
In situ synthesis of crystalline Ag–WO3 nanosheets with enhanced solar photo-electrochemical performance for splitting water†
Puhong
Wen
 *,
Chuanchuan
Wang
,
Yuzhu
Lan
,
Xiaowen
Jiang
and
Lijun
Ren
*,
Chuanchuan
Wang
,
Yuzhu
Lan
,
Xiaowen
Jiang
and
Lijun
Ren
Faculty of Chemistry and Chemical Engineering, Baoji University of Arts and Sciences, 1 Hi-Tech Avenue, Baoji, Shaanxi 721013, PR China. E-mail: wenpuhong@hotmail.com
First published on 2nd December 2019
Abstract
Monoclinic WO3 nanosheets preferentially exposing the (100) facet were synthesized successfully by a hydrothermal topological reaction using orthorhombic H2WO4 disk particles as the precursor. The evolution mechanism of the morphology and crystal structure transformed into WO3 nanosheets from H2WO4 disk particles has been presented in detail. In this process, the H2WO4 disk crystals were firstly split along the direction perpendicular to their b-axis and transformed into thin WO3 nanosheets through an in situ topological dehydration reaction. Then, the thin nanosheets were dissolved further into smaller WO3 nanosheets via a dissolution reaction. After the photoreduction reaction between the WO3 nanosheets and AgNO3 solution, the mainly exposed (100) facet of WO3 nanosheets was homogeneously covered by Ag nanoparticles. The photo-electrochemical performance of the obtained WO3 nanosheets and Ag deposited WO3 (Ag–WO3) nanosheets as photocatalysts was evaluated by splitting water using WO3 monoclinic spherical particles (C-WO3) as a contrast under simulated sunlight. The results show that the photocurrent density generated by the prepared WO3 nanosheets is seven times that of C-WO3, indicating that the (100) facet of monoclinic WO3 is one of the most photocatalytically active surfaces. In addition, the photocurrent density generated by the prepared Ag–WO3 sample is 30 μA cm−2, which is ten times that of the WO3 sample. The results indicate that the solar light utilization efficiency of the WO3 sample has been significantly improved after silver deposition on the surface, which implies that it has potential applications in photocatalysis.
1. Introduction
The production of hydrogen and oxygen by photocatalytic decomposition of water is one of the most promising but challenging technologies for solving environmental problems and satisfying the demand of energy, as it is of great practical value in converting solar energy into chemical and electric energy.1–5 Tungsten trioxide (WO3) is an important n-type semiconductor functional material with orthogonal, monoclinic, cubic, hexagonal and other symmetrical crystal structures, which has received much attention in the past few decades due to its intriguing physiochemical properties and widespread potential applications. Currently, WO3 materials have been used in a wide range of fields, such as in electrochromic materials,6 lithium ion batteries,7 photocatalysts,8–10 gas sensors,11,12 heterogeneous catalysts,13 solar devices,14,15 field electron emission devices,16 superconductors,17 and pyroelectric and ferroelectric materials.18In the photocatalytic reaction, photocatalytic activity depends on the crystallinity, crystal size and surface structure of the photocatalyst.19–25 Furthermore, the crystal structure of the surface has a great influence on its photocatalytic activity, because the photocatalytic reaction occurs on the surface of the photocatalyst.26 However, the effect of the surface crystal structure of WO3 on its photocatalytic activity has rarely been reported, because traditional synthesis routes are difficult to use to control the crystal facet on the surface of particles.
In recent years, we have developed a novel hydrothermal topological synthesis process for the preparation of TiO2 nanocrystals to control the crystal facet on the particle surface.27 Anatase TiO2 nanocrystals dominantly exposing the (010) facet with high photocatalytic activity have been successfully prepared using titanate nanosheets.28,29 Then, orthorhombic Nb2O5 rectangular nanocrystals mainly exposing the (010) facet have been also prepared using K4Nb6O17·4.5H2O nanoparticles as precursors.30 In this study, we firstly successfully obtained pure single crystal monoclinic WO3 nanosheets dominantly exposing the (100) facet using H2WO4 disk particles as precursors by an in situ hydrothermal topological reaction. The evolution mechanism of the morphology and crystal structure transformed into WO3 nanosheets from H2WO4 disk particles is presented in detail. The photocatalytic activity of nanomaterials can be improved by doping metal or non-metal impurities.31,32 Thus, crystalline Ag deposited WO3 (Ag–WO3) nanosheets were prepared by the in-situ photocatalytic reduced reaction between the prepared WO3 nanosheets and AgNO3 aqueous solution. The photo-electrochemical performance of the obtained Ag–WO3 nanosheets is evaluated by decomposing water to H2 under simulated sunlight. We find that the prepared Ag–WO3 nanosheets have high solar photocatalytic activity and high photoelectric conversion efficiency, which indicates that they have a potential application value in the preparation of photocatalysts and clean energies.
