
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic addition of C–H bonds across C–C unsaturated systems promoted by iridium(I) and its group IX congeners
David F.
Fernández†
 a,
José L.
Mascareñas
*a and
Fernando
López
*ab
a,
José L.
Mascareñas
*a and
Fernando
López
*ab
aCentro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: joseluis.mascarenas@usc.es; fernando.lopez@csic.es
bInstituto de Química Orgánica General, CSIC, Juan de la Cierva 3, 28006, Madrid, Spain
First published on 14th September 2020
Abstract
Transition metal-catalyzed hydrocarbonations of unsaturated substrates have emerged as powerful synthetic tools for increasing molecular complexity in an atom-economical manner. Although this field was traditionally dominated by low valent rhodium and ruthenium catalysts, in recent years, there have been many reports based on the use of iridium complexes. In many cases, these reactions have a different course from those of their rhodium homologs, and even allow performing otherwise inviable transformations. In this review we aim to provide an informative journey, from the early pioneering examples in the field, most of them based on other metals than iridium, to the most recent transformations catalyzed by designed Ir(I) complexes. The review is organized by the type of C–H bond that is activated (with C sp2, sp or sp3), as well as by the C–C unsaturated partner that is used as a hydrocarbonation partner (alkyne, allene or alkene). Importantly, we discuss the mechanistic foundations of the methods highlighting the differences from those previously proposed for processes catalyzed by related metals, particularly those of the same group (Co and Rh).
 David F. Fernández | David F. Fernández graduated from the University of Santiago de Compostela in 2014 with an Master's degree in Chemistry. Following this, he conducted his PhD studies under the supervision of José Luis Mascareñas, Fernando López and Moisés Gulías at the Center for Research in Biological Chemistry and Molecular Materials (CiQUS, USC). His postgraduate research centered on the development of Ir-catalyzed intramolecular hydrocarbonations of acrylamides to enable access to high regio- and enantioselective cyclic scaffolds. He also spent four months in 2017 as VSRC at Princeton University with Prof. MacMillan studying metallaphotoredox difluoromethylation reactions and HTE technologies. In 2020, David began a postdoctoral appointment with Professor David MacMillan at Princeton University where he is focused on developing new strategies for biological systems using photoredox catalysis. |
 José L. Mascareñas | José Luis Mascareñas completed his PhD in the University of Santiago in 1988, and carried out postdoctoral studies at Stanford University (1989–1990) and Harvard University (1993 and 1995). He became full professor at the University of Santiago in 2005. He has published over 200 articles, and supervised 35 PhD theses. In 2014 was awarded with an ERC Advanced Grant for his project METBIOCAT (http://metbiocat.eu/) and, more recently, an ERC Proof of Concept (2020). He has been a scientific director of CIQUS since 2014. In 2015 he received the Gold Medal of the Spanish Society of Chemistry and, in 2016, was appointed as a member of the European Academy of Sciences. His current research splits between a synthetic program aimed at discovering novel methods based on metal catalysis and a chemical biology program focused on the development of synthetic and catalytic tools for biological intervention. |
 Fernando López | Fernando López obtained his PhD in 2003 from the University of Santiago de Compostela. He carried out two predoctoral stays with Professors Erick M. Carreira and John F. Hartwig, and postdoctoral studies, as a Marie Curie Fellow (2004–2006), with Prof. Ben L. Feringa (Univ. Groningen). In 2006, he joined the University of Santiago de Compostela (USC) under the Ramón y Cajal programme. In 2008, he was appointed Tenured Scientist at the Spanish National Research Council (CSIC) at the IQOG and, in 2018, he was promoted to Senior Research Scientist. From 2012, he has been assigned as PI at the Center for Research in Biological Chemistry and Molecular Materials (CiQUS, USC), where his research lines are aimed at discovering efficient synthetic methods as well as novel biocompatible tools, both based on transition metal catalysis. |
1 Introduction
Transition metal-catalyzed (TMC) carbon–carbon (C–C)-bond forming reactions are among the most relevant transformations available in the toolbox for synthetic chemistry. This type of reaction is used daily by pharmaceutical and biotech companies for the synthesis of different types of drugs and functional materials.1 Despite their huge impact, many of the methods still present important limitations, such as the requirement of functionalized reactants, or the generation of secondary molecular waste. Therefore, there is a need to develop more efficient and environmentally friendly technologies that could improve our ability to make C–C bonds using transition metal catalysis.2In this context, methods wherein all the atoms of the reactants are transferred into products, which warrant full atom economy, are especially attractive.3
In recent years there has been a boom in the development of transition metal-based technologies for selective activation of relatively inert carbon–hydrogen (C–H) bonds.4 The resulting C–[M] species can then engage in different types of transformations. In terms of atom economy, one of the more attractive options consists of the hydrocarbonation of C–C unsaturated partners, the formal addition of a C–H bond across a C–C double or triple bond, owing to the intrinsic atom economy and ready availability of the reaction partners (e.g. alkenes, alkynes, dienes).5 Key to the success of the reaction is the control of the regio-, diastereo- and, eventually, enantioselectivities of the process. The general mechanistic scenery for these reactions involves an oxidative addition of C–H bonds to the metal, usually aromatic C(sp2)–H bonds, followed by migratory insertion and reductive elimination steps (Scheme 1).
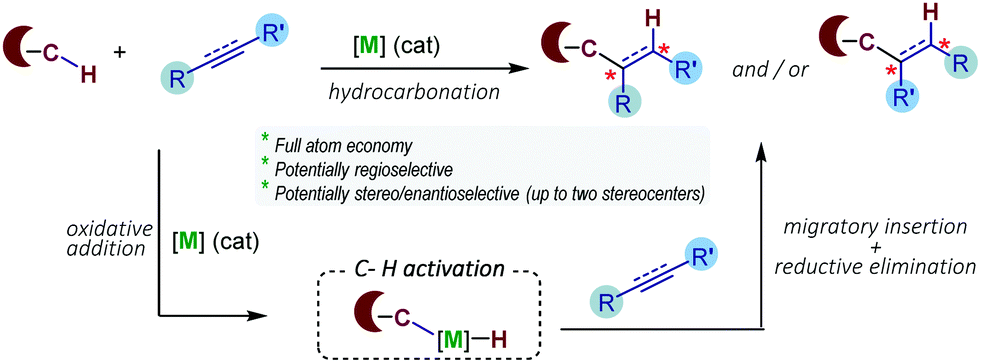 | ||
| Scheme 1 General mechanistic principles of TMC catalyzed hydrocarbonations of C–C unsaturated systems. | ||
This research topic was originally introduced in the early nineties using low-valent Ru complexes.5a Later on, other transition metal catalysts, including rhodium, palladium, nickel, iridium and cobalt complexes, have also found their place in this type of transformation.6
Curiously, low valent iridium complexes were not deeply explored in catalytic hydrocarbonation reactions until quite recently,7 likely because of the notion that iridium is less reactive than other second row metals (i.e. Rh, Pd or Ru).8 In some cases, Ir(I) complexes were used in the context of mechanistic studies, whereas the metal of choice for inducing the catalytic process was its homologous counterpart, rhodium.
This paucity is quite surprising considering that iridium(I) complexes were known to participate in oxidative additions to C–H bonds from the early 80s.9 Indeed, compared to their rhodium analogs, iridium complexes are more nucleophilic and tend to undergo oxidative addition more easily.10
Therefore, in recent years, a significant number of hydrocarbonation reactions enabled by iridium (I) catalysts have been developed.7 Moreover, in some cases, these reactions proceed through pathways that are different from those found for other late transition metals.
Albeit not in an entirely comprehensive manner, we herein discuss the most relevant iridium-catalyzed hydrocarbonation processes, highlighting their particularities with respect to alternative processes based on other low valent metal catalysts, especially their group partners rhodium and cobalt. We will highlight several cases in which the selectivity generally achieved with rhodium (as well as with ruthenium) catalysts is opposite to that obtained with iridium and with cobalt counterparts. To place the discussion into proper context, we also present pioneering examples of hydrocarbonation reactions, regardless of the type of low-valent metal used. Finally, it is also pertinent to underline that high valent Rh(III), Ir(III), and Co(III) catalysts have also been proven to be successful in formal hydrocarbonation reactions. However, from a mechanistic point of view, in these reactions the C–H activation usually proceeds through a carboxylate-assisted concerted metallation deprotonation (CMD), not by oxidative addition. Accordingly, the final step that closes the catalytic cycle is usually a protodemetallation, and therefore they cannot be formally considered as additions of C–H bonds across unsaturated partners. For these reasons, as well as for the sake of conciseness, we do not include these transformations.11
The review is organized by the type of C–H bond that is activated, and by the type of unsaturated partner used in the reaction. Moreover, we discuss inter- and intramolecular processes in a separate manner.
2 TMC addition of C(sp2)–H bonds across C–C unsaturated bonds
2.1 Addition to alkenes
During the last three decades, a number of alkene hydrocarbonations have been described. Not surprisingly, most of them rely on the activation of C(sp2)–H bonds of arenes and heteroarenes. The addition of related C(sp2)–H bonds of alkenes and, especially, of alkane C(sp3)–H bonds, has lagged clearly behind.5d
In 1993, Murai and co-workers presented a seminal contribution describing a Ru-catalyzed addition of aromatic ketones to alkenes.12 In this reaction the ruthenium(II) complex RuH2(CO)(PPh3)3 is transformed in situ into a catalytically active Ru(0) species that undergoes an oxidative addition with the C–H bond of an arene, to generate ruthenated species of type I (Scheme 2A). Key to this activation is the presence of a ketone at the arene precursor that coordinates the Ru complex, directing the C–H insertion at its ortho position. Species I evolves into the linear hydroarylation product 1 through a regioselective alkene migratory insertion into the Ru–H bond (with the formation of intermediate II), and a subsequent C–C reductive elimination.
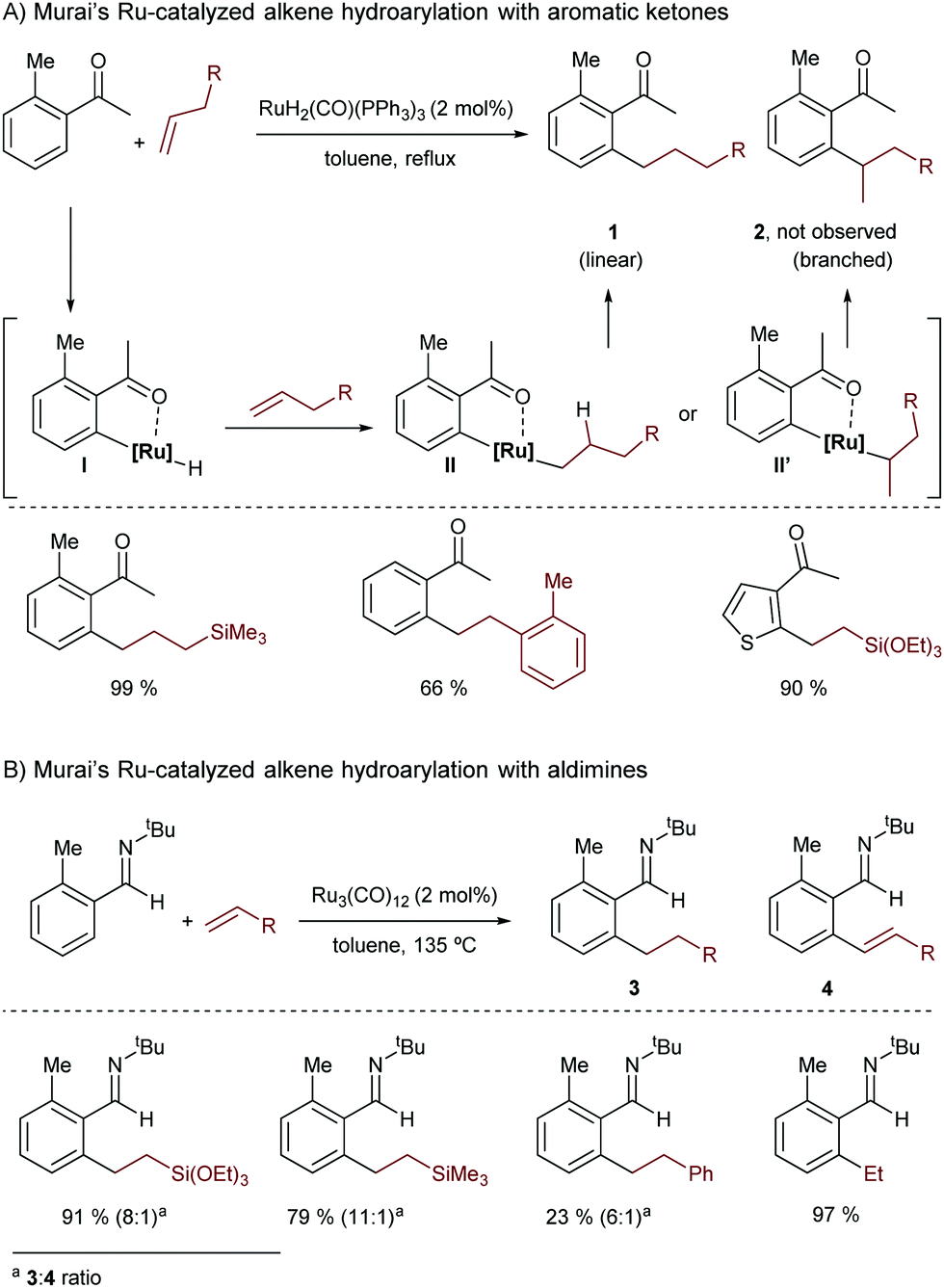 | ||
| Scheme 2 Pioneering ruthenium-catalyzed hydroarylation of alkenes developed by Murai and co-workers. | ||
Remarkably, this method was the first high-yield catalytic C–H bond functionalization in which the substrate containing the reacting C–H bond was used as a limiting reagent, which further highlights the synthetic potential of the reaction.
Three years later, the same group reported a related ruthenium-catalyzed hydroarylation using Ru3(CO)12 as a catalyst, and an imine as a chelation-assisted directing group (Scheme 2B).13 The reaction also produces linear addition products (3), although dehydrogenated derivatives like 4 are also observed.
Overall, these groundbreaking discoveries paved the way for many other related hydrocarbonation reactions, using different transition metals and C–C unsaturated partners.5d
In the year 2000, Jun and co-workers introduced the use of group IX metals in this type of reaction, demonstrating that the Wilkinson catalyst is also competent at promoting the hydroarylation of alkenes with aromatic ketimines (Scheme 3A).14 The reaction tolerates different types of substituted alkenes, although in most of the cases it requires harsh conditions (e.g. heating at 150 °C). The proposed mechanism features the same three key steps of that previously proposed by Murai, namely an oxidative addition assisted by the imine chelating group, the insertion of the olefin into the metal-hydride bond and, finally, a C–C reductive elimination. The resulting ortho-alkylated ketimines are transformed into their respective ketones (5) upon acidic aqueous treatment.
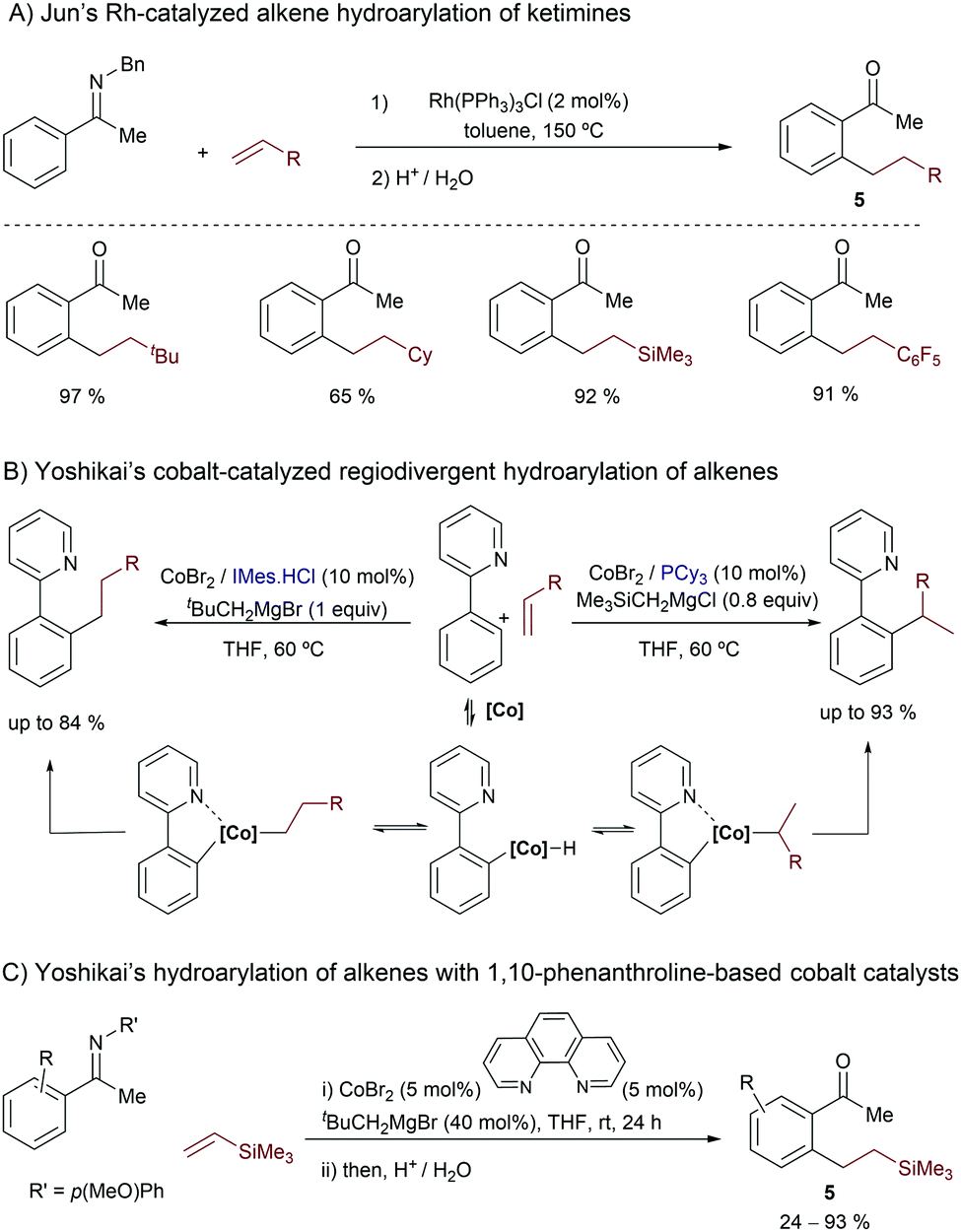 | ||
| Scheme 3 Representative Rh- and Co-catalyzed hydroarylations of alkenes developed by Jun (A) and Yoshikai (B and C). | ||
More than ten years later, in 2011, Yoshikai and coworkers introduced low-valent cobalt catalysts for promoting hydroarylation reactions of alkenes.15 In sharp contrast to the linear selectivity typically achieved with Rh and Ru,5 an appropriate selection of the ancillary ligand at cobalt (i.e. PCy3 or IMes) allowed regiodivergent access to both linear and branched products using arylpyridine substrates (Scheme 3B).15a Mechanistically, the authors proposed that a low-valent cobalt(0) catalyst, generated from the Co(II) precursor and a bulky Grignard reagent, is the species undergoing the oxidative addition of the C–H bond. Extensive deuterium-labeling experiments indicate that this C–H activation and the subsequent migratory insertions are both reversible steps; moreover, the two regioisomeric insertions, leading to either linear or branched alkylcobalt intermediates, would be competing in both the Co–PCy3 and Co–IMes regimes. Thus, the C–C reductive elimination would be the rate and regioselectivity determining step. According to DFT calculations carried out by Fu and coworkers, the ligand-controlled divergence is mainly due to the steric factors, related to the ligand shape and the steric crowding between the ligand and the cobalt center.6c,16
In 2011, the same authors also demonstrated that weaker field ligands such as 1,10-phenanthroline are also suitable for performing linear-selective cobalt-catalyzed hydroarylations of alkenes. In this case, the method makes use of electron rich acetophenone imines, and the reactive low valent cobalt species are likewise generated from Co(II) sources and bulky Grignard reagents (Scheme 3C).15b Deuterium labeling studies also indicate that the C–C reductive elimination is rate determining but, contrary to the above cases (Scheme 3B), the selectivity is mostly determined in the migratory insertion, which selectively provides the linear (aryl)alkylcobalt intermediate.
