
Kinetic and mechanistic analysis of a synthetic reversible CO2/HCO2− electrocatalyst†
Drew W.
Cunningham
 and
Jenny Y.
Yang
*
and
Jenny Y.
Yang
*
Department of Chemistry, University of California, Irvine, 92617, USA. E-mail: j.yang@uci.edu
First published on 30th September 2020
Abstract
[Pt(depe)2](PF6)2 electrocatalyzes the reversible conversion between CO2 and HCO2− with high selectivity and low overpotential but low rates. A comprehensive kinetic analysis indicates the rate determining step for CO2 reduction is the reactivity of a Pt hydride intermediate to produce HCO2−. To accelerate catalysis, the use of cationic and hydrogen-bond donor additives are explored.
Photosynthesis utilizes solar energy to upgrade CO2 to organic structural materials and chemical fuels. The prospect of replicating this feat in a synthetic system has driven interest in bio-inspired systems for carbon conversion schemes.1 The two-electron oxidation of formate (HCO2−) to CO2 is catalyzed in nature by the formate dehydrogenase (FDH) enzyme. A key feature of FDH is that it also catalyzes the reverse reaction, CO2 reduction to formate.2 Reversible catalysis is a hallmark of many redox enzymes and indicates catalysis is occurring with negligible overpotentials.3
We recently reported the reversible conversion between CO2 and HCO2− by the synthetic electrocatalyst, [Pt(depe)2](PF6)2 (1) where depe = 1,2-bis(diethylphosphino)ethane.4 Complex 1 operates with high selectivity in both directions (>95% faradaic efficiency) and <50 mV of overpotential for CO2 reduction. We previously noted the catalytic rate was too slow for reliable measurement by cyclic voltammetry (<0.5 s−1). The slow rate of catalysis precludes 1 from being a faithful functional mimic of FDH.
In this study, detailed kinetic analyses of [Pt(depe)2](PF6)2 (1) using electrochemical methods and stoichiometric reactions are described. The proposed catalytic cycle is shown in Scheme 1: Step A, 2e− reduction of 1 to Pt(depe)2 (2); (B) protonation to generate [HPt(depe)2]+ (3); and (C), CO2 reduction to formate. The rate for a possible competitive side reaction (Step B′) was also considered. We note that we recently published a kinetic analysis on the similar catalyst, [Pt(dmpe)2](PF6)2 (where dmpe = 1,2-bis(dimethylphosphino)ethane).5 Although switching from methyl to ethyl substituents on the phosphine ligand is a subtle change, it results in the free energy for hydride transfer being nearly 4 kcal mol−1 more favorable for the methyl-containing variant [HPt(dmpe)2]+. This minor modification leads to important differences in their reactivities; notably [Pt(dmpe)2](PF6)2 is not a reversible catalyst.
 | ||
| Scheme 1 Proposed catalytic cycle and corresponding rates of reactions for electrocatalytic CO2 reduction to HCO2− by [Pt(depe)2](PF6)2 (1). Conditions: 5 mM 1, 100 mM CH2(TBD)2·HPF6, 1 atm CO2. | ||
The studies on [Pt(depe)2](PF6)2 (1) reveal that Step C, the reaction of [HPt(depe)2]+ (3) with CO2 to regenerate 1 and HCO2− is likely the rate-determining step (RDS) for catalysis. Sluggish reactivity at metal hydride intermediates is common for CO2 to HCO2− electrocatalysts. There are only a few known homogeneous electrocatalysts with high selectivity for HCO2− (>90% faradaic efficiency). In addition to [Pt(depe)2](PF6)2 (1) and [Pt(dmpe)2](PF6)2,6 an Fe carbonyl cluster from Berben et al.,7 two Ir complexes from Brookhart and Meyer et al.8 and Berskoetter, Hazari, Palmore et al.,9 and a Co complex from Artero et al.10 have been reported. For all of these catalysts, the reaction of a metal hydride intermediate and CO2 to produce formate is proposed to be the rate-determining step.
