
Metal-free α-arylation of α-fluoro-α-nitroacetamides employing diaryliodonium salts†
Mohd Khalid
Zaheer
a,
Ekta
Gupta
a,
Ruchir
Kant
b and
Kishor
Mohanan
 *ac
*ac
aMedicinal and Process Chemistry Division, CSIR-Central Drug Research Institute, Lucknow 226031, India. E-mail: kishor.mohanan@cdri.res.in
bMolecular and Structural Biology Division, CSIR-Central Drug Research Institute, Lucknow 226031, India
cAcademy of Scientific and Innovative Research, New Delhi, India
First published on 27th November 2019
Abstract
Herein, we present a mild and efficient metal-free arylation of α-fluoro-α-nitroacetamides employing diaryliodonium salts. A broad range of diaryliodonium salts and α-fluoro-α-nitroacetamides containing sensitive functional groups was successfully employed in this protocol to yield the arylated products in good yields. The synthetic value of this novel protocol was further highlighted by extending the α-arylation to α-cyano-α-fluoroacetamides.
The growing interest in fluorinated aromatic compounds in drug discovery research is attributed to the potential of the fluorine atom to fine tune biological properties such as metabolic stability and lipophilicity when it is strategically placed in a molecule.1,2 Over the past two decades, there has been significant progress in the development of novel methods for the introduction of fluorine and fluoroalkyl groups into arenes.3,4 The most frequently used strategies, particularly in the case of fluorocarbonyl compounds, rely on metal-catalyzed coupling between aryl halides and appropriately functionalized fluoroalkyl compounds.5 Hence, a complementary method to introduce fluoroalkyl groups into arenes remains a relevant task.6
Hypervalent iodine compounds have received great attention over the last few decades as a versatile class of reagents useful for a wide variety of synthetic transformations.7,8 Among them, diaryliodonium salts have emerged as mild, selective and robust reagents for both metal-catalyzed and metal-free electrophilic arylation reactions.9,10 Recent studies have ascertained the versatile use of diaryliodonium salts in the installation of the arene moiety into a multitude of heteroatom nucleophiles such as alcohols, amines, amides, thiols, and enolizable carbon nucleophiles.11,12 In addition to metal-catalyzed C-arylations, metal-free α-arylations of enolizable substrates such as ketones, α-ketoesters, malonates, nitroalkanes, α-nitroketones, and cyanoesters have also been documented in recent years (Scheme 1, eqn (1)).13 However, to our knowledge, this strategy has not yet been exploited for fluorocarbon nucleophiles. Intrigued by the compatibility of diaryliodonium salts with enolizable substrates, we wondered about the possibility of using this strategy for the introduction of arenes into α-substituted fluoroacetamides, given their immense synthetic value as essential motifs in pharmaceuticals.14 Herein, we report a novel metal-free protocol for the direct α-arylation of α-fluoro-α-nitroacetamides under mild basic conditions to generate a fully substituted benzylic fluoroalkyl center (Scheme 1, eqn (2)). Moreover, the process was found to be useful for the arylation of α-cyano-α-fluoroacetamides.
