
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFourfold symmetric MCR's via the tetraisocyanide 1,3-diisocyano-2,2-bis(isocyanomethyl)propane†
Roberto
Butera
 a,
Annadka
Shrinidhi
b,
Katarzyna
Kurpiewska
c,
Justyna
Kalinowska-Tłuścik
c and
Alexander
Dömling
*a
a,
Annadka
Shrinidhi
b,
Katarzyna
Kurpiewska
c,
Justyna
Kalinowska-Tłuścik
c and
Alexander
Dömling
*a
aUniversity of Groningen, Department of Drug Design, A. Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: a.s.s.domling@rug.nl
bUniversity of California, Department of Chemistry & Biochemistry, La Jolla, San Diego, California 92093-0358, USA
cDepartment of Crystal Chemistry and Crystal Physics Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland
First published on 4th August 2020
Abstract
Oligoisocyanides are attractive synthetic targets, however, only a few are known. Here, we describe the smallest stable tetraisocyanide possible, the 1,3-diisocyano-2,2-bis(isocyano-methyl)propane (1) with S4 symmetry. Its four-step synthesis, structure, and reactivity in unprecedented symmetric fourfold Ugi 4CR and fourfold Passerini 3CR are described. Exhibiting high functional group tolerance and moderate to high yields, we foresee multiple applications of 1,3-diisocyano-2,2-bis(isocyanomethyl)propane, for example in MOFs, COFs, dendrimers, or artificial organs.
The isocyanide group is a highly useful functional group in chemistry with applications in organic, inorganic, metalorganic, and medicinal chemistry and also in material science.1–4 While the number of monoisocyanides is very large, the number of biisocyanides is sizable, but the number of oligoisocyanides is minuscule by comparison.5–12 A representative molecule bearing three isocyanide groups is tris(2-isocyanoethyl)amine, providing access to unprecedented complex structures. By implementing this isocyanide in a threefold isocyanide-based multicomponent reaction (IMCR), Wessjohann and Rivera were able to synthesise complex supramolecules, as hybrid peptide–peptoid podands and macromulticycles.7–9 Organic tetraisocyanides are a very rare species of isocyanide bearing compounds; only three synthesised organic tetraisocyanides (2, 3, 4, Fig. 1) are reported to date.10,11,13 And, as far as reported, no successful fourfold IMCR was achieved with these tetraisocyanides.10,11,13,14
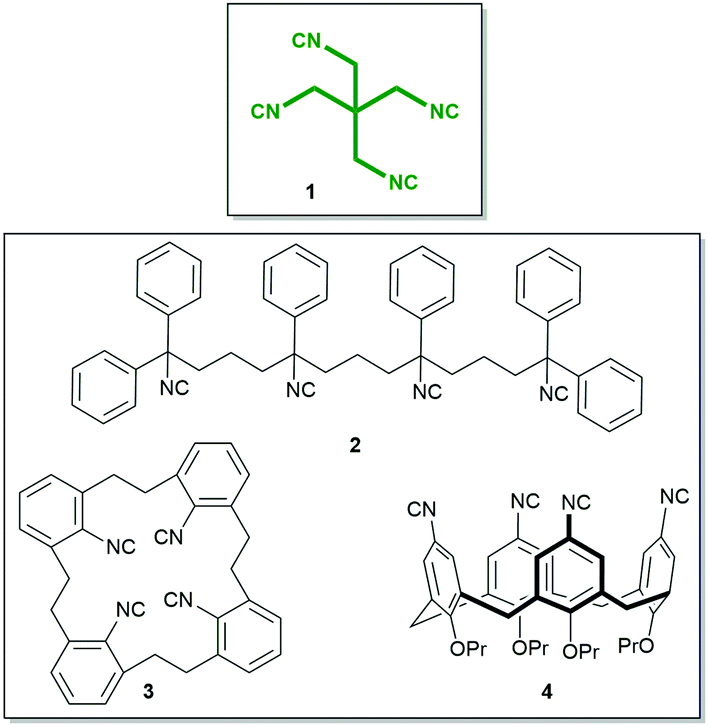 | ||
| Fig. 1 Tetraisocyanide 1 and previously reported tetraisocyanides 2, 3, and 4.10–12 | ||
