
A homochiral 3D framework of mechanically interlocked 1D loops with solvent-dependent spin-state switching behaviors†
Xiao-Peng
Sun
,
Zheng
Tang
,
Zi-Shuo
Yao
 * and
Jun
Tao
*
* and
Jun
Tao
*
Key Laboratory of Cluster Science of Ministry of Education, School of Chemistry and Chemical Engineering, Liangxiang Campus, Beijing Institute of Technology, Beijing 102488, People's Republic of China. E-mail: taojun@bit.edu.cn; zishuoyao@bit.edu.cn
First published on 26th November 2019
Abstract
An atypical homochiral spin-crossover (SCO) framework (1) constructed from mechanically interlocked 1D molecular loops was prepared. Due to the flexibility of the interlocked structure, the guest solvent molecules can be reversibly exchanged. Consequently, its SCO behavior was capable of modulating between one- and two-stepped transitions in response to acetonitrile and methanol.
The self-assembly of mechanically interlocked structures (MISs) based on non-covalent mechanical interactions may allow access to topologically fascinating molecules and potential applications in molecular machines, information storage, and molecular catalysis.1–4 Typical interlocking structures including Hopf link, Solomon knot, star of David catenane, Brunnian braid, Borromean ring (the simplest Brunnian braid), interlocked cage and poly[n]catenane have been gaining momentum in the past few decades.5–13 In these exquisite structures, the isolated components are predominantly held together by various intermolecular interactions, e.g. π⋯π stacking, C–H⋯π interactions, hydrogen bonds, electrostatic interactions or metal–metal interactions. It is worthy of note that rational strategies to synthesize novel MISs are continuously being developed; for example, Loeb et al.14–16 introduced dynamical rotaxane components into metal–organic frameworks and prepared a series of mechanically interlocked frameworks. Very recently, a state-of-the-art coordination-driven mechanical interlocking strategy was employed to successfully fabricate a three-dimensional (3D) covalent organic framework.17 These results indicate that more novel MISs may be synthesized by sophisticated molecule-structure design strategies.
However, the important combination of particular benefit such as chirality within MISs is less explored. Homochiral MISs are highly applicable in biological systems and pharmaceutical industries,18–22 which can be generated by stereoselective synthesis or separated by means of professional techniques from a pair of corresponding enantiomers. The reversible interlocking–unlocking between homochiral [2]catenane and dinuclear helicate, bis[2]catenane and trinuclear double helicates has been realized.23 These results may light up a new method to assemble chiral poly[2]catenane—a special kind of MIS. Furthermore, MISs can be endowed with more interesting functions if a metal centre is incorporated. One of the metal ion-related functions is spin-state switching, i.e. spin crossover (SCO). The occurrence of SCO between high-spin (HS) and low-spin (LS) states is highly sensitive to chemical and physical stimuli;24–28 therefore, the marriage of SCO and MIS may make novel interlocking structures stimuli-responsive multifunctional materials. To the best of our knowledge, a known SCO-active MIS has not yet been reported.
We have recently adopted R-2,2′-bis(methoxymethoxy)-6,6′-bis(4-pyridyl)-1,1′-binaphthyl (R-L1, Scheme 1) to synthesize a non-interlocking structure,29 because the window of the 1D loops resulting from this is not large enough for mechanical interlocking. Hence, the longer linker, R-2,2′-bis(methoxymethoxy)-6,6′-bis(4-pyridylethynyl)-1,1′-binaphthyl (R-L2, Scheme 1), was designed to assemble the MIS. Meanwhile, the Fe(II) ion was introduced for the purpose of combining SCO switching properties. As expected, we demonstrate such a homochiral SCO-active MIS with the formula of [Fe(NCBH3)2(R-L2)2]·6CH3CN (1·CH3CN). Interestingly, compound 1·CH3CN is competent to undergo single-crystal-to-single-crystal (SCSC) transformation upon solvent exchange (1·CH3CN ⇌ 1·CH3OH), which is accompanied by reversible spin-state switching between one-step (1·CH3CN) and two-step (1·CH3OH) SCO behaviors.
