
Highly selective redistribution of primary arylsilanes to secondary arylsilanes catalyzed by Ln(CH2C6H4NMe2-o)3@SBA-15†
Chenjun
Guo
a,
Min
Li
a,
Jue
Chen
*b and
Yunjie
Luo
 *a
*a
aSchool of Material Science and Chemical Engineering, Ningbo University, Ningbo 315211, P. R. China. E-mail: luoyunjie@nbu.edu.cn
bSchool of Biological and Chemical Engineering, Ningbo Institute of Technology, Zhejiang University, Ningbo 315100, P. R. China. E-mail: chj@nit.zju.edu.cn
First published on 26th November 2019
Abstract
Rare-earth metal tris(aminobenzyl) complexes Ln(CH2C6H4NMe2-o)3 (Ln = La, Y) were grafted onto the dehydroxylated periodic mesoporous silica support SBA-15 to generate the organometallic–inorganic hybrid materials Ln(CH2C6H4NMe2-o)3@SBA-15 (Ln = La (2a), Y (2b)), which demonstrated extremely high selectivity (>99%) in catalyzing the redistribution of primary arylsilanes to secondary arylsilanes without the requisition of strict control of the reaction conditions. The hybrid materials still showed a perfect selectivity and activity after three catalytic cycles.
Hydrosilanes are regarded as important organic compounds in organic chemistry, the pharmaceutical industry and materials science.1–3 In this respect, the formation of C–Si bonds has attracted increasing attention in academia and industrial interest.4–6 Redistribution of hydrosilanes is considered to be a straightforward synthetic strategy to access organosilicon compounds; however, poor selectivity and low efficiency hamper its wide applications due to it converting one hydrosilane to two or more compounds through C–Si and Si–H bond cleavage and reformation.7–19 This phenomenon is particularly common in the redistribution of primary silanes because they are ready not only to undergo dehydrocoupling to give oligo- or polysilanes,20,21 but also to experience a redistribution process to produce a mixture of secondary, tertiary and quaternary silanes22–24 (Scheme 1). Moreover, the redistribution of hydrosilanes is usually observed as a side reaction of transition-metal-catalyzed dehydrocoupling of hydrosilanes,8,11 or in metal-mediated stoichiometric transformations,9,10 while the examples of direct catalytic transformation are still limited.7,12–15 Although Hou and co-workers communicated that B(C6F5)3 was able to catalyze the redistribution of hydrosilanes, this approach was only effective for converting tertiary silanes into quaternary silanes.25 To our knowledge, only one example reported by Chen and co-workers in 2019 could promote the selective redistribution of primary silanes to secondary silanes by using the divalent ytterbium complex [MeC(NDipp)CHC(Me)NCH2CH2N(Me)CH2CH2NMe2]YbR (R = CH2SiMe3, CH2C6H4NMe2-o) as the catalyst. However, to obtain the desired reaction outcomes, the reaction conditions should be controlled severely.26 Therefore, it is still a great challenge to redistribute primary hydrosilanes to secondary hydrosilanes with high efficiency by a catalytic process.
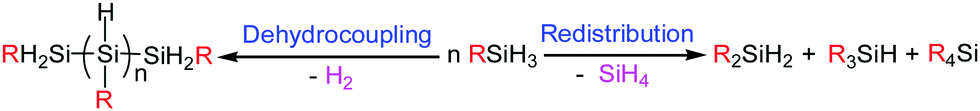 | ||
| Scheme 1 Redistribution and dehydrocoupling of primary silanes. | ||
Meanwhile, to avoid the well-known limitations of homogeneous catalysts such as poor recyclability and catalyst contamination in the products, much effort has been made toward the utilization of surface organometallic chemistry for the precise synthesis of supported catalysts in recent years.27 Among the support materials, the dehydroxylated periodic mesoporous silica support SBA-15 has shown significant attraction due to its high surface area, uniform pore size, and high hydrothermal stability.28 While organo rare-earth metal complexes have exhibited unique and promising catalytic performance in the field of organic transformation and polymerization,29–33 in striking contrast, studies on immobilization of rare-earth metal complexes,34–37 especially rare-earth metal alkyl/benzyl derivatives onto inorganic supports, are still scarce owing to their high reactivity toward protic reagents.38–42
In view of our long-standing interest in developing efficient rare-earth metal catalysts,43–46 we became interested in the supported rare-earth metal catalysts to uncover the nature of their potential reactivity. Here, we describe that rare-earth metal tris(aminobenzyl) complexes Ln(CH2C6H4NMe2-o)3 could be grafted onto SBA-15 to generate the corresponding organometallic–inorganic materials Ln(CH2C6H4NMe2-o)3@SBA-15. Remarkably, it was found that such simple easily accessible hybrid materials Ln(CH2C6H4NMe2-o)3@SBA-15 were able to catalyze the redistribution of primary arylsilanes to secondary arylsilanes with extremely high selectivity and showed reasonable activity after several cycles.