 | ||
| Scheme 1 Electrophilic arylations using diaryliodonium salts. | ||
We selected N-benzyl-2-fluoro-2-nitro-N-phenylacetamide 1a as a suitable enolizable substrate and the unsymmetrical mesityl(phenyl)iodonium salt 2a (1.1 equiv.) as an arylating agent to optimize the reaction conditions. The high chemoselectivity observed in the selective transfer of aryl groups over the mesityl group in the C-arylation of carbon nucleophiles prompted us to choose the mesityl group as the dummy ligand in this study.15 Pleasingly, the reaction conducted in the presence of Cs2CO3 gave the α-arylated product 3a in 65% yield in THF at 50 °C (Table 1, entry 1). In attempts to improve the reaction efficiency, various solvents were screened (entries 2–7), and toluene proved to be an optimal solvent furnishing the expected product in 86% yield (entry 4). Further evaluation revealed that the reactions conducted using TBME, dioxane, DCM, and DMF as solvents were not as efficient as that of toluene, and no product formation was observed with Et2O. We continued the optimization studies by examining various bases for this transformation, and K2CO3 was found to be the best choice giving the product in 88% yield and shortening the reaction time to 2 h (entries 8–12). The triflate and tetrafluoroborate salts were tried subsequently, but with no improvement in the efficiency (entries 13 and 14). Further increase in the stoichiometry of K2CO3 (1.2 equiv.) was found to be beneficial, enhancing the yield of 3a to 90% (entry 15). The use of a higher reaction temperature of 80 °C afforded the product in lower yield (entry 16).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Anion X− | Base | Solvent | Time (h) | Yieldb (%) |
| a General reaction conditions: 1a (0.2 mmol), 2a (0.22 mmol), and base (0.22 mmol) in 2 ml of solvent at 50 °C. b NMR yield using 1,3,5-trimethoxybenzene as an internal standard. c Reaction performed using 0.24 mmol of K2CO3. d Isolated yield after silica gel column chromatography. e Reaction carried out at 80 °C. | |||||
| 1 | OTs | Cs2CO3 | THF | 12 | 65 |
| 2 | OTs | Cs2CO3 | TBME | 12 | 79 |
| 3 | OTs | Cs2CO3 | Dioxane | 12 | 82 |
| 4 | OTs | Cs2CO3 | Toluene | 6 | 86 |
| 5 | OTs | Cs2CO3 | Et2O | 6 | — |
| 6 | OTs | Cs2CO3 | DCM | 6 | 50 |
| 7 | OTs | Cs2CO3 | DMF | 12 | 77 |
| 8 | OTs | K2CO3 | Toluene | 2 | 88 |
| 9 | OTs | t-BuOK | Toluene | 6 | 80 |
| 10 | OTs | KOH | Toluene | 6 | 82 |
| 11 | OTs | NaH | Toluene | 6 | 60 |
| 12 | OTs | DABCO | Toluene | 12 | Trace |
| 13 | OTf | K2CO3 | Toluene | 6 | 80 |
| 14 | BF4 | K2CO3 | Toluene | 6 | 55 |
| 15c | OTs | K2CO3 | Toluene | 2 | 90 (85)d |
| 16e | OTs | K2CO3 | Toluene | 2 | 76 |
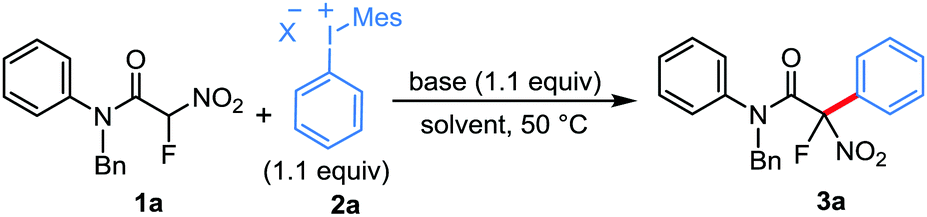
Having identified the optimal conditions, we proceeded with the evaluation of the scope for this arylation strategy. First, we examined the structural diversity possible for unsymmetrical diaryliodonium salts keeping the mesityl group as the dummy ligand to achieve the requisite chemoselectivity. As shown in Table 2, the transformation was effective for diaryliodonium salts containing a broad series of substituents, and the aryl groups were selectively transferred to the fluoroacetamide moiety in excellent yields. Notably, substrates bearing various alkoxy, alkyl and phenyl substituents at the p-position of the aryl group generated the corresponding arylated fluoroacetamides in good to excellent yields (3a–3g). Importantly, diaryliodonium salts containing bromo, fluoro, triflate, carbalkoxyl, trifluoromethyl, and cyano substituents at the p-position were competent for this arylation, illustrating the advantage of this strategy in producing these compounds which are often difficult to achieve through metal-catalyzed coupling reactions (3h–3m). Additionally, the reactions carried out using electronically varied functional groups at the m-position of the aryl ring proceeded smoothly to afford the arylated products in moderate yields (3n–3p). A substituent at the o-position was also tolerated to afford the product, albeit in lower yield (3q). In addition to the monosubstituted diaryliodonium salts, a disubstituted diaryliodonium salt was also found to be viable for this transformation, and product 3r was obtained in 62% yield. Our further evaluation of the scope revealed that a sterically demanding aryl group such as mesityl could not be transferred under the present reaction conditions. Pleasingly, 2-naphthyl and 4-iodobiphenyliodonium salts could also be effectively employed in this conversion (3t and 3u). Regarding the introduction of heteroaryls, 2-chloropyridine-derived diaryliodonium salt was successfully transformed to the corresponding product 3v in excellent yield.