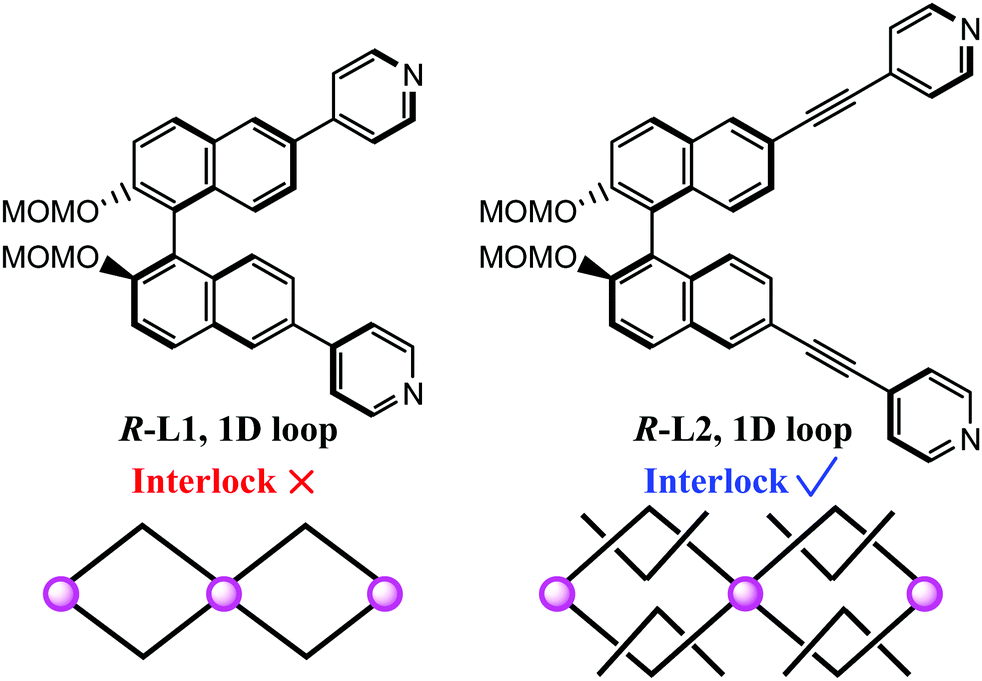 | ||
| Scheme 1 Schematic illustration of the formation of the 1D looped structure (left) and 3D MIS (right). With the longer arm, R-L2 is capable of assembling with [Fe(NCBH3)2] to form a 3D MIS (MOMO = methoxymethoxy). | ||
Slow diffusion of [Fe(NCBH3)2] and R-L2 in acetonitrile generated the orange crystals of 1·CH3CN. Yellow crystals of 1·CH3OH were produced by solvent-treatment through immersion of 1·CH3CN in pure methanol for one day (see Experimental section and Fig. S1, ESI†). Phase purity of the bulk samples was confirmed by powder X-ray diffractometry, scanning electron microscopy and elemental mapping photographs (Fig. S2–S4, ESI†). Six acetonitrile and eight methanol molecules were, respectively, fixed to 1·CH3CN and 1·CH3OH, in agreement with the results of thermogravimetric (Fig. S5, ESI†) and elemental analyses.
Single-crystal data of 1·CH3CN and 1·CH3OH were collected at 240, 160 and 100 K to correlate the spin states of Fe(II) ions uncovered by magnetic measurements (see below). Both 1·CH3CN and 1·CH3OH adopt the isostructural interlocking framework irrespective of the solvent molecules (Fig. 1 and Table S1, ESI†). The Fe(II) center displays a distorted {FeN6} octahedron formed by a pair of axial NCBH3− anions and four equatorial R-L2 ligands. The rhombic unit [(1/2Fe)2(R-L2)2] is defined as a molecular loop (Fig. S6, ESI†), which is not isolated but the Fe(II) nodes are connected to 4 R-L2 linker ligands and form a 1D corner-sharing looped structure (Fig. 1a and b). Due to the longer arm of R-L2, the diagonal dimensions of 22.4 Å × 15.5 Å (1·CH3CN) and 22.2 Å × 15.7 Å (1·CH3OH) in the molecular loop described here are (Table S2, ESI†) remarkably larger than that of 18.8 Å × 12.4 Å in our previous reported 1D looped molecule.29 Consequently, the adjacent R-L2 linkers are capable of threading the loops and the 1D looped structures are mutually interlocked to generate the title 3D MIS (Fig. 1c, d and Fig. S7, ESI†). In the crystal lattice, the parallel loops are separated by ca. 5.5 Å (1·CH3CN) and 5.3 Å (1·CH3OH) (Fig. S8 and Table S3, ESI†), and the interlocked loops run along two different directions with an included angle of 56° (Fig. 1c and Table S4, ESI†), resulting in an open channel of dimensions of 9.7 Å × 6.9 Å (1·CH3CN) and 10.2 Å × 6.5 Å (1·CH3OH) along the c-axis (Fig. 1d, Fig. S9 and Table S5, ESI†).