Here, we report the synthesis, structure, and reactivity of the tetraisocyanide 1,3-diisocyano-2,2-bis(isocyanomethyl)-propane (1). Inspired by its symmetry and anticipating numerous unique applications, our goal was not only to synthesise and characterise this symmetrical tetraisocyanide 1 (Fig. 1) but also to utilise it in isocyanide-based multicomponent reactions.15,16
We envisioned to synthesise the tetraisocyanide 1 from available starting material pentaerythritol tetrabromide (5) via a short sequence involving the Ugi dehydration of the tetraformamide as the last step (Scheme 1). Nucleophilic substitution reaction of pentaerythritol tetrabromide (5) with sodium azide (12 eq.) yielded the 1,3-diazido-2,2-bis(azidomethyl)propane (6, 88%). Reduction of the tetraazide 6 by employing hydrogen and palladium-on-charcoal in a Paar apparatus at a pressure of 3 bar yielded in quantitative conversion of 6 to 2,2-bis(aminomethyl)propane-1,3-diamine (7). The tetraformamide 8 was prepared by refluxing the tetraamine 7 in excess ethyl formate. Despite the unusual long reaction time of 48 h, we were able to generate the compound N,N′-(2,2-bis(formamidomethyl)propane-1,3-diyl)diformamide (8) in 92% yield. The tetraformamide 8 exhibited extremely poor solubility in most organic solvents which opportunely could be exploited by facilitating the purification of 8 by a simple precipitation/filtration/washing procedure (ESI†).
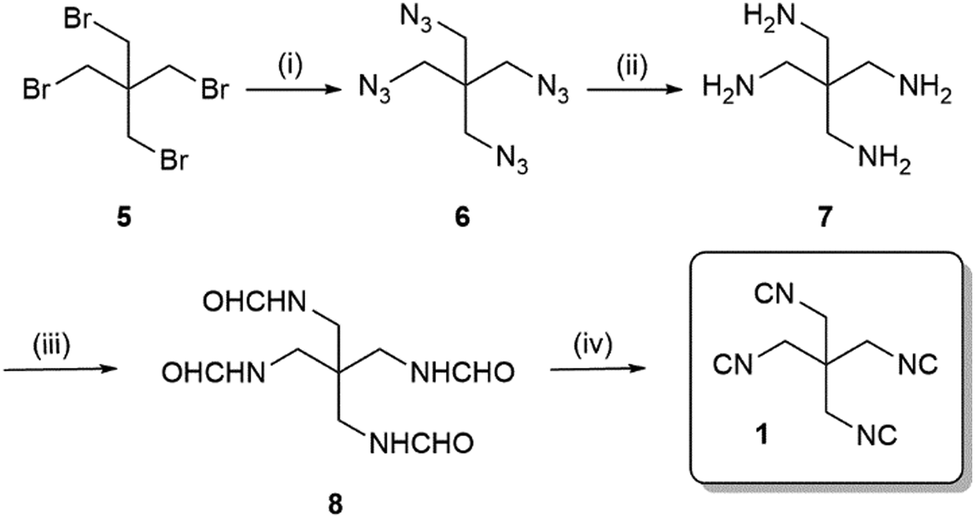 | ||
Scheme 1 Synthesis of 1,3-diisocyano-2,2-bis(isocyanomethyl)propane 1, ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (i) NaN3, DMF, 80 °C, 12 h, 88%; (ii) H2–Pd/C, MeOH, 40 °C, 20 h, 99%; (iii) ethyl formate, 60 °C, 48 h, 92%; (iv) POCl3, Et3N, DCM, −20 °C – rt, 18 h, 55%. (i) NaN3, DMF, 80 °C, 12 h, 88%; (ii) H2–Pd/C, MeOH, 40 °C, 20 h, 99%; (iii) ethyl formate, 60 °C, 48 h, 92%; (iv) POCl3, Et3N, DCM, −20 °C – rt, 18 h, 55%. | ||
Finally, dehydration of the tetraformamide 8 was completed using phosphoryl chloride (Scheme 1). Comparably to the reaction leading to compound 8 the reaction time required an increase to 18 h to reach completion. Since the tetraisocyanide 1 showed a similarly poor solubility in most organic solvents, we exploited this property in the workup of the reaction by filtrating the residue and washing with DCM and water yielding in 1,3-diisocyano-2,2-bis(isocyanomethyl)propane (1). Although the yield of the dehydration reaction was 55%, we achieved the synthesis of the tetraisocyanide 1 with a moderate overall yield of 44% over four steps on a multigram scale.
