
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic dimer–monomer equilibrium in a cycloruthenated complex of [Re(η6-C6H6)2]+†
Daniel
Hernández-Valdés
 ,
Lionel
Wettstein
,
Ricardo
Fernández-Terán
,
Benjamin
Probst
,
Thomas
Fox
,
Bernhard
Spingler
,
Qaisar
Nadeem
and
Roger
Alberto
*
,
Lionel
Wettstein
,
Ricardo
Fernández-Terán
,
Benjamin
Probst
,
Thomas
Fox
,
Bernhard
Spingler
,
Qaisar
Nadeem
and
Roger
Alberto
*
Department of Chemistry, University of Zurich, Winterthurerstr. 190, CH-8057 Zurich, Switzerland. E-mail: ariel@chem.uzh.ch
First published on 7th August 2020
Abstract
Cycloruthenation is a well known process in organometallic ruthenium chemistry. In this work, we report unprecedented cycloruthenated rhenium bis-arene compounds with planar chirality. In a two-step process, the reaction of acetyl-pyridine with [Re(η6-C6H6)2]+ introduced a pyridinyl-methanol ligand at one of the arene rings. Coordination of [Ru(CO)2Cl2] led to cycloruthenation, and the products were obtained as two diastereomeric pairs of enantiomers. Under basic pH conditions, the two pairs of enantiomers undergo spontaneous and reversible dimerization. The cycloruthenated monomers were fully characterized, and the dimerization process was studied by NMR, IR spectroscopy, and DFT calculations.
Organometallic compounds are extensively used in many areas, as catalysts in the synthesis of new pharmaceuticals or natural products, in polymerization processes, but also as medicinal organometallic drugs, and in agriculture or industry.1 In this respect, cyclometalated complexes of transition metals such as Pd, Pt, Ir, Os, Rh and Ru have attracted special attention. Some of them have been identified as powerful anticancer agents, catalysts for C–C or C–Y (Y = heteroatom) coupling reactions or in asymmetric catalysis. Often, they also display useful photochemical properties.2
Cyclometalation offers straight ways to form metal–carbon bonds. For ruthenium, different approaches towards cycloruthenation have been reported; C–H activation via concerted metalation–deprotonation (CMD) being the most common one.3 Other routes can involve a transmetalation by ligand exchange, also known as “C–M activation” or the less commonly found cleavage of a carbon–heteroatom bond “C–Y activation” (Y = F, P).3b,c
In our search for new organometallic building blocks for catalysis, we recently developed a series of new ferrocene-like scaffolds with additional ligands conjugated directly to one or both of the arene rings in [Re(η6-C6H6)2]+ (1+).4 This scaffold has barely been investigated despite being discovered about sixty years ago.5 Complexes of the type [Re(η6-C6H5-L)2]+, with L being a multidentate ligand, are very stable under aerobic conditions, as well as in aqueous solutions for prolonged periods of time.6 The reactivities of the benzene rings in [Re(η6-C6H6)2]+ enable different chemical routes to derivatization, comparable to what was reported for ferrocene.7 Enhanced water solubility and higher redox stabilities are advantages of [Re(η6-C6H5-L)2]+ over ferrocene, which might open a broader field of chemical options for derivatizations for which ferrocene is not so suitable.6
One way for introducing ligands or substituents to [Re(η6-C6H6)2]+ is to first generate in situ a carbanion by deprotonation of the benzene rings with lithium diisopropylamide (LDA) at −78 °C.6a,c In the next step, electrophilic reagents such as organic carbonyls are added, which react rapidly with the carbanions as potent nucleophiles. In this way, the treatment of 1+ with LDA and subsequent reaction with prochiral 2-acetylpyridine leads to the formation of multiple products (Scheme 1). A racemic mixture of the mono-functionalized complex [Re(η6-C6H6)(η6-C6H5C(–CH3)(–OH)(–C5H4N))] ((±)-2+) and the bis-substituted [Re(η6-C6H5C(–CH3)(–OH)(–C5H4N))2] (3+ as SS, RR and meso stereoisomers, see ESI†) represented the main products in 38% and 35% yields respectively. The racemate ((±)-2+) could be separated by preparative HPLC (see ESI†).