Treatment of the white SBA-15 (partially dehydroxylated at 220 °C under high vacuum 10−5 Torr at 220 °C for 12 h) with a colourless toluene solution of Ln(CH2C6H4NMe2-o)3 (Ln = La (1a), Y (1b)) at room temperature for 20 h, after workup, afforded the corresponding orange organometallic–inorganic hybrid materials Ln(CH2C6H4NMe2-o)3@SBA-15 (Ln = La (2a), Y (2b)) via Ln–C bond protonolysis, as shown in Scheme 2.
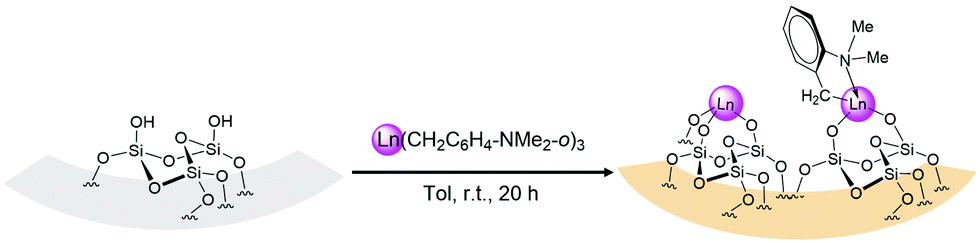 | ||
| Scheme 2 Reaction of Ln(CH2C6H4NMe2-o)3 with SBA-15 to generate Ln(CH2C6H4NMe2-o)3@SBA-15 (Ln = La (2a), Y (2b)) | ||
Elemental analyses indicated Ln/C/N loading of 18.21/8.53/1.04 wt% for 2a and 12.10/8.80/1.08 wt% for 2b. The corresponding C/N/Ln molar ratio was 9.6/1/1.8 in 2a and 9.5/1/1.8 in 2b. This showed the formation of the hybrid materials Ln(CH2C6H4NMe2-o)3@SBA-15. Moreover, the average number of aminobenzyl groups per rare-earth metal was close to 1.8, assignable to a mixture of [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)2Ln(CH2C6H4NMe2-o)] (≈55%) and [(SiO)3Ln] (≈45%) surface species as illustrated in Scheme 1. Infrared spectra of the hybrid materials displayed signals around 2940 cm−1 and 2780 cm−1 for νsp2C–H and νsp3C–H. Bands at about 1590 cm−1 and 1490 cm−1 also supported the presence of C
SiO)2Ln(CH2C6H4NMe2-o)] (≈55%) and [(SiO)3Ln] (≈45%) surface species as illustrated in Scheme 1. Infrared spectra of the hybrid materials displayed signals around 2940 cm−1 and 2780 cm−1 for νsp2C–H and νsp3C–H. Bands at about 1590 cm−1 and 1490 cm−1 also supported the presence of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C aromatic-ring stretching vibrations and methylene δ(C−H) vibration, respectively (Fig. 1). Characterization by nitrogen physisorption and analysis of the Barret–Joyner–Halenda (BJH) pore size distribution revealed a consistent decrease of the pore diameter (Δdp ≈ 0.5–0.9 nm) for the hybrid materials (Table 1). Scanning electron microscopy (SEM) showed that the morphology of SBA-15 was retained after the immobilization of Ln(CH2C6H4NMe2-o)3 on its surface. These results confirmed that the rare-earth metal aminobenzyl species were successfully grafted onto SBA-15 (Fig. S21 in ESI†). XPS spectra showed that the oxidation state of the metal ions did not change after heterogenization over SBA-15, which was consistent with the rare-earth elements that usually adopt the most stable 3+ oxidation state.30
C aromatic-ring stretching vibrations and methylene δ(C−H) vibration, respectively (Fig. 1). Characterization by nitrogen physisorption and analysis of the Barret–Joyner–Halenda (BJH) pore size distribution revealed a consistent decrease of the pore diameter (Δdp ≈ 0.5–0.9 nm) for the hybrid materials (Table 1). Scanning electron microscopy (SEM) showed that the morphology of SBA-15 was retained after the immobilization of Ln(CH2C6H4NMe2-o)3 on its surface. These results confirmed that the rare-earth metal aminobenzyl species were successfully grafted onto SBA-15 (Fig. S21 in ESI†). XPS spectra showed that the oxidation state of the metal ions did not change after heterogenization over SBA-15, which was consistent with the rare-earth elements that usually adopt the most stable 3+ oxidation state.30