Although it is evident that the reaction of a metal hydride with CO2 is a key step in formate production, there have been few experimental studies on the nature of the transition state for electrocatalysts. However, mechanistic studies on CO2 hydrogenation catalysts and the enzyme formate dehydrogenase suggest the addition of secondary sphere cations11 or hydrogen-bond donors12 could accelerate this step. We explore the use of these additives and discuss the information they provide on the transition state.
Electron transfer rate constant. Upon reduction, [Pt(depe)2](PF6)2 (1) exhibits a 2 e− reversible redox event at −1.64 V vs. Fc+/0 (where Fc = Fe(C5H5)2). The electron transfer rate constant, kET, was determined using the Butler–Volmer method,13 which is described in more detail in the ESI.† The scan rate dependent cyclic voltammetry is shown in Fig. S1 and S2 (ESI†). Using this method, the 2e−kET is 0.069(80) cm s−1.
Reactivity of [Pt(depe) 2 ] ( 2 ) with H + vs. CO 2 . Under electrocatalytic conditions, the reduced intermediate [Pt(depe)2] (2) can either react with CO2 or the proton source CH2(TBD)2·HPF6 where TBD = triazabicyclodecene (pKa = 29 in CH3CN).14 The direct reaction of CO2 with reduced metal centers typically leads to a metal carboxylate.15 The metal carboxylate can either protonate twice to generate CO and H2O or disproportionate with another equivalent of CO2 to give CO and CO32−. Conversely, direct protonation at a reduced metal center results in formation of a metal hydride as proposed in our catalytic cycle. To explore both these possibilities, we investigated the independent reactivity of electrochemically generated [Pt(depe)2] with either CO2 or H+.
Protonation of [Pt(depe) 2 ] ( 2 ). In the absence of CO2, the stoichiometric reaction of [Pt(depe)2] (2) with CH2(TBD)2·HPF6 was previously shown by 31P{1H} NMR to give [HPt(depe)2]+ (3) (Scheme 1). Thus, we would expect electrochemically-generated 2 to protonate and generate 3in situ. The cyclic voltammograms of 1 in the presence of H+ under 1 atm N2 retain the expected cathodic peak associated with reduction to 2. However, the anodic peak is only present at higher scan rates (Fig. 1). At more cathodic potentials, a feature appears at ca. −2.9 V vs. Fc+/0 (Fig. S3, ESI† blue trace), which is attributed to the subsequent reduction of [HPt(depe)2]+ (3). The reduction at −2.9 V vs. Fc+/0 is only present with the addition of acid. CVs of independently-isolated 3 have the same irreversible feature (Fig. S3, ESI† red trace), confirming generation of [HPt(depe)2]+ (3) upon reduction in the presence of CH2(TBD)2·HPF6.4
 | ||
| Fig. 1 Variable scan-rate CVs (0.025–10 V s−1) of [Pt(depe)2](PF6)2 (1) (1.06 mM) with CH2(TBD)2·HPF6 (5.00 mM) and (b) linear plot for the change in cathodic peak potential used for calculating kobs,H+. Additional data used for calculation shown in Fig. S1 and S2 (ESI†). | ||
The rate constant for protonation of the electrogenerated reduced platinum species, 2, was investigated using scan-rate dependent cyclic voltammetry (Fig. 1 and Fig. S4, S5, ESI†). As expected for an EEC reaction (2e− transfer followed by a chemical step), the cathodic peak potentials shift to more negative values at higher scan rates (Fig. 1 inset, and Fig. S4, S5, ESI†). Additionally, at slower scan rates, the return oxidation wave is almost completely diminished due to consumption of electrogenerated 2 to form 3. At faster scan rates, almost complete reversibility is achieved as re-oxidation of 2 outcompetes protonation. Analysis of the cathodic peak shift16 (see ESI†) with 10 mM of CH2(TBD)2·H+ results in an observed rate constant for protonation, or kobs of 74(11) s−1.