With respect to the use of iridium, the first examples in the hydroarylation reactions of alkenes can be traced back to the early 2000s, and dealt with symmetric alkenes.17 The most relevant case, developed by Togni and coworkers, involves the reaction of 2-norbornene with phenols, a process that is catalyzed by the chiral Ir(I)-Josiphos complex Ir1.17b The reaction provides the ortho-monoalkylated product 6, together with minor amounts of the corresponding ortho-dialkylated derivatives (7, Scheme 4A). Unfortunately, the product was obtained with negligible enantioselectivity. In 2008, Shibata et al. reported a related hydroarylation of 2-norbornene with acetophenone, promoted by the chiral catalyst resulting from the combination of [Ir(cod)2]BF4 and the bisphosphine MeO-BIPHEP (L1). In this case, the resulting product (8) was obtained with a moderate ee of 70% (Scheme 4B).18
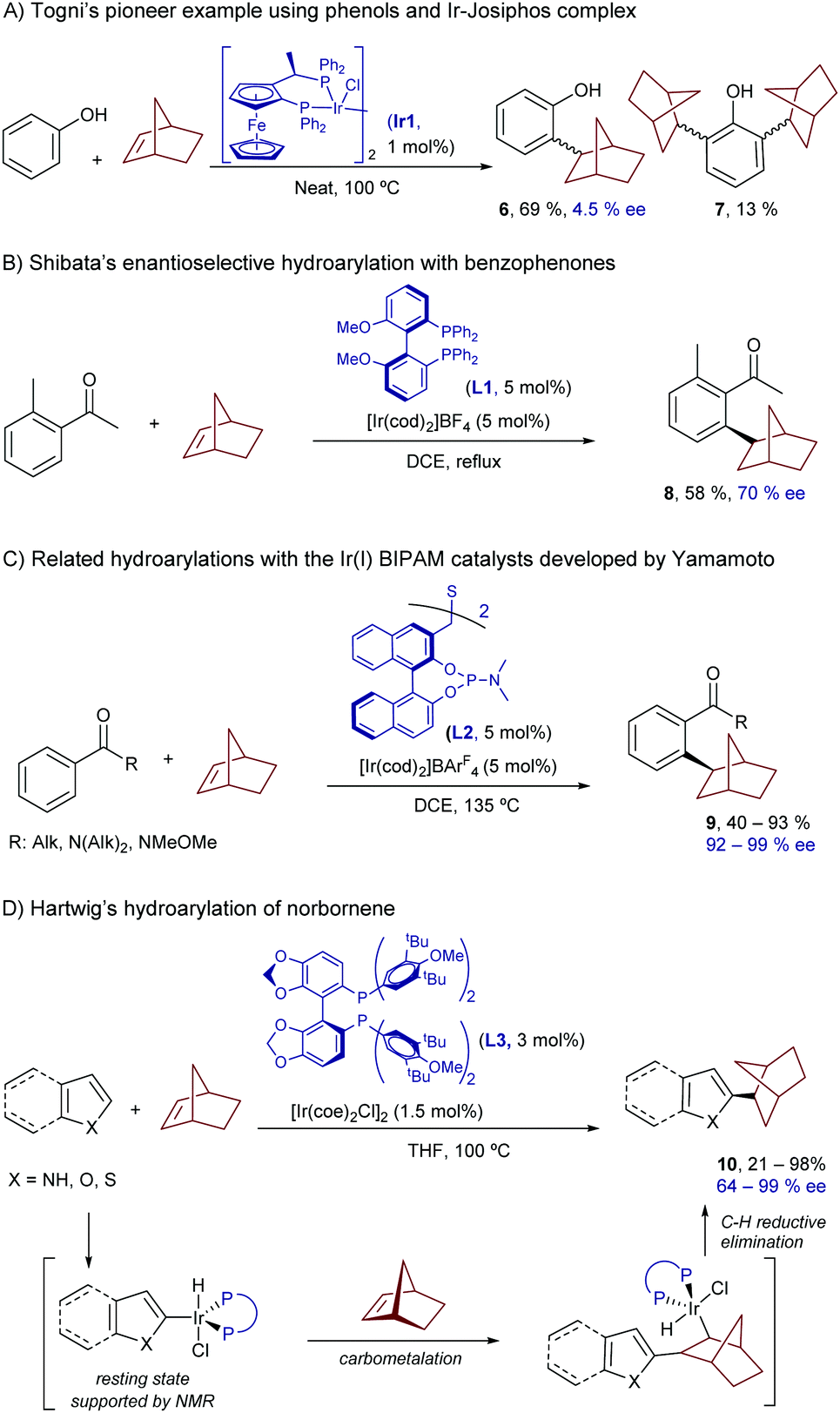 | ||
| Scheme 4 Ir-Catalyzed hydroarylations of 2-norbornene. | ||
It was not until quite recently that an iridium-catalyzed highly enantioselective protocol for the hydroarylation of norbornenes with benzophenones was revealed (Scheme 4C).19 In particular, Yamamoto and coworkers found that the catalyst formed in situ from [Ir(cod)2]BArF4 and the chiral bis-phosphoramidite ligand [(R,R)-S-Me-BIPAM] (L2), developed by the same group, was able to consistently provide the corresponding ortho-alkylated benzophenones (9) with enantioselectivities above 90%.17a
Hartwig and coworkers further expanded the scope of this chemistry disclosing an iridium promoted enantioselective hydroarylation of norbornene and related bicycloalkenes with heteroaromatic systems such as indoles, furans, pyrroles and thiophenes.20 In this case, the method does not require the use of directing groups to assist the C–H insertion, as the electronics of the heteroaromatic system warrants a selective activation of the C(2)–H bond. Thus, under optimal conditions, which involve the neutral iridium complex [Ir(coe)2Cl]2 and the chiral ligand DTBM-SEGPHOS (L3), the corresponding products of type 10 were obtained in good yields and excellent enantioselectivities, of up to 98% (Scheme 4D). These examples showcase the relevance of using iridium for promoting enantioselective processes, owing to the robustness of the corresponding chiral catalysts. Furthermore, the use of iridium as metal was instrumental in obtaining mechanistic information, such as the detection of Ir-deuteride species at room temperature by NMR. Given that reductive eliminations to form carbon–carbon bonds from an Ir center were rare or even unknown, the authors suggested that the most plausible mechanistic pathway might involve a turnover-limiting carbometallation followed by a C–H reductive elimination.
While these reactions of norbornenes and related symmetrical strained alkenes represent excellent proof-of-concept contributions to demonstrate the potential of iridium complexes, including chiral versions, as catalysts for hydroarylation reactions, their synthetic applicability is limited. The extension of the methodology to synthetically more relevant alkene partners is challenging, owing to the lower reactivity of non-strained systems and to regioselectivity issues associated with the use of unsymmetrical alkenes.
In this context, the first examples of iridium catalyzed hydroarylations of unsymmetrical, non-strained alkenes were reported by Shibata and coworkers in 2008.18 In particular, the authors demonstrated that the same class of cationic iridium complexes that promote the hydrocarbonation of norbornene could also promote the homologous addition of o-methoxy-acetophenone to styrene (Scheme 5). However, the reaction gives moderate yields of mixtures of linear (11) and branched (12) products. Moreover, the linear isomer, which is less interesting due to its achiral nature, is the major component. The reaction was best carried out using rac-BINAP instead of the MeO-BIPHEP ligand used in the norbornene hydroarylation (Scheme 4B).
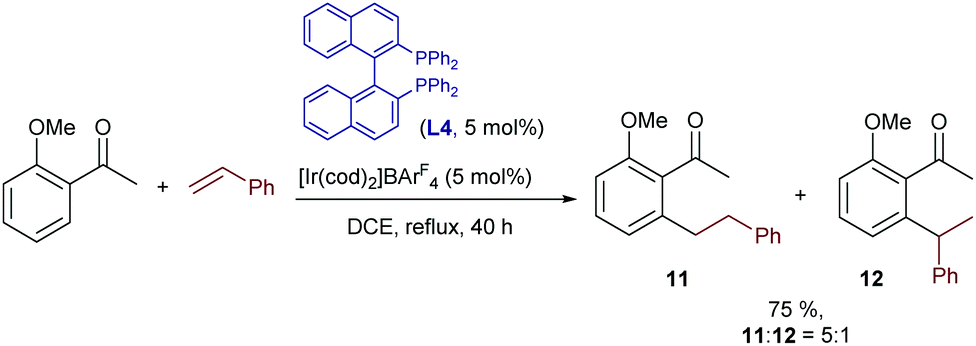 | ||
| Scheme 5 Ir-Catalyzed hydroarylation of styrenes developed by Shibata. | ||
In 2012, the same group extended the scope of the method by reporting that cationic iridium(I)-bisphosphine catalysts were competent at promoting the selective C2-alkylation of N-substituted indoles with styrenes.21 Importantly, depending on the nature of the catalyst and of the directing group attached to the indole nitrogen, linear or branched products of types 13 and 14 could be obtained (Scheme 6). Thus, N-acetyl indoles react with the catalyst resulting from mixing [Ir(cod)2]BF4 and DM-SEGPHOS (L6) to give the linear product 14 (branched:linear ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :95). In contrast, the homologue chiral catalyst generated from [Ir(cod)2]BArF4 and the spirobisphosphine ligand SDP (L5) allowed transforming an N-benzoyl indole into the branched product 13 (branched:linear ratios around 98:2), with moderate to good enantioselectivities. The methodology is mostly limited to the coupling of indoles and styrenes, although the catalyst made from [Ir(cod)2]BF4 and DM-SEGPHOS (L6) also works with electronically activated alkenes such as acrylonitrile and methyl acrylate, to provide the expected linear isomers. Curiously, when an aliphatic alkene such 1-nonene is used, the branched product was predominant, regardless of the directing group and the catalyst.
:95). In contrast, the homologue chiral catalyst generated from [Ir(cod)2]BArF4 and the spirobisphosphine ligand SDP (L5) allowed transforming an N-benzoyl indole into the branched product 13 (branched:linear ratios around 98:2), with moderate to good enantioselectivities. The methodology is mostly limited to the coupling of indoles and styrenes, although the catalyst made from [Ir(cod)2]BF4 and DM-SEGPHOS (L6) also works with electronically activated alkenes such as acrylonitrile and methyl acrylate, to provide the expected linear isomers. Curiously, when an aliphatic alkene such 1-nonene is used, the branched product was predominant, regardless of the directing group and the catalyst.
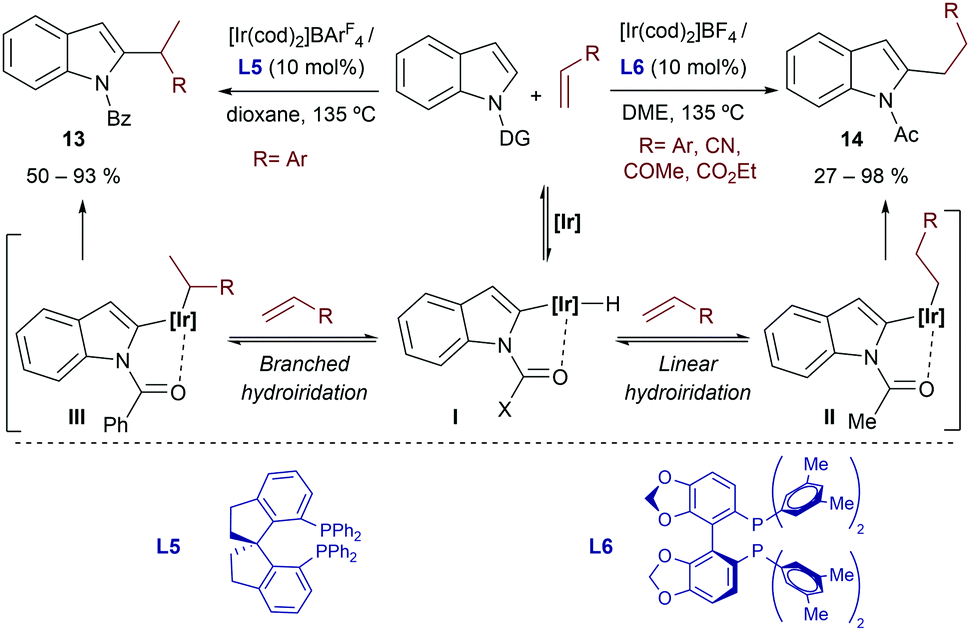 | ||
| Scheme 6 Iridium promoted alkylation at the C2-position of indoles developed by Shibata and coworkers (DG stands for the directing group). | ||
From a mechanistic point of view, the authors proposed an initial C–H activation followed by hydroiridation of the alkene, which would provide regioisomeric intermediates II and III (Scheme 6). Deuterium labeling experiments suggested that these two steps are reversible. Eventually, C–C reductive eliminations from II and III can afford linear (14) or branched (13) products. Unfortunately, to the best of our knowledge, the reasons behind this interesting ligand- and directing group-dependent regioselectivity have not yet been disclosed.
Interestingly, a related intermolecular, branched-selective C–H alkylation of imine derivatives of N-Boc indoles with styrenes was reported by Yoshikai and coworkers in 2015, using low-valent cobalt catalysts and chiral phosphoramidite ligands.22 In this case, the imine located at the C3 position of the indole acts as a directing group and it is hydrolyzed after completion of the reaction, so that chiral indole-3-carboxaldehydes are eventually obtained (Scheme 7), with moderate to good enantioselectivities (58–87% ee). The chiral low valent catalyst is generated in situ from Co(acac)3 (10 mol%), stoichiometric amounts of a Grignard reagent (that acts a reductant) and the phosphoramidite ligand L7 (20 mol%). Based on deuterium labelling studies, the authors suggested that the enantioselectivity is not only influenced by the migratory insertion of the alkene, which proved to be a reversible process, but also by the final C–C reductive elimination.
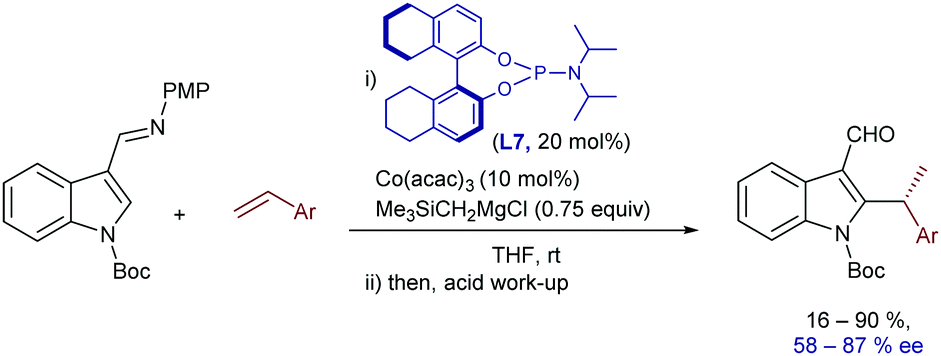 | ||
| Scheme 7 Co-Catalyzed branched-selective indole C–H alkylation developed by Yoshikai and coworkers. | ||
Although chiral phosphorus-based compounds are usually the ligands of choice for the development of enantioselective hydroarylations with Ir, Co and Rh catalysts, chiral dienes, a strategy pioneered by Hayashi (with Rh) and Carreira (with Ir), have also been used in some iridium catalyzed processes.23 In particular, Shibata et al. demonstrated that quinoline-substituted ferrocenes can be desymmetrized by reaction with olefins in the presence of an iridium catalyst generated in situ from [Ir(coe)2Cl]2 and the chiral diene ligand L8. This hydrocarbonation constitutes one of the very few methods that allows the generation of planar chirality via a TMC C–H activation process. Remarkably, its scope includes different types of alkenes, such as styrenes, acrylates and other non-activated alkenes, proceeding in many cases with complete selectivity towards the linear isomers of type 15, which are obtained with good to excellent enantioselectivities (Scheme 8).24
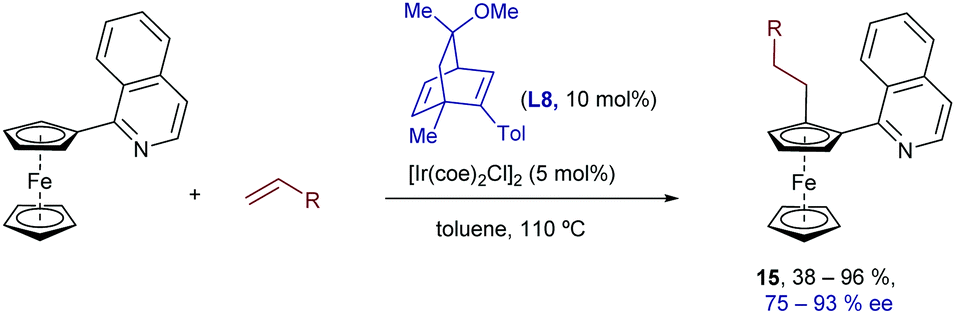 | ||
| Scheme 8 Ir-Promoted alkylation of ferrocenes developed by Shibata. | ||
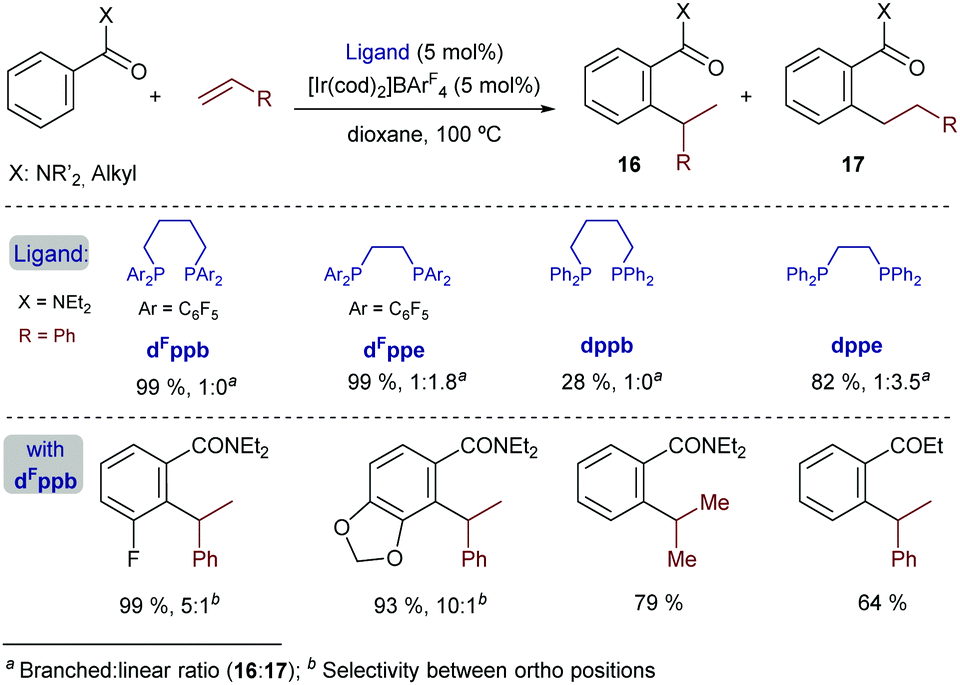
In 2014, Bower and coworkers further vitalized this field by describing a versatile iridium-catalyzed branched-selective hydroarylation of alkenes.25 The method, which is based on the use of aryl dialkylamides and related aryl ketones, is effective with a variety of styrenes, and non-activated olefins (e.g. propene) (Scheme 9). The use of an iridium(I) catalyst containing electron-deficient bisphosphines with a wide bite angle, such as dFppb – the pentafluorinated analogue of bisphosphine dppb, was instrumental in the efficiency and high regioselectivity of the process. Indeed, electronically related bisphosphines with narrower bite angles, like dFppe, led to lower branched (16) to linear (17) ratios, whereas the use of a homologous non-fluorinated bisphosphine ligand, dppb, led to significantly lower yields, while retaining the branched selectivity.
 | ||
| Scheme 9 Ir-Catalyzed branched-selective hydroarylation developed by Bower and coworkers. | ||
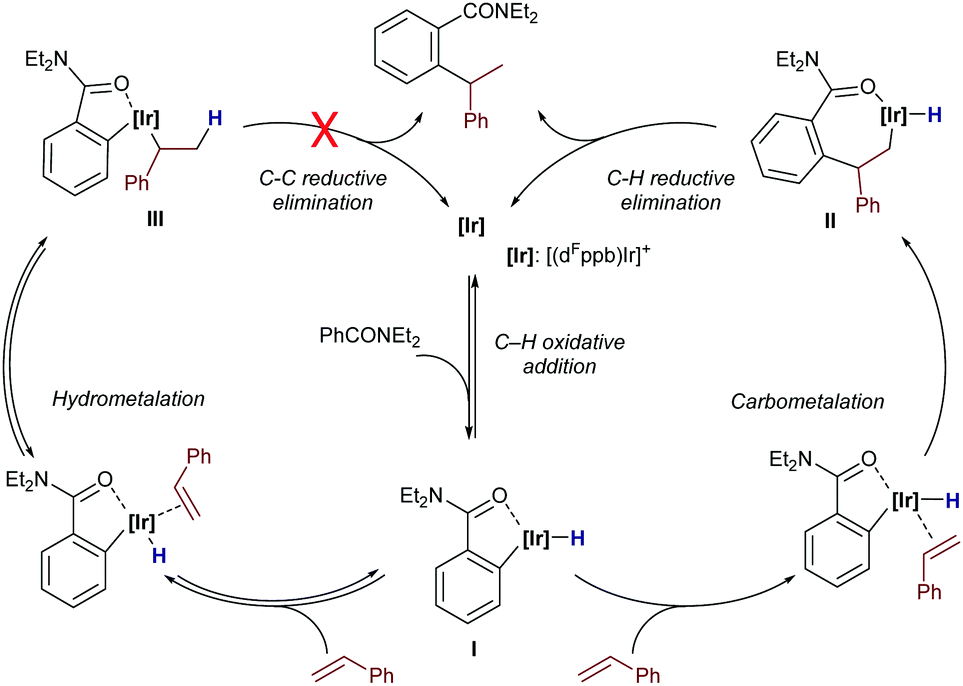
The authors proposed a mechanistic pathway in which the iridium hydride intermediate I evolves by hydrometallation and C–C reductive elimination to the product (Scheme 10). Deuterium labeling experiments revealed that both the oxidative addition and the alkene hydrometallation are reversible steps, so that the C–C reductive elimination was proposed as the product determining step. Interestingly, subsequent DFT calculations reported by Huang and Liu also confirmed that the oxidative addition and hydrometallation steps are reversible; however, the proposed C–C reductive elimination was found to involve an exceedingly high energy barrier.26 These calculations revealed that a pathway based on a carbometallation step – namely a migratory insertion of the olefin into the Ir–C(sp2) bond – followed by a C–H reductive elimination was energetically more feasible. In the latter pathway, the carbometallation is proposed to be the rate and regioselectivity-determining step of the overall process. The computational study also concludes that ligands with large bite angles like dppb and dFppb disfavor the linear selectivity due to the presence of unfavorable steric repulsions between the substrate and the aryl substituents of the ligand in the transition state of the migratory insertion step.