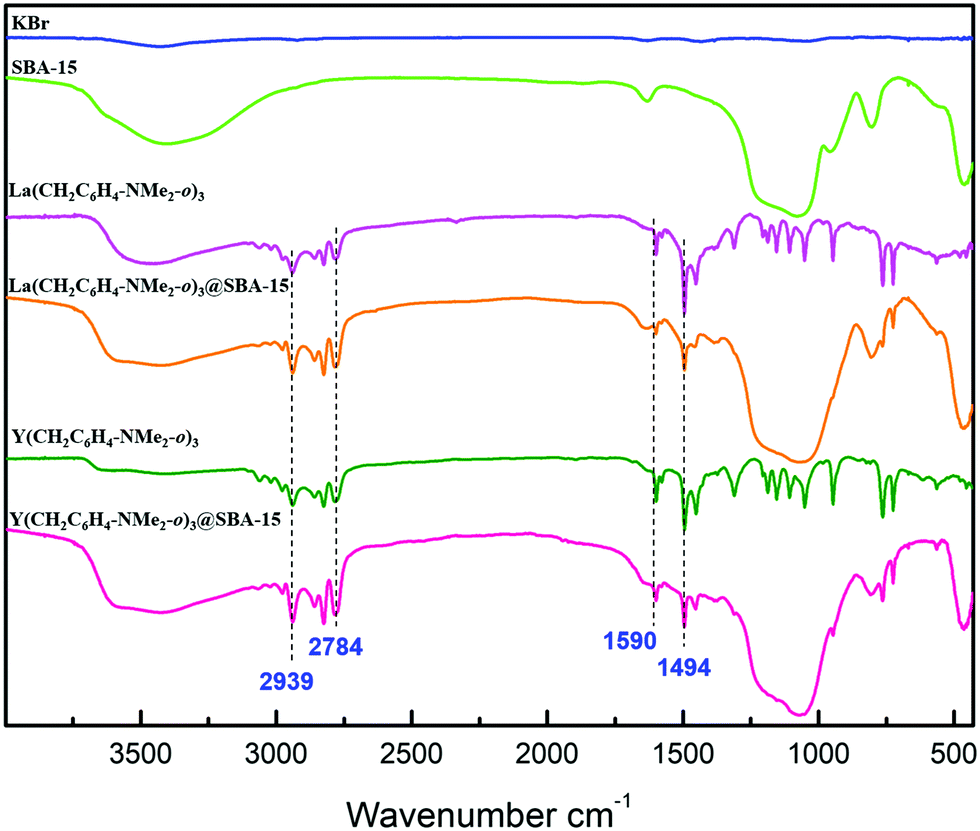 | ||
| Fig. 1 The FT-IR spectra (KBr) of SBA-15, Ln(CH2C6H4NMe2-o)3 (Ln = La (1a), Y (1b)) and Ln(CH2C6H4NMe2-o)3@SBA-15 (Ln = La (2a), Y (2b)). | ||
| Hybrid materiala | wt% Lnb (mmol g−1) | wt% N (Ln/N) | Asc (m2 g−1) | Vpd (cm3 g−1) | d p,des (nm) |
|---|---|---|---|---|---|
| a Pretreatment temperature: 220 °C, 12 h, 10−5 Torr for the PMS material SBA-15; 25 °C, 6 h, 10−3 Torr for 2a and 2b. b Estimated by EDTA titration. c Specific BET surface area. d BJH from desorption branch cumulative pore volume of pores between 1.0 and 25 nm diameter. e Pore diameter according to the maximum BJH pore size distribution calculated from the desorption branch. | |||||
| SBA-15 | — | — | 647 | 1.09 | 5.7 |
| 2a | 1.31 | 1.04 (1.8) | 246 | 0.43 | 4.8 |
| 2b | 1.36 | 1.08 (1.8) | 340 | 0.58 | 5.2 |
With the hybrid materials 2a and 2b in hand, we assessed their catalytic performance in the redistribution of primary arylsilanes. Firstly, the reactions of 1a and 1b with PhSiH3 were carried out at the loading of 5 mol% 1a or 1b in C6D6 at 25 °C. A black precipitate was observed rapidly on the addition of PhSiH3 to a C6D6 solution of 1a and 1b, with the release of PhSiH2CH2C6H4NMe2-o. This could be ascribed to the homogeneous reaction mixture facilitating the fast formation of insoluble rare-earth metal hydride complexes. As shown in Table 2, 1a and 1b showed poor activity toward the redistribution reaction, and only 18–22% PhSiH3 was consumed after the reaction took place at 25 °C for 12 h (entries 1 and 2, Table 2). The low activity of 1a and 1b might be temporarily attributed to the generation of the active intermediate for redistribution which was not favourable compared to that of the rare-earth metal hydride species in this homogeneous reaction. Encouragingly, employing the hybrid materials 2a as the catalysts, the redistribution of PhSiH3 proceeded smoothly at room temperature and the conversion of PhSiH3 reached 82% in 6 h using 5 mol% of 2a as the catalyst (entry 3, Table 2). Raising the reaction temperature accelerated the reaction, and nearly full conversion was achieved in 4 h at 120 °C catalyzed by 2a (entry 10, Table 2).
| Entry | Cat. | ArSiH3 | Temp (°C) | t (h) | Yieldb (%) | Selectivityb (%) |
|---|---|---|---|---|---|---|
| a Reactions were performed in a Schlenk tube with 5 mol% of catalyst and 0.5 mmol ArSiH3 in 1.5 mL C6D6. b Determined by 1H NMR spectra using 1,3,5-trimethylbenzene as the internal standard. | ||||||
| 1 | 1a | PhSiH3 | 25 | 12 | 22 | >99 |
| 2 | 1b | PhSiH3 | 25 | 12 | 18 | >99 |
| 3 | 2a | PhSiH3 | 25 | 6 | 82 | >99 |
| 4 | 2b | PhSiH3 | 25 | 6 | 78 | >99 |