2. Experimental section
2.1. Preparation of the H2WO4 disk particles
H2WO4 disk particles were prepared by a simple hydrothermal route. There are two steps in this process. First, 12 mmol of Na2WO4·2H2O was dissolved in 5 mL of distilled water and then an aqueous solution of 16 mL of 3 mol L−1 HCl was slowly added in a dropwise manner under stirring. The product collected by filtration was washed with distilled water and ethanol several times and dried at room temperature to obtain an amorphous tungstic acid sample. Secondly, 0.2 g of the amorphous sample and 10 mL of distilled water were placed in a Teflon-lined, sealed stainless-steel vessel with an inner volume of 50 mL. After stirring, the pH of the solution was adjusted to about 4.8 with n-propylamine. Then, the solution was hydrothermally treated at 60 °C for 8 h under stirring. After that, the filtered product is washed with distilled water and dried at room temperature to obtain crystal H2WO4 disk particles.2.2. Preparation of the WO3 nanosheets
When WO3 nanosheets were prepared, 30 mL of 4 mg mL−1 H2WO4 disk particle suspension was placed in a Teflon-lined stainless-steel, sealed vessel with an inner volume of 50 mL. The pH value of the suspension was adjusted to 1.5–5.0 with an aqueous solution of 3 mol L−1 HCl, and then hydrothermally treated at 140 °C for 6 h under stirring. The obtained product was centrifuged and washed, and finally dried at room temperature.2.3. Preparation of the Ag–WO3 nanosheets
The crystalline Ag–WO3 nanosheets were fabricated by a photoreduction process. 20 mg of the obtained WO3 nanosheet sample was transferred into a 0.0115 mol L−1 AgNO3 solution, and stirred for 10 min under light emitting diode (LED, China, 30 W, 6000 K) light illumination. Then, the product was centrifuged and washed with distilled water, and then dried at room temperature.2.4. Physical analysis
The crystal structure, size and morphology of the sample particles were investigated by using a powder X-ray diffractometer (XRD, Rigaku D/max-2200PC) with Cu Kα (λ = 0.15418 nm) radiation, a field-emission scanning electron microscope (FE-SEM, Hitachi SU8010) with an accelerating voltage of 5 kV, and a transmission electron microscope (TEM, Tecnai G2F20STWIN). TEM and selected-area electron diffraction (SAED) were performed using a system at 200 kV. The Raman spectra were collected using a Renishaw in via Raman microscope, from a laser operating at 532 nm, 50 times objectives. The laser power at the sample was estimated to be 1% and the nominal laser spot size was 10 μm. The solid-state diffuse reflectance spectra of the samples are measured on a UV-vis spectrophotometer (Varian Cary 100) equipped with an integrating sphere attachment, and a standard BaSO4 plate is used as 100% reflectance standard. Chemical composition analysis was performed with in situ energy dispersive X-ray spectroscopy (EDX) and mapping images using an instrument equipped in the FE-SEM system at an accelerating voltage of 20 kV. The chemical composition of the samples is also analyzed by in situ energy dispersive X-ray photoelectron spectroscopy (XPS). XPS analysis was carried out using a Nexsa multifunctional X-ray photoelectron spectroscopy system, by using Al-Kα radiation as the excitation source. The binding energies of the target elements were determined at a pass energy of 29.35 eV and with a resolution of ±0.3 eV, by using the binding energy of carbon (C 1 s: 284.8 eV) as the reference.2.5. Measurement of photoelectric performance for splitting water
Photoelectrochemical measurement of the samples is carried out on a Zahner XPOT electrochemical analyzer in a three-electrode system consisting of fluorine-doped tin oxide (FTO) glass covered with the sample as the working electrode, a platinum sheet as the counter electrode, and an Hg/Hg2Cl2 electrode (KCl saturated) as the reference electrode. The electrodes are immersed in 1 mol L−1 Na2SO4 solution in a quartz cell. When preparing the working electrode, 5 mg of the sample was dispersed in 1 mL of deionized water to form a suspension of the sample, then 0.25 mL of the suspension was dropped onto clean FTO glass, and then dried under ambient conditions to obtain the working electrode of the sample. The area of the sample on the electrode was ca. 1 cm2. Commercial monoclinic WO3 (C-WO3, Tianjin Lanhua Tungsten Molybdenum Chemical Co, Ltd.) spherical particles with a diameter of about 200 nm were used as a contrast for comparison. C-Ag–WO3 was prepared by a preparation method similar to Ag–WO3. The method for preparing the C-WO3 and C-Ag–WO3 electrodes was the same as the method of preparing the sample WO3 electrode except that the suspension concentration of the sample was 10 mg mL−1. The working electrode was irradiated with a simulated sunlight source, which is a light emitting diode flat downlight (LED, Wolike, China, 30 W, 380–780 nm) during the measurement. The distance between the working electrode and the light source was kept at 10 cm.3. Results and discussion