 | ||
| Scheme 1 Preparation of cycloruthenated bis-arenes. (i) Lithiation/deprotonation 2.5 eq. LDA, 1.5 h, −78 °C, THF; (ii) in situ nucleophilic addition to 2 acetylpyridine; (iii) cycloruthenation reaction, [Ru(CO)2Cl2]n as precursor, microwave, 70 min, 100 °C (L = acetonitrile or trifluoroacetic acid). | ||
In an attempt to synthesize a mixed Ru/Re complex with the [Ru(CO)2Cl2] fragment, the reaction with the racemic mixture ((±)-2+) as ligand and [Ru(CO)2Cl2]n under microwave conditions (100 °C, 70 min) did not give the expected N,O-bidentate coordination but an unexpected cycloruthenation to one of the arene rings in [Re(η6-C6H6)2]+. Taking the diastereotopicity of the positions ortho to the bidentate ligand into account (green and orange in Scheme 1), four different stereoisomers in two sets of enantiomers were obtained (Scheme 1, represented as (±)-4a+/(±)-4b+ together 35% yield). The stereochemical inequality of the two former ortho-hydrogen positions is due to the planar chirality of the rhenium sandwich.8 UPLC-MS analysis of the product mixture evidences that two enantiomeric mixtures were obtained in similar amounts (ESI† and Fig. S1). The mechanism of cycloruthenation is still unclear, but the ortho-C–H activation process certainly has a high activation barrier. Attempting the reaction under standard refluxing conditions in CD3CN/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for more than 18 h at 80 °C did not yield any product.
:1) for more than 18 h at 80 °C did not yield any product.
Very few examples of ortho-metalation of d6 RuII complexes to planar and prochiral ferrocenes or bis-arene complexes of Ru have been reported.9 Djukic et al. coordinated a [RuII(η6-C6H6)]2+ to the π-system of a phenyl–pyridyl unit in an already existing ruthenocycle.9d They found that this was only possible if an electron-donating substituent was present in the preformed metallacycle. Otherwise, the repulsion from the positive charges of both metal centers prevented the reaction, as shown by calculations. In this work, the sequence of reactions is inverted. First, the bis-arene complex of ReI was synthesized, and the cycloruthenation occurred only in the second step. Therefore, electron-donating groups are not necessary. The arene π-coordination in the [Re(η6-C6H5-R)]+ fragment may even facilitate cycloruthenation, by stabilizing the intermediate formal negative charge on the ring prior to ruthenation of the ortho-carbon.
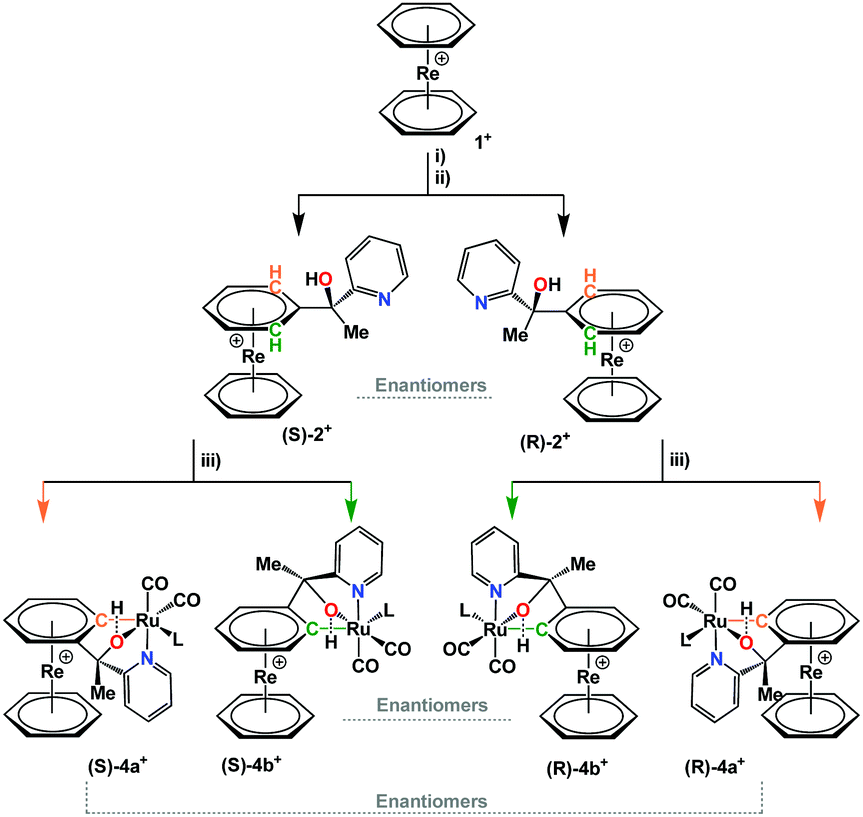
The mixture of complexes (±)-4a+ and (±)-4b+ was separated by preparative HPLC. The racemates were isolated as TFA− salts from an acidic solution by solvent evaporation. Single crystal structures confirmed the cycloruthenation to one of the arene rings in 1+ (Fig. 1). The crystal structure analyses show that the reaction leads to the formation of two five- and one six-membered ruthenocycle systems in a bicyclic type of conformation. It is noteworthy that complexes (±)-4a+ and (±)-4b+ evidence clear differences in the coordination sphere around the Ru center. The most obvious difference is the orientation of the pyridines with respect to the rhenium sandwich. If the derivatized arene ring is taken as reference, the pyridine coordinates from “above” in (±)-4a+, and from above for complex (±)-4b+. TFA completes the coordination sphere to RuII in the crystal but this site can also be occupied by a coordinating solvent such as acetonitrile in solution. UPLC-MS and HR-ESI of both (±)-4a+ and (±)-4b+ support this statement (Fig. S2–S7, ESI†). When “trifluoroacetate” coordinates the RuII center, O1 remains protonated and the cationic nature of the complex is maintained.