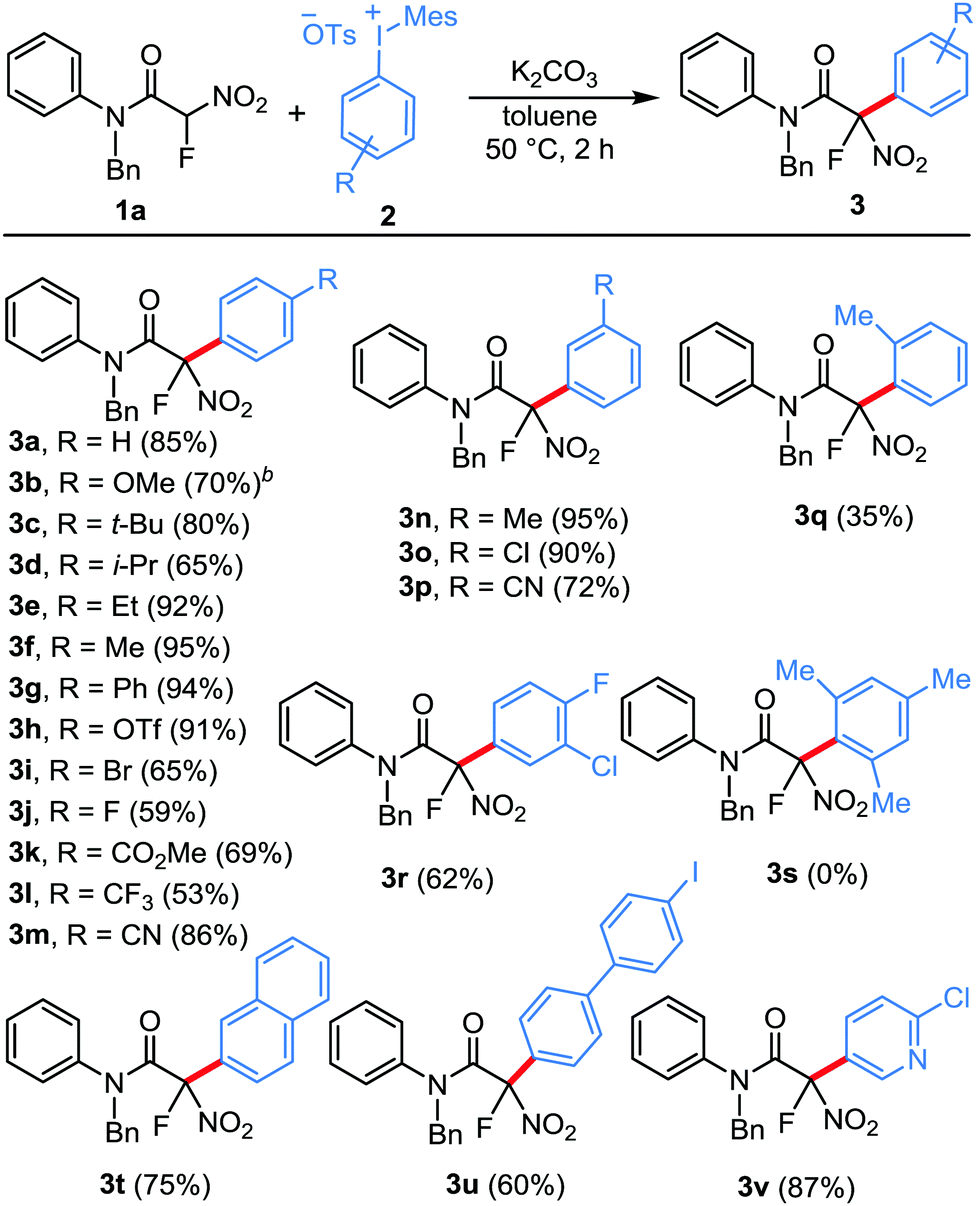
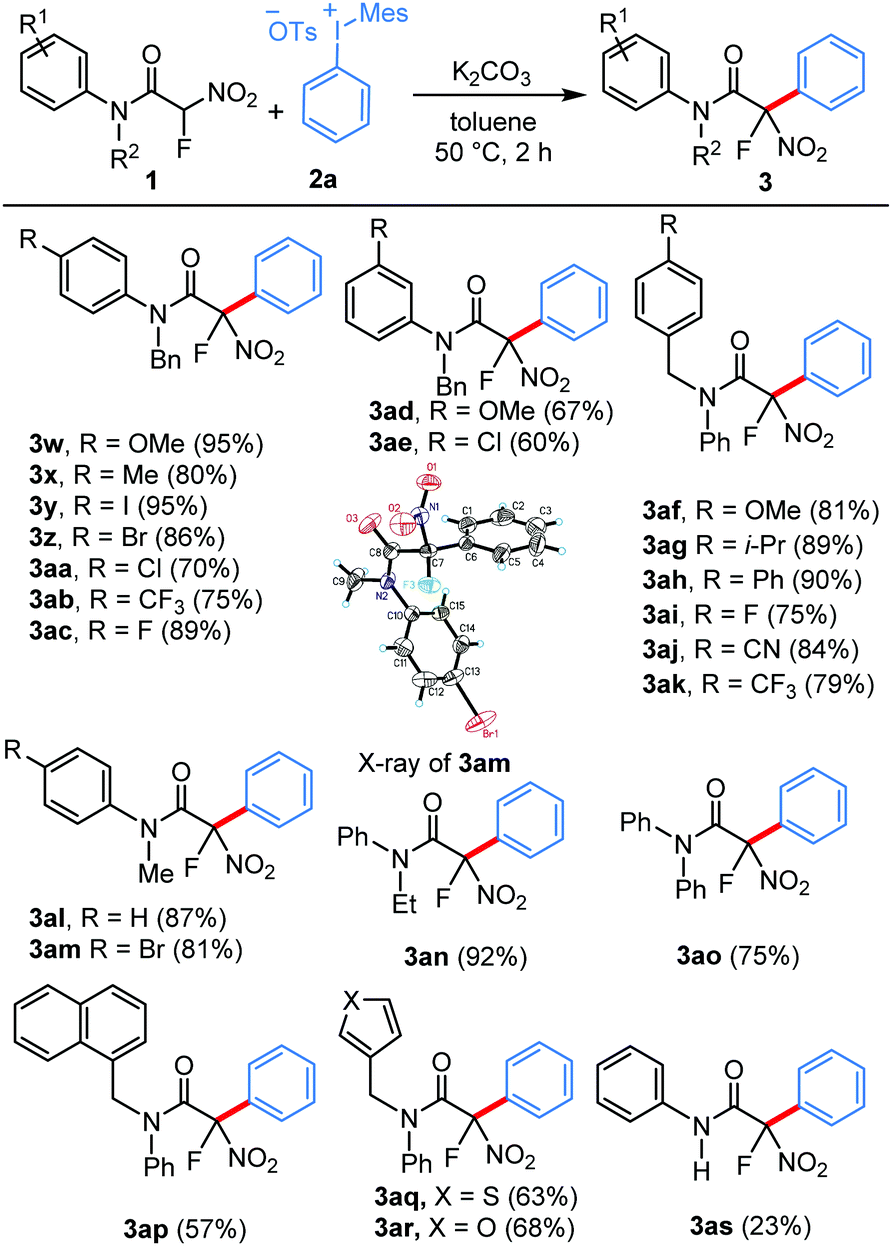
We next turned our attention to the scope of the amide component for this practical arylation strategy. Notably, a diverse range of electronically varied tertiary amides can be used for this new protocol to deliver these arylated products in good to excellent yields (Table 3). Fluoronitroacetamides bearing electron-withdrawing and electron-donating substituents at the p-position of the aryl ring underwent the reaction smoothly to afford the arylated products in excellent yields (3w–3ac). Moreover, these mild metal-free conditions are compatible for substrates bearing methoxy and chloro substituents at the m-position of the aryl ring (3ad and 3ae). Subsequently, we examined the electronic effects of the benzyl substitution of the fluoronitroacetamides, and all the substrates used were found to be competent nucleophiles in this transformation (3af–3ak). Furthermore, the reaction scope was evaluated by varying the N-substituent of the aniline moiety. Pleasingly, the amides having the most commonly used N-substituents such as Me, Et and Ph underwent arylation smoothly to furnish the corresponding products in excellent yields (3al–3ao). The structure of the product was unambiguously confirmed by the X-ray analysis of compound 3am.16 Similarly, α-fluoronitroacetamides bearing N-(naphthalen-1-ylmethyl), N-(thiophen-3-ylmethyl), and N-(furan-3-ylmethyl) groups displayed reasonable efficiency in this reaction (3ap–3ar). Pleasingly, though in a moderate yield, a substrate bearing a secondary amide also participated in this reaction (3as).