The XRD pattern of the amorphous tungstic acid sample prepared by the reaction between Na2WO4·2H2O and HCl is shown in Fig. 1a. It can be observed in Fig. 1a that no diffraction peak appears, indicating that the sample is amorphous. The XRD pattern of the sample obtained from the hydrothermally treated amorphous tungstic acid is shown in Fig. 1b. All of the diffraction peaks are exactly consistent with those of the orthorhombic H2WO4 phase (JCPDS No. 43-0679, Pmnb (62), Z = 4, a = 5.238 Å, b = 10.704 Å, c = 5.12 Å, α = β = γ = 90°), where the diffraction peaks of the (020), (111), and (002) facets appear at 16.5°, 25.7°, and 35.0°, respectively. In addition, there are some weak diffraction peaks, which correspond to (040), (200), (042), etc. The result indicates that the obtained sample is a pure orthorhombic H2WO4 phase. The crystal structure of orthorhombic H2WO4 is given in Fig. 2. It can be seen that the crystal structure of H2WO4 is composed of the basic structure unit of a [WO5–H2O] octahedron, where W is located in the center of an octahedron and connected to six oxygen atoms by a W–O covalent bond. The six oxygen atoms at the six corners of the octahedron can be divided into three types, the oxygen atom in H2O (O1) and the other oxygen atom (O2) form two vertices of the octahedron, and the rest of the four oxygen atoms (O3) are located at four corners of the same plane and form a rectangle perpendicular to the [010] direction. The octahedron with the upper vertex as the O1 atom is connected in a shared corner mode by four O3 atoms to four adjacent octahedra with the upper vertex as the O2 atom, respectively, and forms a two-dimensional sheet structure parallel to the (010) facet. The sheet is interlaced with each other by a hydrogen bond (O1–H⋯O2) and stacked in the direction of [010] to form a three-dimensional network structure.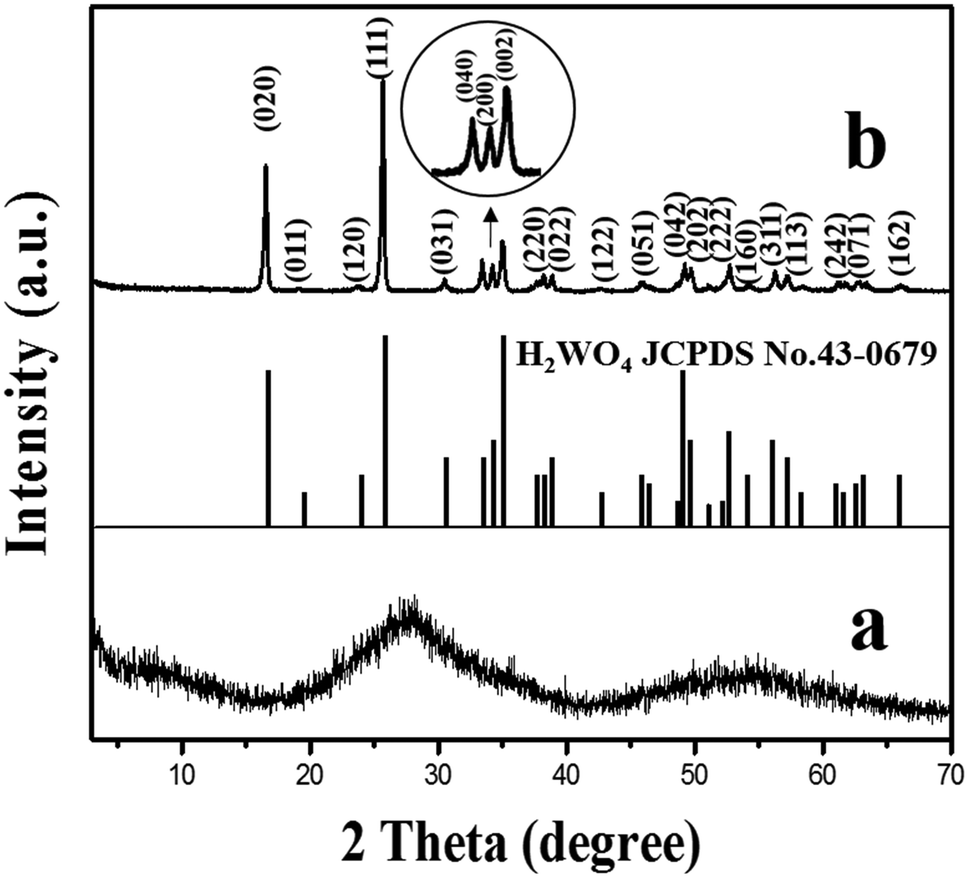 | ||
| Fig. 1 XRD patterns of the obtained samples of amorphous tungstic acid (a) and H2WO4 (b). | ||
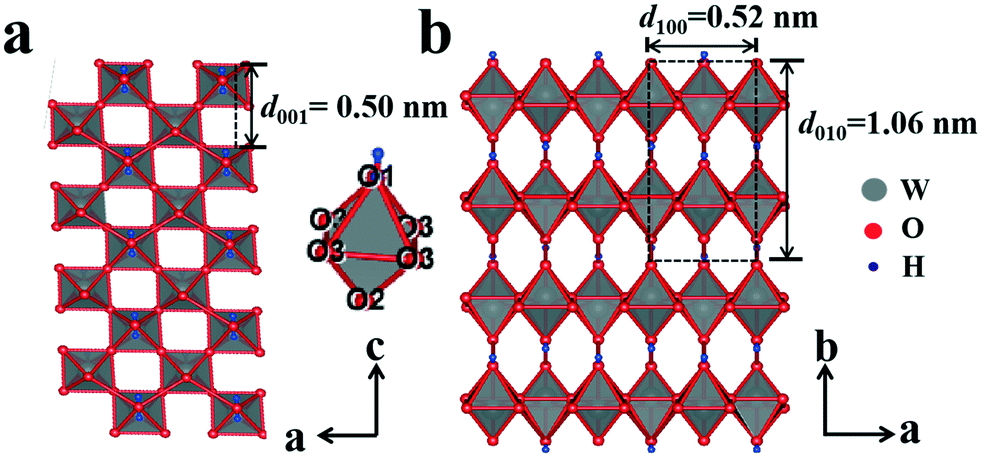 | ||
| Fig. 2 A view of the orthorhombic crystal structure of H2WO4 projected on (010) (a) and (001) (b). | ||
In order to investigate the effect of the reaction temperature on transforming the WO3 nanosheets, the H2WO4 suspension with a pH value of 2.0 was hydrothermally treated for 6 h at 110, 120, 130, 135, 140 and 150 °C, respectively. Fig. 3 shows the XRD patterns of the samples obtained under the above conditions. The XRD pattern of the sample obtained at 110 °C is completely consistent with that of orthorhombic H2WO4, indicating that there is no transformation into WO3 (Fig. 3a). A weak peak of the monoclinic phase WO3 can be observed in the sample obtained at 120 °C (Fig. 3b). The peak intensity of WO3 continuously increases and the peak intensity of H2WO4 ceaselessly decreases with the increase of the hydrothermal temperature (Fig. 3c and d). As the temperature reaches 140 °C, the H2WO4 peak disappears completely and all the peaks can be easily indexed to the monoclinic phase WO3 (JCPDS No. 83-0950, P21/n (14), Z = 8, a = 7.3008 Å, b = 7.5389 Å, c = 7.6896 Å, γ = 90.892°), indicating that H2WO4 has been completely transformed into monoclinic WO3 (Fig. 3e). The XRD pattern of the sample obtained at 150 °C is similar to that at 140 °C, but the intensity of the peak increases, indicating that the WO3 particles with higher crystallization are formed (Fig. 3f).
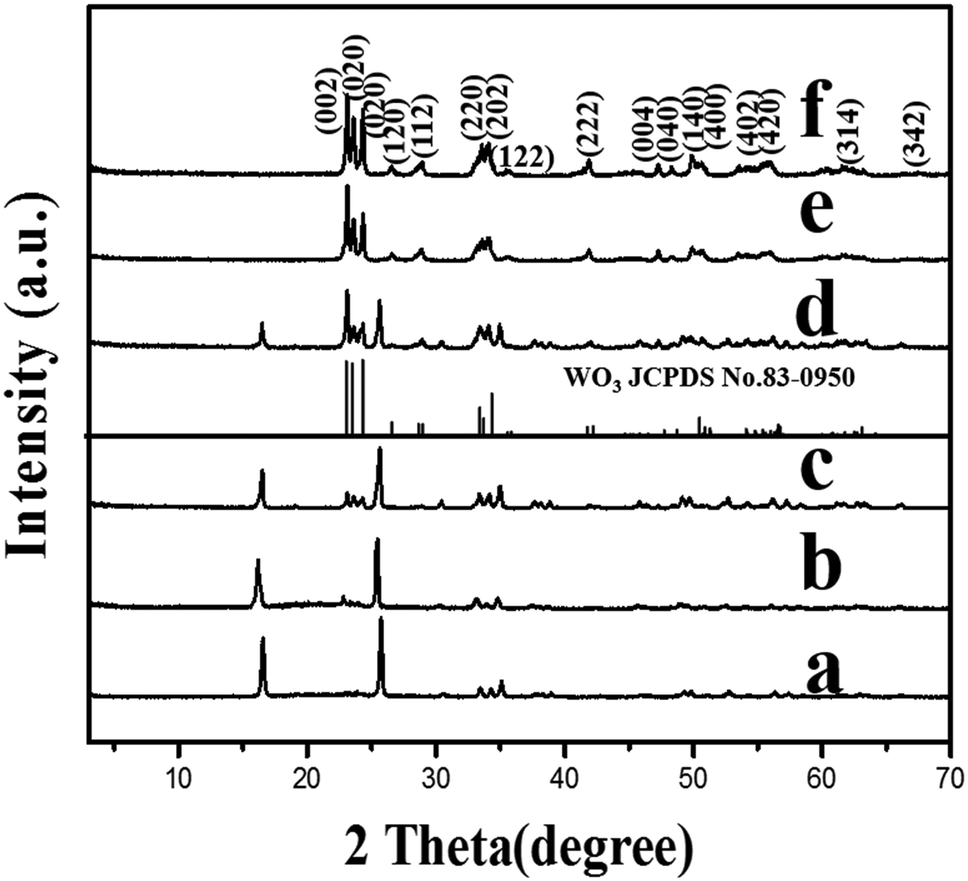 | ||
| Fig. 3 XRD patterns of the samples obtained by hydrothermally treating the H2WO4 suspension with a pH value of 2.0 at different temperatures for 6 h: (a) 110 °C, (b) 120 °C, (c) 130 °C, (d) 135 °C, (e) 140 °C, and (f) 150 °C. | ||
In order to investigate the effect of the reaction time on the transformation of the WO3 nanosheets, the H2WO4 suspension with a pH value of 2.0 was hydrothermally treated at 140 °C for different times, respectively. A weak peak of the monoclinic phase WO3 can be observed in the sample obtained for 3 h (Fig. 4a). The intensities of the peaks for WO3 increase continuously and those for H2WO4 decrease while the reaction time increases (Fig. 4b and c). The peak of H2WO4 completely disappears after the reaction for 6 h, indicating that the H2WO4 phase is completely transformed into the monoclinic phase WO3 (Fig. 4d). The result indicates that the pure monoclinic phase WO3 crystals can be obtained by the hydrothermal treatment of the H2WO4 suspension at 140 °C for 6 h. The XRD pattern of the sample obtained by prolonging the reaction time to 7 h (Fig. 4e) is basically in concordance with that in Fig. 4d, but the intensities of the peaks are higher, indicating that the crystallinity of the obtained WO3 particles can be increased by extending the reaction time.