 | ||
| Fig. 1 Displacement ellipsoid representations of (±)-4a+ (left) and (±)-4b+ (right). Hydrogen atoms have been omitted for clarity; thermal ellipsoids represent 50% probability. Selected bond lengths (Å) for (±)-4a+: Ru1–C1 2.042(5), Ru1–O1 2.140(3), Ru1–C20 1.899(6), Ru1–C21 1.849(6), Ru1–N1 2.113(4), Re1–Centroid1 1.753(3), Re1–Centroid2 1.738(4). For (±)-4b+: Ru1–C1 2.060(4), Ru1–O1 2.096(3), Ru1–C20 1.885(5), Ru1–C21 1.851(5), Ru1–N1 2.130(4), Re1–Centroid1 1.7443(18), Re1–Centroid2 1.738(2). | ||
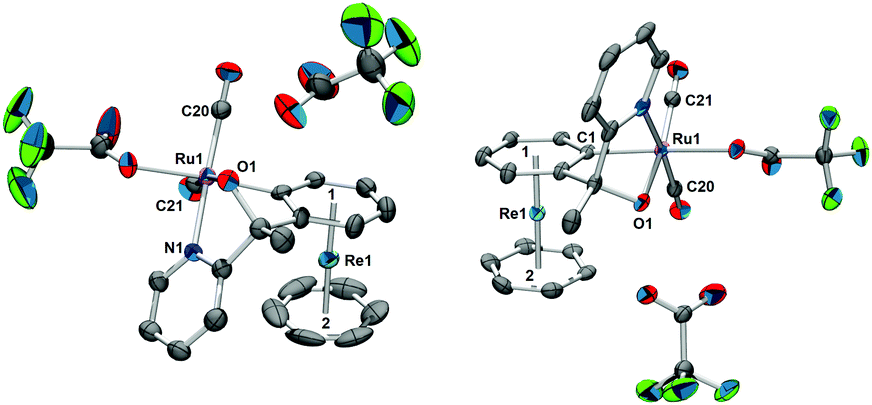
The racemic mixture of (±)-[4a][TFA] was redissolved in water and precipitated with NH4PF6. After washing with water and drying, NMR analysis of (±)-4a+ in acetonitrile-d3 revealed a systematic “impurity” besides the expected signals (Fig. 2). There was a striking resemblance between the peak patterns of (±)-4a+ and this “impurity”; obviously, the multiplicities, the chemical shifts, and the intensities of most of the signals are related to each other. A complete characterization by 2D NMR spectroscopy lead to the clear identification of two different species (Fig. S9–S26, ESI†). The 2D DOSY evidenced different diffusion coefficients and therefore different sizes of the two compounds (Fig. 2), although the differences were modest. These similarities pointed to a relationship between the two species.
 | ||
| Fig. 2 2D DOSY of (±)-4a+ in CD3CN, 278 K, d20 = 55 ms, p30 = 700 μs. There are two sets of signals with different diffusion coefficients. The peak patterns and the chemical shifts are similar, though. As the majority of the signals are found in the range from 9.0 ppm to 4.8 ppm, only this section is shown. | ||
UPLC-MS of the very same sample showed three distinct mass signals; the expected m/z = 620.1 for (±)-4a+, m/z = 661.0 due to the coordination of acetonitrile to the former HTFA site (both at Rt = 1.3 min), and a third mass signal at m/z 620.0 (Rt = 1.7 min). This signal with m/z 620.0 and a different retention time featured a densely packed isotope pattern with mass peaks separated by half-integer values (Fig. S4–S8, ESI†). This appearance inferred a species with a 2+ charge and exactly double the mass of (±)-4a+ as further confirmed by HR-ESI-MS (Fig. S6 and S8, ESI†).