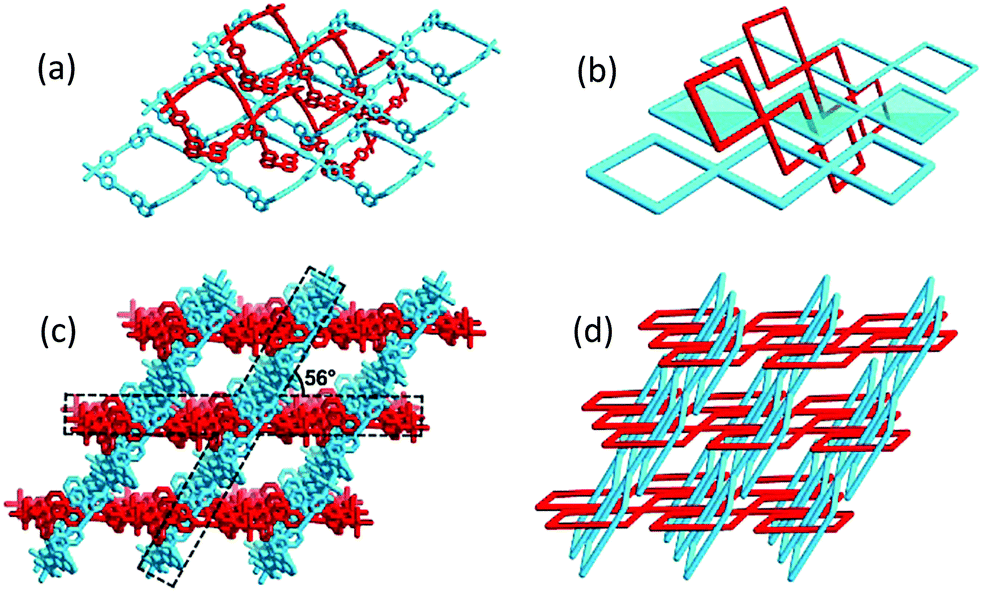 | ||
| Fig. 1 Stick representation (a) and topological view (b) of the interlocking mode of 1D loops in 1, the 3D mechanically interlocked structure of 1 (c), and the 3D topological structure of 1 (d). Identical 1D loops in the structure are marked in red and blue for better view. Solvent molecules and hydrogen atoms are omitted for clarity. | ||
These 3D MISs are stabilized by three kinds of mechanical interlocking interactions, i.e. C–H⋯π interactions (A, 2.948–3.290 Å for 1·CH3CN and 2.827–2.913 Å for 1·CH3OH at 240 K), π⋯π interactions (B, 4.484 Å for 1·CH3CN and 4.096 Å for 1·CH3OH at 240 K) and C–H⋯π interactions (C, 3.166–3.727 Å for 1·CH3CN and 4.072–4.086 Å for 1·CH3OH at 240 K). Detailed structural analyses reveal that these interactions in the subcomponent of linear poly[n]catenane are periodically arrayed in the ABAC⋯manner (Fig. 2, Fig. S10, S11 and Table S6, ESI†). These cooperative intermolecular interactions may enable the entropically unfavourable process of interlocking by providing suitable stabilization energy.11,30–32
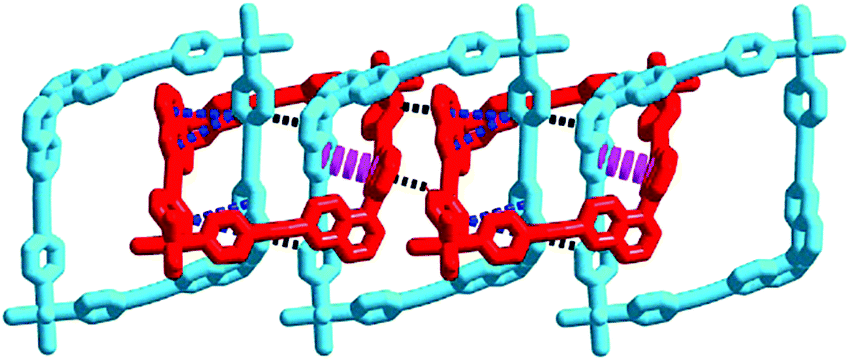 | ||
| Fig. 2 The mechanically interlocked loops in 1 showing three kinds of interactions in the ABAC⋯manner (A: black dots, B: pink dots, and C: blue dots). Hydrogen atoms and the MOMO groups are omitted for clarity. | ||
Due to the homochirality of R-L2, the 1D loops are mechanically interlocked in a Sohncke (chiral) space group C2. As shown in Fig. 3 and Fig. S12 (ESI†), the fabrication of interlocked loops forms an infinite right-handed helical chain running along the b-axis with a helical pitch of 17 Å. Moreover, the neighboring interlocked helical chains give a periodically ordered chiral architecture in the ac plane (Fig. 3c and Fig. S13, ESI†), forming the 3D chiral framework via mechanical interlocking. As