| a General reaction conditions: 1 (0.2 mmol), 2a (0.22 mmol), and K2CO3 (0.24 mmol) in 2 ml of toluene at 50 °C for 2 h. Isolated yield after silica gel column chromatography. |
|---|
|
The cyanide group is one of the most important functionalities in organic synthesis as it can be easily converted to various functional groups such as an amine, acid, aldehyde and ester. Encouraged by the versatile chemistry involving the cyanide moiety, we decided to check whether this new protocol can be extended to a cyano-containing fluoroacetamide nucleophile. In a preliminary experiment, α-cyano-α-fluoroacetamide 4a derived from cyanoacetic acid was tested for its compatibility with diaryliodonium salt 2a under identical reaction conditions, and the arylated product 5a was obtained, albeit only in 37% yield. This result prompted us to carry out a brief optimization study which concluded that t-BuOK/THF at 25 °C suits best for this reaction affording the product in 70% yield.17 As shown in Table 4, a brief survey of the substrate scope was conducted, and it was pleasing to find that electronically varied substituents at the p-position of iodonium salts were well tolerated in the α-arylation of α-cyano-α-fluoroacetamides, affording the products in moderate to good yields (5a–5e). Of note, the o-fluorophenyl group was successfully transformed to the product in 65% yield (5f). A heteroaryliodonium salt was also successfully employed in the transformation to incorporate a heteroarene into a fluoroacetamide substrate (5g). Subsequently, we focused our attention on the variation of α-cyano-α-fluoroacetamides. We were pleased to find that N-benzyl-N-aryl-substituted acetamides bearing methoxy, t-butyl, and fluoro substituents at the p-position of the phenyl ring were readily transformed to the corresponding arylated products (5h–5k). Moreover, the scope of this novel protocol was demonstrated by varying the N-substituent of the tertiary amide (5l–5p). Of note, the arylation of the secondary amide was also feasible under the present reaction conditions (5q).
| a General reaction conditions: 5 (0.2 mmol), 2 (0.22 mmol), and t-BuOK (0.24 mmol) in 2 ml of THF at 25 °C for 30 min. Isolated yield after silica gel column chromatography. |
|---|

|
Generally, the metal-free α-arylation is presumed to take place via a T-shaped intermediate formed by the coordination of the enolate generated by the base.18 Either of the T-shaped intermediates (I or II) formed by the coordination of the enolate through the O–I or C–I bond subsequently undergoes a rapid ligand coupling process to afford the α-arylated α-fluoro-α-nitroacetamide derivative (Scheme 2). Furthermore, the possibility of a radical process was ruled out after performing the reactions in the presence of well-known radical scavengers TEMPO and 1,1-diphenylethylene (DPE). In both the reactions conducted using α-fluoro-α-nitroacetamide 1a, the desired product 3a was obtained in 80% and 74% respectively.
 | ||
| Scheme 2 Reaction mechanism. | ||
In conclusion, the use of diaryliodonium salts as arylating agents has enabled the development of a convenient metal-free protocol for the α-arylation of synthetically valuable α-fluoro-α-nitroacetamides for the creation of a quaternary benzylic fluorocarbon center. Unlike the other metal-free arylation strategies, the present reaction could be used for the selective transfer of a wide range of electronically varied arenes, thereby representing a new complementary approach for sp3 C arylation. The strategy was further shown to be useful for the α-arylation of α-cyano-α-fluoroacetamides.