All these analytical data obtained implied the formation of a dimer [(4a)2]2+ which would explain the close resemblance of the two sets of signals. Dimerization would also be in accordance with the smaller diffusion coefficient found for one of the two components (implying a larger size for the dimer). The ratio between the experimental diffusion coefficients is consistent with the values calculated via the Stokes–Einstein equation taking the sizes of the dimer and the monomer coordinated to acetonitrile into account. Furthermore, 2D 1H ROESY of the sample containing the mixture of (±)-4a+ and its dimer [(4a)2]2+, shows through-space correlations between the six equivalent protons from the non-derivatized benzene ring coordinated to ReI (δ = 5.01 ppm), and the pyridine protons (Fig. S24, ESI†). These correlations are not found for the monomeric species (±)-4a+, where, due to the higher degree of freedom, the pyridine accommodates further away from the bis-arene unit.
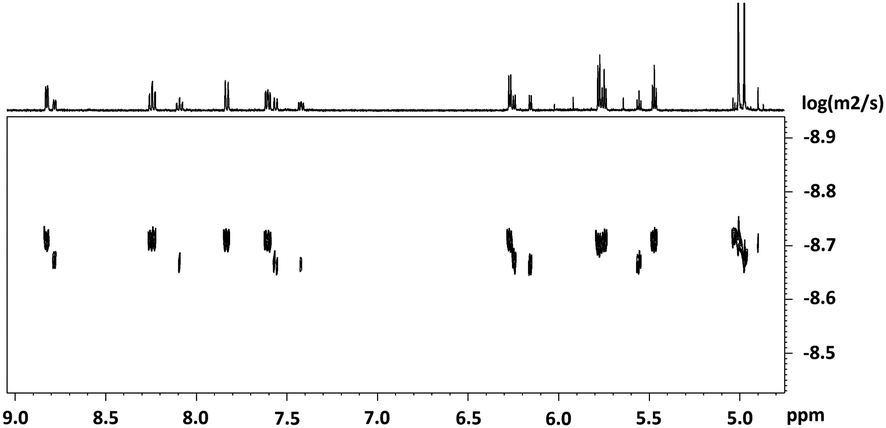
Similar results are obtained when complex (±)-[4b][TFA] was precipitated with NH4PF6 and washed multiple times with water. In this case, the dimer [(4b)2]2+ could be isolated as the only component. The NMR spectra show only one species and the mass pattern detected by HR-ESI corresponds to the dimer (Fig. S8, ESI†). Single crystal structure analysis confirmed the authenticity of [(4b)2]2+ (Fig. 3). The crystal structure shows that the two enantiomers, (R)-4b+ and (S)-4b+ dimerize via two μ2-O bridges. The two RuII centers are located in the corners of a rhomboid with distances Ru(1)–O(1) and Ru(1)–O(2) of 2.097 Å and 2.185 Å, respectively. The two RuII atoms in the dimer have a distorted octahedral coordination sphere, as is the case in the monomeric units. The distances between the RuII atoms and the carbonyls trans to the pyridine are slightly shorter in the dimer [(4b)2]2+ as compared to the monomer (±)-4b+ (1.874 Å for the dimer and 1.885 Å for the monomer). In contrast, the distance between the RuII and the carbonyls cis to the pyridine are longer for [(4b)2]2+ than for (±)-4b+ (1.873 Å and 1.851 Å, respectively). In solution, the wavenumbers of the CO stretching frequencies blue shift by approximately 30 cm−1 as (±)-4b+ dimerize into [(4b)2]2+.