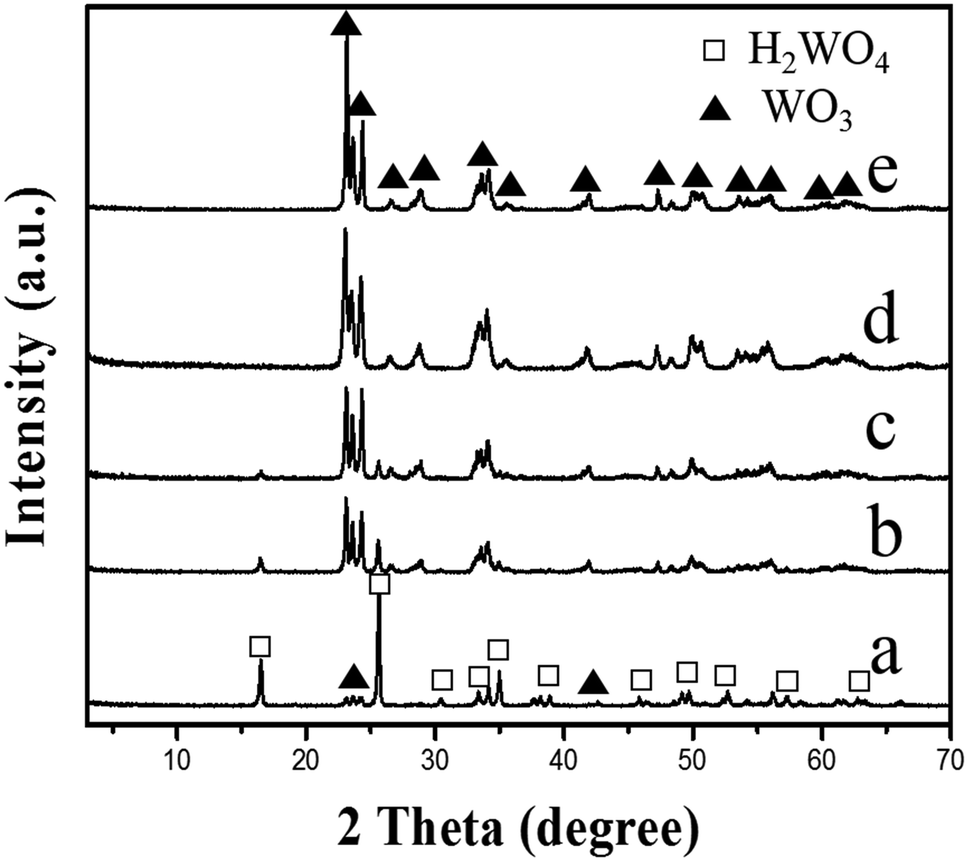 | ||
| Fig. 4 XRD patterns of the samples obtained by hydrothermally treating the H2WO4 suspension with a pH value of 2.0 at different times at 140 °C: (a) 3 h, (b) 4 h, (c) 5 h, (d) 6 h, and (e) 7 h. | ||
The FE-SEM and TEM images and the SAED patterns of the obtained H2WO4 and WO3 samples are shown in Fig. 5. It can be observed that the prepared H2WO4 and WO3 samples have a similar morphology, and both samples display a circular sheet shape, a length of about 1.7 μm, and a width of about 1.5 μm (Fig. 5a and g), but the thickness of the WO3 sample is about 30 nm and is much thinner than that of the H2WO4 sample (about 300 nm) (Fig. 5e and h). In addition, the other difference is that the larger disc WO3 nanosheets are split into smaller rectangle sheets (Fig. 5c and d). The FE-SEM image of the sample with mixed phases of WO3 and H2WO4 shows that WO3 nanosheets split from outside to inside (Fig. 5f). According to the analysis of the SAED pattern in Fig. 5b, the longer axis of H2WO4 disk particles corresponds to the [001] direction and the shorter axis corresponds to the [100] direction, which indicates that the disk particles mainly exposed the (010) facet of the orthorhombic H2WO4 phase.
 | ||
| Fig. 5 TEM images (a–d) and FE-SEM images (e–h) of the obtained samples H2WO4 (a, b and e) and WO3 (c, d, g and h), and the sample obtained by hydrothermally treating the H2WO4 suspension with a pH value of 2.0 at 135 °C for 6 h (f). The inset is the SAED pattern of the corresponding sample. | ||
Fig. 6 shows the HRTEM images and SAED patterns of the obtained H2WO4 and WO3 samples to confirm their detailed structure. The clear lattice fringes can be observed in Fig. 6a. The lattice spacings with d-values of 0.25, 0.26 and 0.36 nm correspond to the (002), (200) and (101) facets of the orthorhombic phase H2WO4 disk crystals, respectively. In addition, the diffraction spots in the SAED pattern of the H2WO4 sample agrees with the above conclusions (Fig. 6c). The results show that the dominant exposed surface of the H2WO4 crystal is the (010) facet. The lattice fringe spacings of d = 0.37 and 0.38 nm correspond to the (020) and (002) facets of the monoclinic phase WO3 crystal, respectively (Fig. 6b). The result is the same as that of the diffraction spots in the SAED pattern (Fig. 6d). These results demonstrate that the exposed facet of the crystalline WO3 nanosheets is mainly the (100) facet, and the [001] direction is the direction of the long axis.