The tetraisocyanide 1 is an off-white and odourless solid with a m.p. range of 215–219 °C (decomp.). 1,3-Diisocyano-2,2-bis(isocyanomethyl)propane (1) belongs to the reduced symmetry group S4 and not as expected to Th, T, or Td. The NMR of 1 is accordingly simple showing only one signal in the 1H NMR for the methylene group at 3.83 ppm (ESI†). The 13C NMR is equally simple showing three signals corresponding to the central quaternary carbon at 39.1 ppm, the methylene carbons at 42.7 ppm, and the terminal isocyanide carbons at 160.9 ppm. Surprisingly, no signal in the carbon NMR spectrum shows a triplet as many other isocyanides do. The corresponding IR spectrum also reveals the for isocyanide groups typical strong absorption at 2148 cm−1.17 The identity of compound 1 was also confirmed by X-ray single-crystal analysis (Fig. 2 & ESI†). The distance of the triple bond between C and N is comparable with many other isocyanides exhibiting a length of 1.16 Å. Interestingly, the negatively polarized isocyanide C is also forming a hydrogen bond to a hydrogen atom of a neighbouring tetraisocyanide molecule with 2.59 Å distance.
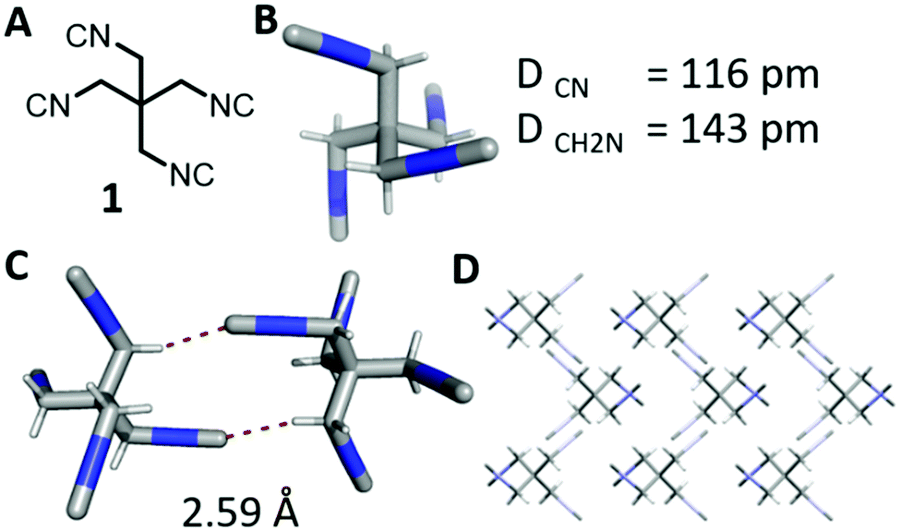 | ||
| Fig. 2 (A) 2D-structure of 1; (B) crystal structure of 1 with corresponding bond lengths; (C) visualisation of intermolecular interactions of 2.59 Å between isocyano group and methylene group; (D) visualisation of the crystal structure of 1 along the X-axis. | ||
Having successfully synthesised the tetraisocyanide 1, we investigated its potential in multicomponent reactions (MCR). For this purpose, we decided to implement it in a fourfold Ugi four-component reaction. For the optimisation studies we chose acetic acid, phenylethylamine, and paraformaldehyde as substrates to not further introduce a symmetry break. The first attempt for the U4CR was performed under conventional conditions. Carrying out the reaction in MeOH (1 M) at room temperature led to the detection of traces of the desired product 12 after 3 days (Table 1, entry 1).