| 5 | 2a | PhSiH3 | 60 | 2 | 74 | >99 |
| 6 | 2a | PhSiH3 | 60 | 4 | 85 | >99 |
| 7 | 2a | PhSiH3 | 60 | 6 | 91 | >99 |
| 8 | 2b | PhSiH3 | 60 | 6 | 90 | >99 |
| 9 | 2a | PhSiH3 | 120 | 2 | 88 | >99 |
| 10 | 2a | PhSiH3 | 120 | 4 | 99 | >99 |
| 11 | 2a | PhSiH3 | 120 | 12 | 99 | >99 |
| 12 | 2a |
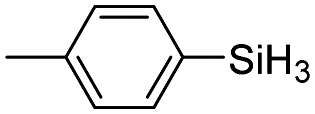
|
60 | 4 | 84 | >99 |
| 13 | 2a |
|
60 | 4 | 75 | >99 |
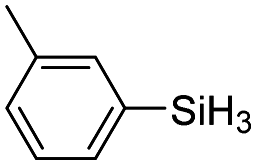
| 14 | 2a |
|
60 | 4 | 43 | >99 |
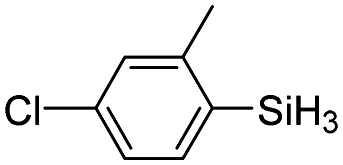
| 15 | 2a |
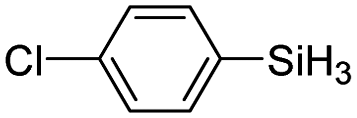
|
60 | 4 | 93 | >99 |
| 16 | 2a |
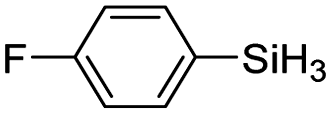
|
60 | 4 | 73 | >99 |
| 17 | 2a |
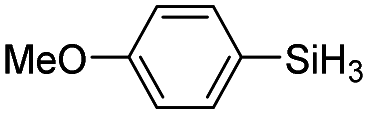
|
60 | 4 | 78 | >99 |
| 18 | 2a |
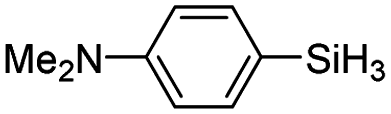
|
60 | 4 | 57 | >99 |
| 19 | 2a | n-C6H13SiH3 | 60 | 4 | 0 | 0 |
The electronic effect of substituents on catalytic reactivity was investigated. The introduction of electron-donating MeO or Me groups on the para-position decreased to some extent the redistribution activity of arylsilanes (entries 6, 12 and 17, Table 2). The redistribution of p-NMe2C6H4SiH3 bearing a strong electron-donating NMe2 group showed a much lower activity (entry 18, Table 2). In comparison, the electron-withdrawing substituted arylsilane, for example p-ClC6H4SiH3, displayed a relatively increased redistribution rate, and a 93% conversion was realized with 5 mol% of 2a in 4 h at 60 °C (entries 6 and 15, Table 2). However, introducing a Me group at the ortho-position of p-ClC6H4SiH3 dramatically hampered the reaction, and only afforded the product in 43% yield (entries 14, Table 2). The reactivity tendency of the substituted arylsilanes Cl > F > H ∼ Me > MeO > NMe2 was nearly in line with the order of the Hammett constants Cl (0.23) > F (0.06) > H (0) > Me (−0.17) > MeO (−0.27).47 Additionally, the redistribution of alkylsilane such as n-C6H13SiH3 was not successful under the same conditions (entry 19, Table 2). These findings were consistent with those reported by Chen.26 The hybrid material 2b could also be applied to catalyze the redistribution of these primary arylsilanes under the same conditions. However, no significant dependence of the reactivity on the metal size was observed.
It was noteworthy that there was only one redistribution product in all these reactions. NMR spectra revealed that only secondary arylsilanes were transformed from primary arylsilanes, and the selectivity reached up to at least 99% in all these cases. Remarkably, employing 2a and 2b as the catalysts, the reaction time and the reaction temperature had no effect on the redistribution selectivity. For example, the redistribution reactions performed in a range of temperatures from 25 to 120 °C, or even at a high temperature (120 °C) for a prolonged time (12 h), produced secondary arylsilanes with high selectivity (entries 10 and 11). The extremely high selectivity of this catalytic system for the