 | ||
| Scheme 10 Mechanistic pathways studied by Huang for a model system of branched-selective hydroarylations reported by Bower et al. (see Scheme 9). | ||
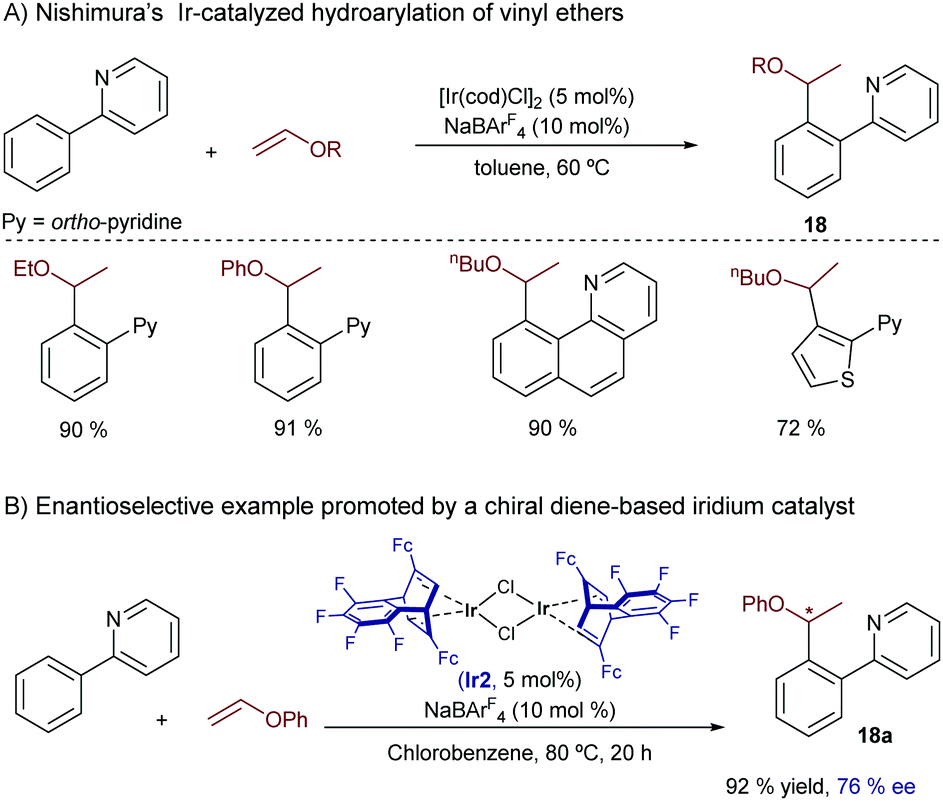
Almost parallel to this work, Nishimura reported an iridium-catalyzed hydroarylation of vinyl ethers using ortho-arylpyridines and related heteroaromatics as substrates.27 The reaction proceeds very efficiently under mild conditions for a variety of precursors and provides the ortho-alkylated products 18 with complete branched selectivity (Scheme 11). Remarkably, the optimal catalyst [IrCl(cod)]2/NaBArF4 does not require the use of bisphosphines like in previous cases. Thus, a preliminary enantioselective version was explored using an iridium complex (Ir2) wherein the 1,5-cyclooctadiene ligand (cod) is replaced with the chiral diene (S,S)-Fc-tfb*. An excellent yield (92% yield) and a moderate ee (76%) were obtained in a model case (18a). Mechanistic experiments with deuterium-labeled substrates indicated that the C–H activation assisted by the pyridine is reversible. Moreover, computational studies by Huang and coworkers suggested that, similarly to the previous reaction by Bower (see Scheme 10), the productive pathway involves a rate and selectivity determining carbometallation step, which is followed by an easier C–H reductive elimination.28 The authors also indicated that the good regioselectivity of the process is related to the electron-donating character of the alkoxy group in the alkene reacting partner.
 | ||
| Scheme 11 Iridium-catalyzed hydroarylation of vinyl ethers developed by Nishimura. | ||
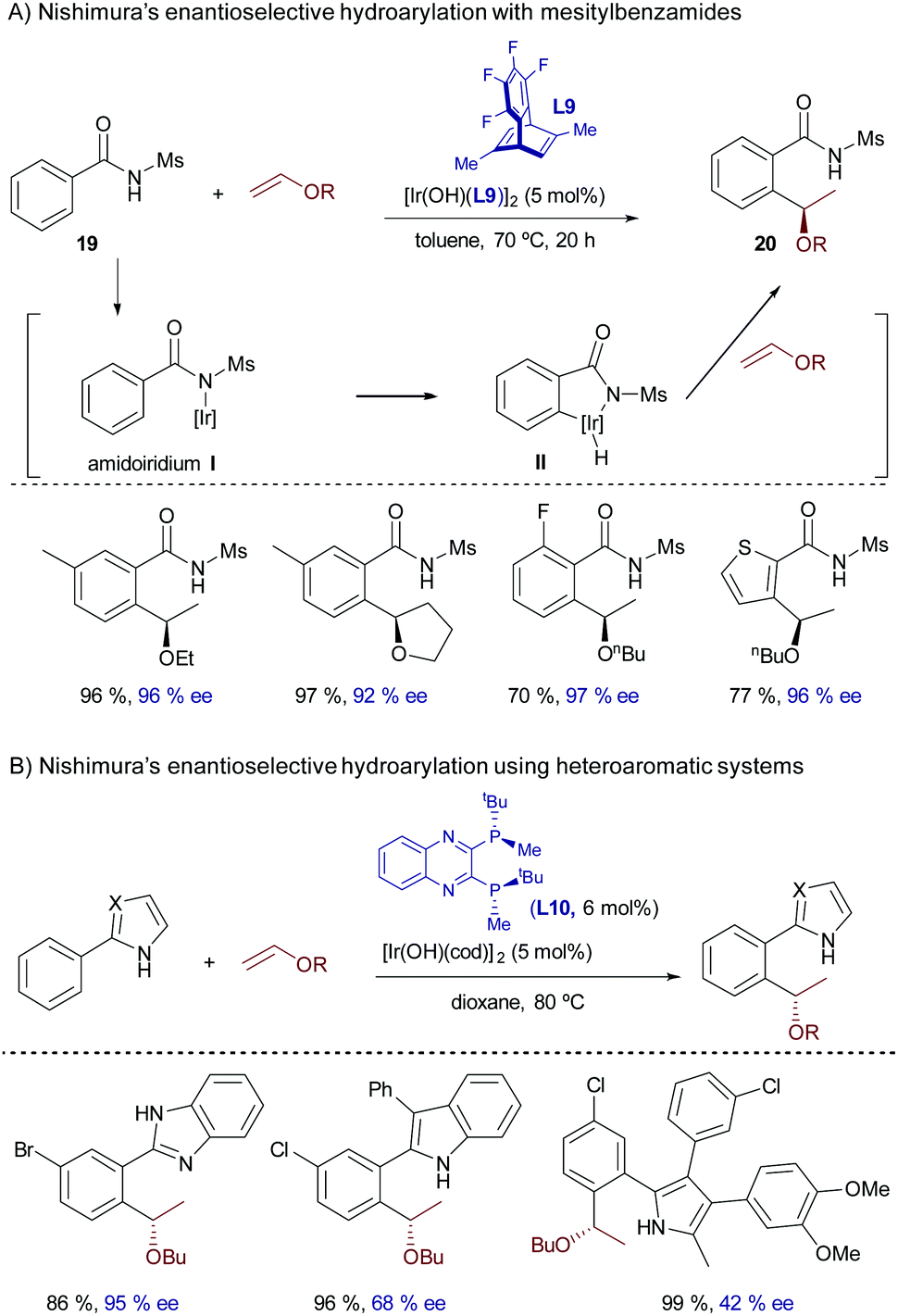
In 2016, Nishimura and coworkers extended the scope of this branched-selective hydroarylation reaction by introducing mesitylbenzamides of type 19 as C(sp2)–H donors.29 The reaction was catalyzed by a hydroxoiridium(I) complex featuring the chiral diene (S,S)-Me-tfb* (L9), and provided the corresponding products with complete branched selectivity, good yields and excellent enantioselectivities. From a mechanistic perspective, the reaction was proposed to be initiated by deprotonation of the amide by the hydroxoiridium catalyst to yield an amidoiridium species, I, that undergoes a reversible oxidative insertion with the ortho C–H bond to yield intermediate II (Scheme 12). Subsequent carbometallation and C–H reductive elimination yield the ortho-alkylated amide 20 (Scheme 12A). In consonance with the role of the hydroxide in the iridium reagent, the chloride complex [Ir(cod)Cl]2/NaBArF4 failed to promote the reaction. Remarkably, the analogous hydroxorhodium complex [Rh(OH)(cod)]2 was completely ineffective under otherwise identical reaction conditions. Although the precise factors responsible for this dissimilar reactivity were not explored, this result underscores the advantages of iridium catalysts in these types of hydrocarbonation reactions that are based on N–H deprotonations.
 | ||
| Scheme 12 Enantioselective hydroarylation of vinyl ethers developed by Nishimura. | ||
The addition reaction is not restricted to benzamides of type 19. Indeed, in a related strategy, the same group reported a highly enantioselective hydroarylation of vinyl ethers using heteroaromatic systems that contain appropriately located N–H groups to anchor the iridium complex (e.g. azoles, pyrroles, imidazoles, indoles and benzimidazoles, Scheme 12B).30 In this case, the optimal catalyst consisted of a hydroxoiridium(I) complex prepared from [Ir(OH)(cod)]2 and the chiral C2-symmetric bisphosphine (R,R)-QuinoxP* (L10).
More recently, the group extended this method to the hydroarylation of 1,3-conjugated dienes, using [Ir(OH)(cod)]2 as a catalyst.31 As in the above reaction, [Rh(OH)(cod)]2 was not effective. Importantly, species II (Scheme 13), generated after C–H insertion and hydrometallation, was detected by NMR and even isolated. Moreover, it was found to be an intermediate rather than an off-cycle resting state of the process, since a crossover experiment based on the reaction of the isolated intermediate IIa with the tosyl amide 21 led to the exclusive formation of the non-crossed product 22a (Scheme 13, bottom). Therefore, the authors proposed a mechanistic pathway involving the reaction of the hydridoiridium(III) intermediate I with the diene to afford the observed π-allyliridium(III) species II. The C–C reductive elimination was proposed to be the turnover-limiting step of the process (Scheme 13).
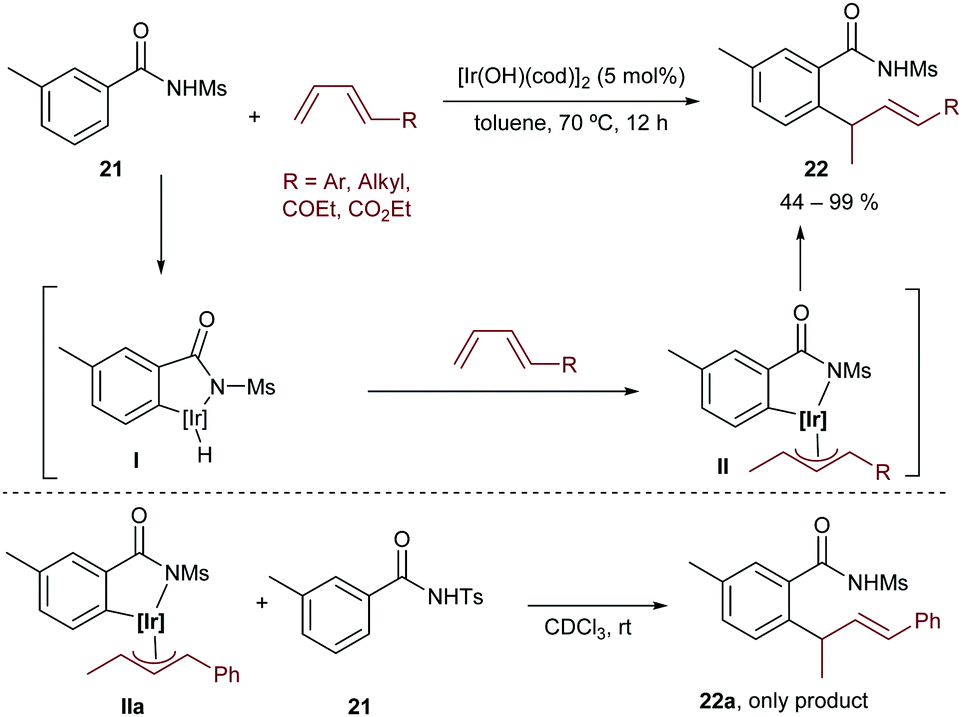 | ||
| Scheme 13 Hydroarylation of conjugated dienes developed by Nishimura. | ||
Besides vinyl ethers and conjugated dienes, Nishimura and coworkers also demonstrated that the hydroarylation chemistry can be extended to allyl ethers like 23, using substrates containing neutral directing groups, like pyridines (Scheme 14).32 In particular, the catalyst generated in situ from [IrCl(cod)]2, BINAP and NaBArF4 promoted the reaction of these ethers with 2-aryl pyridines, generating enantioenriched products of type 24 in good yields, complete regioselectivities and enantioselectivities typically above the 90% level (Scheme 14A).
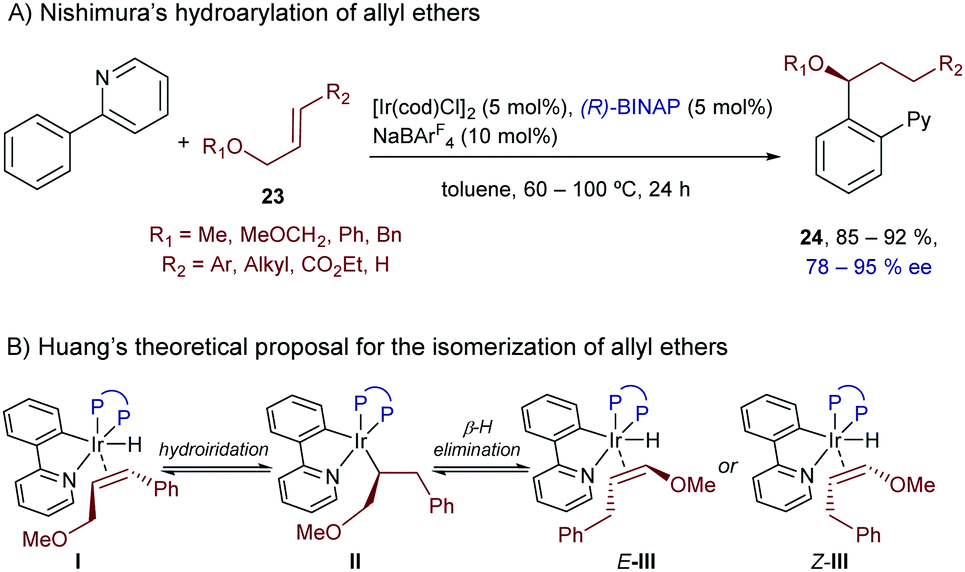 | ||
| Scheme 14 Ir-Catalyzed enantioselective hydroarylation of alkenes bearing an ether at an allylic position, developed by Nishimura. | ||
The reaction was proposed to involve an initial isomerization of the allyl ether 23 into the corresponding vinyl ether, through a hydrometallation/β-hydride elimination sequence promoted by the initially formed aryl(hydrido)iridium species I (Scheme 14B). Moreover, the authors observed H/D scrambling in both reagents, the phenylpyridine and the allyl ether, which suggests that the C–H activation and insertion steps are reversible.
Computational studies performed recently by Huang and coworkers further confirmed this reversible isomerization pathway, and suggested that both the Z- and E-isomers of the vinyl ether should be obtained after the β-hydride elimination step. Moreover, calculations proposed that the reaction of the Z and E isomers would lead to opposite enantiomers of product 24. Therefore, the excellent enantioselectivities observed experimentally are likely associated with unfavorable migratory insertions of the Z-alkenyl isomers (Z-III), for steric reasons (Scheme 14B).33
Acetanilide derivatives have also been shown to participate in iridium-catalyzed hydroarylation reactions. Indeed, in 2014, Shibata and coworkers reported an Ir(I)-catalyzed direct C-7 alkylation of N-acetyl indolines with electron deficient alkenes like acrylates.34 The catalyst [Ir(cod)2]BF4/BINAP afforded preferentially linear products of type 25 in good yields (Scheme 15), typically under harsh reaction conditions (e.g. heating at 135 °C for 24 h). Other alkenes such as styrene derivatives were also tolerated, although mixtures of linear (25) and branched (26) isomers were obtained. From a mechanistic perspective, the authors assumed a hydroiridation/C–C reductive elimination pathway, although the proposal was not validated by experimental data or computational investigations.
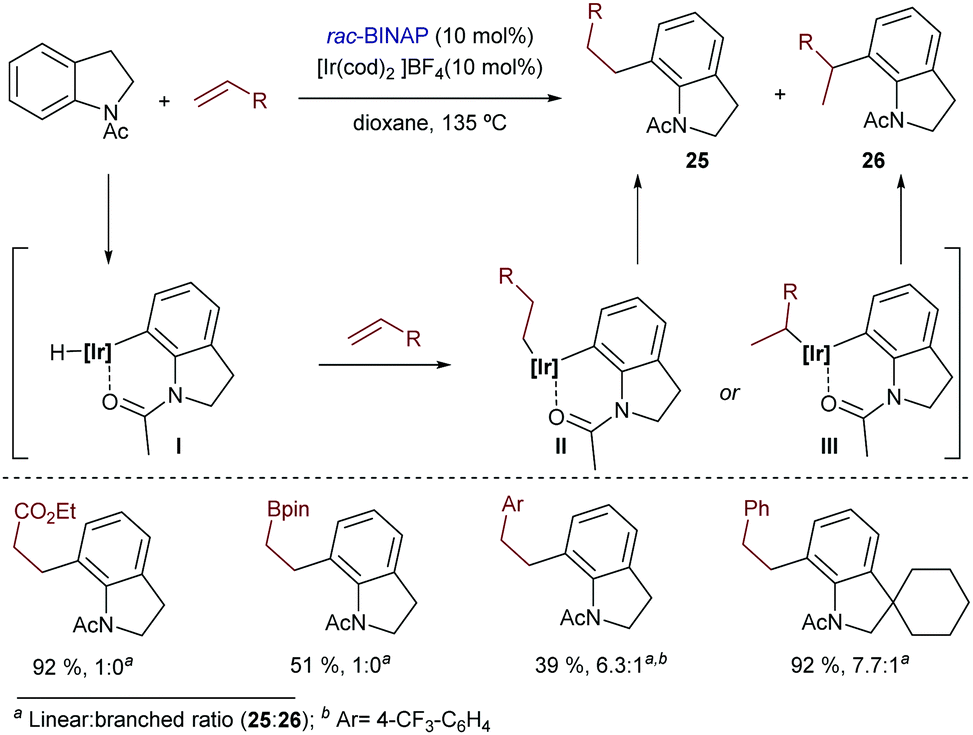 | ||
| Scheme 15 Ir-Catalyzed hydrocarbonation of acetyl indolines developed by Shibata. | ||
In this context, Bower and coworkers described a branched-selective ortho-alkylation of acetanilides with aryl- or alkyl-substituted alkenes.35 Like in the hydroarylation of alkenes with benzamides (Scheme 9), an evolution from low to high branched-to-linear selectivity was observed as the bidentate ligand changed from a narrow (e.g. dppm) to a wide bite angle (e.g. dppb). The use of pentafluorinated aryl bisphosphines (see Scheme 16A) was also key to obtain high conversions and improved branched selectivity. Thus, the best results were achieved with the iridium complex generated in situ from [Ir(cod)2]OTf and the wide-bite-angle and electron-deficient bisphosphine dFppb.
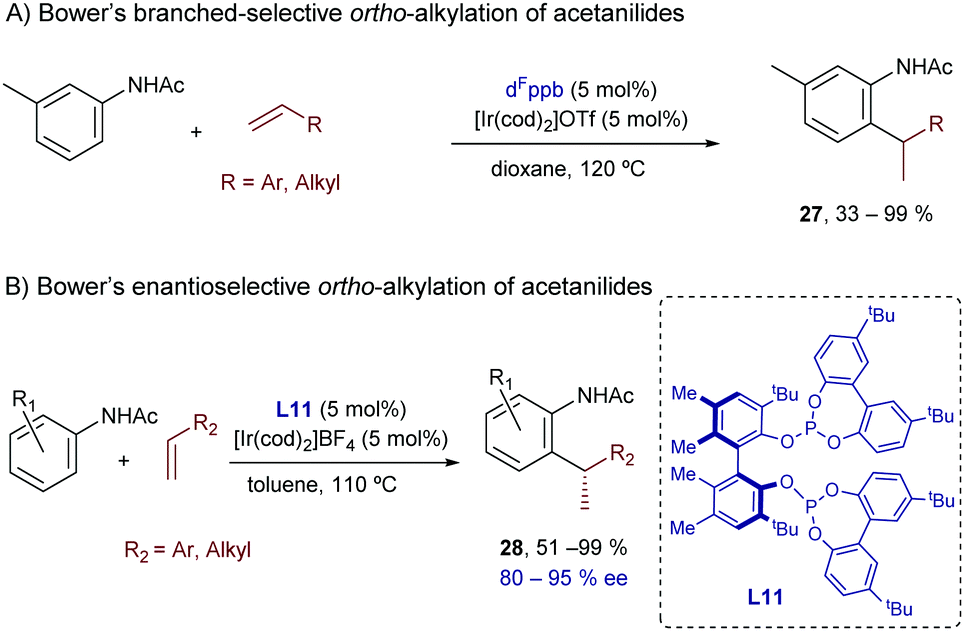 | ||
| Scheme 16 Ir-Catalyzed branched-selective hydrocarbonation of alkenes with acetanilides developed by Bower. | ||
Very recently, Bower and collaborators reported an enantioselective variant of this hydroarylation process.36 The optimal conditions were achieved by the combination of [Ir(cod)2]BF4 and the chiral bisphosphite ligand L11 (Scheme 16B). The authors carried out natural abundance 13C KIE experiments that allowed discarding a mechanism involving a C–C reductive elimination. Thus, a face-selective carbometallation, followed by an irreversible and turnover-limiting C–H reductive elimination, was proposed as the most plausible pathway.
In 2017, Shibata and coworkers developed an enantioselective hydroarylation of α,β-unsaturated carbonyl compounds with acetanilides.37 Iridium cationic catalysts featuring the chiral bisphosphines Chiraphos (L12) or Difluorophos (L13) enabled excellent enantioselectivities in favor of the β-arylated carbonyl product 29. Interestingly, and contrary to most iridium-catalyzed hydroarylation reactions, this method only works with β-substituted olefins, and proceeds with complete regioselectivity. H/D exchange experiments in the presence of D2O allowed the authors to suggest that the initial C–H oxidative addition and the hydrometallation are reversible (Scheme 17).