 | ||
| Fig. 6 HRTEM images (a and b) and SAED patterns (c and d) of the obtained samples H2WO4 (a and c) and WO3 (b and d). | ||
The Raman spectra of the obtained H2WO4 and WO3 samples are shown in Fig. S1.† For the monoclinic WO3 samples, the Raman bands at 807.2 and 712.0 cm−1 are attributed to stretching vibration of the tungsten atom with neighboring oxygen atoms [ν (O–W–O)], and two obvious peaks at 328.8 and 275.5 cm−1 are identified as [σ (O–W–O)] bending vibrations.33 The sharp and symmetrical Raman peaks disclose that WO3 nanostructures possess high crystal quality and are chemically pure. Compared with the Raman spectrum of the WO3 sample, it is found that the two strong Raman bands at 807.2 and 712.0 cm−1, which appear for WO3, are absent in the Raman spectrum of H2WO4, while two strong peaks at 945.8 and 639.4 cm−1 are present, which correspond to stretching modes of terminal W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and bridging O–W–O bonds, respectively.34,35
O and bridging O–W–O bonds, respectively.34,35
Based on the above results, we propose a formation mechanism of the WO3 nanosheets from the H2WO4 disk particles, as shown in Fig. 7. The formation mechanism of the WO3 nanosheets mainly includes two processes, topological structure transformation and dissolution splitting. Firstly, the H2WO4 disk particles are topologically transformed into WO3 nanosheets by a hydrothermal process. In the topological reaction, the two-dimensional layer formed by an octahedral common angle on the (010) facet of the orthogonal phase H2WO4 is separated with a water molecule, that is, the loss of hydrogen bonds and O1, and is connected by sharing O2 with the staggered adjacent layers, so that the layers are neatly superimposed and transformed into the structure of the monoclinic phase WO3 crystal. In the crystal structure of the monoclinic phase WO3, the octahedral structure unit is [WO6], and each octahedron is connected to each other in a co-angular manner. As a result of the topological dehydration reaction, each octahedron rotates horizontally about 17° clockwise on the (010) facet of orthorhombic H2WO4, forming the (100) facet of the monoclinic phase WO3, which indicates that the (010) facet of the orthorhombic H2WO4 crystal is transformed into the (100) facet of the monoclinic WO3 crystal, and the b axis direction of the monoclinic phase WO3 crystal is 45° different from the c axis of the orthorhombic phase H2WO4 crystal. At the same time, the disk particles are split into thin circular nanosheets along the direction perpendicular to the b axis of the H2WO4 crystal. Secondly, due to the dissolution reaction, the WO3 circular sheet crystal dissolves and splits along its b axis and c axis directions, forming a rectangle first, and then the rectangle dissolves again, becoming a variety of flake WO3 nanocrystals.
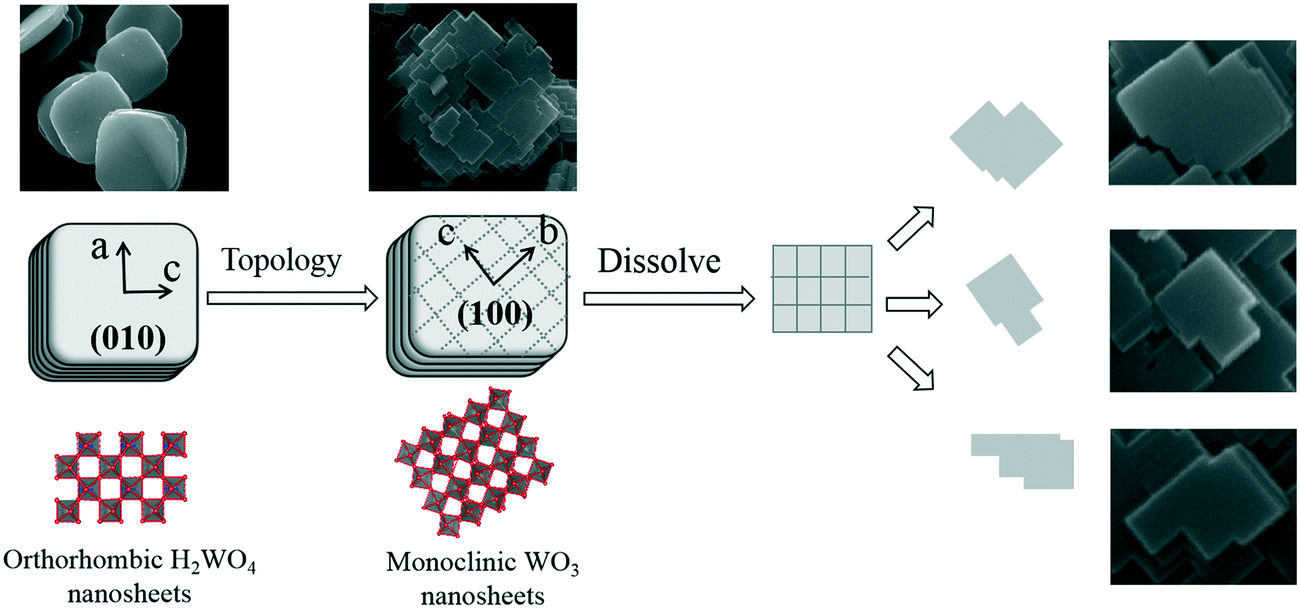 | ||
| Fig. 7 Crystal structure evolution from the H2WO4 disk crystal into a WO3 nanosheet crystal. | ||
Since the deposition of Ag on the surface of two-dimensional nanomaterials introduces mid-gap energy levels, the nanomaterials enhance visible light spectrum properties and improve visible light photocatalytic performance.36 For example, after depositing Ag on the (001) facet of anatase TiO2 nanosheets, its visible light photocatalytic activity is significantly improved and superior to the deposition of Zn or Al.37 In order to improve the solar energy utilization rate of the WO3 nanosheets, the visible light catalytic activity of the material is remarkably improved by increasing the visible light absorption of the material, for example, Ag ions are in situ photo-reduced and deposited on the surface of the sample in AgNO3 solution. The formation mechanism of Ag+ to Ag0 on the WO3 nanosheet under LED light irradiation in AgNO3 solution is similar to that of Ag deposited on TiO2 under UV light irradiation.38 The Ag content on the surface of the sample was determined by the EDX method (Fig. S2 and S3†). The result shows that the Ag content is 1.5% and the Ag nanoparticles mainly deposited on the (100) facet of the WO3 nanosheet crystals, indicating that the (100) facet is one of the highest photocatalytically active surfaces. Furthermore, the sample was probed by XPS. The XPS spectra of Ag 3d, W 4f and O 1s of the Ag–WO3 sample are shown, calibrated using C 1s (BE = 284.8 eV) as a reference in Fig. S4a.† The Ag content in the Ag–WO3 sample determined by XPS is 1.4%, which is basically consistent with the result of EDX analysis. The O 1s peak located at 532.5 eV is associated with the lattice oxygen in WO3. Two peaks located at 367.8 and 373.8 eV can be assigned to 3d5/2 (367.78 eV) and 3d3/2 (373.68 eV) of metallic Ag in the high resolution XPS spectrum of Ag 3d, respectively (Fig. S4b†).39,40 The high resolution XPS spectrum of W 4f reveals two obvious peaks at 37.98 and 35.78 eV corresponding to W 4f5/2 and W 4f7/2, respectively, which evidently demonstrates the valence state of tungsten element at +6 in the Ag–WO3 sample (Fig. S4c†).41
The visible light photo-electrochemical performance of the obtained WO3 and Ag–WO3 nanosheets as photocatalysts was evaluated by decomposing water to H2 under simulated sunlight and measuring the accompanying photocurrent. The structure and morphology of commercial C-WO3 are shown in Fig. S5,† and it was used for comparison. Fig. 8 shows the typical current–time curves obtained by irradiating intermittently the nanosheets (WO3, Ag–WO3, contrast C-WO3 and C-Ag–WO3 electrodes) using LED light at zero applied voltage, respectively. The obtained data of the photo-electrochemical performance for the samples are listed in Table 1. It can be found that the value of the photocurrent density is 3 μA cm−2 for the WO3 sample, 30 μA cm−2 for the Ag–WO3 sample, 0.4 μA cm−2 for contrast C-WO3 and 1.0 μA cm−2 for C-Ag–WO3, respectively. Simultaneously, the amount of H2 evolved by the photocatalyst (μmol h−1 g−1) is calculated from the current–time curve by eqn (1):
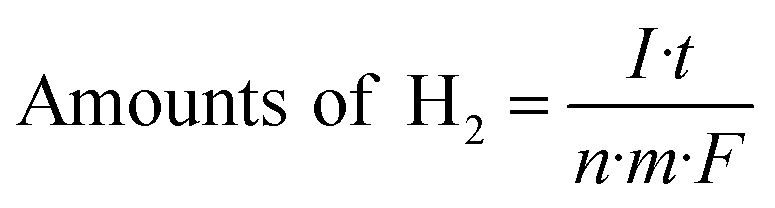 | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 487 C mol−1. It can be seen from Fig. 8 that the amounts of H2 evolved by the photocatalysts under LED light irradiation are 43.2 (the WO3), 448 (the Ag–WO3), 2.98 (contrast C-WO3) and 7.45 (C-Ag–WO3) μmol h−1 g−1, respectively. The results show that the photocurrent density generated by the prepared WO3 nanosheets is seven times that of the contrast C-WO3, which again indicates that the (100) facet of monoclinic WO3 is one of the most photocatalytically active surfaces. In addition, the photocurrent density generated by the prepared Ag–WO3 sample is ten times that of the WO3 sample and thirty times that of C-Ag–WO3, and the amount of H2 evolved by the Ag–WO3 sample is sixty times that of C-Ag–WO3. The results indicate that the Ag–WO3 nanosheet crystals have higher photo-electrochemical conversion efficiency, which is attributed to the fact that the morphology of the prepared WO3 nanosheet sample and its surface crystal structure are more advantageous for improving the performance of the photocatalyst. This reveals that the solar light utilization efficiency of the WO3 sample has been significantly improved after silver deposition on the surface, which suggests that it has potential application values in photocatalysis.