The dependence of acid concentration on observed rate confirmed that protonation is first order in acid (Fig. S6, ESI†). The observed rate constants for protonation of 2 are 36(4) and 54(10) s−1 for 5.0 and 7.5 mM CH2(TBD)2·H+, respectively, and the 2nd order rate constant is 8.2(7) × 103 M−1 s−1. Thus, the kobs under catalytic conditions with 100 mM CH2(TBD)2·H+ is 820(7) s−1, indicating protonation of 2 is relatively facile.
Reactivity of [Pt(depe) 2 ] ( 2 ) and CO 2 . Reduction of [Pt(depe)2](PF6)2 (1) under 1 atm CO2 and aprotic conditions leads to the loss of reversibility of the Pt(II/0) couple, suggesting the electrogenerated [Pt(depe)2] (2) can also react with CO2. We observed similar reactivity with the related compound [Pt(dmpe)2]. The loss of reversibility is scan-rate dependent (Fig. S7–S9, ESI†); at faster scan rates the current associated with re-oxidation of 2 to 1 increases. The electrochemical peak current analysis used to derive the rate of protonation (vide supra) could not be used for this reaction because the cathodic shifts are too small (indicating a slow rate). An alternative method was used to determine the rate. Analysis of the ratio between the anodic return current vs. the initial cathodic current (ipa/ipc)17 at different scan rates provides a half-life for the reaction of 26 s−1, which corresponds to a pseudo first-order CO2 binding rate constant, kCO2, of 0.027 s−1. The data indicate that while the reaction between 2 and CO2 is favorable in the absence of protons, the rate constant is 105 slower than the rate at which 2 is protonated under catalytic conditions. Thus the direct reactivity of CO2 with 2 is negligible during catalysis.
Hydride transfer step. [HPt(depe)2]+ (3) was independently synthesized and isolated. We previously demonstrated that hydride transfer from 3 to CO2 proceeds cleanly without any side products or reactions. The generation of HCO2− was quantified using 1H NMR spectroscopy. The quantity of HCO2− at each time point was subtracted from the initial concentration of 3 to deduce the concentration of 3 at each time point (Fig. 2). The rate constant, kobs, was calculated to be 2.78(17) × 10−4 s−1 using initial rates (see ESI†). Initial rates were used because the reaction has a Keq of 1.05(7) and approaches equilibrium over time.4
 | ||
| Fig. 2 For the reaction of [HPt(depe)2]+ (3) (21.9 mM) with CO2 (1.0 atm) (top left) concentrations of 3 over time at various temperatures, (top right) corresponding Eyring plot, and (bottom) activation parameters. | ||
To determine more information about the nature of the transition state, the activation parameters were calculated by measuring the rates at variable temperatures (Fig. 2). The observed rate constants for the reactions at 5 and −13 °C are 6.52(21) × 10−5 s−1 and 1.23(4) × 10−5 s−1, respectively. As expected, the rates are significantly slower than the reactions at 25 °C. From the Eyring plot (Fig. 2b) the activation parameters are: ΔH‡ = 12.2(4) kcal mol−1, ΔS‡ = −34(1) cal mol−1 K, and 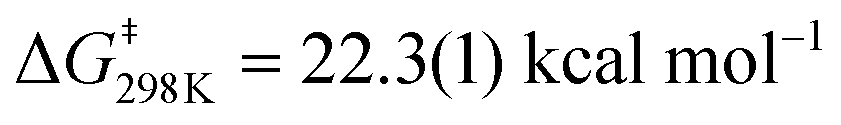 . As anticipated, the large
. As anticipated, the large  term suggests sluggish kinetics at room temperature. The entropic term suggests a large degree of order in the transition state and is consistent with a bimolecular reaction between two species and is in agreement with the few reported activation parameters for hydride transfer to CO2.11