far as we know, SCO compounds with chirality as bulk materials are still rare33–37 and the helical chain constructed from components that are connected via mechanical interlock is unprecedented in molecular materials. The homochirality reported here is determined by solid-state circular dichroism (CD) spectra with dried KBr pellets (1·CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KBr = 1:800, 1·CH3OH:KBr = 1:600, R-L2: KBr = 1:600) featuring strong Cotton effects of two positive signals centered at 232 and 340 nm and a negative one at 291 nm (Fig. 3d).38 In addition, the non-centrosymmetric structures arising from chirality are further confirmed by the second-harmonic-generation measurement (Fig. S14, ESI†).
:KBr = 1:800, 1·CH3OH:KBr = 1:600, R-L2: KBr = 1:600) featuring strong Cotton effects of two positive signals centered at 232 and 340 nm and a negative one at 291 nm (Fig. 3d).38 In addition, the non-centrosymmetric structures arising from chirality are further confirmed by the second-harmonic-generation measurement (Fig. S14, ESI†).
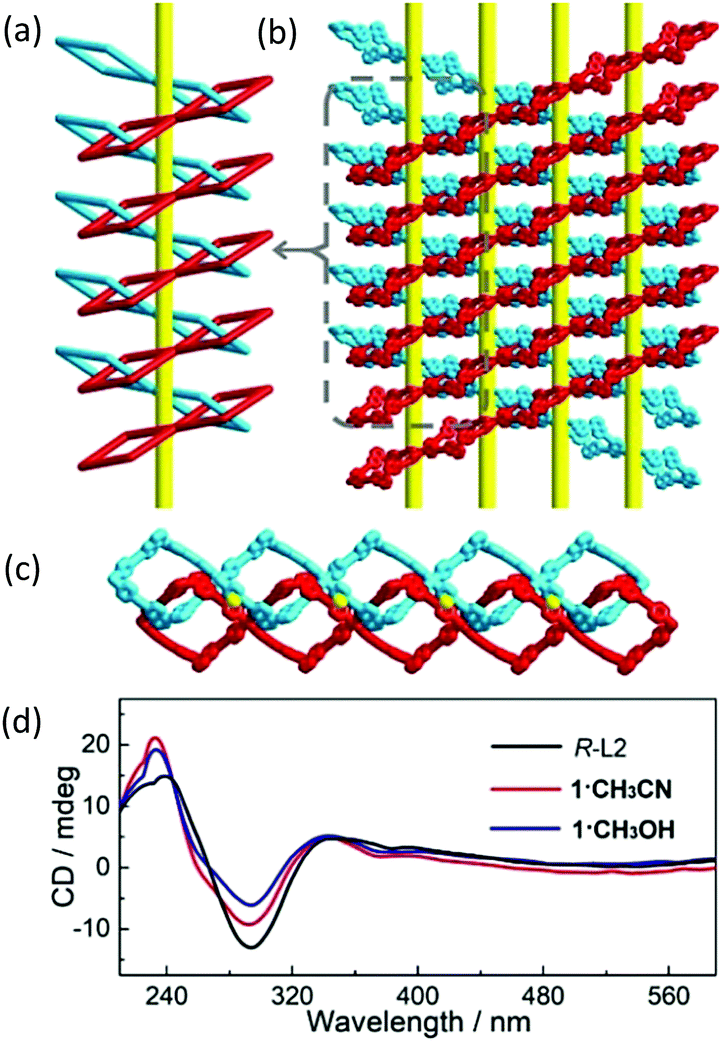 | ||
| Fig. 3 Topological (a) and stick (b) representations of the interlocked right-handed helical chains viewed along the b-axis, and down the b-axis (c) in 1 (the yellow tubes represent the helical axes and the identical 1D loops are marked in red and blue for better view), and the solid-state circular dichroism spectra of R-L2, 1·CH3CN and 1·CH3OH (d). | ||
Magnetic susceptibility data on a polycrystalline sample show that 1·CH3CN exhibits an abrupt one-step SCO behavior without hysteresis (Fig. 4a). At 240 K, χMT is 3.80 cm3 K mol−1 and typical of HS-Fe(II) ions. χMT rapidly decreases to 0.16 cm3 K mol−1 below 100 K with the transition temperature (Tc) of 180 K, indicating that all the HS-Fe(II) ions switch to the LS state. Accordingly, the continuous contraction of the average Fe–N bond length upon spin change is confirmed by structural analyses (2.153 Å at 240 K, 1.990 Å at 160 K, 1.976 Å at 100 K, Table S7, ESI†).