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time | Yield (%) |
| a All reactions were performed using 1 (0.25 mmol), 9 (1 mmol), 10 (1 mmol), 11 (1 mmol) and solvent (1 mL). b No/conventional heat source. c Microwave-assisted heating. d 5 mol% ZnCl2. e 8 eq. of 9, 10, and 11. f Beforehand stirring of 10 and 11 for 30 min at rt. g Beforehand stirring of 10 and 11 for 30 min at rt and using 16 eq. of 9. nP: no product formed. | ||||
| 1 | MeOH | rt | 3 d | Tracesb |
| 2 | MeOH | 60 | 3 d | Tracesb |
| 3 | MeOH | rt | 21 d | Tracesb |
| 4 | MeOH | 100 | 1 h | Tracesc |
| 5 | MeOH | 120 | 2 h | Tracesc |
| 6 | MeOH | 100 | 1 h | nPcd |
| 7 | MeOH | 100 | 1 h | Tracese |
| 8 | H2O | 120 | 1 h | Tracesc |
| 9 | Toluene | 120 | 1 h | 6c |
| 10 | TFE | 120 | 1 h | 11c |
| 11 | TFE | 120 | 2 h | 58 |
| 12 | TFE | 150 | 2 h | 50cf |
| 13 | TFE | 120 | 2 h | nPcg |
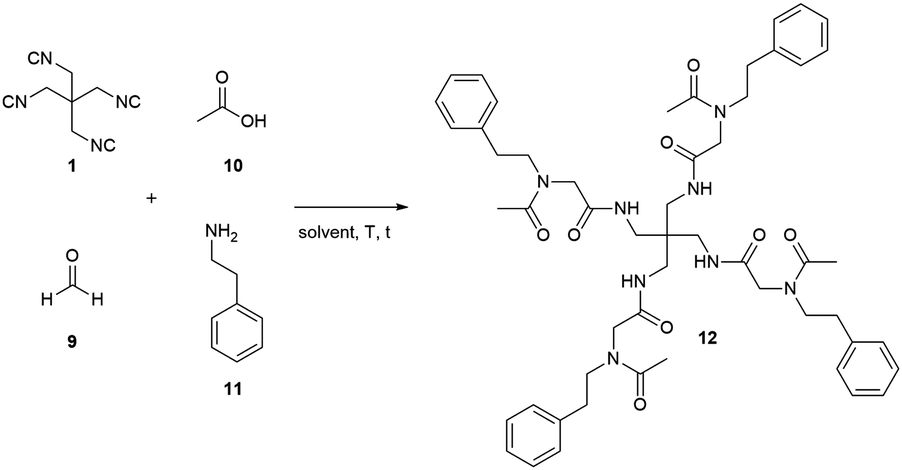
Increasing the reaction temperature to 60 °C (Table 1, entry 2) or the reaction time to 21 days (Table 1, entry 3) did not show any improvement. Therefore, we decided to apply microwave radiation for further optimisation reactions (Table 1, entries 4–13). The first attempts with microwave heating (Table 1, entries 4 and 5) demonstrated a diminished presence of unidentified side products compared to entries 1–3. The addition of catalytic amount of zinc chloride surprisingly eliminated any presence of the desired product 12 (Table 1, entry 6) and consequently, we continued optimisation without additional catalyst. The attempt to improve product formation by increasing equivalents of all substrates except the isocyanide resulted in no improvement compared to previous reactions. Based on the poor solubility of the isocyanide we decided to explore the effect of different solvents on the reaction employing water, toluene, and 2,2,2-trifluoroethanol (TFE) as solvents. Water could not achieve any improvement as opposed to toluene and TFE. Both, toluene and TFE demonstrated a significant improvement in product formation with yields for the isolated compound 12 of 6% and 11%, respectively (Table 1, entries 9 and 10). Based on these findings we chose the sole use of TFE for further optimisation of the fourfold Ugi four-component reaction.
During the optimisation process, we formulated the hypothesis that the isocyanide is not stable in the presence of free acid and therefore degrades in the reaction mixture.18,19 Consequently, we changed the addition order to confirm indirectly this hypothesis. We mixed the amine substrate 11, the acid substrate 10, and TFE (0.5 mL) in advance and stir the reaction mixture at room temperature for 30 min expecting an acid–base reaction and hence reducing the amount of free acid. Adding paraformaldehyde (9) and the tetraisocyanide 1 afterwards and carrying out the reaction with microwave-assisted heating yielded the desired product 12 in 58% (Table 1, entry 11). Further attempts of improvement by increasing the reaction temperature to 150 °C (Table 1, entry 12) or by increasing the equivalents of paraformaldehyde (9) (Table 1, entry 13) led to reduced yields or even no product formation at all.