term suggests sluggish kinetics at room temperature. The entropic term suggests a large degree of order in the transition state and is consistent with a bimolecular reaction between two species and is in agreement with the few reported activation parameters for hydride transfer to CO2.11
Rate determining step. Understanding the heterogenous electron transfer rate constant with respect to chemical steps is not inherently intuitive. However, we can compare overall electrocatalytic rates and proposed RDS of catalysts that have slower rates of electron transfer than 1. [Fe4N(CO)12]− is an electrocatalyst for CO2 reduction to HCO2− and operates with a fast turnover frequency (TOF) of 210 s−1.7d The RDS for this catalyst is hydride transfer to CO2, not electron transfer. A Co diphosphine H2 evolution catalyst that operates with a TOF of 160 s−1 has a kET = 0.003 cm s−1.18 For this particular catalyst, protonation of the reduced Co(I) species to generate the metal hydride is proposed to be rate-limiting.18b The measured kET values in these examples are an order of magnitude slower than 1 and have TOFs in the 100s. The rates of our last proposed chemical step, C, has a kobs of 2.78(17) × 10−4 s−1. From these comparisons, we conclude that the 2 e− reduction of 1 to generate 2 is not rate-limiting. Step C is also significantly slower than our measured rate for step B (protonation to generate the Pt hydride), and is therefore likely the rate-determining step. The slow rate measured for step C is consistent with the small catalytic current we observe for CO2 reduction to HCO2−.4
Accelerating proposed RDS. To date, intimate mechanistic details for the insertion of CO2 into metal hydrides bonds are lacking and mostly based on computational studies.19 Two of the most commonly proposed mechanisms for CO2 insertion are shown in Scheme 2a and b.11 In the (a) outer-sphere mechanism, a hydride is delivered directly to CO2 with concomitant cleavage of the M–H and C–H bond formation. In the (b) inner-sphere mechanism, M–H and CO2 join in a side-on fashion giving rising to a metallocyclic transition state. Both mechanisms have a large negative entropy of activation, so they are not distinguishable by our Eyring parameters. Hazari and colleagues studied the kinetics of CO2 insertion into various metal hydrides.11,20 They found the rate of the reaction between CO2 and an Ir pincer trihydride complex and [HRu(tpy)(bpy)](PF6) can be accelerated by up to a factor of ×103 upon addition of salts, particularly Lewis acidic (LA) cations.11 In these cases, it is believed hydride transfer follows an outer-sphere mechanism, and the acceleration in rate is due to stabilization of the negative charge on the carboxylate moiety in the transition state. In the case of a Ni pincer hydride complex, no enhancement was observed, leading the authors to conclude the reaction likely follows an inner-sphere mechanism.
 | ||
| Scheme 2 (a and b) Common mechanisms for CO2 Insertion into metal hydrides described in ref. 11, (c) proposed mechanism for hydride transfer in formate dehydrogenase described in ref. 12. | ||
In the most recent proposed mechanism of the enzyme formate dehydrogenase, hydride transfer resembles an outer-sphere approach (Scheme 2c).12 In metal-dependent formate dehydrogenation enzymes, a conserved arginine residue near the active site is critical for catalytic activity. With a pKa of 12.5, arginine is expected to be protonated under biological conditions. In this mechanism, a cationic protonated arginine in the secondary coordination sphere is proposed to stabilize the carboxylate species through hydrogen-bonding and/or electrostatic interactions.