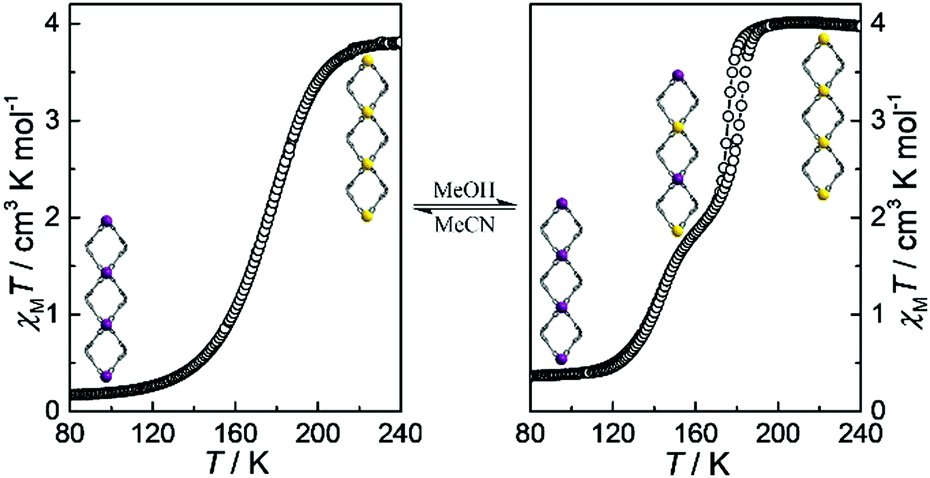 | ||
| Fig. 4 Plots of χMT versus T for 1·CH3CN (a) and 1·CH3OH (b). Inset: View of the spin-state ordering in 1D loops. LS and HS-Fe sites are, respectively, marked in purple and yellow balls. | ||
The SCO character is known to be effectively modulated by solvent molecules,39–43 which are adopted to construct solvent-responsive SCO materials. Herein, the SCSC adduct, 1·CH3OH, undergoes a two-step SCO process with a hysteresis loop of 6 K width (Fig. 4b). The χMT value of 4.00 cm3 mol−1 K above 190 K agrees well with the expected value for one HS-Fe(II) ion. Upon cooling, this value decreases to 1.99 cm3 mol−1 K at 160 K with a narrow inclined plateau, indicating that 50% HS-Fe(II) ions convert to the LS-Fe(II) state (Fig. S15, ESI†). The residual 50% HS-Fe(II) ions are switched to the LS state on further cooling to 100 K (χMT = 0.36 cm3 K mol−1). Therefore, the two-step SCO behaviour in 1·CH3OH presents a spin-state sequence of HS1.0 ↔ HS0.5LS0.5 ↔ LS1.0 (Fig. S16, ESI†), in which the first-step is accompanied by a hysteresis loop of 6 K width (TC1↓ = 176 K, and TC1↑ = 182 K) while the second one is nonhysteretic (TC2↓ = 143 K) (Fig. S17, ESI†). The average Fe–N bond length is also consistent with the spin-state stages (2.175 Å at 240 K, 2.152 Å for Fe1 and 1.984 Å for Fe2 at 160 K, and 2.009 Å at 100 K), which further proves the two-step SCO behaviors (Table S8, ESI†). During this spin-state switching process, two independent Fe(II) centres were found in the asymmetric unit at 160 K. Correspondingly, the crystallographic space group of 1·CH3OH changed from C2 (in LS1.0 and HS1.0 phases) to P21 (in HS0.5LS0.5 phase).