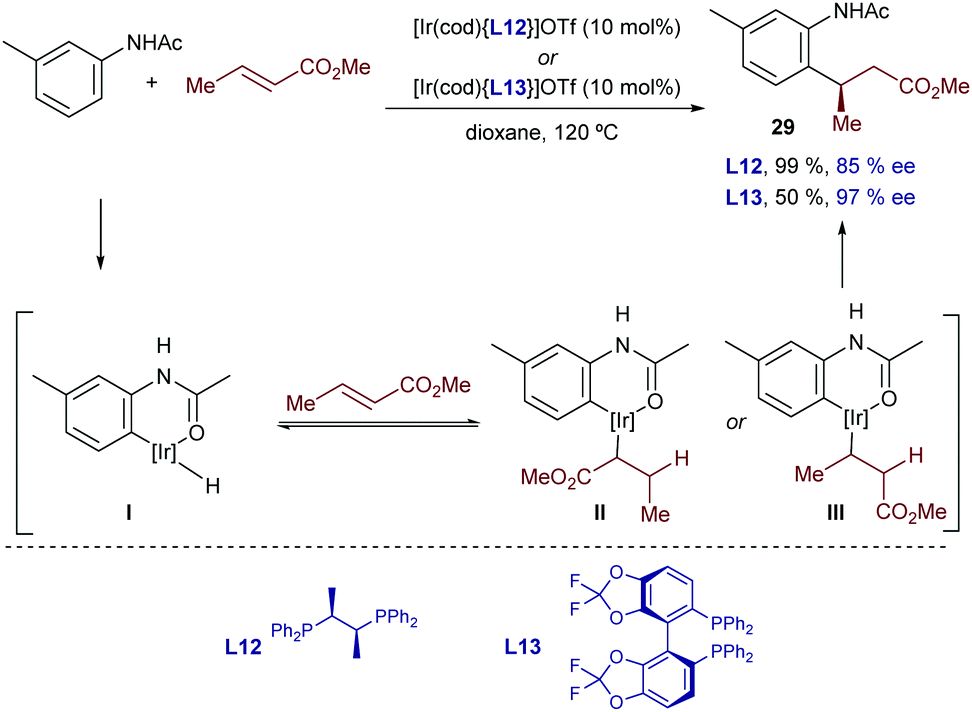 | ||
| Scheme 17 Ir-Catalyzed enantioselective hydrocarbonation of α,β unsaturated carbonyl compounds developed by Shibata and co-workers. | ||
Very recently, Yamamoto demonstrated that a chiral iridium catalyst featuring as a ligand the bisphosphoramidite (R,R)-S-Me-BIPAM (L2), previously used for the enantioselective hydroarylation of norbornene with benzamides (Scheme 4C), is also able to promote the hydroarylation of this alkene using acetanilides (Scheme 18).38 Unfortunately, despite the high yields and enantioselectivities achieved for several acetanilides, the reaction seems to be limited to norbornene as an alkene partner.
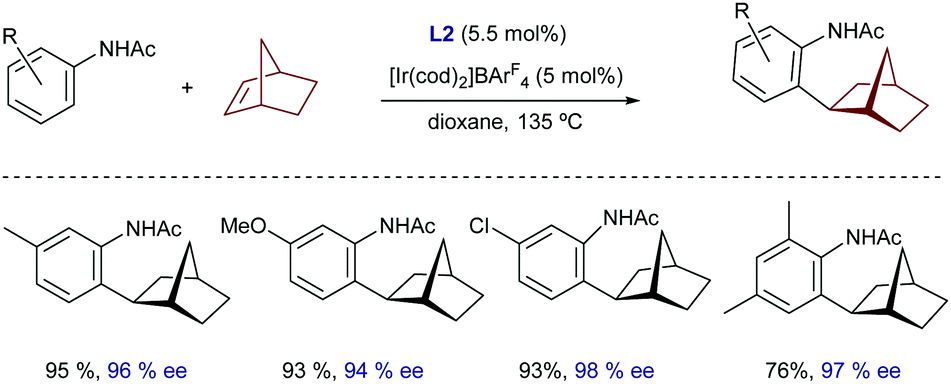 | ||
| Scheme 18 The Ir-BIPAM system for the enantioselective hydroarylation of norbornene, developed by Yamamoto. | ||
Besides the above-mentioned aromatic and heteroaromatic systems, Shibata and coworkers introduced in 2015 the use of pyridine N-oxides as substrates for Ir-catalyzed hydroarylation reactions.39 The catalyst derived from [Ir(cod)2]BArF4 and BINAP afforded the best results when ethyl acrylate was used as a partner. Other alkenes like methyl vinyl ketone, phenyl vinyl sulfone and styrene led to lower yields of the addition products (30, Scheme 19).
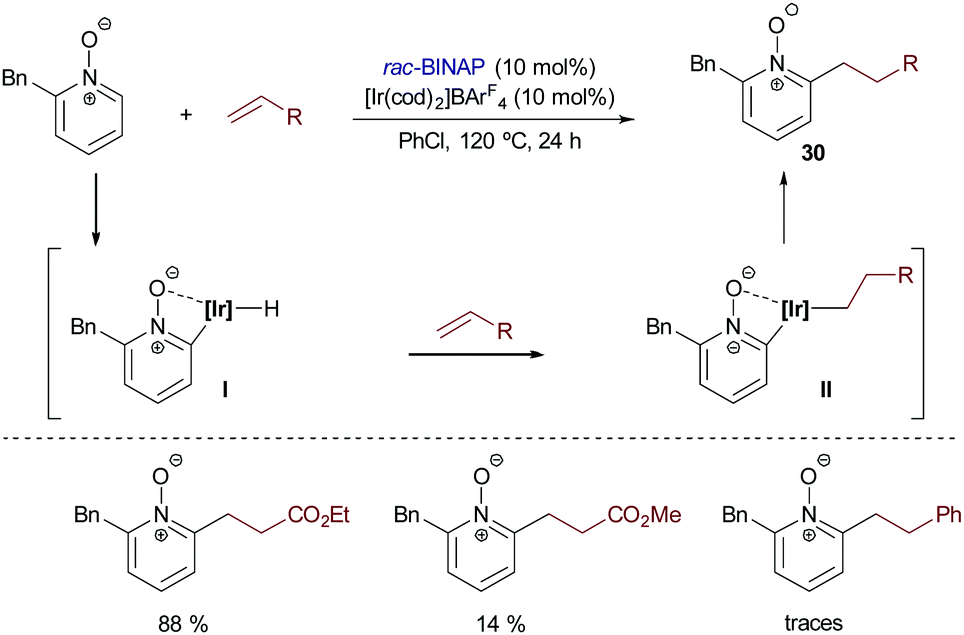 | ||
| Scheme 19 Ir-Catalyzed hydroarylation of pyridine N-oxides developed by Shibata and coworkers. | ||
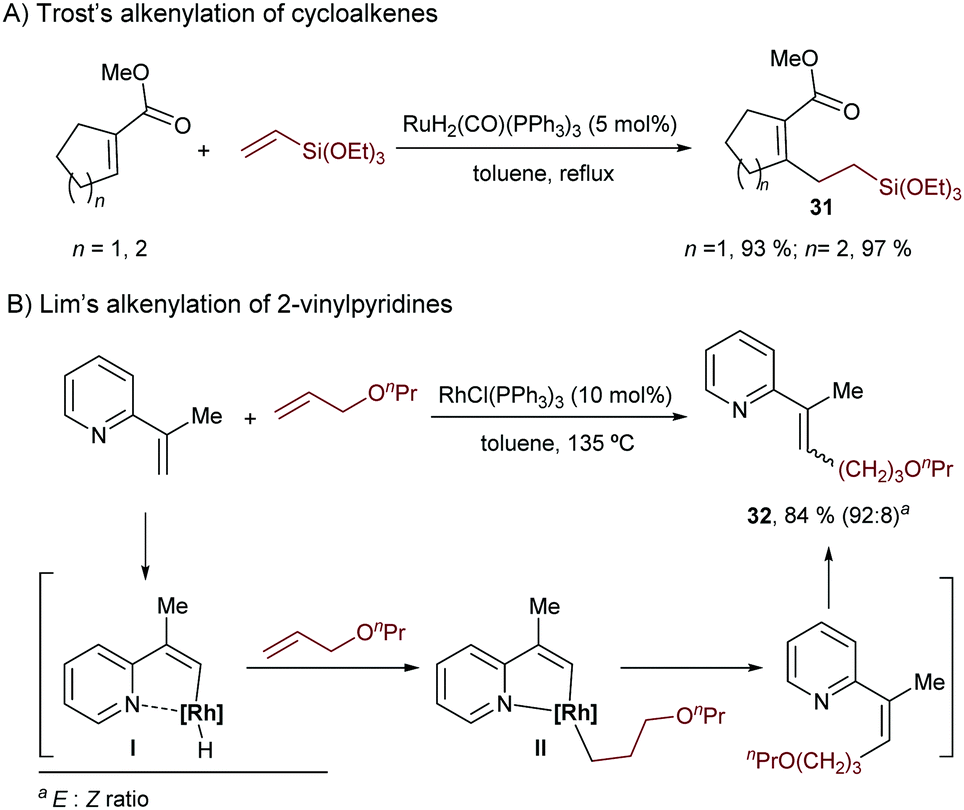 | ||
| Scheme 20 Seminal reports on the hydroalkenylation of alkenes. | ||
A few years later, in 1999, Lim and coworkers published a rhodium-catalyzed variant using 2-vinylpyridines and allyl ethers. The methodology relies on the use of Wilkinson's complex in toluene and led to the alkenes 32, as Z/E mixtures (Scheme 20B).41 Mechanistically, the authors proposed an oxidative addition to generate the rhodium hydride complex I, followed by hydrometallation of the alkene and the final C–C reductive elimination to generate the Z-isomer of the product, which would isomerize to the E-counterpart in the reaction media.
More recently, in 2014, Dong and coworkers demonstrated in an elegant study that transient enamines of type 32 (Scheme 21A), generated in situ from a ketone precursor and 7-azaindoline (25 mol%), can undergo Rh(I)-promoted activations of their C(sp2)–H bond, to subsequently insert a non-activated olefin. In particular, the reaction proved to be highly efficient when using cyclopentanones and ethylene as partners, whereas alternative ketones and olefins (e.g. propene, styrene) required the use of the ketone reactant as solvent. The overall process, after reductive elimination and enamine hydrolysis, constitutes a TMC formal addition of ketone α-C–H bonds across non-activated olefins, and provides exclusively the linear products (Scheme 21A).42 Preliminary mechanistic studies by the authors, as well as detailed theoretical investigations carried out by Wang and coworkers,43 confirmed that the 7-azaindoline generates a relatively stable enamine intermediate (32) that can undergo a facile C–H activation process. Moreover, this step is favored by a C–H agostic interaction with the metal. After the oxidative addition, the reaction would proceed via a hydrometallation and a final C–C reductive elimination step which was found to be the rate-determining step of the overall process.
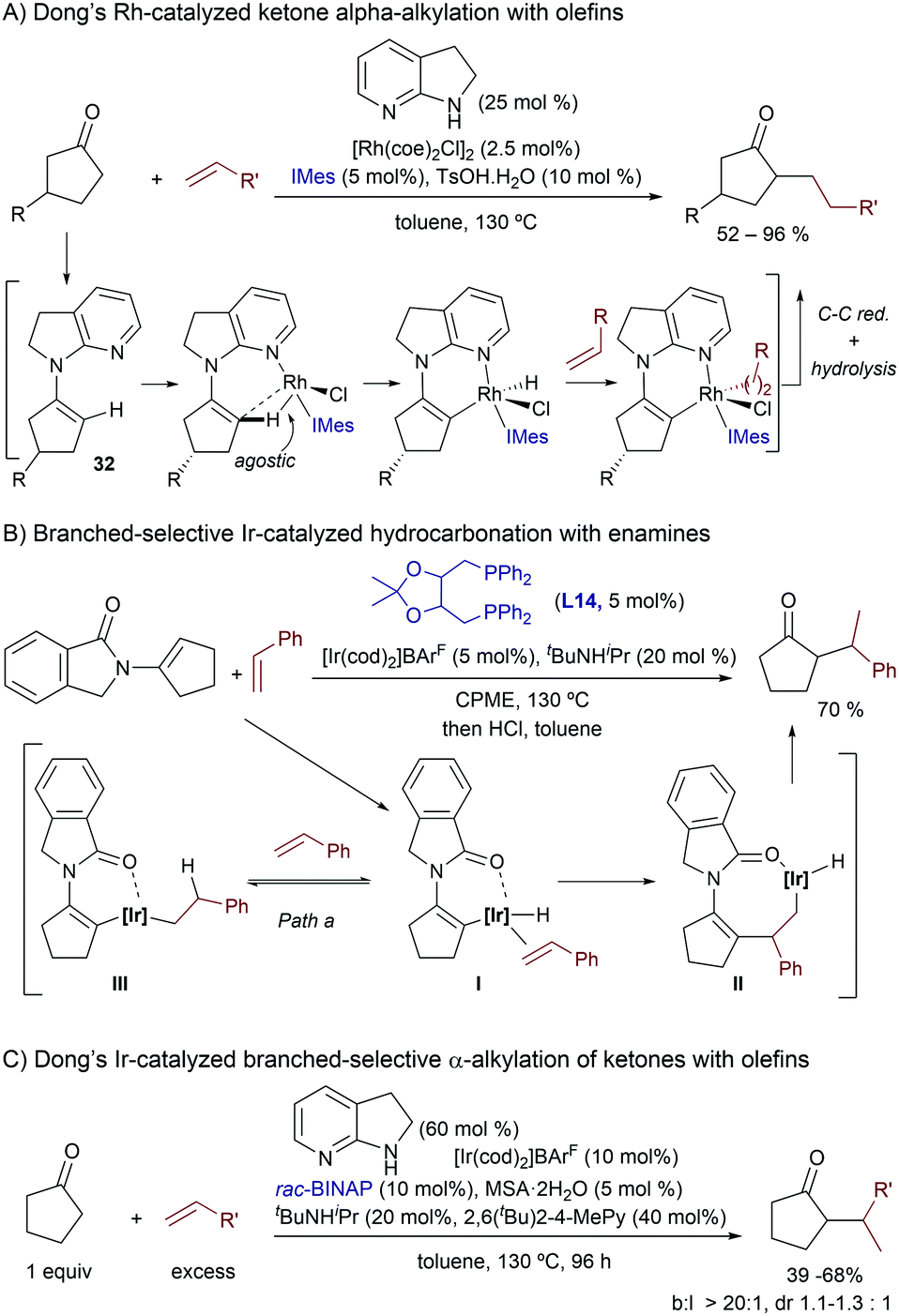 | ||
| Scheme 21 Ir-Catalyzed branched-selective ketone α-alkylation by Dong and co-workers. | ||
A few years later, the same group described a related hydrocarbonation process using iridium instead of rhodium catalysis, which renders branched alkylation products (Scheme 21B). The transformation is promoted by the cationic iridium-bisphosphine complex [Ir(cod)2]BArF4/DIOP (L14) and, contrary to the previously developed Rh-based method, the pre-installation of the enamide moiety, by condensation between the ketone precursor and isoindolin-1-one (instead of the previously used 7-azaindoline), is required (Scheme 21B).44 The carbonyl moiety of the isoindolinone group enables the activation of the C–H bond at the enamide β-position. Importantly, besides styrenes, monoalkyl substituted alkenes are also suitable coupling partners, generating α-substituted ketones bearing two consecutive stereocenters after enamide hydrolysis. Unfortunately, despite using chiral catalysts, neither the absolute nor the relative stereochemistry could be controlled, so diastereoselectivities were typically below the 1.5 to 1 ratio. Remarkably, the use of the homologous rhodium, instead of the iridium complex, under otherwise identical reaction conditions, resulted in nearly no conversion of the enamide; a result that again highlights the advantages of iridium in some of these transformations.
To gain insight into the mechanism, the authors performed deuterium labeling experiments that confirmed the reversibility of the hydroiridation step (addition of the Ir–H bond across the alkene, path a, Scheme 21B). Taking into account all the experimental observations, as well as related DFT studies,26 the authors suggested that the productive pathway would involve a carboiridation followed by a C–H reductive elimination. Moreover, subsequent DFT calculations carried out by Huang and coworkers further confirmed this hypothesis and shed light on the differences between Rh(I) and Ir(I) catalysts, as they analyzed the energy profiles of both model catalysts, [Ir(DIOP)(COD)]+ and [Rh(DIOP)(COD)]+.45 As a result, the authors proposed that the lower catalytic efficiency of Rh in this case is related to its weaker relativistic effects compared to the iridium, which leads to a significantly more endergonic C–H oxidative addition process (ΔΔG ≈ 8 kcal mol−1).
Moreover, the authors indicated that the different regioselectivity observed with Rh (typically linear) and Ir (branched) is related to the low stability of the Rh-hydride intermediate, which makes the overall energy barrier of the carbometallation/C–H reductive elimination pathway much higher for Rh than for Ir. On the other hand, the authors also found that the Rh catalyst provided significantly lower energy barriers for the C–C reductive eliminations, a result that was rationalized on the basis of the weaker nature of the C(sp3)–Rh bonds, compared to their homologous C(sp3)–Ir counterparts.45
Finally, in a new tour de force, Dong, Liu and coworkers reported in 2019 an Ir-catalyzed version of the ketone alkylation reaction, which gives branched instead of linear products.46 The reaction is promoted by [Ir(COD)2]BArF/BINAP, in the presence of a protic acid (2-mesitylenesulfonic acid), a bulky secondary amine (tBuNHiPr) and 7-azaindoline as transient directing groups, and provides the branched products with complete selectivity. The reaction works for a wide range of cyclic ketones and monosubstituted alkenes, but it is only very efficient when large excesses of both ketone and alkene reactants are used (neat). However, if the bulky 2,6-di-tBu-4-methylpyridine is used as an additive (40 mol%), the ketone can be used as a limiting reagent, although the reaction yields are usually below 60% (Scheme 21C).
In line with previous mechanistic studies, DFT calculations carried out by the authors allowed discarding a pathway based on a hydroiridation/C–C reductive elimination sequence (energy barriers of the latter step >30 kcal mol−1). Moreover, of the two possible regioisomeric carboiridation processes, the one leading to the linear product is proposed to be disfavored by steric contacts with BINAP, which are avoided throughout a branched insertion.
Finally, it is also worth highlighting that iridium catalysis can also promote intermolecular hydrocarbonations of allenes. Indeed, already in 2009, Krische demonstrated that α,β-unsaturated dialkyl carboxamides react with 1,1-dimethyl allene, in the presence of the cationic iridium complex [Ir(cod)2]BArF4/BINAP to give the corresponding prenylated product in excellent yield, as a single isomer (Scheme 22).47 Aromatic precursors do also work, and deuterium labeling experiments corroborate the hydrogen migration process. Unfortunately, despite the relevance of the transformation, the scope only included this specific allene.
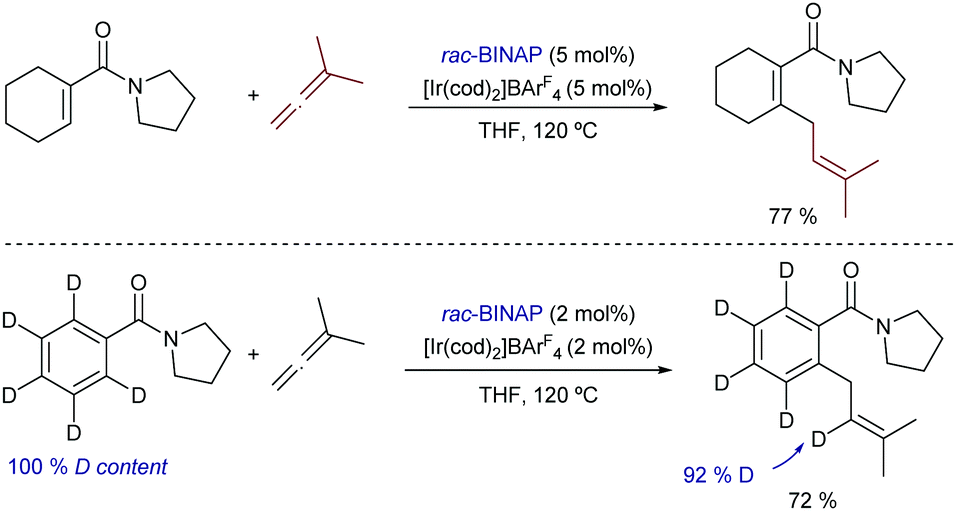 | ||
| Scheme 22 Iridium-catalyzed hydroarylation of carboxamides with allenes developed by Krische and co-workers. | ||
In contrast to the case of aromatic precursors, to the best of our knowledge, cobalt catalysts have not yet been described for intermolecular hydroalkenylations of alkenes (or allenes). This might be related to the compatibility problems of the starting precursors with the Grignard reagents which are usually used for generating the active low valent Co species. This might be solved in the future by using alternative, softer conditions to generate low-valent cobalt species, as recently demonstrated in the context of hydroalkynylation reactions (vide infra, Scheme 46).