In an attempt to accelerate the rate of CO2 insertion into 3, lithium bis(trifluoromethane)sulfonimide (LiNTf2) was added to the reaction. We selected LiNTf2 because of its relatively high solubility in acetonitrile and because it exhibited the highest rate acceleration for the previously reported iridium pincer trihydride complex.11b UV-Visible spectroscopy (UV-Vis) was used to quantify the rate instead of 1H NMR measurements in the presence of LiNTf2 due to precipitation of lithium formate at the mM concentrations required for NMR (Fig. S10, ESI†). Using the method of initial rates (data for t < 20 min) and a lower concentration of 1 (0.100 mM) the rate was 6.50(7) × 10−5 s−1 without LiNTf2 and 7.23(6) × 10−5 s−1 with 10 mM LiNTf2. Thus, the addition of LiNTf2 does not significantly increase the rate of CO2 reactivity with 3.
To mimic the role of a protonated arginine, guanidinium tetraphenylborate was also tested as an additive to accelerate reactivity. Guanidinium (pKa of 23.3 in CH3CN) is a stronger proton donor than the CH2(TBD)2·HPF6 we normally use for catalysis.21 Fig. S12 (ESI†) shows the CVs with addition of guanidinium·BPh4. Upon addition of guanidinium·BPh4, the anodic (return) wave diminishes after reduction, consistent with protonation of [Pt(depe)2] (2) to form [HPt(depe)2]+ (3). The addition of CO2 leads to a reduction in cathodic current, which is recovered with addition of excess guanidinium·BPh4. However, the peak current does not increase with the addition of guanidinium·BPh4.
As neither LiNTf2 nor guanidinium·BPh4 increase the rate of the proposed RDS or catalysis, we conclude that Step C likely proceed through an inner-sphere mechanism (Scheme 2b). An inner-sphere mechanism was previously attributed to a Ni pincer hydride complex as various additives did not significantly affect the rate of CO2 insertion.11a This mechanism may consist of several elementary steps to produce HCO2−, although we expect any potential intermediates are likely short-lived. We have observed no catalyst-based intermediates from 3 to 2.
Currently, all of the evidence for reported CO2 reduction to HCO2− electrocatalysts indicate the rate limiting step is the reaction of a metal hydride with CO2. Understanding the different possible mechanisms, the metal hydride properties that make each mechanism favorable, and how each mechanism can be accelerated through synthetic design or reaction conditions would make a profound impact on developing new catalysts. As our understanding of metal hydride reactivity progresses, we can aspire to new synthetic catalysts that replicate the selectivity, low overpotential, and speed of enzymes.
This study was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences Award DE-SC0012150 and DE-0000243266. J. Y. Y. acknowledges support as a Sloan Foundation Fellow, Canadian Institute for Advanced Research (CIFAR) Azrieli Global Scholar in the Bio-Inspired Solar Energy Program, and a Camille Dreyfus Teacher-Scholar.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) H. B. Gray, Nat. Chem., 2009, 1, 7 Search PubMed; (b) N. S. Lewis, Science, 2007, 315, 798–801 Search PubMed; (c) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 Search PubMed.
- T. Reda, C. M. Plugge, N. J. Abram and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10654–10658 Search PubMed.
- (a) J.-M. Savéant, ACS Catal., 2018, 8, 7608–7611 Search PubMed; (b) F. A. Armstrong and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14049–14054 Search PubMed.
- D. W. Cunningham, J. M. Barlow, R. S. Velazquez and J. Y. Yang, Angew. Chem., Int. Ed., 2020, 59, 4443–4447 Search PubMed.
- B. M. Ceballos and J. Y. Yang, Organometallics, 2020, 39, 1491–1496 Search PubMed.
- B. M. Ceballos and J. Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12686–12691 Search PubMed.
- (a) A. Taheri, E. J. Thompson, J. C. Fettinger and L. A. Berben, ACS Catal., 2015, 5, 7140–7151 Search PubMed; (b) N. D. Loewen, T. V. Neelakantan and L. A. Berben, Acc. Chem. Res., 2017, 50, 2362–2370 Search PubMed; (c) A. Taheri and L. A. Berben, Inorg. Chem., 2016, 55, 378–385 Search PubMed; (d) A. Taheri, C. R. Carr and L. A. Berben, ACS Catal., 2018, 8, 5787–5793 Search PubMed.