More interestingly, the crystals of 1·CH3OH can be reversibly transformed to 1·CH3CN by treating with acetonitrile, resulting in a reversible solvent modulation of one- and two-stepped SCO behaviors. In addition, the suitable solvent molecule is the key point to activate spin-state transition in this framework. The Tc values of ethanol- and n-propanol-modulated samples shift to 177 K and 159 K, respectively (Fig. S18, ESI†). However, both the solvent-free and water-solvated samples show inconsistent PXRD patterns with the simulated signals (Fig. S19, ESI†) and, so, they persist in the high-spin state in the measured temperature range (Fig. S20 and S21, ESI†).
In fact, several complexes assembled by 1D loops have been synthesized;44–47 however, we prepare the first homochiral SCO-MIS by finely modulating the arm length of the ligands, whose 1D loops are held together via mechanical interlocking. A possible interlocking process is presented (Fig. S7, ESI†): the 1D loops assembled by R-L2 and [Fe(NCBH3)2] could be opened reversibly, and the interlocking of these closed 1D loops with the opened ones gives rise to the title MIS. Besides, the subunit of linear poly[n]catenane could directly reflect the specific interlocking mode described here (Fig. 2 and Fig. S10, ESI†).48,49 Compared to the interpenetrated SCO frameworks with the feature of the assembly of 2D networks,50–52 such a 3D chiral SCO-MIS constructed from intertwinement of 1D loops represents a novel structural model for developing new molecular functional materials.17
In summary, we construct a unique homochiral MIS with SCO behavior response to temperature and solvent molecules. The mutual interlocking of 1D loops via a catenane-like mode is revealed by detailed single-crystal X-ray diffraction analyses, and the homochirality of bulk samples is confirmed by CD spectra. Due to the flexibility of the interlocked framework, reversible switching between one- and two-step SCO behaviors has been realized. The achievement of such homochiral SCO-MISs provides a means to design multifunctional materials with versatile interlocking structures and highlights the scientific significance of mechanical entanglement combined with chirality and SCO properties.
This work was financially supported by the National Natural Science Foundation of China (Grants 21671161, 21701013, 21701014 and 21971016). We thank Professors Pengtao Ma and Jingyang Niu at Henan University for their help in refining the crystallographic data.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Liu, Y. Ma, Y. Zhao, X. Sun, F. Gándara, H. Furukawa, Z. Liu, H. Zhu, C. Zhu, K. Suenaga, P. Oleynikov, A. S. Alshammari, X. Zhang, O. Terasaki and O. M. Yaghi, Science, 2016, 351, 365–369 CrossRef CAS PubMed.
- M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879–889 CrossRef CAS.
- C. S. Wood, T. K. Ronson, A. M. Belenguer, J. J. Holstein and J. R. Nitschke, Nat. Chem., 2015, 7, 354–358 CrossRef CAS PubMed.
- L.-L. Dang, Z.-B. Sun, W.-L. Shan, Y.-J. Lin, Z.-H. Li and G.-X. Jin, Nat. Commun., 2019, 10, 2057 CrossRef PubMed.
- Y. Liu, M. O’Keeffe, M. M. J. Treacy and O. M. Yaghi, Chem. Soc. Rev., 2018, 47, 4642–4664 RSC.
- J. J. Danon, A. Krüger, D. A. Leigh, J.-F. Lemonnier, A. J. Stephens, I. J. Vitorica-Yrezabal and S. L. Woltering, Science, 2017, 355, 159–162 CrossRef CAS PubMed.
- L. Zhang, A. J. Stephens, J.-F. Lemonnier, L. Pirvu, I. J. Vitorica-Yrezabal, C. J. Robinson and D. A. Leigh, J. Am. Chem. Soc., 2019, 141, 3952–3958 CrossRef CAS PubMed.
- D. A. Leigh, R. G. Pritchard and A. J. Stephens, Nat. Chem., 2014, 6, 978–982 CrossRef CAS.
- Y.-G. Huang, S.-Q. Wu, W.-H. Deng, G. Xu, F.-L. Hu, J. P. Hill, W. Wei, S.-Q. Su, L. K. Shrestha, O. Sato, M.-Y. Wu, M.-C. Hong and K. Ariga, Adv. Mater., 2017, 29, 1703301 CrossRef.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S.-H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308–1312 CrossRef CAS.
- L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS.
- Y. Lu, H.-N. Zhang and G.-X. Jin, Acc. Chem. Res., 2018, 51, 2148–2158 CrossRef CAS.
- D. J. Hoffart and S. J. Loeb, Angew. Chem., Int. Ed., 2005, 44, 901–904 CrossRef CAS PubMed.
- S. J. Loeb, Chem. Commun., 2005, 1511–1518 RSC.
- V. N. Vukotic, K. J. Harris, K. Zhu, R. W. Schurko and S. J. Loeb, Nat. Chem., 2012, 4, 456–460 CrossRef CAS PubMed.
- Y. Liu, C. S. Diercks, Y. Ma, H. Lyu, C. Zhu, S. A. Alshmimri, S. Alshihri and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 677–683 CrossRef CAS PubMed.
- S. Corra, C. de Vet, J. Groppi, M. La Rosa, S. Silvi, M. Baroncini and A. Credi, J. Am. Chem. Soc., 2019, 141, 9129–9133 CrossRef CAS PubMed.
- M. Denis, J. E. M. Lewis, F. Modicom and S. M. Goldup, Chem, 2019, 5, 1512–1520 CAS.
- C.-Z. Liu, S. Koppireddi, H. Wang, D.-W. Zhang and Z.-T. Li, Angew. Chem., Int. Ed., 2019, 58, 226–230 CrossRef CAS.
- Y. Cui, S. J. Lee and W. Lin, J. Am. Chem. Soc., 2003, 125, 6014–6015 CrossRef CAS PubMed.
- R. Hovorka, G. Meyer-Eppler, T. Piehler, S. Hytteballe, M. Engeser, F. Topić, K. Rissanen and A. Lützen, Chem. – Eur. J., 2014, 20, 13253–13258 CrossRef CAS.
- O. Gidron, M. Jirásek, N. Trapp, M.-O. Ebert, X. Zhang and F. Diederich, J. Am. Chem. Soc., 2015, 137, 12502–12505 CrossRef CAS.
- O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef CAS PubMed.
- J. A. Real, E. Andres, M. C. Munoz, M. Julve, T. Granier, A. Bousseksou and F. Varret, Science, 1995, 268, 265–267 CrossRef CAS PubMed.
- M. J. Murphy, K. A. Zenere, F. Ragon, P. D. Southon, C. J. Kepert and S. M. Neville, J. Am. Chem. Soc., 2017, 139, 1330–1335 CrossRef CAS PubMed.
- W. Liu, Y.-Y. Peng, S.-G. Wu, Y.-C. Chen, Md. N. Hoque, Z.-P. Ni, X.-M. Chen and M.-L. Tong, Angew. Chem., Int. Ed., 2017, 56, 14982–14986 CrossRef CAS PubMed.
- J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703–737 RSC.
- W.-T. Deng, H. Qu, Z.-Y. Huang, L. Shi, Z.-Y. Tang, X.-Y. Cao and J. Tao, Chem. Commun., 2019, 55, 2210–2213 RSC.