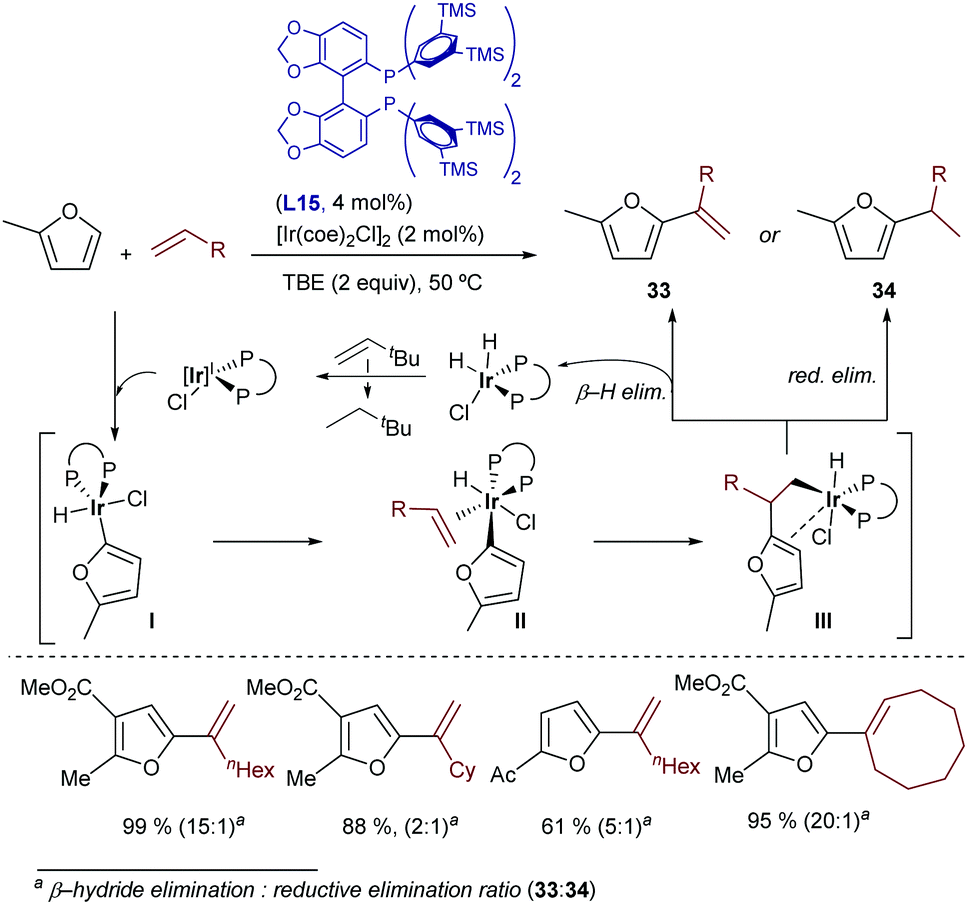 | ||
| Scheme 23 Ir-Catalyzed alkenylation of furans developed by Hartwig and coworkers. | ||
More recently, Bower disclosed a related oxidative hydroarylation that allows installing styrene moieties at the ortho position of anilides.49 The catalyst generated from [Ir(cod)2OTf] and the bisphosphine-ferrocenyl ligand L16 afforded the desired products 35 in good yields and with excellent regioselectivities, although variable amounts of hydroarylated products of type 36, resulting from a C–H reductive elimination, were typically observed [35:36 ratio from 2:1 to >20:1, depending on the properties of the acetanilide and alkene (Scheme 24)]. TBE was also used as a hydrogen acceptor to regenerate the Ir(I) catalyst. Remarkably, heteroaromatic systems such as pyrrole-1-carboximides can also be used as substrates.
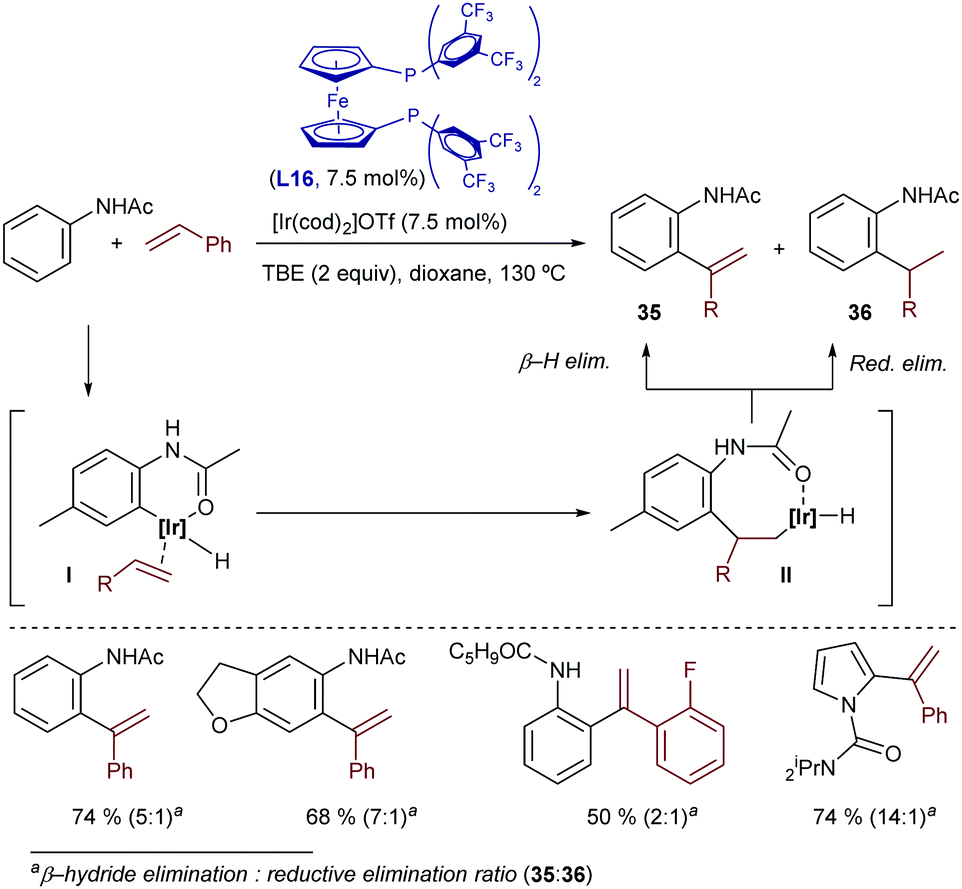 | ||
| Scheme 24 Ir-Catalyzed oxidative alkenylation of anilides developed by Bower and coworkers. | ||
Murai and coworkers also pioneered this type of intramolecular reaction, using substrates like 37, which contain a pyridine as a directing group (Scheme 25A).50 Both ruthenium and rhodium complexes were shown to be competent catalysts for this transformation, although the best results were achieved using Wilkinson's catalyst in THF at 120 °C. The authors proposed an initial activation of the C(sp2)–H bond of the alkene, assisted by coordination of the pyridine to the metal, to give a metallacyclic intermediate of type II. The olefin insertion can then take place through a hydrometallation process (III). A subsequent reductive elimination yields the hydroalkenylated product 38.
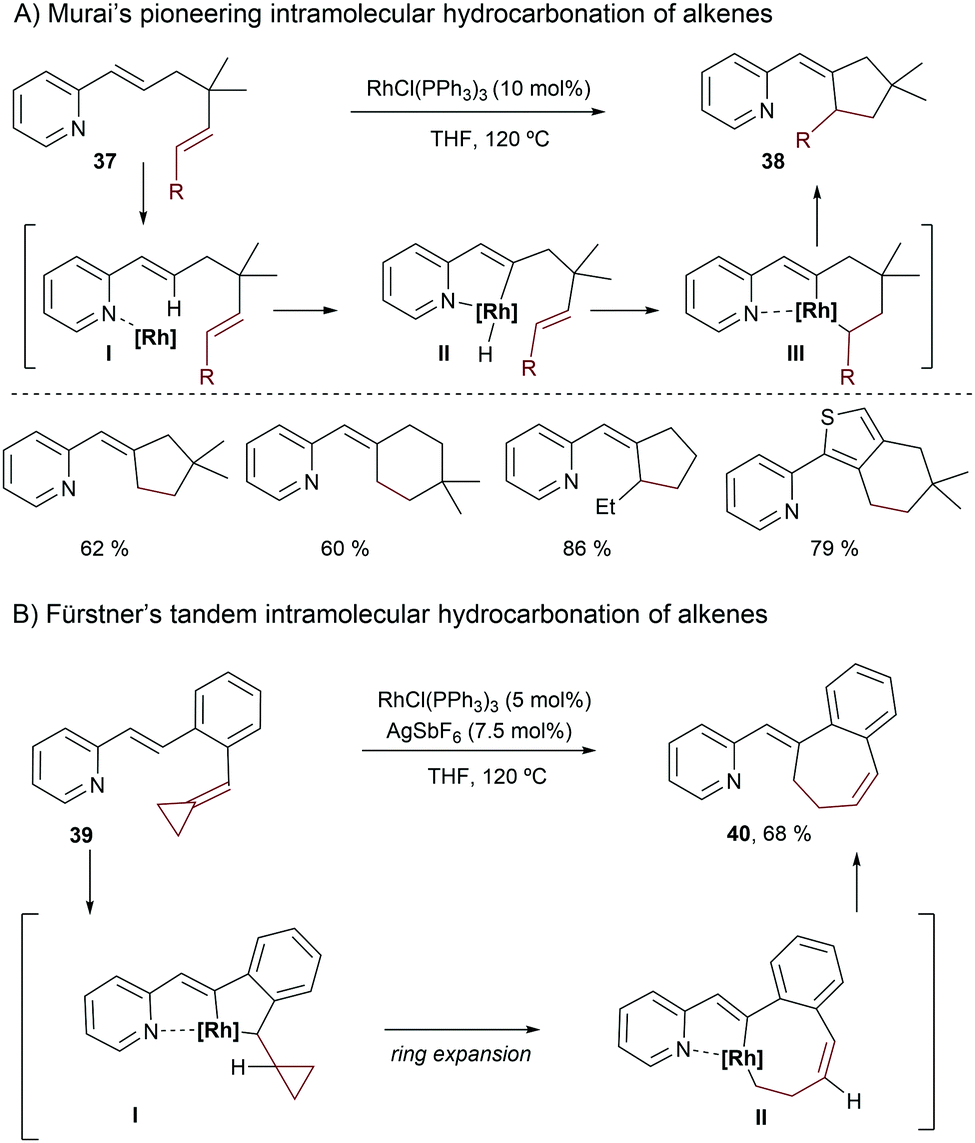 | ||
| Scheme 25 Rhodium-catalyzed intramolecular hydroalkenylation reactions using pyridine as a directing group. | ||
The group of Fürstner also reported alternative intramolecular Rh-catalyzed hydrocarbonation reactions, using substrates 39 (Scheme 25B).51 The cationic catalyst derived from treatment of Wilkinson's complex with AgSbF6 provided the best results. Remarkably, due to the presence of the alkylidenecyclopropyl moiety, a ring expansion took place after the migratory insertion to give II, which undergoes a reductive elimination to provide the seven-membered carbocycle 40.
Another landmark study in this field was reported by Ellman and Bergman in 2001, and consisted of a rhodium-catalyzed intramolecular cyclization of aromatic ketimines bearing a pendant alkene at their meta position.52 The reaction provides expedient access to useful indane, tetraline, dihydrobenzofuran and dihydroindole scaffolds (41, Scheme 26A). The optimal conditions involved the use of Wilkinson's catalyst in toluene at 150 °C and a benzyl imine as a directing group. Using the rhodium catalyst prepared in situ from [RhCl(coe)2]2 and the chiral phosphoramidite ligand (S,R,R)-L17, the reaction can be carried out in an enantioselective fashion (Scheme 26B).53 The reaction likely starts by coordination of the imine to the rhodium, followed by C–H bond insertion at the imine ortho position, to generate a rhodium hydride intermediate (I) that follows the hydrometallation/C–C reductive elimination sequence previously proposed by Murai.
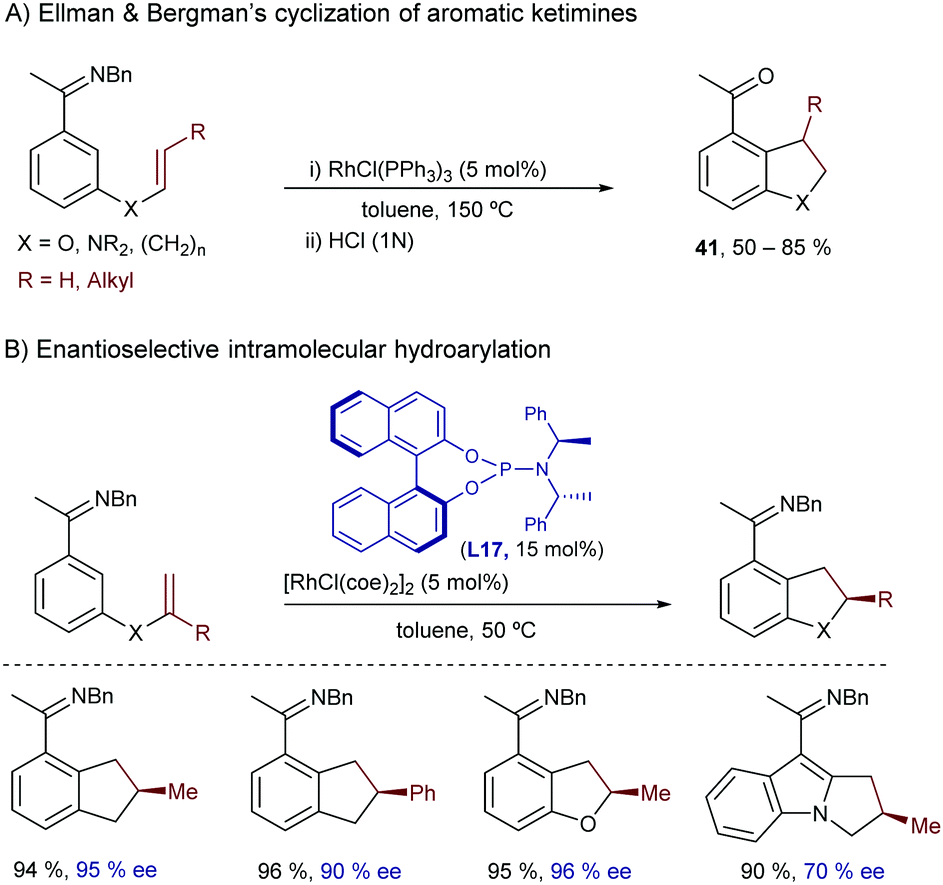 | ||
| Scheme 26 Rhodium-catalyzed intramolecular hydroarylation reactions of imines developed by Ellman. | ||
Despite the relevance of these two methods, their scopes are mostly limited to the assembly of five-membered rings. Moreover, these endo-type cyclizations have only been proven successful for the generation of products with tertiary carbon stereocenters.
Besides Rh catalysts, during the last few years, some other transition metals were also found to promote intramolecular hydrocarbonations of alkenes. For instance, Yoshikai and collaborators reported in 2013 a cobalt-catalyzed intramolecular alkene hydroarylation using indole substrates.54 The optimal catalyst was generated from CoBr2, a N-heterocyclic carbene ligand (SIMes or IPr) and the Grignard reagent Me3SiCH2MgCl in THF. Like in the intermolecular cases shown in Scheme 3B, different selectivity was also obtained depending on the source of ancillary ligand employed. Thus, in this case, using SIMes·HCl, the exo-adducts 42, containing a tertiary carbon center, are preferentially generated with good selectivity, while the bulkier IPr ligand favored the endo cyclization, to deliver products like 43 (Scheme 27). Unfortunately, the transformation is limited to indole precursors and the regioselectivity of the process (endo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) : exo ratios) also depends on the structure of the olefin tether. Deuterium labeling experiments led the authors to propose a hydrometallation-based pathway like that shown in Scheme 27. It is worth noting that, contrary to most Rh-catalyzed related processes, which usually involve reversible alkene migratory insertions and rate-determining C–C reductive eliminations, in this case, the mechanistic experiments point to the cleavage of the C–H bond as the turnover-limiting step. Indeed, the lack of H/D scrambling allowed the authors to propose that both the C–H oxidative addition and the olefin insertion steps are irreversible.
: exo ratios) also depends on the structure of the olefin tether. Deuterium labeling experiments led the authors to propose a hydrometallation-based pathway like that shown in Scheme 27. It is worth noting that, contrary to most Rh-catalyzed related processes, which usually involve reversible alkene migratory insertions and rate-determining C–C reductive eliminations, in this case, the mechanistic experiments point to the cleavage of the C–H bond as the turnover-limiting step. Indeed, the lack of H/D scrambling allowed the authors to propose that both the C–H oxidative addition and the olefin insertion steps are irreversible.
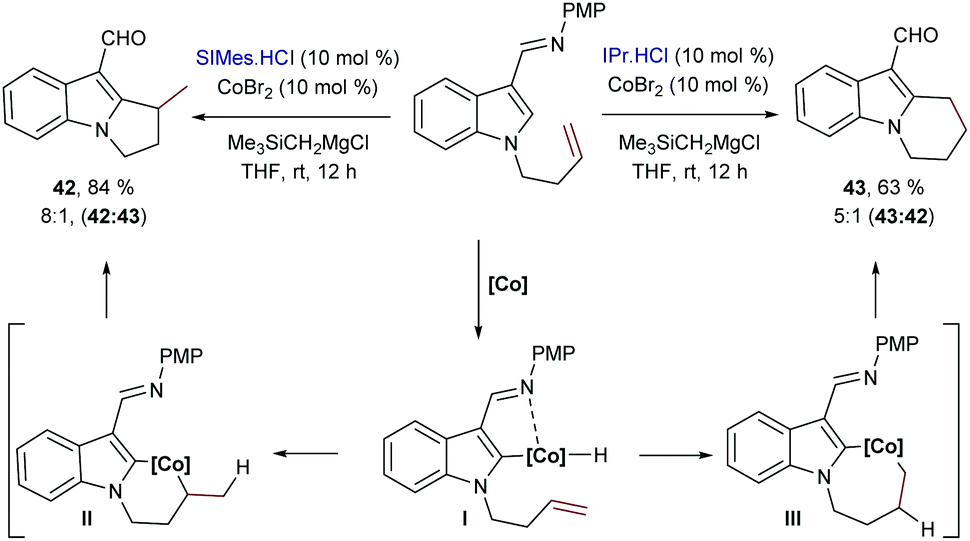 | ||
| Scheme 27 Cobalt catalyzed intramolecular cyclization of indoles developed by Yoshikai and co-workers. | ||
Iridium catalysts were not explored until quite recently. Thus, in 2015, Shibata reported a catalytic intramolecular enantioselective hydroarylation using N-tethered alkenyl indoles and a catalyst generated in situ from [Ir(cod)2]OTf and a chiral bisphosphine, Xyl-BINAP (L18) or SEGPHOS (L19, Scheme 28).55 Interestingly, depending on the substitution pattern of the alkene moiety, and the type of bisphosphine used as the ancillary ligand (L18, L19), either 5-exo or 6-endo cyclization products can be obtained. Thus, terminally unsubstituted and alkyl substituted olefins favor an exo pathway to create dihydropyrroloindoles like 45, bearing a tertiary stereocenter (Scheme 28, right). In contrast, when the terminal substituent of the alkene is an aryl group, and L18 is used as a ligand (Scheme 28, left), a 6-endo-selective cyclization occurs, delivering tetrahydropyridoindoles (44). Despite this interesting divergence, whose origin is not clear, the method is limited to indoles and tolerates a limited number of substituents in the alkene partner.
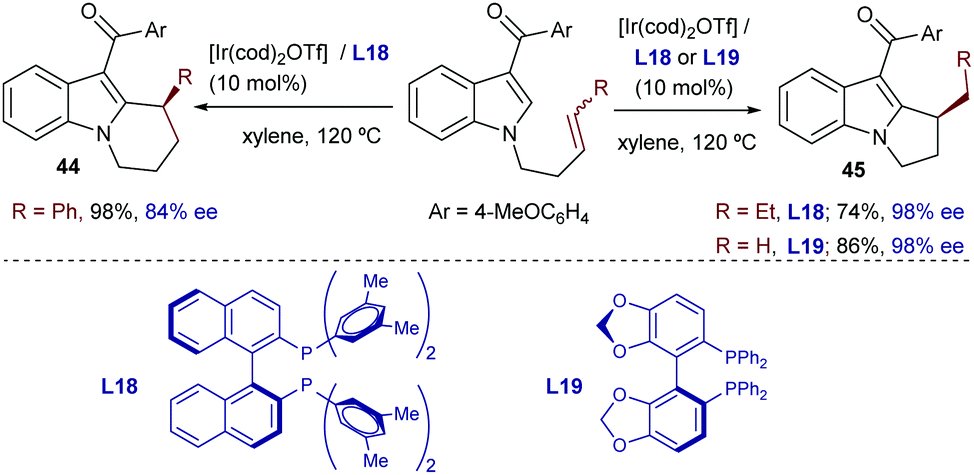 | ||
| Scheme 28 Ir-Catalyzed cyclization of indole derivatives developed by Shibata and coworkers. | ||
Moreover, neither this method nor the related cobalt-catalyzed transformation (Scheme 27) was proven to be successful for the generation of cyclic products bearing quaternary stereocenters.
In this regard, in 2017 our group reported an iridium(I)-catalyzed intramolecular hydrocarbonation reaction of alkenes that allows access to cyclic systems bearing quaternary carbon stereocenters.56 The method relies on the synergistic ability of the iridium/bisphosphine catalyst and a dialkyl carboxamide directing group to selectively activate an adjacent C(sp2)–H bond and direct the selective exo-insertion of a tethered olefin (Scheme 29A). From all the different rhodium and iridium complexes tested, we found that the catalyst generated from Ir(cod)2BArF4 and the electron deficient bisphosphine ligand dFppe was the most efficient to give the cyclic product 46. The transformation works very well for a variety of α,β-unsaturated amides as well as for heteroaromatic and aromatic amides.