redistribution of primary arylsilanes to secondary arylsilanes was unique and was different to that reported by Chen, in which to obtain the desired reaction outcomes, the reaction conditions, such as reaction time, should be severely controlled, otherwise, the desired products would be further redistributed to give a mixture of products.26 The formation of triarylsilanes was efficiently suppressed in this catalytic process. This could be ascribed to SBA-15 possibly also acting as a steric hindrance ligand set, which hampered the further redistribution reaction. To validate the practical utility of this procedure to get secondary arylsilanes, gram-scale reaction employing 5 mol% of 2a in the redistribution of PhSiH3 was conducted by refluxing n-hexane for 8 h, which also afforded only one product Ph2SiH2 in 93% isolated yield.
Remarkably, the hybrid materials 2a and 2b could be recovered and reused. After each cycle, 2a and 2b were separated by simple filtration, washed several times using toluene, and dried under vacuum. In every cycle, there was no change in the selectivity (>99%). However, after three cycles (fresh + two cycles), the hybrid materials showed a rather low activity toward the redistribution of arylsilanes (Table 3). The ICP-AES analysis revealed a gradual metal loss from the fresh catalyst (Table S1 in ESI†). In contrast, Sheldon's hot filtration tests indicated that the metals leaching into the reaction solution showed hardly any activity toward the redistribution reaction (Fig. S24 in ESI†). These results suggested that the instability of the catalyst hetereogeneity might account for the poor activity after several cycles. Besides, the hybrid materials demonstrated poor selectivity for the cross-coupling of two different primary arylsilanes, producing a mixture of two homo-coupling and one cross-coupling reaction outcomes. For example, in the presence of 5 mol% 2a at 60 °C for 4 h, the redistribution of p-MeC6H4SiH3 and p-ClC6H4SiH3 afforded only 55% cross-coupling product (Fig. S16 in ESI†), indicative of a strikingly different reaction pattern to the rare-earth metal catalysts reported by Hou and Chen.25,26
1H NMR spectra showed the release of SiH3(CH2C6H4NMe2-o) on treatment of the hybrid materials with PhSiH3, suggestive of the generation of an intermediate (SiO)2LnPh from Si–C bond activation of PhSiH3 (Fig. S17 in ESI†). This intermediate has been considered to play a crucial role in catalyzing the redistribution process.26,48 Therefore, the mechanism for the redistribution in this context was proposed as follows. Treatment of the hybrid materials [(SiO)2Ln(CH2C6H4NMe2-o)] with PhSiH3 generated the intermediate (SiO)2LnPh, which would react with Si–H bond of PhSiH3 to produce the redistribution outcome Ph2SiH2 and the rare-earth metal hydride (SiO)2LnH. The hydride species would further react with PhSiH3 to regenerate (SiO)2LnPh to promote the catalytic cycle (Scheme S1 in the ESI†).