- W.-L. Shan, Y.-J. Lin, F. E. Hahn and G.-X. Jin, Angew. Chem., Int. Ed., 2019, 58, 5882–5886 CrossRef CAS PubMed.
- Q. Chen, F. Jiang, D. Yuan, L. Chen, G. Lyu and M. Hong, Chem. Commun., 2013, 49, 719–721 RSC.
- H. Xue, F. Jiang, Q. Chen, D. Yuan, J. Pang, G. Lv, X. Wan, L. Liang and M. Hong, Chem. Commun., 2015, 51, 13706–13709 RSC.
- L.-F. Qin, C.-Y. Pang, W.-K. Han, F.-L. Zhang, L. Tian, Z.-G. Gu, X. Ren and Z. Li, CrystEngComm, 2015, 17, 7956–7963 RSC.
- K. E. Burrows, S. E. McGrath, R. Kulmaczewski, O. Cespedes, S. A. Barrett and M. A. Halcrow, Chem. – Eur. J., 2017, 23, 9067–9075 CrossRef CAS PubMed.
- I. A. Guralskiy, O. I. Kucheriv, S. I. Shylin, V. Ksenofontov, R. A. Polunin and I. O. Fritsky, Chem. – Eur. J., 2015, 21, 18076–18079 CrossRef CAS PubMed.
- W.-Q. Gao, Y.-S. Meng, C.-H. Liu, Y. Pan, T. Liu and Y.-Y. Zhu, Dalton Trans., 2019, 48, 6323–6327 RSC.
- W.-K. Han, L.-F. Qin, C.-Y. Pang, C.-K. Cheng, W. Zhu, Z.-H. Li, Z. Li, X. Ren and Z.-G. Gu, Dalton Trans., 2017, 46, 8004–8008 RSC.
- C. Gütz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topić, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef PubMed.
- J. Yuan, S.-Q. Wu, M.-J. Liu, O. Sato and H.-Z. Kou, J. Am. Chem. Soc., 2018, 140, 9426–9433 CrossRef CAS PubMed.
- O. Sato, J. Tao and Y.-Z. Zhang, Angew. Chem., Int. Ed., 2007, 46, 2152–2187 CrossRef CAS.
- B. Li, R.-J. Wei, J. Tao, R.-B. Huang, L.-S. Zheng and Z. Zheng, J. Am. Chem. Soc., 2010, 132, 1558–1566 CrossRef CAS PubMed.
- Z.-P. Ni, J.-L. Liu, Md. N. Hoque, W. Liu, J.-Y. Li, Y.-C. Chen and M.-L. Tong, Coord. Chem. Rev., 2017, 335, 28–43 CrossRef CAS.
- J.-Y. Ge, Z. Chen, L. Zhang, X. Liang, J. Su, M. Kurmoo and J.-L. Zuo, Angew. Chem., Int. Ed., 2019, 58, 8789–8793 CrossRef CAS PubMed.
- C.-Y. Su, A. M. Goforth, M. D. Smith and H.-C. zur Loye, Chem. Commun., 2004, 2158–2159 RSC.
- H.-F. Zhu, W. Zhao, T. Okamura, J. Fan, W.-Y. Sun and N. Ueyama, New J. Chem., 2004, 28, 1010–1018 RSC.
- H.-F. Zhu, J. Fan, T. Okamura, W.-Y. Sun and N. Ueyama, Cryst. Growth Des., 2005, 5, 289–294 CrossRef CAS.
- L. Sun, R.-Y. Guo, X.-D. Yang, S. Ma and J. Zhang, CrystEngComm, 2018, 20, 3297–3301 RSC.
- C.-M. Jin, H. Lu, L.-Y. Wu and J. Huang, Chem. Commun., 2006, 5039–5041 RSC.
- Q. Wu, P. M. Rauscher, X. Lang, R. J. Wojtecki, J. J. de Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- G. J. Halder, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS PubMed.
- G. J. Halder, K. W. Chapman, S. M. Neville, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2008, 130, 17552–17562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, characterizations, and additional tables, figures and supplementary movie. CCDC 1942449, 1942451–1942454 and 1966026. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc09063k |
| This journal is © The Royal Society of Chemistry 2020 |