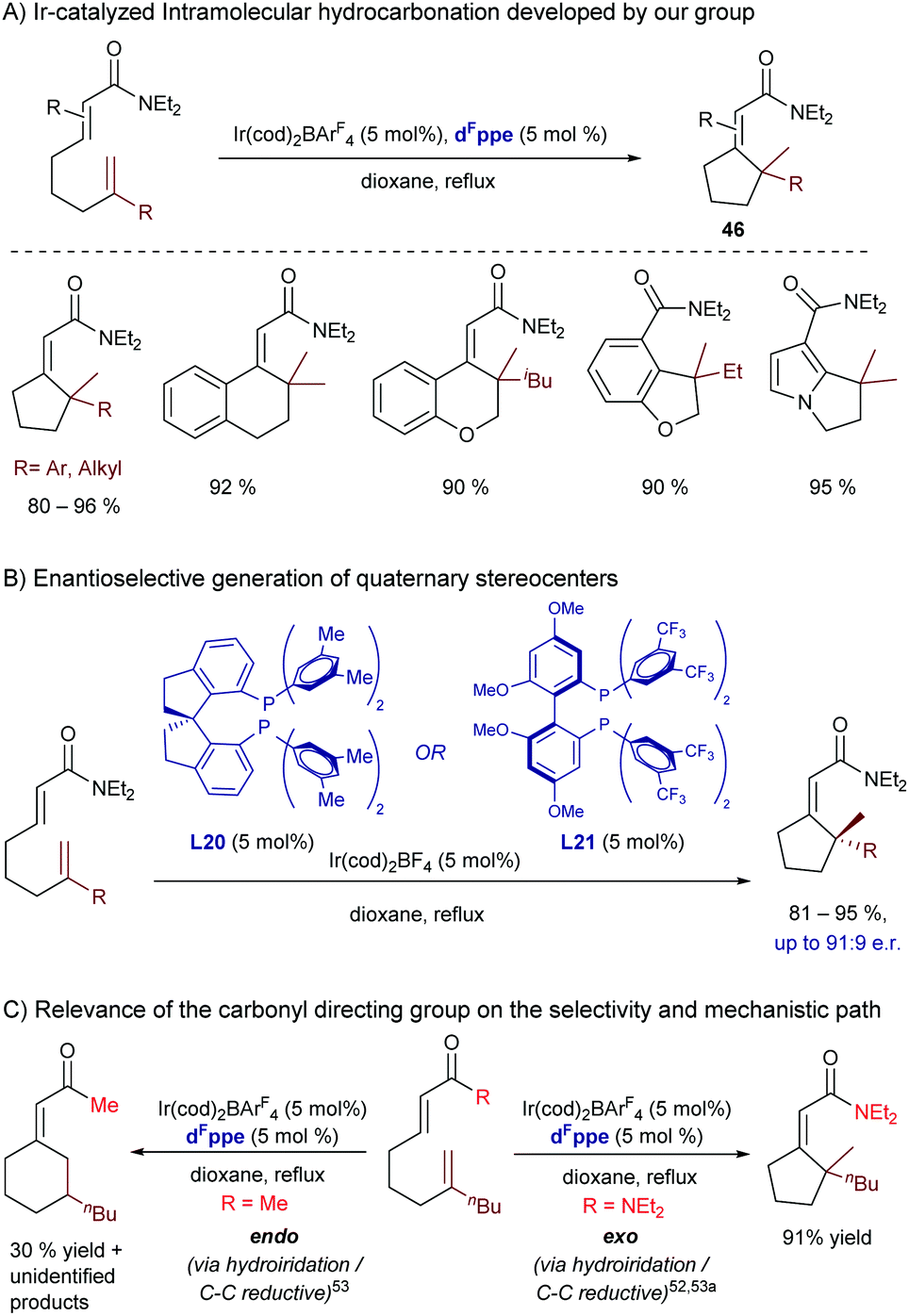 | ||
| Scheme 29 Ir-Catalyzed intramolecular hydroalkenylation developed by our group, which allows building cycles with quaternary stereocenters. | ||
Interestingly, we also reported a preliminary enantioselective version (Scheme 29B), using the chiral bisphosphines BTFM-Garphos (L21) and DM-SDP (L20). These reactions led to the highest enantiomeric ratios so far reported for a metal catalyzed hydroalkenylation process. Interestingly, the products can be manipulated in a straightforward manner to obtain synthetically challenging cyclic ketones bearing α-quaternary chiral stereocenters.
Deuterium labelling studies confirmed that the reaction starts by oxidative addition to generate an iridium-hydride species. DFT calculations carried out by Huang and coworkers suggested that after this C–H activation the exo-cyclization product is generated by migratory insertion of the alkene into the iridium–carbon bond, followed by C–H reductive elimination.57 Importantly, these DFT calculations corroborated our experimental results which indicated that the nature of the carbonyl directing group has a significant influence on the type of migratory insertion. Thus, they confirmed that the use of a dialkyl amide is key to obtaining the exo-selective cyclization product via the above-mentioned path, whereas the endo-cyclization counterpart, which was experimentally observed when using a methyl ketone as a directing group (Scheme 29C), would be the result of an alternative hydrometallation/C–C reductive elimination sequence. In this regard, the calculations pointed out that the use of the ketone as a directing group decreases the energy barrier of the C–C reductive elimination by ≈4 kcal mol−1, which eventually enables the selectivity switch. The weaker coordinating ability of the ketone, compared to that of the dialkyl amide, would indeed provide an electron poorer metal center that is more prone to suffer C–C reductive eliminations.58
In 2018, Shibata and coworkers reported an intramolecular hydroarylation of α,β-unsaturated acrylates with acetanilides to yield chiral γ-lactones of type 47, featuring quaternary carbon stereocenters (Scheme 30).59 The optimal conditions involved the use of [Ir(cod)2OTf] and BINAP. Although the scope is modest, the chiral γ-lactones can be obtained in good yields, and with moderate to good enantioselectivities.
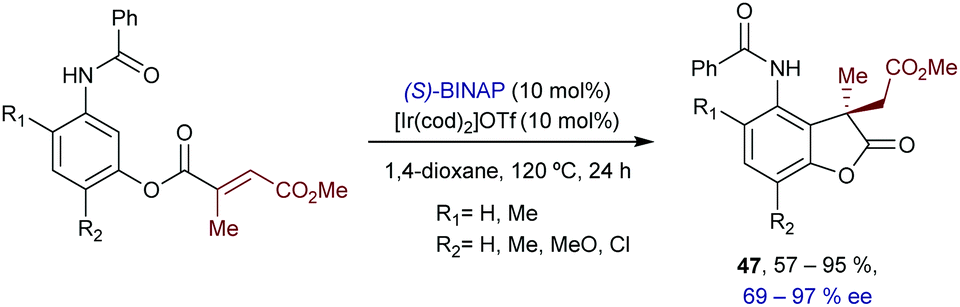 | ||
| Scheme 30 Ir-Catalyzed intramolecular hydroarylation of acrylates developed by Shibata. | ||
To sum up Section 2.1, in recent years there have been important advances in the development of hydrocarbonation reactions of alkenes with different sources of C(sp2)–H bonds, using group IX transition metals as catalysts. Albeit initially overlooked, iridium catalysts have demonstrated to be quite powerful, and to present advantages over the rhodium counterparts in terms of reactivity control and selectivity, including enantioselectivity. Nonetheless, there is a need to develop catalysts that present a broader scope and improved regio-, diastereo- and enantioselectivity profiles, specially to build products bearing chiral quaternary carbon stereocenters. Moreover, addition reactions with substrates that do not need the presence of a directing group for the C–H activation are of clear interest.
2.2 Additions to alkynes
In 1995, shortly after describing the hydrocarbonation of alkenes, Murai and coworkers described a ruthenium catalyzed intermolecular hydroarylation of alkynes using arenes and heteroarenes that bear a ketone to direct the C–H insertion of the metal (Scheme 31).60 Although the reaction requires high temperatures, it allows access to a variety of β-vinyl substituted ketones (48) in good yields and moderate to excellent E/Z selectivities. They proposed a mechanism involving a hydrometallation and a C–C reductive elimination.
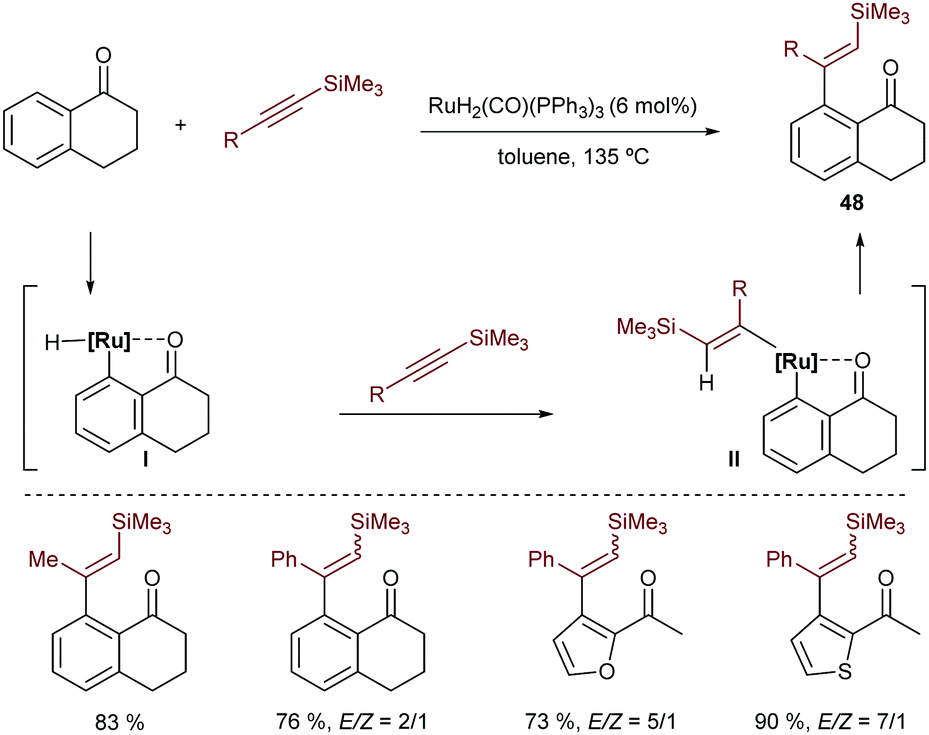 | ||
| Scheme 31 Ru-Catalyzed regioselective hydroarylation of alkynes developed by Murai and coworkers. | ||
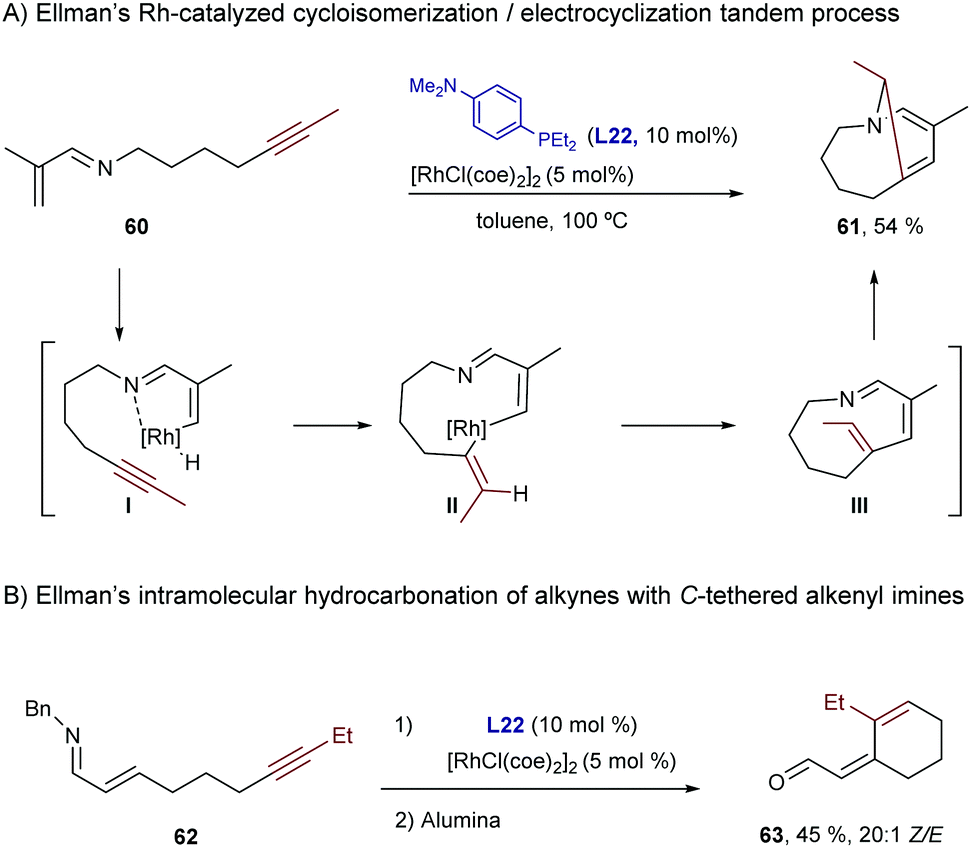
In 2006, Ellman and Bergman described a Rh-catalyzed hydrocarbonation reaction of alkynes with α,β-unsaturated imines.61 Curiously, when disubstituted alkynes were used, the resulting hydrocarbonated products (49) evolved further through an electrocyclic ring closure to afford dihydropyridines (50, Scheme 32).62
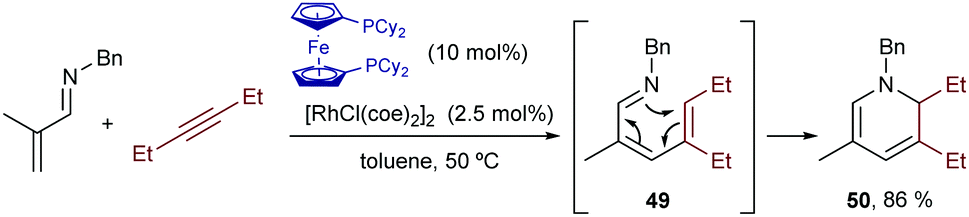 | ||
| Scheme 32 Rh-Catalyzed addition to alkynes followed by an electrocyclic ring closure, developed by Ellman and coworkers. | ||
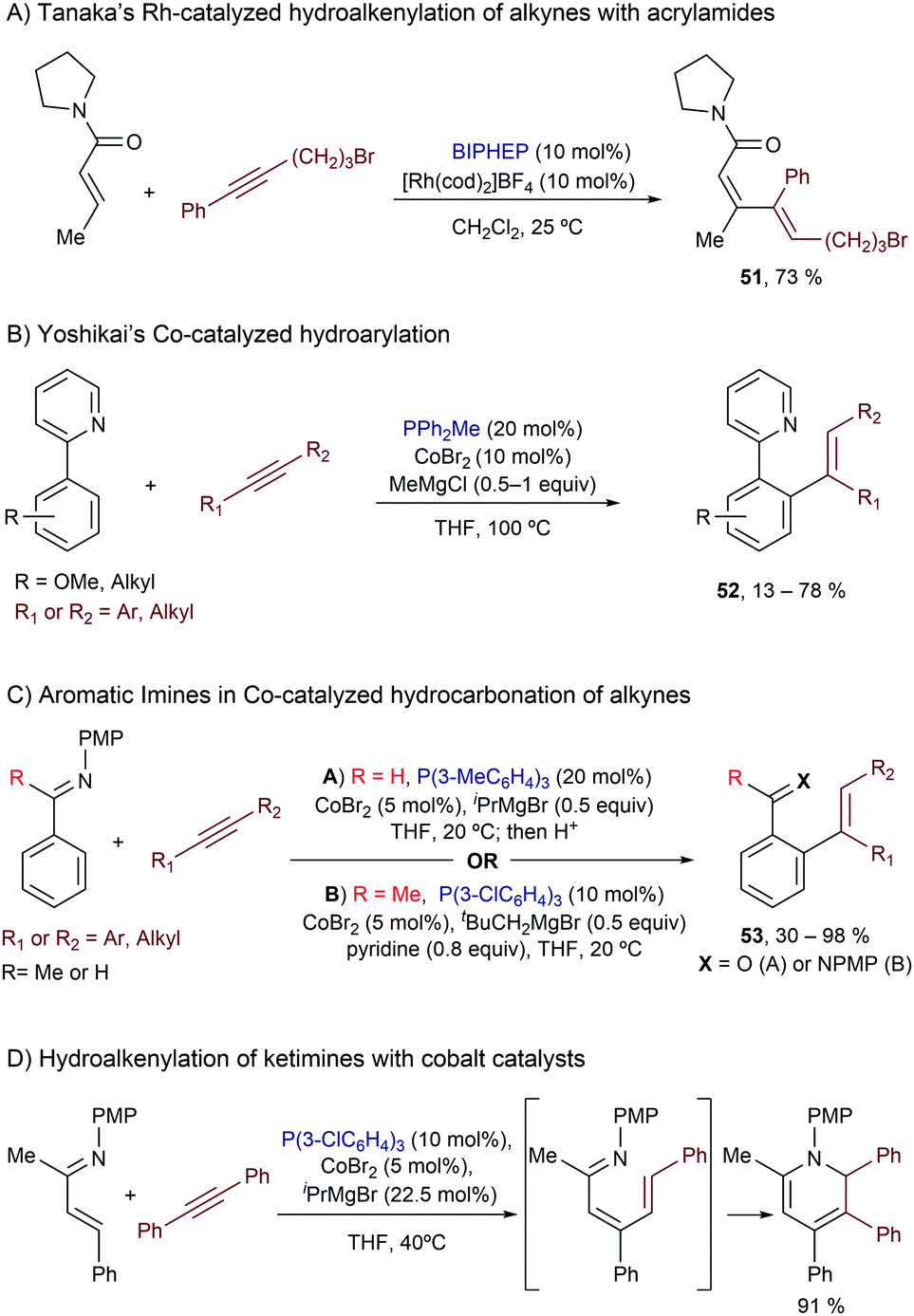
Related intermolecular processes with low valent Rh catalysts were also described by Tanaka and coworkers, but dialkyl carboxamides were used as directing groups for C–H activation.63 The optimal conditions involved the use of [Rh(cod)2]BF4 and BIPHEP in dichloromethane at room temperature, and the reactions provided dienes of type 51 with almost complete selectivity (Scheme 33A). Curiously, the use of a 1-pyrrolidinecarbonyl group was essential for this transformation, as dimethyl amides, carboxylic esters and ketones were less effective.
 | ||
| Scheme 33 Examples of intermolecular Rh- and Co-catalyzed hydrocarbonations of alkynes. | ||
Cobalt complexes can also be used for related catalytic transformations. Thus, in a series of papers from 2010 onwards, Yoshikai and coworkers demonstrated that ortho-aryl pyridines as well as aromatic ketimines and aldimines (Scheme 33B and C) participated in hydroarylation reactions of alkynes, promoted by cobalt-phosphine catalysts.64 Each of these reactions required the careful choice of ligand and of Grignard reagent used to generate a low-valent cobalt catalyst; however, under optimized conditions, the method proved to have a considerable scope and required much lower temperatures than those involving rhodium catalysts. On the other hand, the use of α,β-unsaturated imines is also possible, and similarly to the Rh-catalyzed reactions, the hydroalkenylation is followed by a 6π-electrocyclization to afford a dihydropyridine derivative (Scheme 33D).65
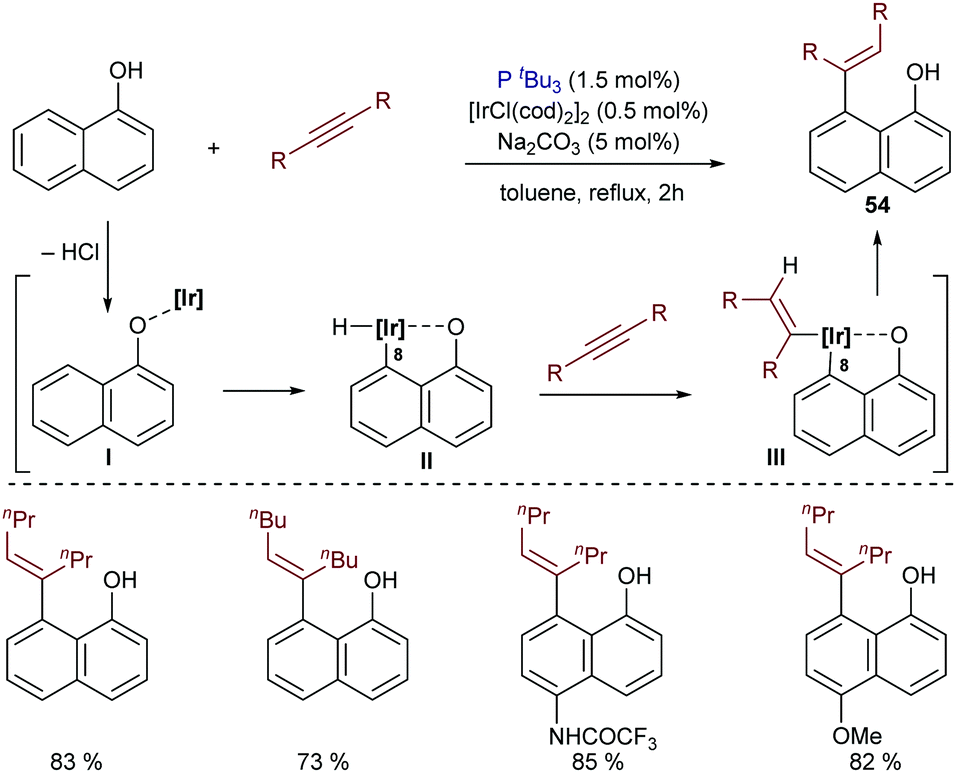
The application of iridium catalysts in this type of hydrocarbonation of alkynes can be traced back to 1999. In particular, Satoh and Miura reported a pioneer case of an iridium-catalyzed hydroarylation of alkynes with 1-naphthols, mediated by the catalyst generated in situ from [IrCl(cod)2]2 and P(tBu)3. Catalytic amounts of Na2CO3 were also employed. The authors suggested a mechanistic pathway that involves the initial formation of a naphtholate complex (I) by liberation of HCl (Scheme 34). Then, an oxidative addition of the C(8)–H bond to the iridium center generates II, which undergoes alkyne hydrometallation followed by a C–C reductive elimination to deliver the cis-hydroarylated product 54.66,67
 | ||
| Scheme 34 Iridium-catalyzed hydroarylation of alkynes with naphthols developed by Miura and coworkers. | ||
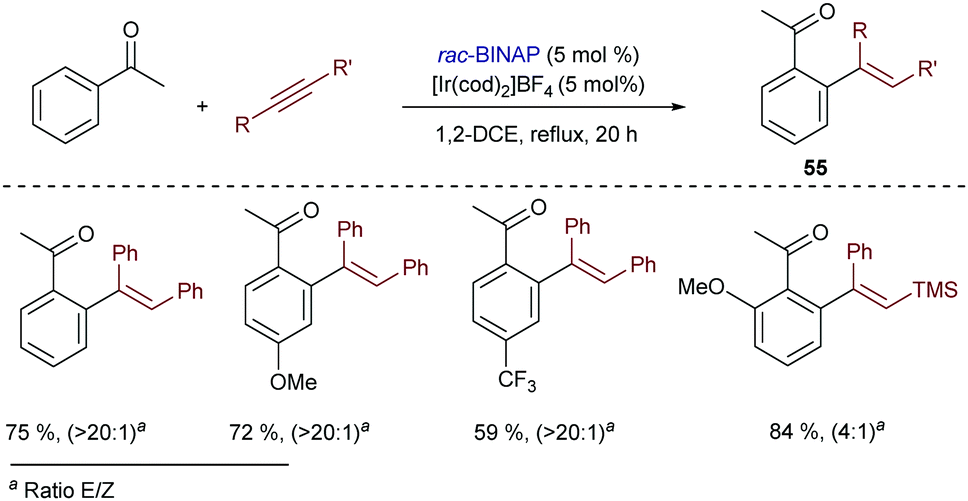
Iridium catalysts were also effective in the addition of aryl ketones to alkynes, to give the corresponding ortho-alkenylated products (55) in good yields (Scheme 35).68 The best results were obtained with the Ir catalyst generated from [Ir(cod)2]BF4 and BINAP, similarly to the hydroarylation of styrene (Scheme 5). The analogous rhodium complexes provided the same products but with lower efficiency (40% lower yield in a model reaction). Contrary to the conditions previously found by Satoh and Miura for the functionalization of naphthols (Scheme 34), monodentate ligands such as PPh3 and PCy3 were not suitable for this transformation.
