin and B. R. Ware, J. Lab. Autom., 2015, 20, 216–250 CrossRef CAS PubMed
- Y. Takahashi, Y. Hori, T. Yamamoto, T. Urashima, Y. Ohara and H. Tanaka, Biosci. Rep., 2015, 35, e00208 CrossRef PubMed
- Z. Wang, X. Luo, C. Anene-Nzelu, Y. Yu, X. Hong, N. H. Singh, L. Xia, S. Liu and H. Yu, J. Appl. Toxicol., 2015, 35, 909–917 CrossRef CAS PubMed
- D. Mueller, L. Krämer, E. Hoffmann, S. Klein and F. Noor, Toxicol. In Vitro, 2014, 28, 104–112 CrossRef CAS
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef CAS PubMed
- S. F. Wong, D. Y. No, Y. Y. Choi, D. S. Kim, B. G. Chung and S.-H. Lee, Biomaterials, 2011, 32, 8087–8096 CrossRef CAS PubMed
- S. J. Fey and K. Wrzesinski, Toxicol. Sci., 2012, 127, 403–411 CrossRef CAS PubMed
- M. Bokhari, R. J. Carnachan, N. R. Cameron and S. A. Przyborski, J. Anat., 2007, 211, 567–576 CAS
- K. Wrzesinski, M. C. Magnone, L. V. Hansen, M. E. Kruse, T. Bergauer, M. Bobadilla, M. Gubler, J. Mizrahi, K. Zhang, C. M. Andreasen, K. E. Joensen, S. M. Andersen, J. B. Olesen, O. B. Schaffalitzky de Muckadell and S. J. Fey, Toxicol. Res., 2013, 2, 163 RSC
- K. Wrzesinski and S. J. Fey, Toxicol. Res., 2013, 2, 123–135 RSC
- S. R. Khetani and S. N. Bhatia, Nat. Biotechnol., 2008, 26, 120–126 CrossRef CAS PubMed
- H. Gaskell, P. Sharma, H. E. Colley, C. Murdoch, D. P. Williams and S. D. Webb, Toxicol. Res., 2016, 5, 1053–1065 RSC
- A. U. Yumoto, S. Watanabe, M. Hirose, T. Kitamura, Y. Yamaguchi and N. Sato, Liver, 2008, 16, 61–66 CrossRef PubMed
- R. M. Power and J. Huisken, Nat. Methods, 2017, 14, 360–373 CrossRef CAS PubMed
- K. Narayan and S. Subramaniam, Nat. Methods, 2015, 12, 1021 CrossRef CAS PubMed
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed
-
C. Sommer, C. Straehle, U. Kothe and F. A. Hamprecht, in 2011 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, IEEE, Chicago, IL, USA, 2011, pp. 230–233
-
P. Cignoni, M. Callieri, M. Corsini, M. Dellepiane, F. Ganovelli and G. Ranzuglia, Eurographics Ital. Chapter Conf., 2008, 129–136
-
J. Ahrens, B. Geveci and C. Law, in The visualization handbook, 2005, vol. 717 Search PubMed
- A. M. Cozzolino, V. Noce, C. Battistelli, A. Marchetti, G. Grassi, C. Cicchini, M. Tripodi and L. Amicone, Stem Cells Int., 2016, 2016, 1–12 CrossRef PubMed
- E. V. Polishchuk, M. Concilli, S. Iacobacci, G. Chesi, N. Pastore, P. Piccolo, S. Paladino, D. Baldantoni, S. C. D. van IJzendoorn, J. Chan, C. J. Chang, A. Amoresano, F. Pane, P. Pucci, A. Tarallo, G. Parenti, N. Brunetti-Pierri, C. Settembre, A. Ballabio and R. S. Polishchuk, Dev. Cell, 2014, 29, 686–700 CrossRef CAS PubMed
- G. Lukinavičius, L. Reymond, E. D'Este, A. Masharina, F. Göttfert, H. Ta, A. Güther, M. Fournier, S. Rizzo, H. Waldmann, C. Blaukopf, C. Sommer, D. W. Gerlich, H.-D. Arndt, S. W. Hell and K. Johnsson, Nat. Methods, 2014, 11, 731–733 CrossRef PubMed
- Y. Guo, D. Li, S. Zhang, Y. Yang, J.-J. Liu, X. Wang, C. Liu, D. E. Milkie, R. P. Moore, U. S. Tulu, D. P. Kiehart, J. Hu, J. Lippincott-Schwartz, E. Betzig and D. Li, Cell, 2018, 175, 1430–1442 CrossRef CAS PubMed
- A. Deniaud, O. Sharaf el dein, E. Maillier, D. Poncet, G. Kroemer, C. Lemaire and C. Brenner, Oncogene, 2008, 27, 285–299 CrossRef CAS PubMed
- Y. Zhang, R. de Mets, C. Monzel, P. Toh, N. Van Hul, S. S. Ng, S. Tamir Rashid and V. Viasnoff, bioRxiv, DOI: DOI:10.1101/636654.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9bm01143a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |