
Ruthenium porphyrin catalysed intermolecular amino-oxyarylation of alkenes to give primary amines via a ruthenium nitrido intermediate†
Daohong
Yu
 ab,
Ka-Pan
Shing
cd,
Yungen
Liu
a,
Haiyang
Liu
b and
Chi-Ming
Che
*acd
ab,
Ka-Pan
Shing
cd,
Yungen
Liu
a,
Haiyang
Liu
b and
Chi-Ming
Che
*acd
aDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China
bDepartment of Chemistry, South China University of Technology, Guangzhou, Guangdong 510641, P. R. China
cHKU Shenzhen Institute of Research and Innovation, Shenzhen, Guangdong 518057, P. R. China. E-mail: cmche@hku.hk
dDepartment of Chemistry and State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Hong Kong, P. R. China
First published on 22nd November 2019
Abstract
Ruthenium porphyrin catalysed direct intermolecular amino-oxyarylation of alkenes including styrenes and 1,3-dienes to give primary amines with O-(2,4-dinitrophenyl)hydroxylamine as the amine source was achieved in moderate to good yields under mild reaction conditions. Spectroscopic analyses revealed that a ruthenium nitrido complex was the key reaction intermediate for the amino-oxyarylation reaction.
Amines are among the most important chemicals commonly encountered in natural products, agrochemicals and industrial fine chemicals.1 For instance, 2-phenylethanamine is an important sub-structure found in natural products and in pharmaceuticals marketed globally (Fig. 1). Classical access to primary amines necessitates pre-functionalisation, and they are synthesised by further organic transformation reactions such as the Gabriel reaction, the Leuckart reaction, the Curtius arrangement, the Schmidt reaction, and reduction of nitro, cyano or azido groups.2 In view of high atom economy, catalytic group transfer reactions are very efficient in the preparation of (protected) primary amines directly from alkenes,3 as exemplified by the Sharpless asymmetric amino-hydroxylation for the synthesis of amino alcohols.4
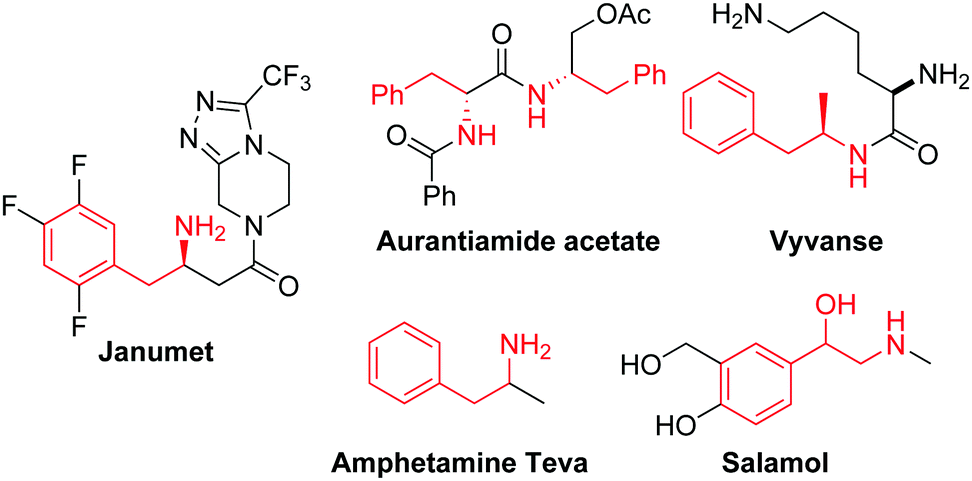 | ||
| Fig. 1 Pharmaceutical products containing the 2-phenylethanamine unit. | ||
To date there have been only a few reports of the direct synthesis of unprotected primary amines.3g,j,5 Morandi and co-worker reported the synthesis of 2-amino-1-phenylethanols from alkenes using PivONH3OTf as the aminating reagent.3j Flack, Kürti, Ess and co-workers reported the dirhodium catalysed functionalisation of alkenes using O-(2,4-dinitro-phenyl)hydroxylamine (DPH)6 as the nitrogen source,3g with styrene amino-oxyarylation being the major reaction pathway. They found that other metal complexes, including those of copper, cobalt, iridium, nickel, gold and ruthenium, could not catalyse the same or similar reactions to give the desired products. Recently, they have realised the dirhodium-catalysed amination of aromatic C–H bonds in the presence of a Brønsted acid,7 and suggested that a highly reactive protonated rhodium-nitrene species was the reaction intermediate.
We are interested in reactive metal nitrido complexes that could react with simple alkenes. We previously reported some structurally defined ruthenium porphyrin imido/nitrene complexes for stoichiometric and catalytic C–N bond formation reactions.8 Here, we report that ruthenium porphyrins are also competent for the direct intermolecular amino-oxyarylation of alkenes with DPH as the nitrogen source. High-resolution electrospray ionisation mass spectrometry (ESI-HRMS) and infrared (IR) and ultraviolet-visible (UV-vis) spectroscopic analysis revealed a ruthenium nitrido intermediate to be responsible for the amino-oxyarylation reaction. In this context, Lau and co-workers previously reported9 that ruthenium nitride can stoichiometrically aminate alkenes in the presence of a strong electrophile (trifluoroacetic anhydride or p-toluenesulfonic anhydride).10
At the outset, the reaction of 4-methoxy styrene 1a and DPH (2) with a panel of metal porphyrin catalysts was screened for reaction optimisation. Interestingly, the ruthenium porphyrin complexes [Ru(TPP)(CO)] (H2TPP = meso-tetraphenylporphyrin), [Ru(TDCPP)(CO)] (H2TDCPP = meso-tetrakis(2,6-dichlorophenyl)porphyrin) and [Ru(F20TPP)(CO)] (H2F20TPP = meso-tetrakis(pentafluorophenyl)porphyrin) showed high catalytic activity and 3a was obtained in 33–78% yields (Table S1, entries 1–3, ESI†). After further optimisation of the reaction conditions (Table S1, entries 4–15, ESI†), we found that the reaction without inert atmosphere protection ([Ru(F20TPP)(CO)]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1a:2a = 0.05:1:1.5) gave 3a in 86% yield. However, other ruthenium porphyrin complexes [Ru(TPP)Cl2], [Ru(TDCPP)Cl2], and [Ru(TMP)(CO)] (H2TMP = meso-tetramesitylporphyrin) gave 3a in ≤43% yields (Table S1, entries 17–19, ESI†). The non-porphyrin ruthenium complexes Ru(PPh3)3Cl2, [Ru(p-cymene)Cl2]2 and [Ru(tBu-Salen)(PPh3)2] exhibited low activity, with 3a being obtained in ≤28% yields (Table S1, entries 20–22, ESI†).
:1a:2a = 0.05:1:1.5) gave 3a in 86% yield. However, other ruthenium porphyrin complexes [Ru(TPP)Cl2], [Ru(TDCPP)Cl2], and [Ru(TMP)(CO)] (H2TMP = meso-tetramesitylporphyrin) gave 3a in ≤43% yields (Table S1, entries 17–19, ESI†). The non-porphyrin ruthenium complexes Ru(PPh3)3Cl2, [Ru(p-cymene)Cl2]2 and [Ru(tBu-Salen)(PPh3)2] exhibited low activity, with 3a being obtained in ≤28% yields (Table S1, entries 20–22, ESI†).
Under the optimised reaction conditions, we examined the substrate scope of ruthenium porphyrin catalysed amino-oxyarylation (Scheme 1). In general, styrenes with the electron-donating 4-methoxy or 4-phenyl group gave the corresponding products in high yields whereas those with electron-withdrawing groups such as 4-CO2Me or a ketone moiety gave the corresponding products in low yields. The reactions of sterically encumbered styrenes such as o-methoxy, α-methyl and α-phenyl styrenes also proceeded smoothly, albeit in 22–52% yields for the corresponding products (3j, 3m, 3t and 3u). Di- or trisubstituted styrenes also afforded products in 52–72% yields (Scheme 1, 3k–m). Electron-rich heterocyclic alkenes, including 2-vinylbenzofuran and 2-vinylnaphthalene, also reacted efficiently to give products in 73% and 75% yields, respectively. Additionally, the reaction of β-methyl styrene yielded the isomers 3s and 3s′ in a ratio of 2.4:1 with an 84% overall yield.
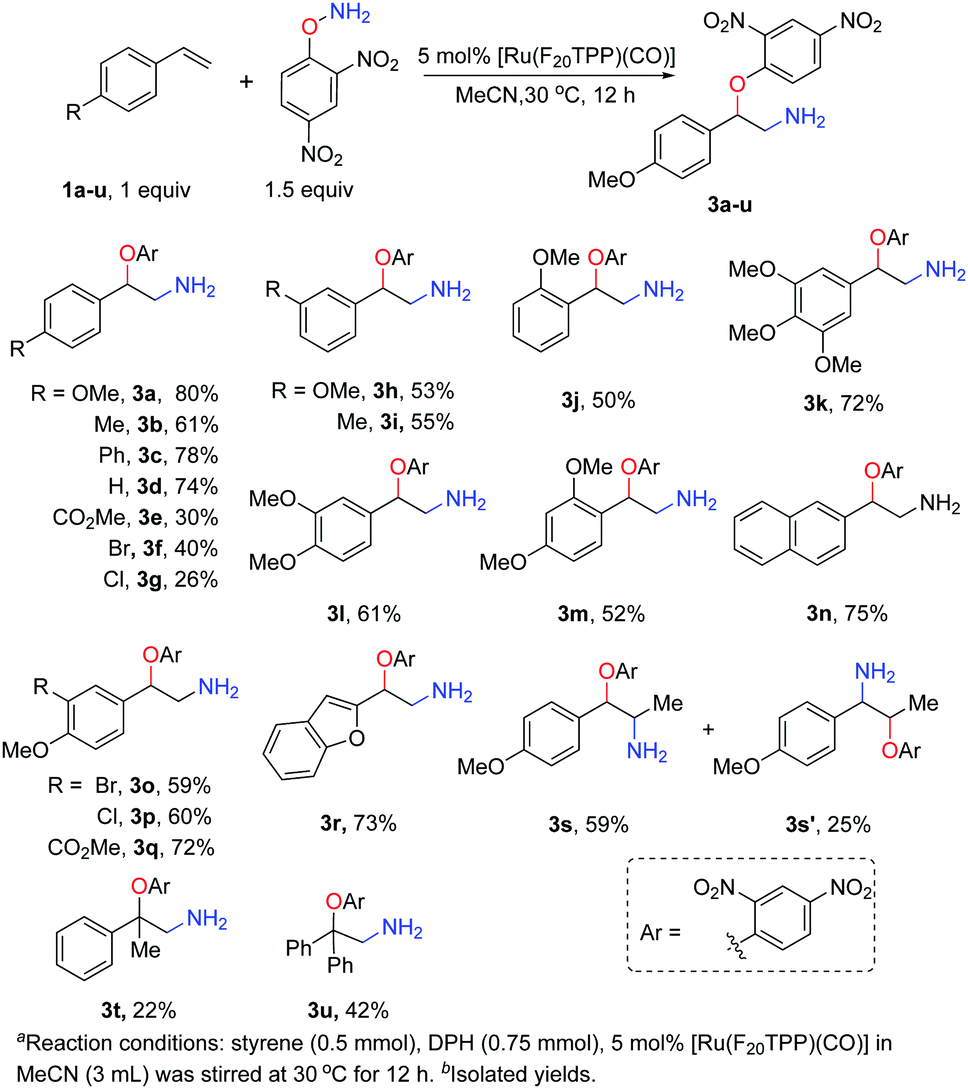 | ||
| Scheme 1 [Ru(F20TPP)(CO)] catalysed amino-oxyarylation of styrenes. | ||
The reaction of 1,3-dienes 4 and DPH was also studied (Scheme 2). Trans-Allylic amino oxyarylation products 5 were exclusively obtained irrespective of the trans-, cis- or mixed trans-/cis-dienes as the substrate. Specifically, both (E)-4-(3′,4′,5′-trimethylphenyl)-buta-1,3-diene (trans:cis = 10:1) and (Z)-4-(3′,4′,5′-trimethylphenyl)-buta-1,3-diene (trans:cis = 1:7.5) gave trans-allylic amino-oxyarylation product 5a as the sole product in 67% and 69% yields, respectively. The other nine diene mixtures (5b–j, trans:cis = 1.7:1–2.1:1) with an electron-rich or electron-deficient substituent afforded the corresponding trans-allylic products in 30–88% yields. Notably, the tolerance of functional groups such as ester, halogen and alkoxy groups is beneficial for further synthetic manipulation. We speculated that the cis-dienes were isomerised to trans-dienes under the reaction conditions, as only residual trans-dienes were detected in the reaction mixture by 1H NMR analysis even when (Z)-4-(3′,4′,5′-trimethylphenyl)-buta-1,3-diene (trans:cis = 1:7.5) was employed.
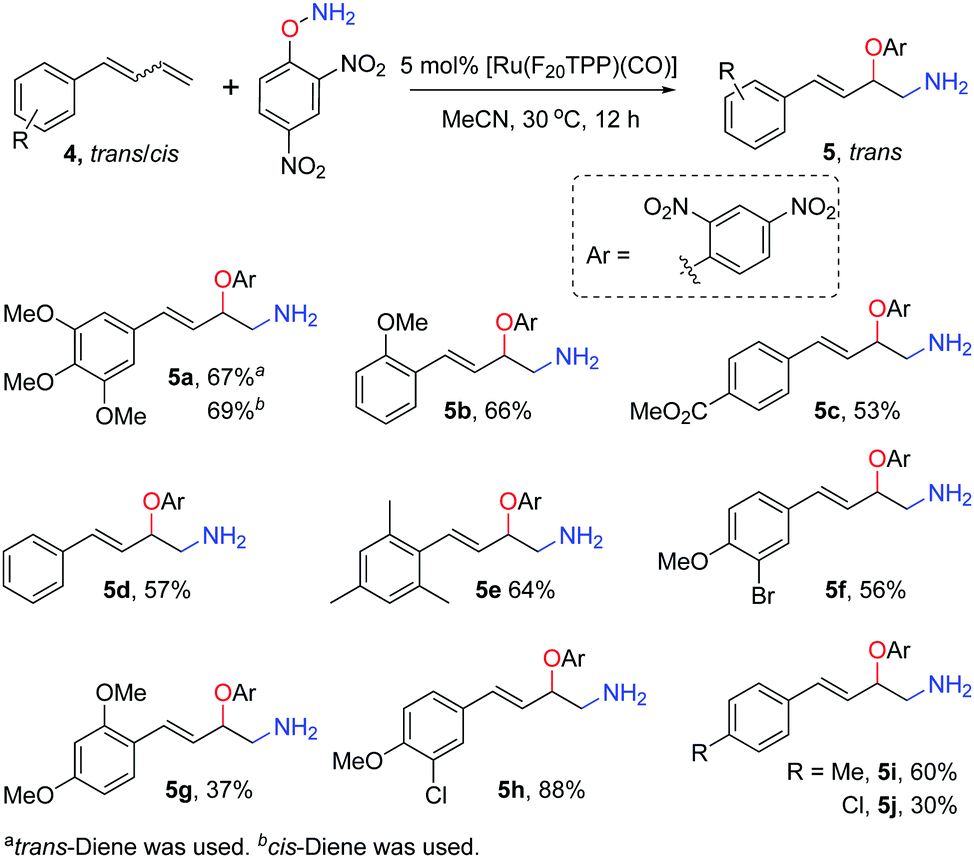 | ||
| Scheme 2 [Ru(F20TPP)(CO)] catalysed amino-oxyarylation of 1,3-dienes. | ||
To understand the mechanism of the nitrogen atom transfer reaction, we examined the reaction of [Ru(F20TPP)(CO)], DPH and styrene in MeCN by ESI-HRMS (Fig. 2). The mixture containing 10 equiv. of DPH and 100 equiv. of styrene was added to an MeCN solution of [Ru(F20TPP)(CO)]. After 1 min, three new and intense cluster peaks at m/z 1088, 1132 and 1156 (C) were detected. The m/z 1088 ion signal was attributed to the ruthenium nitride ion [Ru(F20TPP)(N)]+ (A) by comparing the experimental signal with a simulated pattern (for details, see the ESI,† Fig. S1 and S2). The collision-induced dissociation experiment further substantiated this assignment. The m/z 1132 ion could be attributed to [Ru(F20TPP)(N)(MeCN)]+ (B) and the m/z 1156 ion could be attributed to [Ru(F20TPP)(N)(MeCN)2]+ (C) (Fig. S3 and S4, ESI†). The m/z 1088 signal decreased significantly after prolonging the reaction time to 6.5 h. This implied that m/z 1088 corresponded to a reactive species tentatively assigned as [Ru(F20TPP)(N)]+. After 1.5 h, a new signal at m/z 1282 was detected, which was assigned to [Ru(F20TPP)(MeCN)(amino alcohols)]+ (D) (Fig. S5 and S6, ESI†). We proposed that water served as a nucleophile to attack the ruthenium-aziridino intermediate, to form aminoalcohols. As a result, we speculated that a ruthenium terminal nitride was one of the reactive intermediates. On the other hand, IR spectroscopic studies (Fig. S7 and S8, ESI†) revealed that the CO stretching band (1965 cm−1) vanished immediately after addition of DPH. Removal of the π-acid (CO) is a prerequisite to obtain ruthenium high-valent species. Regarding the nature of this species, we gained insight using UV-vis spectroscopy (Fig. S9–S11, ESI†). Over 10 min, gradual decay of [RuII(F20TPP)(CO)] at 403, 524 and 556 nm (Soret and Q bands) was observed in the presence of excess DPH. However, further spectral changes were also observed after another 15 min (simultaneous decay and growth of the bands at 403 and 418 nm), suggesting that there should be at least two precursors to the ruthenium high-valent species, [Ru(Por)(CO)] (precursor 1) → ‘precursor 2’ → [Ru(Por)(N)(L)] (L = ligand or vacant). The band at 418 nm was likely to have originated from the well-documented RuVI terminal nitrido porphyrin ([RuVI(Por)(N)(L)]),10a (e.g. [RuVI(TMP)(N)(OH)], 416, 485 nm; [RuVI(3,4,5-MeO-TPP)(N)(OH)], 421, 488 nm).
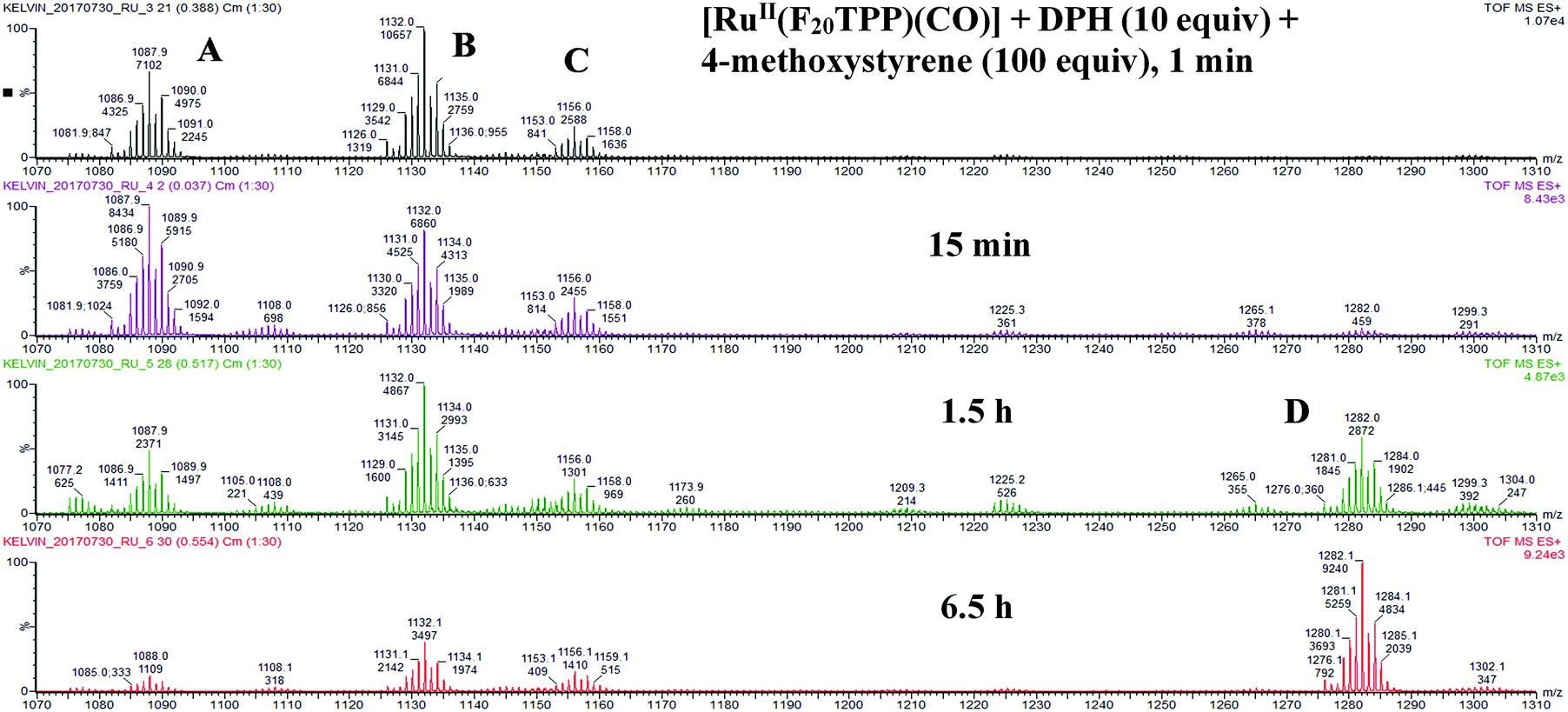 | ||
| Fig. 2 ESI-MS study of the reaction intermediate. | ||
In the presence of [Ru(F20TPP)(CO)], adding 2,2,6,6-tetramethylpiperidine nitroxide (TEMPO, 1.5 equiv.) to the reaction of 4-methoxy styrene 1a and DPH (1.5 equiv.) in MeCN at 30 °C decreased the product yield of 3a from 78% to 38%; adding NaN3 as an extra nucleophile or using a mixed solvent MeCN/MeOH (9:1) could not achieve the azido- or methoxylated products, and 3a was obtained as the sole product but with decreased yields (Scheme 3).
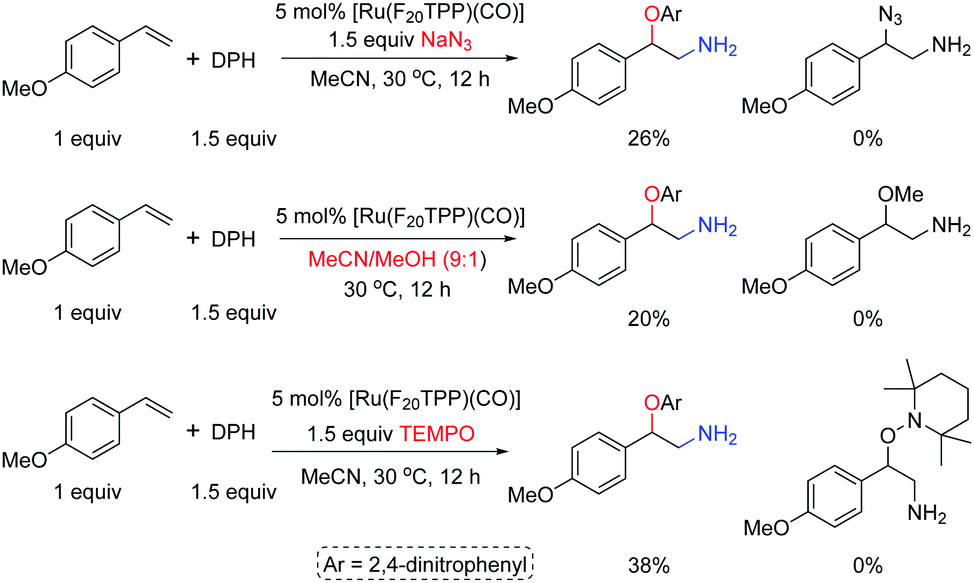 | ||
| Scheme 3 Amino-oxyarylation of 4-methoxy styrene in the presence of NaN3, MeOH or TEMPO. | ||
Based on the aforementioned experiments, we surmise that [Ru(Por)(NH)(X)] (X = MeCN or vacant) was generated by adding DPH immediately, and then [Ru(Por)(NH)(X)] was transformed to [Ru(Por)(N)(X)], the key reactive intermediate, by deprotonation (Scheme 4). The reactive intermediate ([Ru(Por)(N)(X)]) was trapped by alkenes (styrenes or 1,3-dienes) to give an aziridino intermediate E. E was unstable and readily attacked by phenolate to form the final amino-oxyarylation product. The formation of 3s and 3s′ simultaneously (Scheme 1) lends support to the involvement of the aziridino intermediate E.
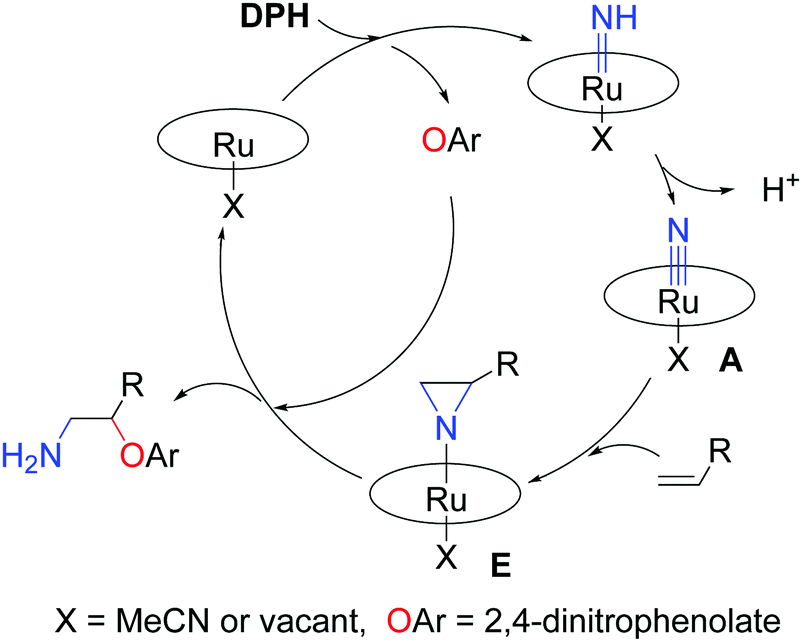 | ||
| Scheme 4 Proposed reaction pathway. | ||
In the literature, most studies on nitrogen atom transfer reactions of metal nitrido complexes are confined to nitrido complexes of manganese and ruthenium.10a,11 The aziridination/amination reactions of metal nitrido complexes typically require an acid anhydride, such as trifluoroacetic anhydride or 4-methylbenzenesulfonic anhydride, as a strong electrophile to elevate nitrido electrophilicity.9a–c,e,10b–d,12 A few metal nitrido complexes were reported to directly undergo a nitrogen atom transfer reaction to nucleophiles such as alkenes in the absence of an acid anhydride or other strong electron-withdrawing reagents.9a,f,13 In this study, we proposed that ruthenium nitrido complexes ([Ru(Por)(N)]) bearing a fluorinated porphyrin ligand (F20TPP) could directly transfer a nitrogen atom to styrenes and terminal 1,3-dienes. We found that the in situ generated [Ru(TMP)(N)(OH)] (from the reaction of [Ru(TMP)(O)2] with HN = CtBu2)10a did not react with styrene to give the corresponding product, which was consistent with the findings that [Ru(TMP)(CO)] could not catalyse such reactions (Table S1, entry 19, ESI†) because the TMP ligand is more electron rich.
In summary, our catalytic and mechanistic studies revealed that ruthenium porphyrins can be developed as efficient catalysts for amino-oxyarylation of alkenes to obtain primary amines. The reaction can be performed under mild conditions without protection by an inert gas, and affords the desired products in good yields.
This work was supported by the China Postdoctoral Science Foundation (2018M633043) and the Shenzhen Science and Technology Innovation Commission (JCYJ20170307104939529 and JCYJ20170412140251576). We thank the Southern University of Science and Technology for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. A. Lawrence, Amines. Synthesis, Properties and Applications, Cambridge University Press, Cambridge, 2004 Search PubMed; (b) L. Legnani, B. N. Bhawal and B. Morandi, Synthesis, 2017, 776 CAS.
- M. B. Smith, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, Hoboken, 7th edn, 2013 Search PubMed.
- (a) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 7179 CrossRef CAS; (b) D. J. Michaelis, C. J. Shaffer and T. P. Yoon, J. Am. Chem. Soc., 2007, 129, 1866 CrossRef CAS; (c) M. Nakanishi, C. Minard, P. Retailleau, K. Cariou and R. H. Dodd, Org. Lett., 2011, 13, 5792 CrossRef CAS; (d) G. S. Liu, Y. Q. Zhang, Y. A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343 CrossRef CAS; (e) B. Zhang and A. Studer, Org. Lett., 2013, 15, 4548 CrossRef CAS; (f) G. Dequirez, J. Ciesielski, P. Retailleau and P. Dauban, Chem. – Eur. J., 2014, 20, 8929 CAS; (g) J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Science, 2014, 343, 61 CrossRef CAS; (h) D.-F. Lu, C.-L. Zhu, Z.-X. Jia and H. Xu, J. Am. Chem. Soc., 2014, 136, 13186 CrossRef CAS; (i) X. Sun, X. Li, S. Song, Y. Zhu, Y.-F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059 CrossRef CAS; (j) L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248 CrossRef CAS.
- (a) O. Reiser, Angew. Chem., Int. Ed. Engl., 1996, 35, 1308 CrossRef CAS; (b) M. A. Andersson, R. Epple, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 490 CrossRef; (c) V. Nesterenko, J. T. Byers and P. J. Hergenrother, Org. Lett., 2003, 5, 281 CrossRef CAS; (d) K. Muniz, Chem. Soc. Rev., 2004, 33, 166 RSC.
- F. Minisci and R. Galli, Tetrahedron Lett., 1965, 6, 1679 CrossRef.
- (a) T. Sheradsky, Tetrahedron Lett., 1968, 9, 1909 CrossRef; (b) F. Yamamoto and S. Oae, Bull. Chem. Soc. Jpn., 1975, 48, 77 CrossRef CAS; (c) C. Legault and A. B. Charette, J. Org. Chem., 2003, 68, 7119 CrossRef CAS; (d) J.-J. Wang, J.-H. Lao, X.-M. Li, R.-J. Lu, H. Miao and M. Yan, Synth. Commun., 2012, 42, 1577 CrossRef CAS; (e) Z. Yang, Synlett, 2014, 1186 CAS.
- M. P. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Falck, Science, 2016, 353, 1144 CrossRef CAS.
- (a) C.-M. Che, V. K.-Y. Lo, C.-Y. Zhou and J.-S. Huang, Chem. Soc. Rev., 2011, 40, 1950 RSC; (b) J.-L. Renaud and S. Gaillard, in Iron Catalysis II, ed. E. Bauer, Springer International Publishing, Cham, 2015, p. 83 Search PubMed.
- (a) W.-L. Man, W. W. Y. Lam, S.-M. Yiu, T.-C. Lau and S.-M. Peng, J. Am. Chem. Soc., 2004, 126, 15336 CrossRef CAS; (b) W.-L. Man, T.-M. Tang, T.-W. Wong, T.-C. Lau, S.-M. Peng and W.-T. Wong, J. Am. Chem. Soc., 2004, 126, 478 CrossRef CAS PubMed; (c) W.-L. Man, W. W. Y. Lam, H.-K. Kwong, S.-M. Yiu and T.-C. Lau, Angew. Chem., Int. Ed., 2012, 51, 9101 CrossRef CAS; (d) W.-L. Man, J. Xie, Y. Pan, W. W. Y. Lam, H.-K. Kwong, K.-W. Ip, S.-M. Yiu, K.-C. Lau and T.-C. Lau, J. Am. Chem. Soc., 2013, 135, 5533 CrossRef CAS; (e) W.-L. Man, W. W. Y. Lam and T.-C. Lau, Acc. Chem. Res., 2014, 47, 427 CrossRef CAS; (f) J. Xie, W.-L. Man, C.-Y. Wong, X. Chang, C.-M. Che and T.-C. Lau, J. Am. Chem. Soc., 2016, 138, 5817 CrossRef CAS.
- (a) S. K.-Y. Leung, J.-S. Huang, J.-L. Liang, C.-M. Che and Z.-Y. Zhou, Angew. Chem., Int. Ed., 2003, 42, 340 CrossRef CAS; (b) G.-S. Fang, J.-S. Huang, N. Zhu and C.-M. Che, Eur. J. Inorg. Chem., 2004, 1341 CrossRef CAS; (c) C. M. Che, V. K. Y. Lo and C. Y. Zhou, Comprehensive Organic Synthesis II, Elsevier, Amsterdam, 2nd edn, 2014, p. 26 Search PubMed; (d) J. Wang, L. Huang, X. Yang and H. Wei, Organometallics, 2015, 34, 212 CrossRef CAS.
- (a) J. T. Groves and T. Takahashi, J. Am. Chem. Soc., 1983, 105, 2073 CrossRef CAS; (b) J. Du Bois, J. Hong, E. M. Carreira and M. W. Day, J. Am. Chem. Soc., 1996, 118, 915 CrossRef CAS; (c) J. Du Bois, C. S. Tomooka, J. Hong and E. M. Carreira, Acc. Chem. Res., 1997, 30, 364 CrossRef CAS; (d) S. Minakata, T. Ando, M. Nishimura, I. Ryu and M. Komatsu, Angew. Chem., Int. Ed., 1998, 37, 3392 CrossRef CAS.
- (a) W. P. Griffith and D. Pawson, J. Chem. Soc., Dalton Trans., 1973, 1315 RSC; (b) P.-M. Chan, W.-Y. Yu, C.-M. Che and K.-K. Cheung, J. Chem. Soc., Dalton Trans., 1998, 3183 RSC; (c) H.-Y. Ng, N.-M. Lam, M. Yang, X.-Y. Yi, I. D. Williams and W.-H. Leung, Inorg. Chim. Acta, 2013, 394, 171 CrossRef CAS.
- (a) S. B. Muñoz, W.-T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Angew. Chem., Int. Ed., 2015, 54, 10600 CrossRef PubMed; (b) G. Sabenya, L. Lázaro, I. Gamba, V. Martin-Diaconescu, E. Andris, T. Weyhermüller, F. Neese, J. Roithova, E. Bill, J. Lloret-Fillol and M. Costas, J. Am. Chem. Soc., 2017, 139, 9168 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experiments and additional figures. See DOI: 10.1039/c9cc08043k |
| This journal is © The Royal Society of Chemistry 2020 |