M. K. Z. and E. G. are grateful to CSIR and UGC-New Delhi for the awards of Research Fellowships. We thank Dr Tejender S. Thakur, MSB Division, CSIR-CDRI, for supervising the X-ray data collection and structure determination. We thank the SAIF Division for analytical support. CDRI Communication No. 10006.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) T. Yamazaki, T. Taguchi and I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley, Hoboken, 2009, pp. 1–46 Search PubMed; (b) J. Wang, M. Sanchez-Rosello’, J. L. Aceňa, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS; (c) R. E. Banks, B. E. Smart and J. C. Tatlow, Organofluorine Compounds: Principles and Commercial Applications, Plenum, New York, 1994 CrossRef.
- (a) D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (c) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS; (d) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed.
- (a) S.-Q. Zhu, Y.-L. Liu, H. Li, X.-H. Xu and F.-L. Qing, J. Am. Chem. Soc., 2018, 140, 11613 CrossRef CAS PubMed; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS; (d) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929 RSC; (e) T. Besset, C. Schneider and D. Cahard, Angew. Chem., Int. Ed., 2012, 51, 5048 CrossRef CAS.
- (a) M. G. Campbell and T. Ritter, Chem. Rev., 2015, 115, 612 CrossRef CAS; (b) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS; (c) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS.
- (a) H. Yin, J. Sheng, K.-F. Zhang, Z.-Q. Zhang, K.-J. Biana and X.-S. Wang, Chem. Commun., 2019, 55, 7635 RSC; (b) Y. Zhao, C. Ni, F. Jiang, B. Gao, X. Shen and J. Hu, ACS Catal., 2013, 3, 631 CrossRef CAS; (c) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS; (d) J. Zhu, W. Zhang, L. Zhang, J. Liu, J. Zheng and J. Hu, J. Org. Chem., 2010, 75, 5505 CrossRef CAS.
- (a) Y. Wu, H.-R. Zhang, Y.-X. Cao, Q. Lan and X.-S. Wang, Org. Lett., 2016, 18, 5564 CrossRef CAS; (b) J. Hu, B. Gao, L. Li, C. Ni and J. Hu, Org. Lett., 2015, 17, 3086 CrossRef CAS; (c) Y.-L. Xiao, W.-H. Guo, G.-Z. He, Q. Pan and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 9909 CrossRef CAS; (d) S. Ge, W. Chaladaj and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4149 CrossRef CAS.
- (a) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure and Synthetic Application of Polyvalent Iodine Compounds, John Wiley & Sons Ltd, New York, 2014 Search PubMed; (b) V. V. Zhdankin, Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds, Wiley, Chichester, 2013 CrossRef.
- (a) V. V. Zhdankin and K. Muniz, J. Org. Chem., 2017, 82, 11667 CrossRef CAS; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS PubMed; (c) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS.
- (a) B. Olofsson, Top. Curr. Chem., 2015, 373, 135 CrossRef; (b) N. Yamaoka, Synlett, 2012, 478 CrossRef; (c) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS.
- (a) Z. Zhang, X. Wua, J. Han, W. Wua and L. Wang, Tetrahedron Lett., 2018, 59, 1737 CrossRef CAS; (b) M. Fañanás-Mastral, Synthesis, 2017, 1905 CrossRef; (c) C. J. Teskey, S. M. A. Sohel, D. L. Bunting, S. G. Modha and M. F. Greaney, Angew. Chem., Int. Ed., 2017, 56, 5263 CrossRef CAS; (d) K. Aradi, B. L. Tóth, G. L. Tolnai and Z. Novák, Synlett, 2016, 1456 CAS.