 | ||
| Scheme 35 Ir-Catalyzed hydroarylation of alkynes with aryl ketones developed by Shibata and coworkers. | ||
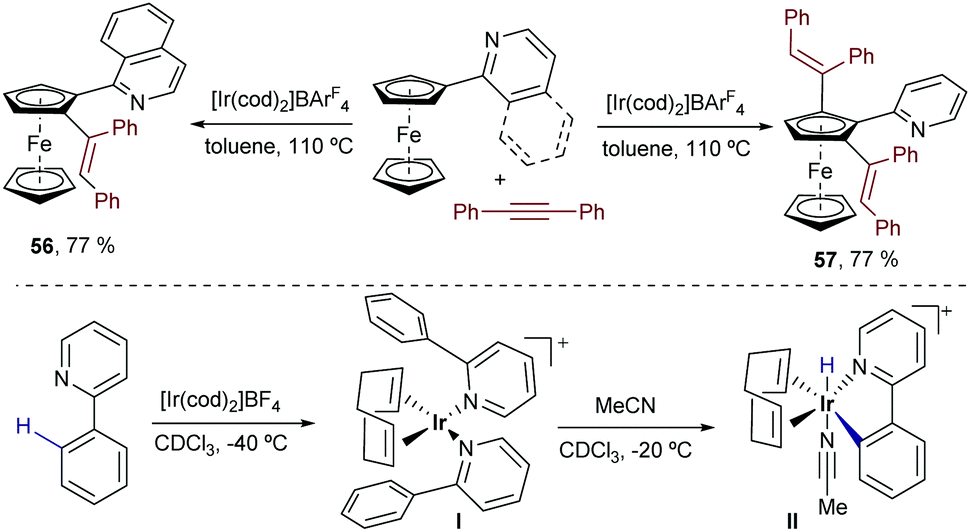
In 2012, Shibata and coworkers described a catalytic alkenylation of ferrocenes, promoted by [Ir(cod)2]BArF in refluxing toluene.69 Curiously, the reaction can give mono- or dialkenylated products depending on the bulkiness of the directing group. Thus, while a pyridine moiety makes the reaction selective towards the ortho-dialkylated product 57 (Scheme 36, right), a quinoline directing group yields almost exclusively the monoalkylated ferrocene 56 (Scheme 36, left).
 | ||
| Scheme 36 Ir-Catalyzed alkenylation and alkylation of ferrocenes developed by Shibata and coworkers. | ||
As expected, the reaction is initiated by chelation-assisted oxidative addition of a C–H bond of the cyclopentadienyl (Cp) ring to the iridium(I) complex. Indeed, by using ortho-phenyl pyridine instead of the ferrocenyl derivative, the authors were able to isolate an Ir(III)–hydride complex (II) that is generated by adding MeCN to the initially formed iridium(I) complex I (Scheme 36, bottom).
N-Sulfonylbenzamides are also appropriate substrates for the addition to alkynes, in a reaction catalyzed by the hydroxoiridium complex [Ir(OH)(cod)]2 (Scheme 37).70 Similarly to the hydroarylation of dienes with the same substrates (Scheme 13), this methodology does not require the use of phosphines or basic additives. The authors proposed that the catalytic cycle starts with the deprotonation of the acidic benzamide 58 by the iridium-hydroxide species to generate an amidoiridium species (I) that promotes an ortho C–H activation to yield a hydridoiridium metallacycle, II (Scheme 37). An alkyne migratory insertion (hydrometallation) and a turnover limiting C–C reductive elimination yield the addition product 59.
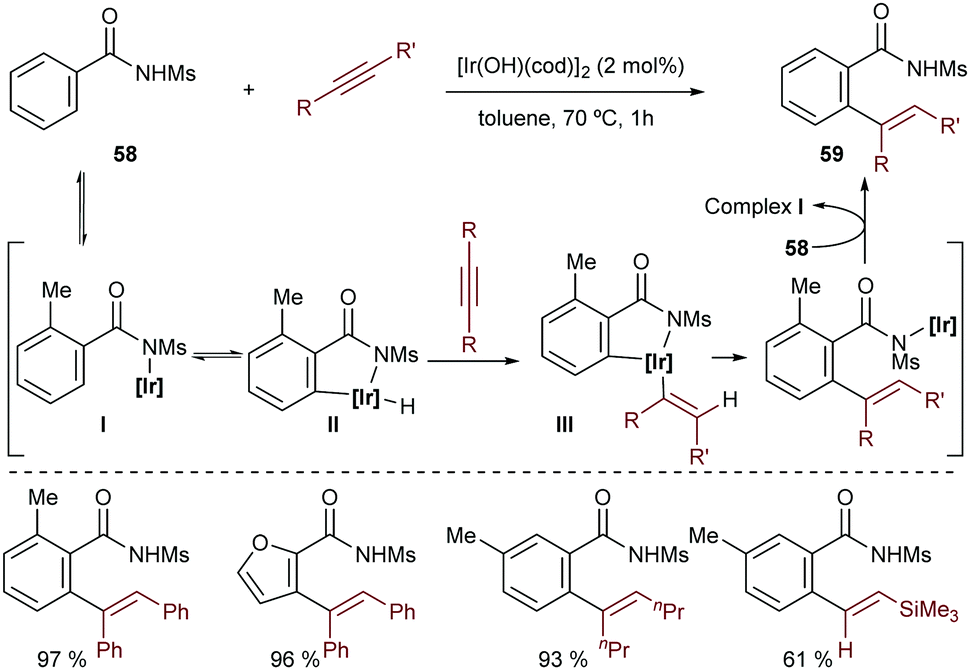 | ||
| Scheme 37 Hydroarylation of alkynes by a hydroxoiridium complex, by Nishimura and co-workers. | ||
 | ||
| Scheme 38 Rh-Catalyzed intramolecular alkyne addition reactions developed by Ellman and coworkers. | ||
In the case of N-tethered alkynyl imines, the authors proposed a mechanism involving an imine-directed C–H activation at the β-position of the alkene to form the vinyl rhodium intermediate I. A regioselective hydrometallation followed by C–C reductive elimination yields the cyclic imine III, which undergoes a concomitant electrocyclization to give the observed product (Scheme 38A).
Iridium catalysts have been scarcely used in this type of annulation. In 2017 Shibata and co-workers described an iridium-catalyzed intramolecular addition of N-tethered alkynyl indoles to alkynes.72 Remarkably, the selectivity of the cyclization could be tuned by the choice of metal catalyst. Thus, while an iridium catalyst generated from [Ir(cod)2]OTf and BINAP promoted a 6-exo-dig cyclization to yield the tricyclic product 65, a homologous Rh catalyst induced an alternative 7-endo-dig cyclization that delivers the seven-membered cycle 64 (Scheme 39, left). On these bases, the authors proposed that, after an initial chelation-assisted C–H activation to give I, a trans hydrometallation would occur in the case of the rhodium catalyst, to yield an eight-membered rhodacyclic intermediate, III. However, with the iridium catalyst, an exo-selective hydrometallation to afford intermediate II was proposed. Then, after the corresponding C–C reductive eliminations, the six- and seven-membered fused cycles were generated.73 The precise reasons for these metal-dependent regioselectivities were not discussed.
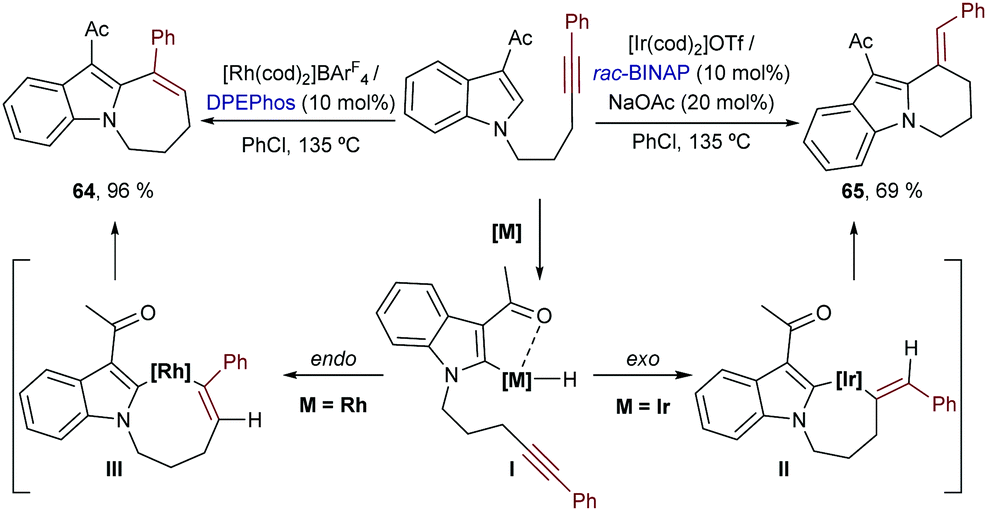 | ||
| Scheme 39 Metal-dependent intramolecular hydroarylation of alkynes developed by Shibata and co-workers. | ||
Shortly after this report, our group disclosed a more general intramolecular Ir-catalyzed exo-cyclization that is not restricted to the activation of (hetero)aromatic C–H bonds, as it also allows the participation of more challenging C–H bonds from alkenes bearing a diethylcarboxamide as the directing group. In this latter case, the process generates exocyclic dienes 66 with complete control of the stereoselectivity (Scheme 40A).74 Remarkably, using ketones or carboxylic esters as directing groups the reactions did not occur. The synthetic power of the methodology was further exemplified by coupling the cycloisomerization reaction with a [4+2] cycloaddition between the generated diene and a dienophile in a one-pot tandem process. Thus, stereochemically rich polycarbocyclic products 67 with up to four stereocenters were readily assembled from simple acyclic precursors (Scheme 40B).
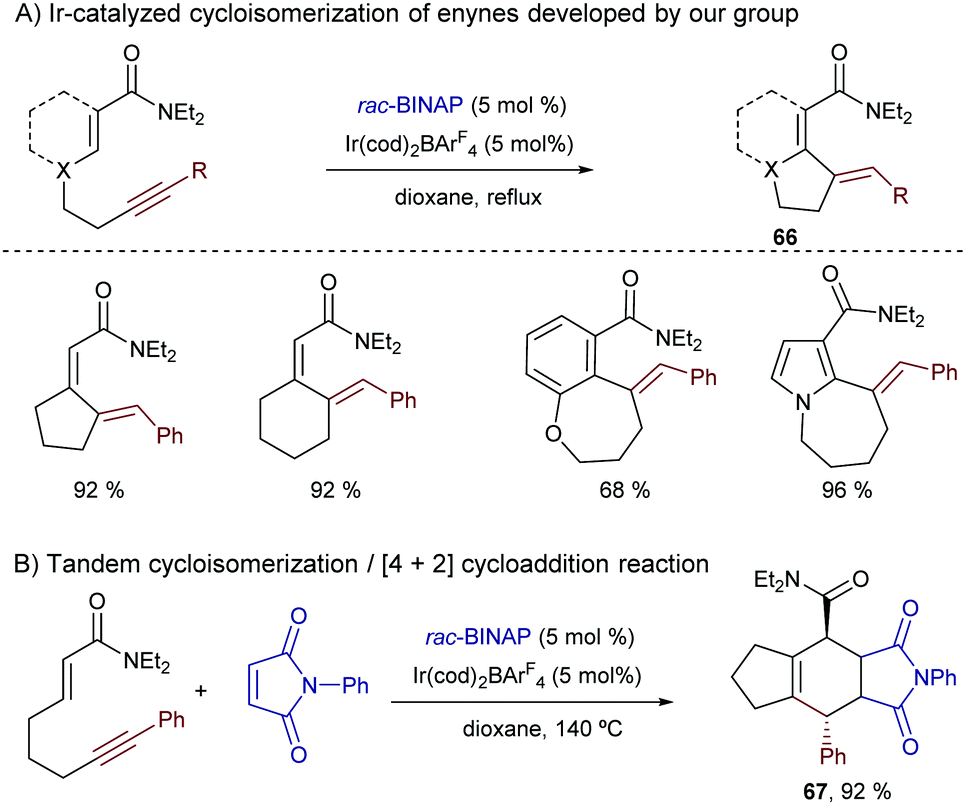 | ||
| Scheme 40 Ir-Catalyzed intramolecular hydrocarbonation of alkynes, by our group. | ||
Mechanistic experiments with a deuterated probe demonstrated the exclusive deuterium incorporation at the vinylic position, confirming that an Ir(III)–H intermediate is involved. DFT calculations revealed that the initial oxidative addition presents a higher energy barrier, and that both the carbometallation and hydrometallation pathways are viable. Nonetheless, the path based on a carboiridation involves a significantly less costly C–H reductive elimination.
To sum up, although the hydrocarbonation reaction of alkynes with C(sp2)–H bond sources had been initially less explored than the homologous reactions with alkenes, recent examples have demonstrated that several TM catalysts, and particularly iridium(I) cationic complexes, can be used to generate cyclic and acyclic dienyl products with complete or almost complete stereoselectivity. This type of cyclization reaction based on C–H activations is highly appealing in terms of increasing structural complexity as well as because their superb atom-economy.
3 Addition of C(sp)–H bonds across unsaturated systems
Hydroalkynylation strategies involving the activation of the relatively acidic C(sp)–H bond of terminal alkynes are appealing methods to build C–C bonds and generate synthetically valuable products.75 In this regard, a significant number of methodologies have been successfully developed based on deprotonations to generate metal alkynylides (e.g. Ni, Pd, Ru, Rh, Co).76 These methods, which do not engage a C–H oxidative addition process, will not be discussed in this review. We will only present systems for which the participation of alkynyl metal hydrides [C(sp)–M–H] has been proved or proposed, with a focus on iridium catalysis and related group IX metals.Especially relevant are the transformations enabling enantioselective processes, which usually need highly strained alkene partners.77 In 2012 Fan, Kwong and coworkers disclosed an Ir-promoted asymmetric hydroalkynylation of norbornadienes promoted by the catalyst generated from [Ir(cod)Cl]2 and (R)-Synphos (L23) in 1,2-dichloroethane.77a The catalyst works with differently monosubstituted alkynes (Scheme 41A), and can also be used to perform hydroalkynylations of oxabenzonorbornadienes (Scheme 41B).77b More recently, the same group described an enantioselective hydroalkynylation of 9-substituted norbornadiene derivatives of type 68 with a similar catalytic system.77c Remarkably, when the analogous rhodium complex was used instead of [Ir(cod)Cl]2, the yield decreased from 92 to 7% (Scheme 41C), a result that comes again to emphasize the superiority of iridium catalysts in some hydrocarbonation reactions.
 | ||
| Scheme 41 Iridium-catalyzed hydroalkynylation of strained alkenes. | ||
Similar reactions with less strained alkene acceptors are not easy. A relevant contribution was described by Li and coworkers in 2016, using β-monosubstituted Z-enamides as reaction partners.78 Remarkably, the reaction shows a complete regiocontrol for the formation of the C–C bond at the β-position when the [Ir(cod)(Ph-BPE)]OTf catalyst (Ir3) is used. Thus, enantioenriched homopropargyl amides can be synthesized under mild conditions (Scheme 42). Based on deuterium-labelling experiments, the authors proposed a mechanism wherein the oxidative addition of silylacetylene generates the iridium(III) hydride intermediate I, which reacts with the enamide to give intermediate II, in which the iridium atom is coordinated to the carbonyl group. Finally, a C–H reductive elimination provides the homopropargyl amides 69.
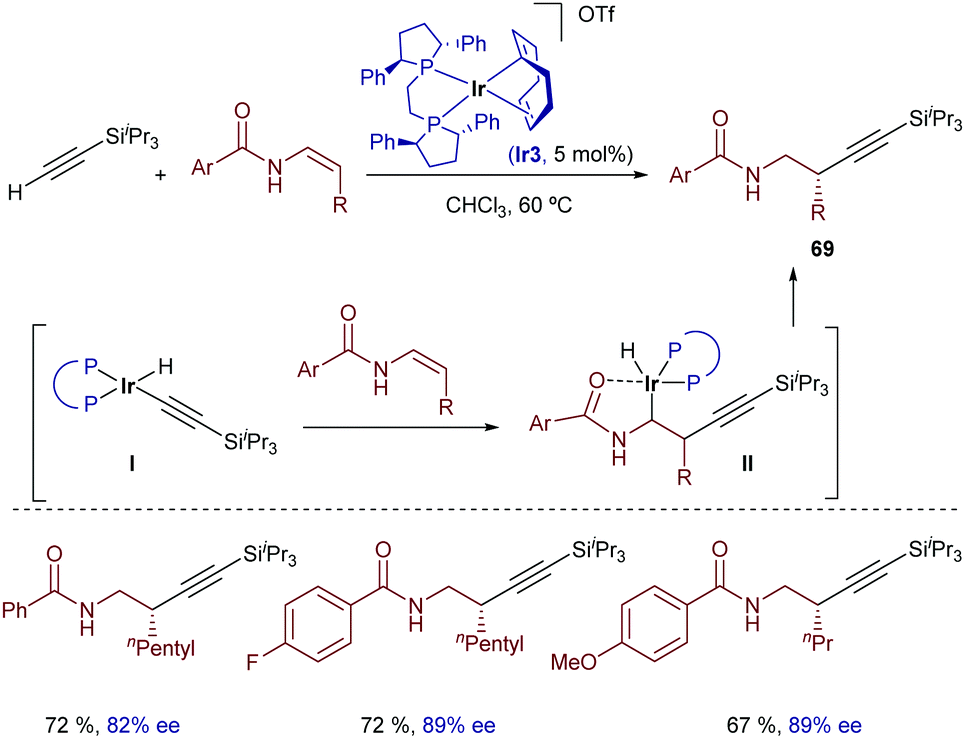 | ||
| Scheme 42 Iridium-catalyzed enantioselective hydroalkynylation of enamides developed by Li and coworkers. | ||
Two years later, the same group demonstrated that the Rh catalysts generated from [Rh(cod)2]OTf and the bisphosphine (R)-iPr-MeO-BIPHEP (L24) in THF could also promote the hydroalkynylation of these enamides.79 Curiously, the regioselectivity of the addition is reversed with respect to the iridium-promoted reaction, and the α-alkyl propargylamides 70 are obtained (Scheme 43). The regioselectivity can be explained assuming a hydro- instead of carbo-metallation (proposed in the case of the iridium catalyst).