In conclusion, Ln(CH2C6H4NMe2-o)3 (Ln = La, Y) could be grafted onto the dehydroxylated periodic mesoporous silica support SBA-15 to give the corresponding organometallic–inorganic hybrid materials Ln(CH2C6H4NMe2-o)3@SBA-15 (Ln = La, Y), which showed a promising catalytic activity in the redistribution of a wide range of primary arylsilanes to secondary arylsilanes with extremely high selectivity (>99%) without the requisition of controlling the reaction conditions. These catalysts could be recovered and reused without the loss in selectivity, albeit the activity decreased dramatically after three recycling steps. Such kind of rare-earth metal materials might find a practical application in organic transformation owing to their easy preparation, high catalytic efficiency and recyclable property. Further study on exploiting the potential applications in catalysis of these hybrid materials is ongoing.
We acknowledge financial support from the National Natural Science Foundation of China (21572205 and 21971130), the Natural Science Foundation of Zhejiang Province (LY19B040002), the Natural Science Foundation of Ningbo Municipal (2019A610030 and 2019A610129), and K. C. Wong Magna Fund in Ningbo University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. A. Brook, Silicon in Organic, Organometallic, and Polymer Chemistry, Wiley, New York, 2000 Search PubMed.
- The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, 1998, vol. 2 Search PubMed.
- The Chemistry of Organic Silicon Compounds, ed. S. Patai and Z. Rappoport, Wiley, Chichester, 1989, vol. 1 Search PubMed.
- T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS.
- E. Langkopf and D. Schinzer, Chem. Rev., 1995, 95, 1375–1408 CrossRef CAS.
- M. D. Curtis and P. S. Epstein, Adv. Organomet. Chem., 1981, 19, 213–255 CrossRef CAS.
- K. A. Brown-Wensley, Organometallics, 1987, 6, 1590–1591 CrossRef CAS.
- N. S. Radu, F. J. Hollander, T. D. Tilley and A. L. Rheingold, Chem. Commun., 1996, 2459–2460 RSC.
- I. Castillo and T. D. Tilley, Organometallics, 2001, 20, 5598–5605 CrossRef CAS.
- L. Rosenberg, C. W. Davis and J. Z. Yao, J. Am. Chem. Soc., 2001, 123, 5120–5121 CrossRef CAS.
- S. Park, B. G. Kim, I. Göttker-Schnetmann and M. Brookhart, ACS Catal., 2012, 2, 307–316 Search PubMed.
- M. Khandelwal and R. J. Wehmschulte, Angew. Chem., Int. Ed., 2012, 51, 7323–7326 CrossRef CAS.
- J. W. Chen and E. Y. X. Chen, Angew. Chem., Int. Ed., 2015, 54, 6842–6846 CrossRef CAS.
- A. Feigl, I. Chiorescu, K. Deller, S. U. H. Heidsieck, M. R. Buchner, V. Karttunen, A. Bockholt, A. Genest, N. Rösch and B. Rieger, Chem. – Eur. J., 2013, 19, 12526–12536 CrossRef CAS.
- N. Lühmann, H. Hirao, S. Shaik and T. Müller, Organometallics, 2011, 30, 4087–4096 CrossRef.
- A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef PubMed.
- K. Müther, P. Hrobárik, V. Hrobáriková, M. Kaupp and M. Oestreich, Chem. – Eur. J., 2013, 19, 16579–16594 CrossRef PubMed.