- (a) P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 Search PubMed; (b) P. Kang, T. J. Meyer and M. Brookhart, Chem. Sci., 2013, 4, 3497–3502 Search PubMed.
- S. T. Ahn, E. A. Bielinski, E. M. Lane, Y. Chen, W. H. Bernskoetter, N. Hazari and G. T. R. Palmore, Chem. Commun., 2015, 51, 5947–5950 Search PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 Search PubMed.
- (a) J. E. Heimann, W. H. Bernskoetter and N. Hazari, and James M. Mayer, Chem. Sci., 2018, 9, 6629–6638 Search PubMed; (b) J. E. Heimann, W. H. Bernskoetter and N. Hazari, J. Am. Chem. Soc., 2019, 141, 10520–10529 Search PubMed.
- L. B. Maia, L. Fonseca, I. Moura and J. J. G. Moura, J. Am. Chem. Soc., 2016, 138, 8834–8846 Search PubMed.
- J. M. Savéant, Investigation of Rates and Mechanisms of Reactions, Wiley, 1986, ch. 7 Search PubMed.
- I. Kaljurand, J. Saame, T. Rodima, I. Koppel, I. A. Koppel, J. F. Kögel, J. Sundermeyer, U. Köhn, M. P. Coles and I. Leito, J. Phys. Chem. A, 2016, 120, 2591–2604 Search PubMed.
- J. M. Barlow and J. Y. Yang, ACS Cent. Sci., 2019, 5, 580–588 Search PubMed.
- J. M. Savéant, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, Wiley, 2006 Search PubMed.
- (a) J. Y. Yang, R. M. Bullock, W. J. Shaw, B. Twamley, K. Fraze, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2009, 131, 5935–5945 Search PubMed; (b) J. M. Saveant, Investigation of Rates and Mechanisms of Reactions, Wiley, 1986 Search PubMed.
- (a) E. S. Wiedner, J. Y. Yang, W. G. Dougherty, W. S. Kassel, R. M. Bullock, M. R. DuBois and D. L. DuBois, Organometallics, 2010, 29, 5390–5401 Search PubMed; (b) E. S. Wiedner and R. M. Bullock, J. Am. Chem. Soc., 2016, 138, 8309–8318 Search PubMed.
- (a) W. H. Bernskoetter and N. Hazari, Eur. J. Inorg. Chem., 2013, 4032–4041 Search PubMed; (b) F. Huang, C. Zhang, J. Jiang, Z.-X. Wang and H. Guan, Inorg. Chem., 2011, 50, 3816–3825 Search PubMed; (c) Q.-Q. Ma, T. Liu, A. Adhikary, J. Zhang, J. A. Krause and H. Guan, Organometallics, 2016, 35, 4077–4082 Search PubMed; (d) I. Osadchuk, T. Tamm and M. S. G. Ahlquist, Organometallics, 2015, 34, 4932–4940 Search PubMed; (e) P. Ríos, A. Rodríguez and J. López-Serrano, ACS Catal., 2016, 6, 5715–5723 Search PubMed; (f) T. J. Schmeier, G. E. Dobereiner, R. H. Crabtree and N. Hazari, J. Am. Chem. Soc., 2011, 133, 9274–9277 Search PubMed; (g) T. J. Schmeier, N. Hazari, C. D. Incarvito and J. A. Raskatov, Chem. Commun., 2011, 47, 1824–1826 Search PubMed; (h) H.-W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 Search PubMed.
- N. Hazari and J. E. Heimann, Inorg. Chem., 2017, 56, 13655–13678 Search PubMed.
- Z. Glasovac, M. Eckert-Maksić and Z. B. Maksić, New J. Chem., 2009, 33, 588–597 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc05556e |
| This journal is © The Royal Society of Chemistry 2020 |