 | ||
| Fig. 3 Displacement ellipsoid representations of [(4b)2]2+. Hydrogen atoms have been omitted for clarity; thermal ellipsoids represent 50% probability. Selected bond lengths (Å) for [(4b)2]2+: Ru1–C1 1.874(4), Ru1–C20 1.873(5), Ru1–C21 1.874(4), Ru1–O1 2.097(3), Ru–O1_i 2.185(2), Ru–N1 2.130(3), C20–O2 1.139(6), C21–O3 1.141(6), Re1–C1 2.330(4), Re1–C2 2.241(3), Re1–C3 2.219(4), R1–C4 2.241(4), Re1–C5 2.231(4), Re1–C6 2.259(4), Re1 –C7 2.235(4), Re1–C8 2.236(5), Re1–C9 2.230(5), Re1–C10 2.212(5), Re1–C11 2.237(4), Re1–C12 2.228(5). | ||
DFT calculations are in support of these data. In Fig. 4, the calculated and the experimentally obtained vibrational frequencies of the coordinated CO groups of (±)-4b+ and its dimer [(4b)2]2+ are plotted against the normalized absorbance. Since the dimer displays only two CO bands, it must contain a center of inversion. This excludes dimer formation between two individual enantiomers to either [(S)-4b2]2+ or [(R)-4b2]2+. Only (S,R) type of dimers are allowed by symmetry.
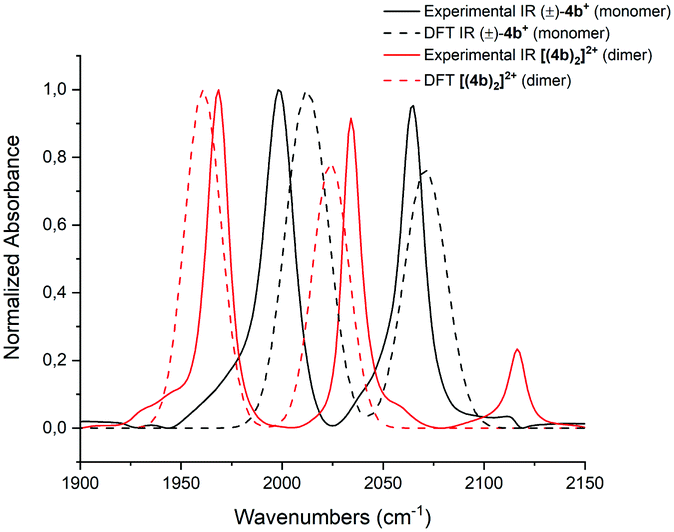 | ||
| Fig. 4 Experimentally obtained IR spectra of (±)-4b+ (black) and [(4b)2]2+ (red) are shown as continuous lines. IR absorptions calculated by DFT for the monomers are shown as dashed lines. The frequencies are plotted against the normalized absorbance. | ||
Complex (±)-4b+ and its dimer [(4b)2]2+ are in an equilibrium, controlled by protonation–deprotonation of the OH groups (Scheme 2). To assess the kinetics of this process, [(4b)2]2+ was dissolved in ACN-d3, and the rate of equilibration was evaluated. After the addition of HTFA to the solution, a new set of peaks evolved with the chemical shifts of the monomer 4b+. The formation kinetics of 4b+ can be quantified either by monitoring the appearance of the six-proton signal at 5.93 ppm or the disappearance of the respective signal from the dimer at 5.64 ppm. The rate of formation was studied under pseudo first-order conditions with HTFA in high excess (100 eq.) over the dimer. The integrals of the resonances at 5.93 ppm (monomer) and 5.64 ppm (dimer) were plotted against time and fitted to a first-order exponential (Fig. 5). Waterfall plots showing the decrease of the [(4b)2]2+ signal and the rise of 4b+ and linear correlations of kobs values vs. concentrations are shown in Fig. S27–S35 and Table S1 (ESI†). A second-order rate constant k1 at 298 K of 0.89 ± 0.10 × 10−2 M−1 s−1 was obtained. The monomer–dimer interconversion is dynamic and reversible: after the addition of 10 eq. of Et3N to the monomer 4b+, dimerization reoccurred (Fig. S34 and S35, ESI†). Addition of a strongly coordinating ligand such as pyridine to the dimer [(4b)2]2+ shifts the equilibrium towards the corresponding monomer as well (Fig. S36 and S37, ESI†).