- R. Labbow, F. Reiß, A. Schulz and A. Villinger, Organometallics, 2014, 33, 3223–3226 CrossRef CAS.
- T. D. Tilley, Acc. Chem. Res., 1995, 26, 22–29 CrossRef.
- J. Y. Corey, Adv. Organomet. Chem., 2004, 51, 1–52 CrossRef CAS.
- J. J. Hao, B. Vabre and D. Zargarian, J. Am. Chem. Soc., 2015, 137, 15287–15298 CrossRef CAS.
- P. Sangtrirutnugul and T. D. Tilley, Organometallics, 2007, 26, 5557–5568 CrossRef CAS.
- J. L. Speier and R. E. Zimmerman, J. Am. Chem. Soc., 1955, 77, 6395–6396 CrossRef CAS.
- Y. H. Ma, L. Zhang, Y. Luo, M. Nishiura and Z. M. Hou, J. Am. Chem. Soc., 2017, 139, 12434–12437 CrossRef CAS.
- X. J. Liu, L. Xiang, E. Louyriac, L. Maron, X. B. Leng and Y. F. Chen, J. Am. Chem. Soc., 2019, 141, 138–142 CrossRef CAS.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 2018, 370, 129–223 CrossRef CAS.
- M. Nishiura, F. Guo and Z. M. Hou, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS.
- M. Zimmermann and R. Anwander, Chem. Rev., 2010, 110, 6194–6259 CrossRef CAS.
- P. M. Zeimentz, S. Arndt, B. R. Elvidge and J. Okuda, Chem. Rev., 2006, 106, 2404–2433 CrossRef CAS.
- G. A. Molander and J. A. C. Romero, Chem. Rev., 2002, 102, 2161–2186 CrossRef CAS PubMed.
- R. Anwander, Chem. Mater., 2001, 13, 4419–4438 CrossRef CAS.
- R. M. Gauvin, L. Delevoye, R. A. Hassan, J. Keldenich and A. Mortreux, Inorg. Chem., 2007, 46, 1062–1070 CrossRef CAS.
- E. L. Roux, Y. C. Liang, M. P. Storz and R. Anwander, J. Am. Chem. Soc., 2010, 132, 16368–16371 CrossRef.
- C. Coperet, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Nunez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef CAS.
- M. P. Conley, G. Lapadula, K. Sanders, D. Gajan, A. Lesage, I. D. Rosal, L. Maron, W. W. Lukens, C. Copéret and R. A. Andersen, J. Am. Chem. Soc., 2016, 138, 3831–3843 CrossRef CAS.
- I. Nagl, M. Widenmeyer, E. Herdtweck, G. Raudaschl-Sieber and R. Anwander, Microporous Mesoporous Mater., 2001, 44–45, 311–319 CrossRef CAS.
- T. Vancompernolle, A. Valenta, T. Chenal, P. Zinck, I. D. Rosal, L. Maron, M. Taoufik, S. Harder and R. M. Gauvin, Organometallics, 2017, 36, 3912–3920 CrossRef CAS.
- A. Fischbach, M. G. Klimpel, M. Widenmeyer, E. Herdtweck, W. Scherer and R. Anwander, Angew. Chem., Int. Ed., 2004, 43, 2234–2239 CrossRef CAS.
- E. L. Roux, Y. C. Liang and R. Anwander, Chem. Cat. Chem., 2018, 10, 1905–1911 Search PubMed.
- M. Li, C. P. Wang, J. Chen, Z. Y. Guo, Z. H. Mou and Y. J. Luo, Dalton Trans., 2018, 47, 15967–15976 RSC.
- Z. X. Zhuo, C. Zhang, Y. J. Luo, Y. R. Wang, Y. M. Yao, D. Yuan and D. M. Cui, Chem. Commun., 2018, 54, 11998–12001 RSC.
- L. Q. Shi, Q. Su, J. Chen, X. N. Li and Y. J. Luo, Dalton Trans., 2016, 45, 1391–1397 RSC.
- F. Chen, S. M. Fan, Y. B. Wang, J. Chen and Y. J. Luo, Organometallics, 2012, 31, 3730–3735 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- L. Perrin, L. Maron, O. Eisenstein and T. D. Tilley, Organometallics, 2009, 28, 3767–3775 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc07493g |
| This journal is © The Royal Society of Chemistry 2020 |