487 C mol−1. It can be seen from Fig. 8 that the amounts of H2 evolved by the photocatalysts under LED light irradiation are 43.2 (the WO3), 448 (the Ag–WO3), 2.98 (contrast C-WO3) and 7.45 (C-Ag–WO3) μmol h−1 g−1, respectively. The results show that the photocurrent density generated by the prepared WO3 nanosheets is seven times that of the contrast C-WO3, which again indicates that the (100) facet of monoclinic WO3 is one of the most photocatalytically active surfaces. In addition, the photocurrent density generated by the prepared Ag–WO3 sample is ten times that of the WO3 sample and thirty times that of C-Ag–WO3, and the amount of H2 evolved by the Ag–WO3 sample is sixty times that of C-Ag–WO3. The results indicate that the Ag–WO3 nanosheet crystals have higher photo-electrochemical conversion efficiency, which is attributed to the fact that the morphology of the prepared WO3 nanosheet sample and its surface crystal structure are more advantageous for improving the performance of the photocatalyst. This reveals that the solar light utilization efficiency of the WO3 sample has been significantly improved after silver deposition on the surface, which suggests that it has potential application values in photocatalysis.
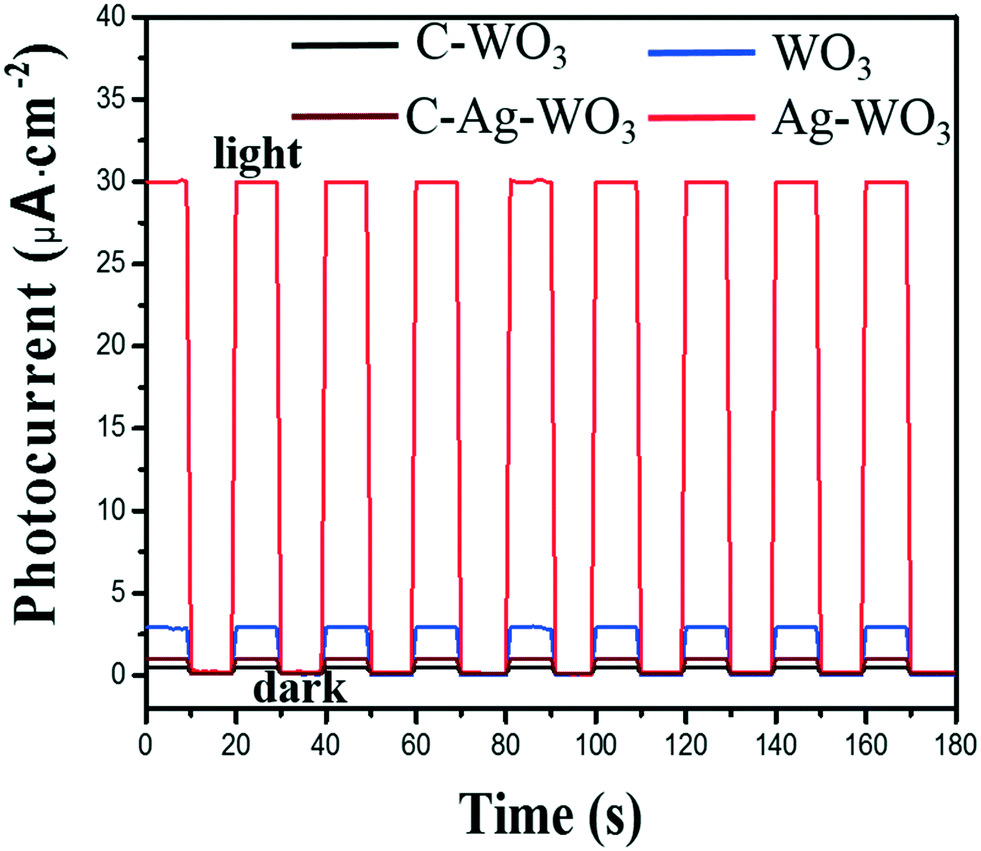 | ||
| Fig. 8 Current–time curves of the obtained samples WO3 and Ag–WO3, contrast C-WO3 and C-Ag–WO3 in the dark and under irradiation with the simulated sunlight (LED, 30 W, 380–780 nm), and the radiometric quantity reaching to the electrode surface is 16.8 mW cm−2 (at 555 nm). | ||
| Sample | Morphology | Photocurrent density (μA cm−2) | Amount of H2 evolved (μmol h−1 g−1) |
|---|---|---|---|
| WO3 | Sheet | 3.0 | 43.2 |
| Ag–WO3 | Sheet | 30 | 448 |
| C-WO3 | Spherical | 0.4 | 2.98 |
| C-Ag–WO3 | Spherical | 1.0 | 7.45 |
In order to explain the difference in visible light photoelectric conversion efficiency of the samples, we investigate the surface electronic band structure of WO3 and Ag–WO3. The UV-vis diffuse reflectance spectra (DRS) of the WO3 and Ag–WO3 samples are shown in Fig. 9. The absorption edges can be evaluated from the spectra as 475 nm for the WO3 nanosheets and 510 nm for the Ag–WO3 nanosheets. The result indicates that Ag–WO3 nanosheets enhance the absorption of sunlight compared with the WO3 nanosheets. The relationship between absorption coefficient (a) and incident photon energy (hν) can be represented by the Kubelka–Munk function a = B (hν − Eg)2/(hν), where B and Eg are the absorption constant and bandgap energy, respectively. The bandgap energy is estimated by the transformed Kubelka–Munk function versus the energy of light as shown in Fig. 9. According to the value of the absorption edge, the bandgap value is 2.87 eV for the WO3 sample and 2.75 eV for the Ag–WO3 sample, respectively. The reduction of the bandgap of the sample is beneficial to the absorption of visible light. The enhanced visible light photoelectric conversion efficiency can be attributed to the synergistic action between