Having accomplished and optimised this fourfold multicomponent reaction with satisfactory yields, we explored the scope and limitations of the U4CR using the tetraisocyanide 1, paraformaldehyde (9), and diverse acid (A) and amine (B) components, as shown in Table 2.
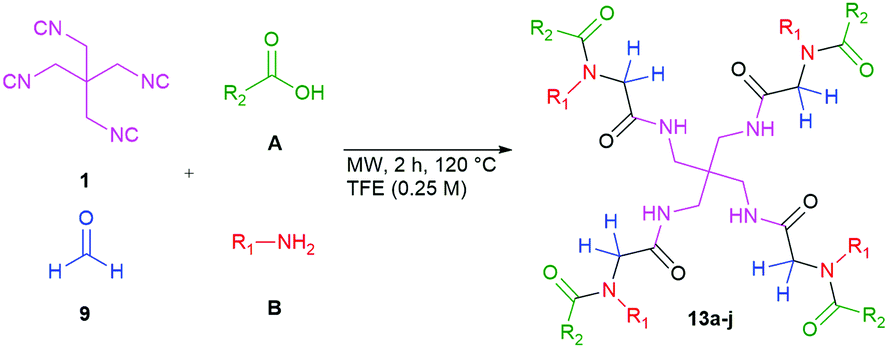
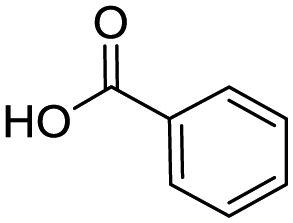
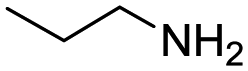
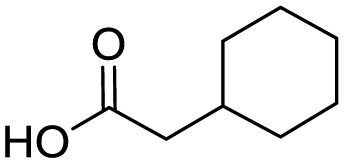
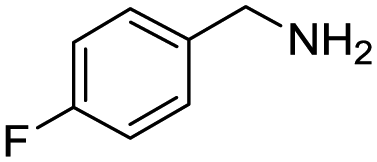
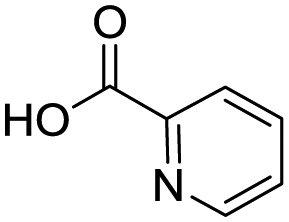
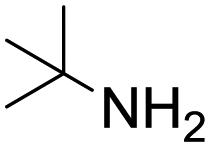
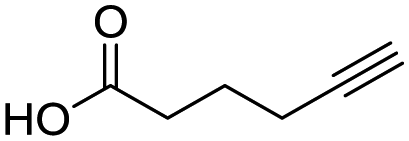
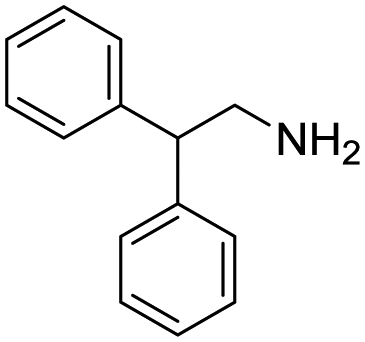
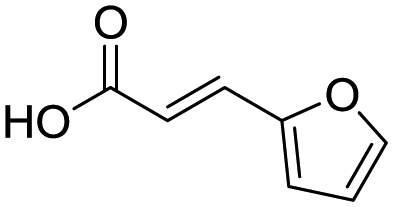
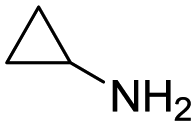
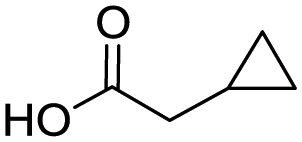
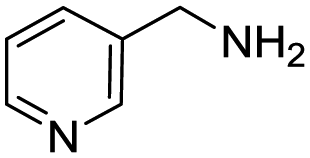
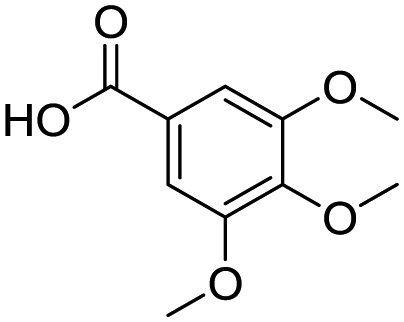
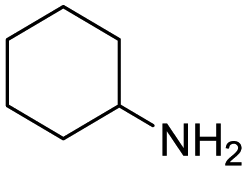
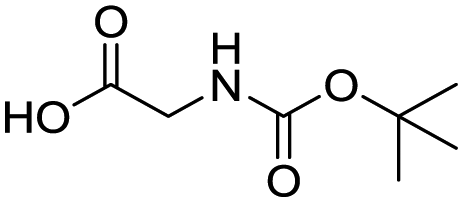
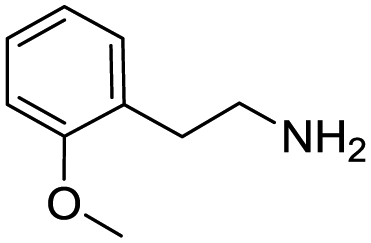
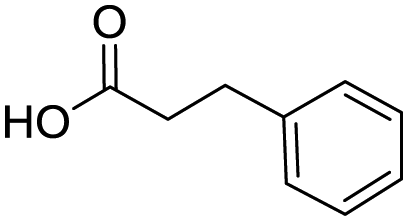
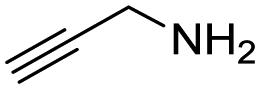
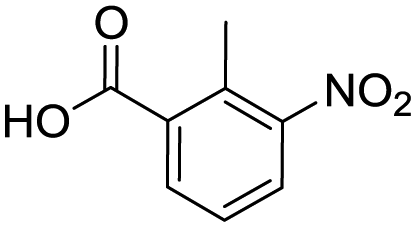
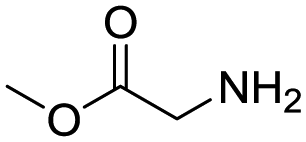