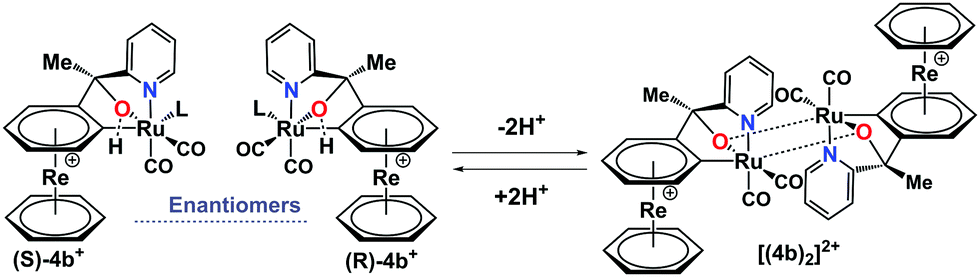 | ||
| Scheme 2 Monomer–dimer equilibrium in acetonitrile controlled by protonation–deprotonation of the OH group (L = acetonitrile). | ||
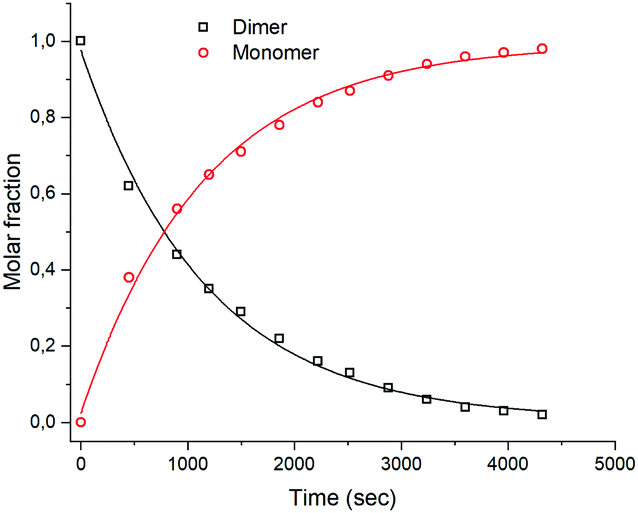 | ||
| Fig. 5 Plot of time-dependent integrals of [(4b)2]2+ (1.11 mM) and (±)-4b+ recorded at 298 K (bottom). Concentration of HTFA, 111 mM, solvent: CD3CN. | ||
In conclusion, the conjugation of ligands to one (or both) of the two benzene rings in [Re(C6H6)2]+ allows for the preparation of hetero-bimetallic complexes. With ruthenium, four ReI–RuII binuclear compounds were obtained as two diastereomeric sets of enantiomers. These complexes feature unexpected cycloruthenation at the positions ortho to the site to which the ligand is conjugated. Since the individual Ru complex is known to be catalytically active, the influence of the coordination of the cationic rhenium sandwich on the electronic properties of the RuII metal center in e.g. enantioselective catalysis might be of interest. The two sets of enantiomers dimerize spontaneously in solution under basic conditions. This process is favorable due to the availability of a labile coordination site, occupied in the monomer by a solvent molecule and by formation of two μ2-O bridges. Dimerization was assessed in detail by NMR spectroscopy, kinetics as well as DFT calculations. We emphasize that this kind of dynamic processes are rare and is an option for the application of cyclometalated compounds in catalysis.
The authors thank the joint Swiss National Science Foundation SNSF and the National Research Foundation of South Africa NRF for financial support through project IZLSZ2_170856, and the University of Zurich for further support.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926–5927 CrossRef CAS PubMed
; (b) C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC
-
(a) J. Sun, J. Zhao, H. Guo and W. Wu, Chem. Commun., 2012, 48, 4169–4171 RSC
-
(a) B. Li, C. Darcel, T. Roisnel and P. H. Dixneuf, J. Organomet. Chem., 2015, 793, 200–209 CrossRef CAS
- D. Hernandez-Valdes, F. Avignon, P. Müller, G. Meola, B. Probst, T. Fox, B. Spingler and R. Alberto, Dalton Trans., 2020, 49, 5250–5256 RSC
-
(a) E. O. Fischer and A. Wirzmüller, Chem. Ber., 1957, 90, 1725–1730 CrossRef CAS
-
(a) G. Meola, H. Braband, D. Hernández-Valdés, C. Gotzmann, T. Fox, B. Spingler and R. Alberto, Inorg. Chem., 2017, 56, 6297–6301 CrossRef CAS PubMed
-
(a) M. R. Das, M. Wang, S. Szunerits, L. Gengembre and R. Boukherroub, Chem. Commun., 2009, 2753–2755 RSC
-
(a) J. Xu, Y. Liu, J. Zhang, X. Xu and Z. Jin, Chem. Commun., 2018, 54, 689–692 RSC
-
(a) A. Hijazi, J.-P. Djukic, M. Pfeffer, L. Ricard, N. Kyritsakas-Gruber, J. Raya, P. Bertani and A. de Cian, Inorg. Chem., 2006, 45, 4589–4591 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2007155–2007157. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04180g |
| This journal is © The Royal Society of Chemistry 2020 |