the Ag nanoparticle and the WO3 nanosheet. This action does not only deepen the color of the sample and increase the absorption of visible light, but also makes the photogenerated electrons rapidly transfer from the WO3 nanosheet to metal Ag, promotes the separation of photogenerated e−–h+ pairs and greatly improves the photoelectric conversion efficiency of the Ag–WO3 nanosheet sample. The main reason for the effective separation of photogenerated e−–h+ pairs is that in Ag–WO3 nanosheets, when Ag nanoparticles are deposited on the surface of WO3 nanosheets, a Schottky barrier formed between the Ag nanoparticles and WO3 due to the Fermi level of Ag is much smaller than WO3 nanosheets,42–45 which will drive the e− from the CB of WO3 to transfer to the Ag nanoparticles. This electron transfer process is beneficial to the separation of photogenerated e−–h+ pairs and inhibits the recombination. However, excess Ag nanoparticles on the surface will inhibit the absorption of sunlight, and excess Ag nanoparticles will also play the role of trapping sites in turn increasing the recombination rate of charges.46,47
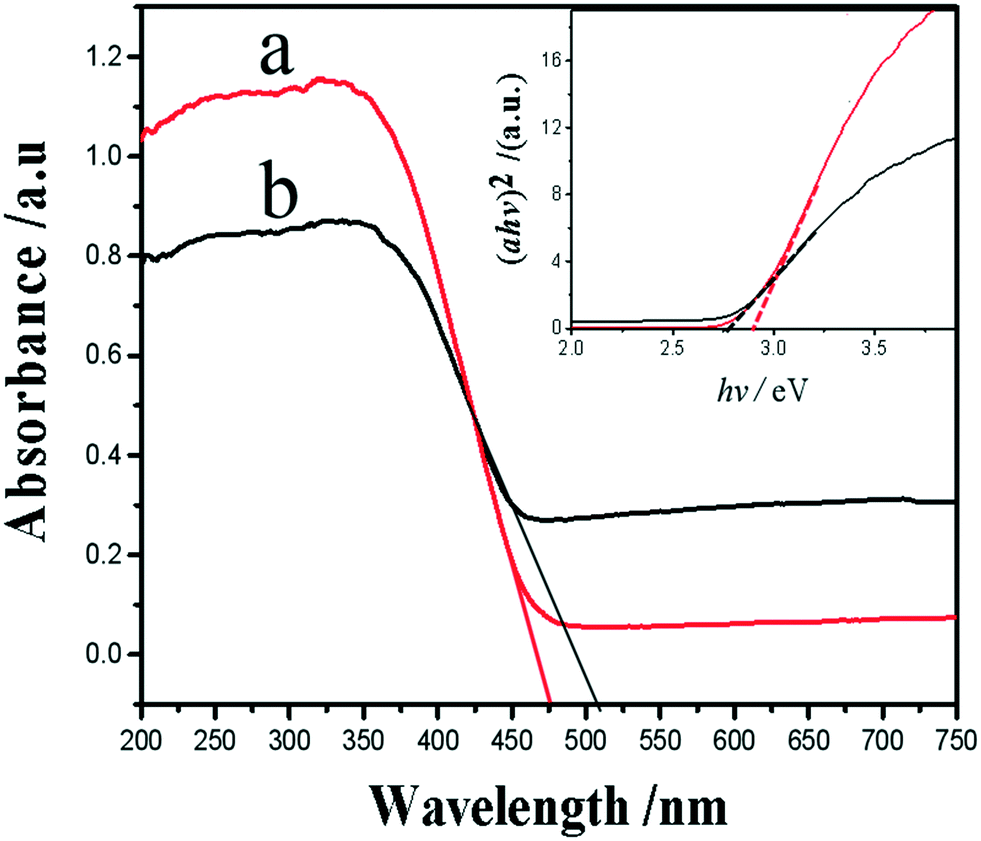 | ||
| Fig. 9 UV-vis DRS of the samples WO3 (a) and Ag–WO3 (b). Inset shows the corresponding plots of the transformed Kubelka–Munk function versus the energy of a photon. | ||
4. Conclusions
Monoclinic phase WO3 circular nanosheet crystals mainly exposing the (100) facet were prepared by a soft chemical in situ topotactic synthesis process with orthogonal H2WO4 disk particles as precursors. Simultaneously, the mechanism of transforming into the WO3 nanosheets from the H2WO4 particles has been presented in detail by analyzing the samples obtained in the process using XRD, FE-SEM, TEM, HRTEM, SAED, UV-vis DRS, EDX, XPS and Raman spectroscopy. Furthermore, Ag–WO3 nanosheets were prepared by an in situ photoreduction reaction between WO3 nanosheets and AgNO3 solution, and Ag nanoparticles are mainly deposited on the exposed (100) facet of WO3 nanosheets, suggesting that the (100) facet is one of the highest photocatalytically active surfaces. The visible light photo-electrochemical performance of the obtained Ag–WO3 nanosheets was evaluated by splitting water under simulated sunlight illumination. The result shows that the generated photocurrent density and the amount of H2 evolved are 30 μA cm−2 and 448 μmol h−1 g−1, respectively, which are ten times those of the WO3 sample. And the amount of H2 evolved by the Ag–WO3 sample is sixty times that of C-Ag–WO3. The results indicate that the Ag–WO3 nanosheets have high visible light photo-electrochemical performance and excellent visible light photoelectric conversion efficiency. Therefore, the Ag–WO3 nanosheets can be used as anticipated active materials for visible light photocatalysts and the preparation of clean energies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Natural Science Foundation of China (No. 21173003).References
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC
.