Remarkably, many different amine and acid sources were compatible in the fourfold U4CR with yields of 35–83% showing a great functional group tolerance and generally disregarding steric or electronic factors. Various aliphatic amines, including propargylamine, 3-picolylamine, tert-butylamine, and 2,2-diphenyl ethylamine, as well as aliphatic acids, including 3-(2-furyl)acrylic acid, 2-cyclohexylacetic acid, and 5-hexynoic acid, yielded the corresponding fourfold U4CR product. Moreover, aromatic acids, for example, picolinic acid, 3,4,5-trimethoxybenzoic acid, and 2-methyl-3-nitrobenzoic acid, were able to react in the fourfold U4CR reaction. Despite the high functional group tolerance, certain substrates did not result in product formation. Utilising any sort of anilines and substrates including an indole or pyrrole function did not generate any traces of product. Noteworthy, the reaction with N-(tert-butoxycarbonyl) glycine (Table 2, entry 13h) as acid source showed the lowest yield overall. The combination of fluorinated solvents and microwave radiation, as demonstrated by Choy et al., is capable of Boc-deprotection which explains the atypical low yield.20 Overall, 11 fourfold Ugi 4CR compounds were successfully synthesised. Besides, we were able to produce two single-crystals from the products of entry 13b and 13e confirming by X-ray single-crystal analyses the fourfold U4CR products (Fig. 3).