- (a) H. Chen, J. Han and L. Wang, Angew. Chem., Int. Ed., 2018, 57, 12313 CrossRef CAS PubMed; (b) B. Xiong, X. Feng, L. Zhu, T. Chen, Y. Zhou, C.-T. Au and S.-F. Yin, ACS Catal., 2015, 5, 537 CrossRef CAS; (c) L. Chan, A. McNally, Q. Y. Toh, A. Mendoza and M. J. Gaunt, Chem. Sci., 2015, 6, 1277 RSC; (d) R. Ghosh, E. Lindstedt, N. Jalalian and B. Olofsson, ChemistryOpen, 2014, 3, 54 CrossRef CAS; (e) K. P. Landge, K. S. Jang, S. Y. Lee and D. Y. Chi, J. Org. Chem., 2012, 77, 5705 CrossRef CAS.
- (a) W. Lecroq, P. Bazille, F. Morlet-Savary, M. Breugst, J. Lalevee, A.-C. Gaumont and S. Lakhdar, Org. Lett., 2018, 20, 4164 CrossRef CAS; (b) P. Villo, G. Kervefors and B. Olofsson, Chem. Commun., 2018, 54, 8810 RSC; (c) Z. Chai, B. Wang, J.-N. Chen and G. Yang, Adv. Synth. Catal., 2014, 356, 2714 CrossRef CAS; (d) A. M. Wagner and M. S. Sanford, J. Org. Chem., 2014, 79, 2263 CrossRef CAS; (e) N. Umierski and G. Manolikakes, Org. Lett., 2013, 15, 188 CrossRef CAS.
- (a) Y. An, X.-M. Zhang, Z.-Y. Li, W.-H. Xiong, R.-D. Yu and F.-M. Zhang, Chem. Commun., 2019, 55, 119 RSC; (b) S. Mao, X. Geng, Y. Yang, X. Qian, S. Wu, J. Han and L. Wang, RSC Adv., 2015, 5, 36390 RSC; (c) C. Dey, E. Lindstedt and B. Olofsson, Org. Lett., 2015, 17, 4554 CrossRef CAS; (d) A. Monastyrskyi, N. Namelikonda and R. Manetsch, J. Org. Chem., 2015, 80, 2513 CrossRef CAS; (e) K. Matsuzaki, K. Okuyama, E. Tokunaga, M. Shiro and N. Shibata, ChemistryOpen, 2014, 3, 233 CrossRef CAS; (f) M. Ito, I. Itani, Y. Toyoda, K. Morimoto, T. Dohi and Y. Kita, Angew. Chem., Int. Ed., 2012, 51, 12555 CrossRef CAS; (g) C. H. Oh, J. S. Kim and H. H. Jung, J. Org. Chem., 1999, 64, 1338 CrossRef CAS; (h) K. Chen and G. F. Koser, J. Org. Chem., 1991, 56, 5764 CrossRef CAS.
- (a) X. Chen, Y. Li, J. Zhao, B. Zheng, Q. Lu and X. Ren, Adv. Synth. Catal., 2017, 359, 3057 CrossRef CAS; (b) S. I. Arlow and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 4567 CrossRef CAS; (c) W. J. Middleton and E. M. Bingham, J. Am. Chem. Soc., 1980, 102, 4845 CrossRef CAS.
- (a) D. R. Stuart, Chem. – Eur. J., 2017, 23, 15852 CrossRef CAS; (b) S. K. Sundalam and D. R. Stuart, J. Org. Chem., 2015, 80, 6456 CrossRef CAS PubMed; (c) F. Tinnis, E. Stridfeldt, H. Lundberg, H. Adolfsson and B. Olofsson, Org. Lett., 2015, 17, 2688 CrossRef CAS PubMed.
- Crystallographic data for 3am: CCDC 1940141 (3am) contains the supplementary crystallographic data for this paper†.
- For details, see the ESI†.
- (a) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334 CrossRef CAS; (b) P.-O. Norrby, T. B. Petersen, M. Bielawski and B. Olofsson, Chem. – Eur. J., 2010, 16, 8251 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, complete characterization data, CIF for 3am and copies of NMR spectra. CCDC 1940141. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc07859b |
| This journal is © The Royal Society of Chemistry 2020 |