- D. Fabian, S. Hu, N. Singh, F. Houle, T. Hisatomi, K. Domen, F. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC
- Y. Yan, B. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC
- Y. Li, Y. L. Li, B. Sa and R. Ahuja, Catal. Sci. Technol., 2017, 7, 545–559 RSC
- P. Wen, T. Liu, F. Wei, L. Ai and F. Yao, CrystEngComm, 2016, 18, 8880–8886 RSC
- J. Zhu, S. Wei, M. Alexander, T. Dang, T. Ho and Z. Guo, Adv. Funct. Mater., 2010, 20, 3076–3084 CrossRef CAS
- W. Li and Z. Fu, Appl. Surf. Sci., 2010, 256, 2447–2452 CrossRef CAS
- Y. Miseki, H. Kusama, H. Sugihara and K. Sayama, J. Phys. Chem. Lett., 2010, 1, 1196–1200 CrossRef CAS
- K. Sayama, H. Hayashi, T. Arai, M. Yanagida, T. Gunji and H. Sugihara, Appl. Catal., B, 2010, 94, 150–157 CrossRef CAS
- L. Li, Y. Zhang, X. Fang, T. Zhai, M. Liao, X. Sun, Y. Koide, Y. Bando and D. Golberg, J. Mater. Chem., 2011, 21, 6525–6530 RSC
- S. Ardizzone, G. Cappelletti, C. Ricci and A. Sin, J. Nanosci. Nanotechnol., 2010, 10, 8367–8374 CrossRef CAS PubMed
- D. Chen, X. Hou, H. Wen, Y. Wang, H. Wang, X. Li, R. Zhang, H. Lu, H. Xu, S. Guan, J. Sun and L. Gao, Nanotechnology, 2010, 21, 035501 CrossRef
- H. Liu, S. Huang, S. Zhang, L. Zhang, S. Liu, W. Xin and L. Xu, Catal. Commun., 2009, 10, 544–548 CrossRef CAS
- H. Zheng, Y. Tachibana and K. Kalantar-zadeh, Langmuir, 2010, 26, 19148–19152 CrossRef CAS
- L. Meda, G. Tozzola, A. Tacca, G. Marra, S. Caramori, V. Cristino and C. Bignozzi, Sol. Energy Mater. Sol. Cells, 2010, 94, 788–796 CrossRef CAS
- J. Liu, Z. Zhang, Y. Zhao, X. Su, S. Liu and E. Wang, Small, 2005, 1, 310–313 CrossRef CAS
- H. Zheng, J. Z. Ou, M. S. Strano, R. B. Kaner, A. Mitchell and K. Kalantar-Zadeh, Adv. Funct. Mater., 2011, 21, 2175–2196 CrossRef CAS
- P. Thakur, A. Kool, B. Bagchi, N. A. Hoque, S. Das and P. Nandya, RSC Adv., 2015, 5, 62819–62827 RSC
- J. Pan, G. Liu, G. Lu and H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 2133–2137 CrossRef CAS
- N. Wu, J. Wang, D. Tafen, H. Wang, J. Zheng, J. Lewis, X. Liu, S. Leonard and A. Manivannan, J. Am. Chem. Soc., 2010, 132, 6679–6685 CrossRef CAS
- C. Li, C. Koenigsmann, W. Ding, B. Rudshteyn, K. Yang, K. Regan, S. Konezny, V. Batista, G. Brudvig, C. Schmuttenmaer and J. Kim, J. Am. Chem. Soc., 2015, 137, 1520–1529 CrossRef CAS
- X. Liu, G. Dong, S. Li, G. Lu and Y. Bi, J. Am. Chem. Soc., 2016, 138, 2917–2920 CrossRef CAS
- Y. Zhao, C. Eley, J. Hu, J. Foord, L. Ye, H. He and S. Tsang, Angew. Chem., Int. Ed., 2012, 51, 3846–3849 CrossRef CAS
- Y. Chiba, D. Koizumi, M. Saito and T. Motohashi, CrystEngComm, 2019, 21, 3223–3231 RSC
- W. Ouyang, F. Teng, J. H. He and X. Fang, Adv. Funct. Mater., 2019, 29, 1807672 CrossRef
- P. Wen, Y. Ishikawa, H. Itoh and Q. Feng, J. Phys. Chem. C, 2009, 113, 20275–20280 CrossRef CAS
- P. Wen, H. Itoh, W. Tang and Q. Feng, Langmuir, 2007, 23, 11782–11790 CrossRef CAS
- P. Wen, H. Itoh, W. Tang and Q. Feng, Microporous Mesoporous Mater., 2008, 116, 147–156 CrossRef CAS
- P. Wen, M. Xue, Y. Ishikawa, H. Itoh and Q. Feng, ACS Appl. Mater. Interfaces, 2012, 4, 1928–1934 CrossRef CAS
- P. Wen, L. Ai, T. Liu, D. Hu and F. Yao, Mater. Des., 2017, 117, 346–352 CrossRef CAS
- A. Alwrawi, V. Ramalingam, H. Fu, P. Varadhan, R. Yang and J. He, Opt. Express, 2019, 27, A352–A363 CrossRef
- P. Wen, L. Ai, F. Wei, D. Hu. J. Guo, F. Yao and T. Liu, Mater. Des., 2017, 131, 219–225 CrossRef CAS
- G. Xi, B. Yue, J. Cao and J. Ye, Chem. – Eur. J., 2011, 17, 5145–5154 CrossRef CAS
- J. Dai, W. Zhong, W. Yi, M. Liu, L. Mao, Q. Xu and D. Yin, Appl. Catal., B, 2016, 192, 325–341 CrossRef CAS
- J. Zheng, G. Song, J. Hong, T. Van, A. Pawar, D. Kim, C. Kim, Z. Haider and Y. Kang, Cryst. Growth Des., 2014, 14, 6057–6066 CrossRef CAS
- L. Zheng, S. Han, J. Liu, P. Yu and X. Fang, Small, 2016, 12, 1527–1536 CrossRef CAS
- K. M. S. Siti, A. U. Akrajas, I. A. U. Marjoni, T. Masahiko, Y. A. R. Mohd, M. S. Muhamad and O. Munetaka, ACS Omega, 2018, 3, 2579–2587 CrossRef
- E. Stathatos, P. Lianos, P. Falaras and A. Siokou, Langmuir, 2000, 16, 2398–2400 CrossRef CAS
- C. Ren, B. Yang, M. Wu, J. Xu, Z. Fu, T. Guo, Y. Zhao and C. Zhu, J. Hazard. Mater., 2010, 182, 123–129 CrossRef CAS PubMed
- Y. Li, Q. Wang, H. Wang, J. Tian and H. Cui, J. Colloid Interface Sci., 2019, 537, 206–214 CrossRef CAS PubMed
- B. Weng, J. Wu, N. Zhang and Y. Xu, Langmuir, 2014, 30, 5574–5584 CrossRef CAS
- M. A. Butler, J. Appl. Phys., 1977, 48, 1914–1920 CrossRef CAS
- Y. Zheng, C. Chen, Y. Zhan, X. Lin, Q. Zheng, K. Wei and J. Zhu, J. Phys. Chem. C, 2008, 112, 10773–10777 CrossRef CAS
- D. Lin, H. Wu, R. Zhang and W. Pan, Chem. Mater., 2009, 21, 3479–3484 CrossRef CAS
- Z. Hosseini, N. Taghavinia, N. Sharifi, M. Chavoshi and M. Rahman, J. Phys. Chem. C, 2008, 112, 18686–18689 CrossRef CAS
- L. Ren, Y. Zeng and D. Jiang, Catal. Commun., 2009, 10, 645–646 CrossRef CAS
- C. Su, L. Liu, M. Zhang, Y. Zhang and C. Shao, CrystEngComm, 2012, 14, 3989–3999 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ce01392j |
| This journal is © The Royal Society of Chemistry 2020 |