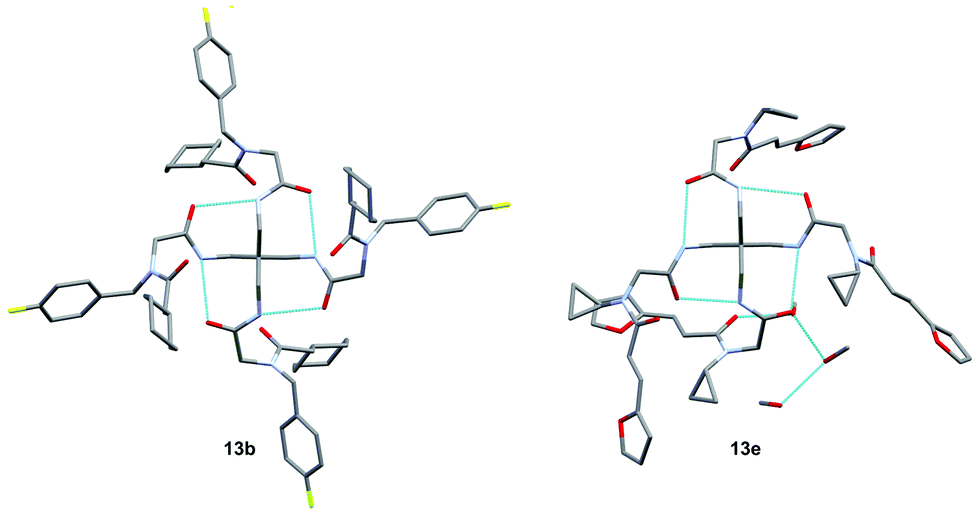 | ||
| Fig. 3 Crystal structures of 13b and 13e; shown in blue are the existing hydrogen bonds with lengths of 2.86 Å (13b) and 2.87 Å (13e) between the core amide bonds, and a length of 2.68 Å between one amide bond of 13e and a residual methanol molecule. | ||
Both crystal structures (Fig. 3) show intramolecular hydrogen bonds depicted in a ring motif around the central carbon; these interactions consist of hydrogen bonds between the core amides. Additionally, 13e shows intermolecular hydrogen bonds with a residual methanol molecule breaking the S4 symmetry exhibited by 1 & 13b and reducing it to C1 symmetry. Following the U4CR, we implemented the tetraisocyanide 1 also in a fourfold Passerini three-component reaction. The optimised reaction conditions of the U4CR were adjusted for the P3CR by switching the solvent from TFE to 1,2-dichloroethane. The absence of an amine component was compensated by the addition of four equivalents of triethylamine to account for the acid lability of 1.
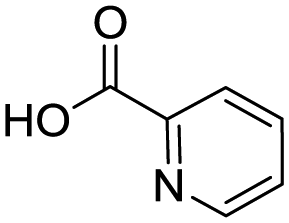
To evaluate the scope and limitations of the P3CR with the tetraisocyanide 1, phenylacetic acid, 3,4,5-trimethoxybenzoic acid, 2-methyl-3-nitrobenzoic acid, picolinic acid, and 5-hexynoic acid were chosen as representative acids. All the engaged acids yielded in the corresponding fourfold Passerini product with 35–77% representing a high functional group tolerance. Only electron-poor aromatic acids, i.e. 2-methyl-3-nitrobenzoic acid, generated low yields in the P3CR reaction for product 14c (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Acid (A) | Isolated yield (%) | Entry | Acid (A) | Isolated yield (%) |
| a The Passerini reactions were performed with adapted optimised reaction conditions of entry 11 of Table 1 using 1 (0.25 mmol), 9 (1 mmol), acid A (1 mmol), triethylamine (1 mmol) and DCE as solvent (1 mL). | |||||
| 14a |
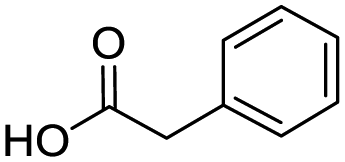
|
77 | 14d |
|
68 |
| 14b |
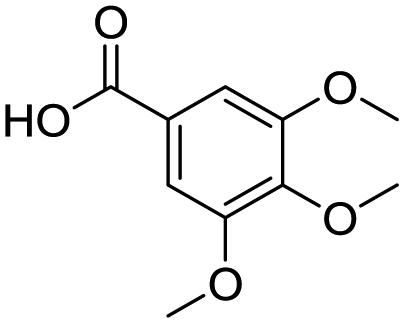
|
60 | 14e |
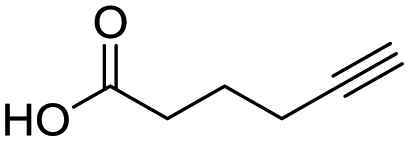
|
71 |
| 14c |
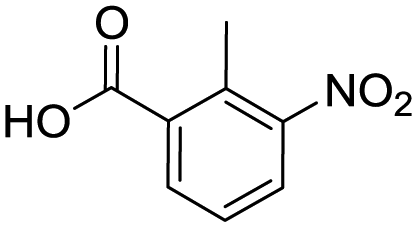
|
35 | |||
In essence, we were able to introduce 1,3-diisocyano-2,2-bis(isocyano-methyl)propane (1) of S4 symmetry by a short four – step synthesis route showing great scalability with an overall yield of 44%. Furthermore, the synthesis of 16 fourfold IMCR compounds employing the tetraisocyanide 1 were accomplished and confirmed by X-ray single-crystal analyses. Showing great compatibility with the U4CR and the P3CR, we were able to generate unique novel four-legged scaffolds proving the great potential of the tetraisocyanide 1. By demonstrating high functional group tolerance and moderate to high yields, the tetraisocyanide 1 can be envisioned for numerous exciting novel applications such as MOFs, COFs, IMCR-based dendrimers, and other materials. Corresponding investigations are ongoing in our lab and will be reported in due course.
This research has been supported by the National Institute of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) (grant agreement number 115489), the Qatar National Research Foundation (NPRP6-065-3-012), COFUNDs ALERT (grant agreement no. 665250), Prominent (grant agreement no. 754425) and KWF Kankerbestrijding grant (grant agreement no. 10504). K. K. and J. K.-T. acknowledge the financial support of the European Regional Development Fund in the framework of the Polish Innovation Economy Operational Program (contract no. POIG.02.01.00-12-023/08) for equipment purchase.
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed.
- J. W. Collet, T. R. Roose, E. Ruijter, B. U. W. Maes and R. V. A. Orru, Angew. Chem., Int. Ed., 2020, 59, 540–558 CrossRef CAS PubMed.
- M. Ayaz, F. De Moliner, J. Dietrich and C. Hulme, Isocyanide Chem., 2012, 335–384 CAS.
- I. Ugi, et al., Organic Chemistry, Volume 20: Isonitrile Chemistry, Academic Press, Inc., London, New York, 1971 Search PubMed.
- C. Cordovilla, S. Coco, P. Espinet and B. Donnio, J. Am. Chem. Soc., 2010, 132, 1424–1431 CrossRef CAS PubMed.
- S. Krawielitzki and W. Beck, Chem. Ber., 1997, 130, 1659–1662 CrossRef CAS.
- D. G. Rivera, F. León, O. Concepción, F. E. Morales and L. A. Wessjohann, Chem. – Eur. J., 2013, 19, 6417–6428 CrossRef CAS PubMed.
- D. G. Rivera and L. A. Wessjohann, J. Am. Chem. Soc., 2009, 131, 3721–3732 CrossRef CAS PubMed.
- D. G. Rivera and L. A. Wessjohann, J. Am. Chem. Soc., 2006, 128, 7122–7123 CrossRef CAS PubMed.
- M. G. Hill, M. C. Comstock and K. R. Mann, J. Org. Chem., 1990, 55, 4950–4951 CrossRef CAS.
- J. Gagnon, M. Drouin and P. D. Harvey, Inorg. Chem., 2001, 40, 6052–6056 CrossRef CAS PubMed.
- N. Elders, D. van der Born, L. J. D. Hendrickx, B. J. J. Timmer, A. Krause, E. Janssen, F. J. J. de Kanter, E. Ruijter and R. V. A. Orru, Angew. Chem., Int. Ed., 2009, 48, 5856–5859 CrossRef CAS.
- Y. Ito, K. Kobayashi and T. Saegusa, J. Organomet. Chem., 1986, 303, 301–308 CrossRef CAS.
- M. Toriyama, T. R. Maher, T. C. Holovics, K. Vanka, V. W. Day, C. L. Berrie, W. H. Thompson and M. V. Barybin, Inorg. Chem., 2008, 47, 3284–3291 CrossRef CAS PubMed.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- T. Zarganes-Tzitzikas, A. L. Chandgude and A. Dömling, Chem. Rec., 2015, 15, 981–996 CrossRef CAS PubMed.
- R. W. Stephany, M. J. A. de Bie and W. Drenth, Org. Magn. Reson., 1974, 6, 45–47 CrossRef CAS.
- A. M. van Leusen, B. E. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 13, 2369–2372 CrossRef.
- F. Millich, Chem. Rev., 1972, 72, 101–113 CrossRef CAS.
- J. Choy, S. Jaime-Figueroa, L. Jiang and P. Wagner, Synth. Commun., 2008, 38, 3840–3853 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2001285, 2001286 and 2002330. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc04522e |
| This journal is © The Royal Society of Chemistry 2020 |