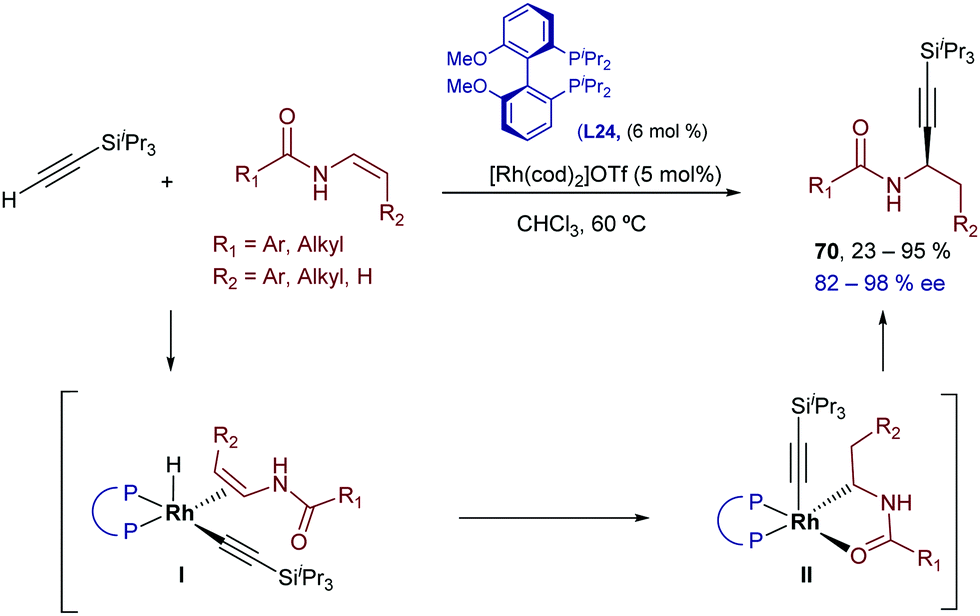 | ||
| Scheme 43 Rhodium-catalyzed enantioselective hydroalkynylation of enamides developed by Li and coworkers. | ||
Very recently, Lin and co-workers studied the origin of the regioselectivity in the above transformations using DFT analyses and the reactions partners indicated in Scheme 44.80 The authors explored all the possible scenarios for the iridium-catalyzed hydroalkynylation process, concluding that the proposed pathway involving a migratory insertion into the Ir–C bond followed by C–H bond reductive elimination to give the β-isomer 69a is the most favoured (Scheme 44, left). In contrast, Rh-catalyzed hydroalkynylation shows a preference for the migratory insertion into the Rh–H bond, which eventually leads to the regioisomeric α-product 70a through a C–C reductive elimination (Scheme 44, right). To explain this divergence, the authors recalled that, in view of their relative positions in the periodic table, Rh is expected to prefer an oxidation state of 1+ in comparison to an oxidation state of 3+, while Ir behaves in an opposite manner. In addition, C–Rh(III) bonds are generally weaker than C–Ir(III) bonds, so that the C–C reductive eliminations from a Rh(III) center are significantly more favoured (ΔΔG = 12 kcal mol−1 in the current case). Therefore, since the C–C reductive elimination from Ir(III) species has an inaccessible energy barrier (>31 kcal mol−1), the Ir-catalyzed reaction takes advantage of the reversibility of the migratory insertion into the Ir–H bond and evolves via the less favourable migratory insertion into the Ir–C bond and an ensuing C–H reductive elimination. The latter process is significantly less demanding than the respective C–C reductive elimination (ΔΔG ≈ 10 kcal mol−1)
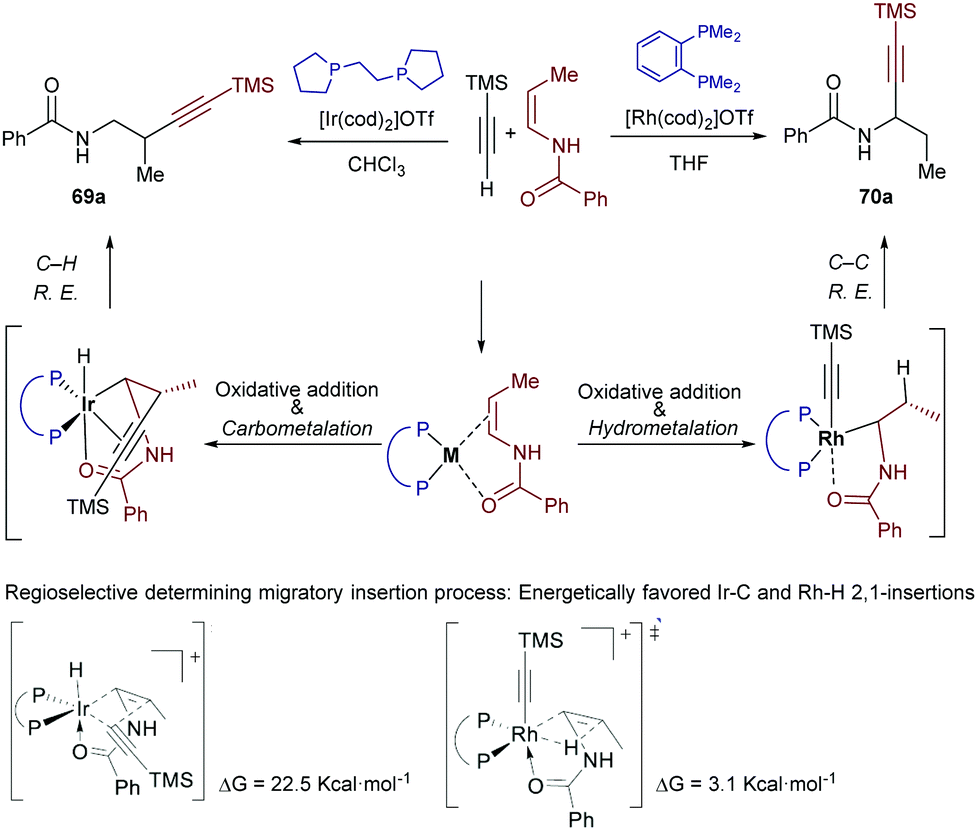 | ||
| Scheme 44 Iridium- and rhodium-catalyzed hydroalkynylation of enamides: results from a DFT study developed by Lin and coworkers. | ||
The migratory insertions into Rh(I)–H and Ir(I)–C bonds present the same regioselectivity, likely because of the coordination of the carbonyl to the metal to form a five-membered metallacycle.
Interestingly, Li and coworkers demonstrated that related hydroalkynylations can be carried out using non-activated trisubstituted alkenes that hold a dialkyl amide at the homoallylic position (Scheme 45A).81 The optimal conditions involve the use of Ir(cod)2OTf and the bisphosphine CTH-(R)-P-Phos (L25). Strikingly, the catalytic alkynylation occurs at the most hindered position of the alkene, allowing the synthesis of γ-alkynylamides bearing quaternary carbon stereocenters in good yields and enantioselectivities.
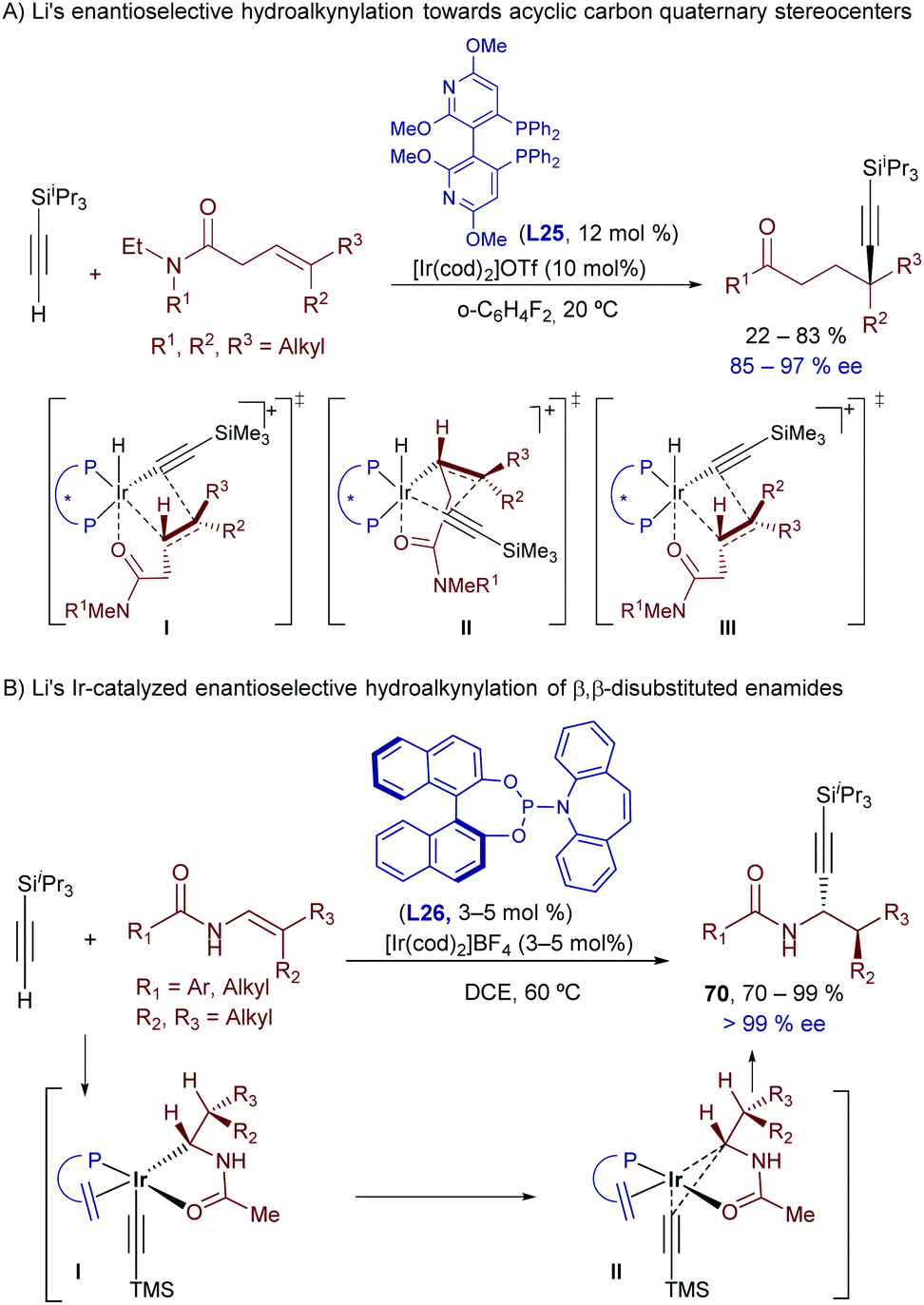 | ||
| Scheme 45 An Ir-phosphoramidite catalyst for the hydroalkynylation of enamides. | ||
DFT computational studies shed light on the origin of their regio- and enantioselectivity. The authors located the two most favored transition states for the migratory insertion of the E-alkene, leading to both enantiomers (I and II, Scheme 45A). In both cases, the carbonyl group of the alkenylamide is coordinated to the iridium center, which determines the regioselectivity. Interestingly the high energy difference observed between both enantiotopic transition states (>5 kcal mol−1) was attributed to the presence of steric repulsions in II, and attractive C−H···O interactions of the carbonyl group with aryl C−H bonds on the ligand in I. Eventually, the authors found that the formation of the minor enantiomer could be better explained on the basis of a marginal E–Z isomerization of the starting alkenylamide, since the most favored migratory insertion of the Z alkene leads to the opposite enantiomer (III).
Finally, in a new tour de force, the group of Li recently disclosed an Ir-catalyzed hydroalkynylation of β,β-disubstituted enamides that affords propargyl amines bearing two vicinal stereogenic centers, in a highly regio-, diastereo- and enantioselective fashion (Scheme 45B).82 Strikingly, the regioselectivity of this hydroalkynylation, which exclusively delivers the α-addition product, is opposite to that previously observed with alternative Ir catalysts (Schemes 45Bvs.42). Insightfully, DFT calculations shed light on this puzzle, proposing that in this case the carbometallation step is energetically more difficult due to the sterically hindered nature of the trisubstituted alkene. Moreover, the pathway based on a hydrometallation/C–C reductive elimination strongly benefits from the electron deficient nature of the phosphoramidite ligand (L26), which reduces the electron density at the metal center. Indeed, the use of this ligand is critical for obtaining both good yields and excellent stereoselectivities.83
Low-valent cobalt catalysts have been scarcely used in these types of hydroalkynylations. The most relevant examples so far involve cross-dimerizations of alkynes, while more challenging hydroalkynylations of alkenes have not yet been described. Thus, Okamoto and coworkers reported in 2013 the first examples of a cobalt catalyzed hydroalkynylation of internal alkynes, by using a Co(I) catalyst generated from a Co(II) precursor and Zn as a reducing agent. Control experiments using deuterated silylacetylene revealed that the reaction involves as the initial step a direct oxidative addition of the cobalt to the acetylenic C–H bond (Scheme 46A).84
 | ||
| Scheme 46 Cobalt-catalyzed hydroalkynylation reactions of alkynes [46C: SET stands for single electron transfer and 4CzIPN for 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene; [Si] = iPr3Si]. | ||
More recently, Mashima and coworkers expanded the scope of these processes reporting a highly E-selective cross-dimerization of two different terminal alkynes, a process that is catalyzed by a Co(0) complex generated by the reaction of EtMgBr with a sterically demanding 1,10-phenanthroline cobalt(II) dichloride precursor like Co1 (Scheme 46B). The independent preparation of the Co(0) precursor allowed the authors to confirm its participation as a catalyst.85
Overall, these two papers nicely show a key characteristic of low-valent cobalt catalysts, namely their ability to participate either via Co(I) or Co(0) species, as well as the possibility to use both strong- and weak-field ancillary ligands. On the downside, the requirement of strong reductants to avoid handling highly air-sensitive cobalt species hampers a wider use of these methods, as functional group tolerance might be compromised.
In this regard, it is pertinent to note that Collins and coworkers have recently provided a novel alternative for achieving related cross-dimerization processes based on the synergistic use of a photoredox catalyst (the carbazole-based organic dye 4CzIPN), and a cobalt(II) source (Scheme 46C).86 Mechanistically, the authors proposed an electron transfer from the excited organic dye to the initial Co(II) to give a Co(0)-bisphosphine complex, which is the species that suffers the oxidative addition of the terminal alkyne. After migratory insertion of a second unit of alkyne, a subsequent oxidation by the cationic dye delivers a Co(III) intermediate, which reductively eliminates to a Co(I) counterpart, with concomitant delivery of the product. A new SET process restores the initial Co(0) species. The method constitutes the first photocatalytic hydroalkynylation process, and the resulting 1,3-enynes are obtained in good to high yields and E-selectivity.
In conclusion, catalytic hydroalkynylations represent powerful tools for accessing synthetically valuable alkynyl-containing products. Although the initial seminal approaches relied on highly strained alkenes, the use of enamides as partners allowed building propargylic or homopropargylic amines in a very effective way. However, the scope of the reactions with respect to the alkene component is yet very limited. The recent demonstration that combining transition metal and photoredox catalysts can drive otherwise difficult hydrocarbonations opens an interesting research avenue.
4 Addition of C(sp3)–H bonds across unsaturated systems
The above-mentioned hydrocarbonation processes involve the activation of C(sp2)–H or C(sp)–H bonds. Extending this chemistry to C(sp3)–H bonds is very attractive, as it could open impressive opportunities for the formation of C–C bonds and of chiral centers from simple precursors. However, achieving a selective functionalization of C(sp3)–H bonds is highly challenging, not only because of their low reactivity but also in terms of regioselectivity.In fact, the entire field of metal-catalyzed C(sp3)–H activation/C–C functionalization can be considered to be yet in its infancy. Usually the activation and cleavage of C–H bonds requires the presence of a heteroatom directing group, and the formation of relatively stable five or six-membered metallacyclic intermediates. The most frequently used transition metals for the activation of C(sp3)–H bonds are Pd, Ru, Rh, Co and Ir.87
While there have been important advances in the development of metal-promoted C(sp3)–H arylations, aminations and oxygenation reactions,88 addition reactions that result in the formation of C–C bonds using alkynes or alkenes as reaction partners remain scarce, and most of them are initiated by CMD mechanisms instead of oxidative additions to the C–H bonds. Only in recent years several examples demonstrating the viability of these hydrocarbonation processes enabled by the metal activation of C(sp3)–H bonds have been published.
In 1998, Jun and coworkers made a seminal contribution to the field.89 They disclosed a ruthenium-catalyzed addition of a benzylic C(sp3)–H bond across alkenes, using 2-(alkylamino)pyridines of type 71 as substrates. The presence of the chelating nitrogen group of the pyridine is key to the success of the reaction (Scheme 47A).
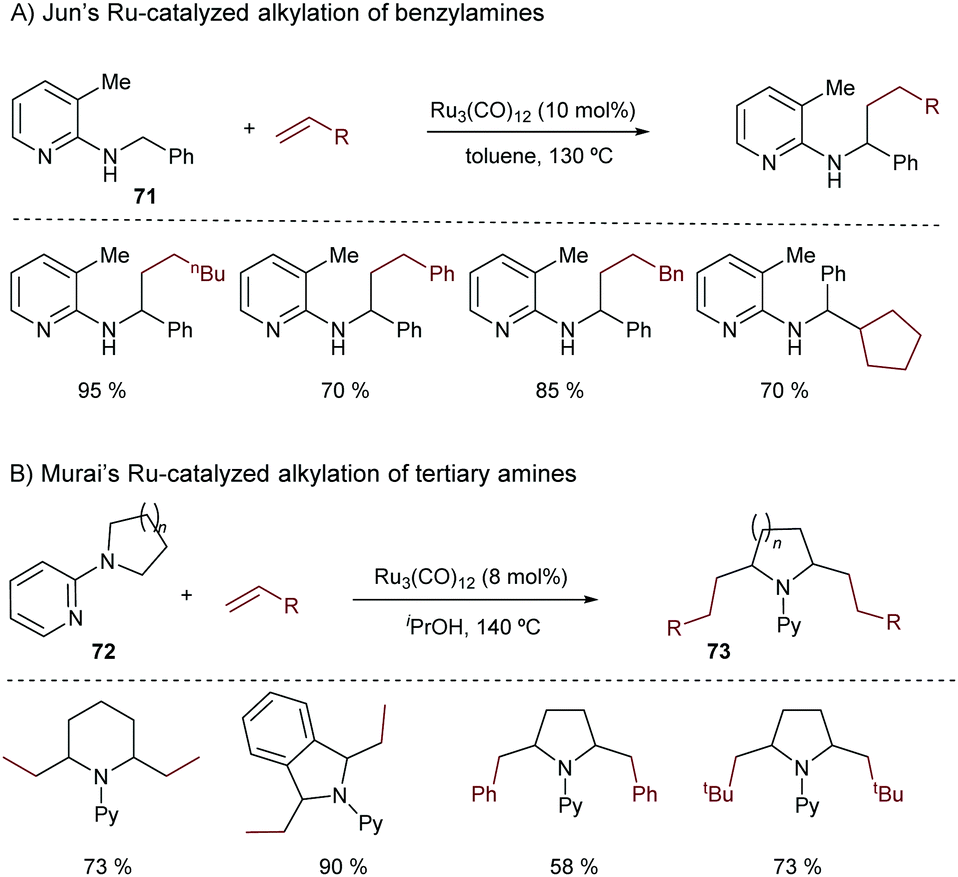 | ||
| Scheme 47 Seminal reports on Ru-catalyzed hydrocarbonation of alkenes by activating C(sp3)–H bonds. | ||
In 2001, Murai disclosed a related Ru-catalyzed methodology to promote the addition of alkenes to a C(sp3)–H bond in N-2-pyridyl cyclic amines of type 72.90 Again, this approach requires harsh conditions and high catalytic loadings of Ru3(CO)12 (Scheme 47B). Remarkably, the reaction works with different types of alkenes, and enables the preparation of dialkylation products of type 73.
In 2007, Brookhart and coworkers reported a Co-catalyzed intramolecular hydrogen-transfer reaction of cyclic amines of type 2.91 Although the process doesn’t generate new C–C bonds, the reaction is worth mentioning as it involves an initial C(sp3)–H activation by oxidative addition of the nitrogen-adjacent C–H bond to a Cp*Co(I) catalyst (Co2). Subsequent hydrometallation and β-hydride elimination deliver a new Co(III)-hydride species that evolved into the isomerized product by C–H reductive elimination (Scheme 48). Interestingly, this cobalt catalyst proved to be more reactive than the analogous Rh counterpart.
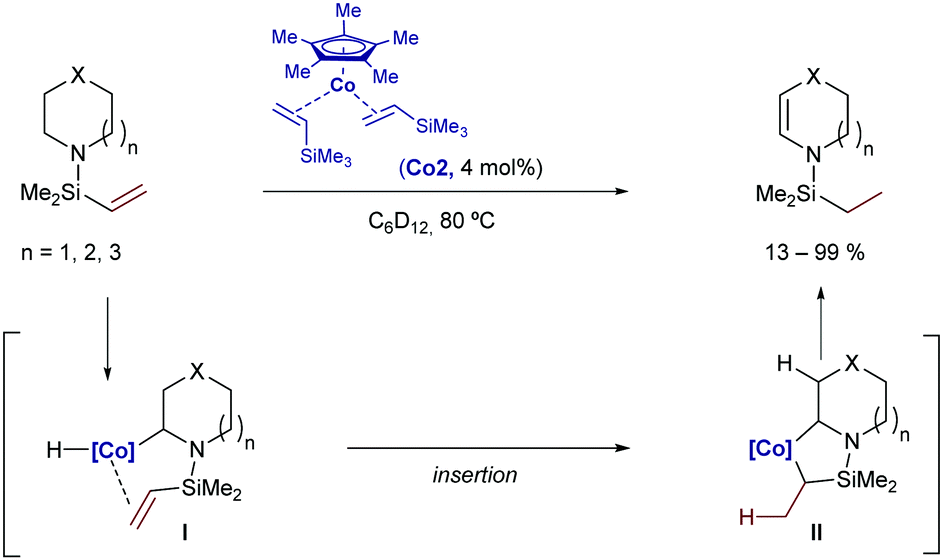 | ||
| Scheme 48 Co-Catalyzed C(sp3)–H activation/intramolecular hydrogen transfer developed by Brookhart and co-workers. | ||
Curiously, despite the efficiency of these and related cobalt(I)-catalysts for promoting C(sp3)–H bond activations,92 their application to hydrocarbonation reactions (formation of C–C bonds) has not yet been accomplished.
In 2010, Yu et al. described an intramolecular cycloisomerization of ene-2-diene substrates like 74 involving a Rh-catalyzed activation of an allylic C(sp3)–H bond and the subsequent addition of the C–Rh bond across the tethered diene. The reactions, which are promoted by RhCl(PPh3)3/AgSbF6, provide tetrahydropyrroles of type 75 in good yields (Scheme 49A).93 Remarkably, the presence of a strongly chelating group is not required, as the diene plays the role of directing the C–H activation to the most reactive allylic C–H bond. DFT studies indicated that the presence of the extra double bond in the alkene acceptor is also key to facilitating the reductive elimination step (from I to II, Scheme 49A).94 In 2011, the same authors reported an enantioselective version using a cationic catalyst generated in situ from [Rh(coe)2Cl]2, AgOTf and the chiral phosphoramidite ligand L27.95 The corresponding chiral tetrahydropyrroles are thus obtained with both good enantioselectivities and diastereoisomeric ratios (Scheme 49B).
 | ||
| Scheme 49 Rh-Catalyzed intramolecular sp3 hydrocarbonation with dienes by Yu and coworkers. | ||
The first reports with iridium didn’t come until 2004 when Sames and coworkers described an intramolecular cyclization of amide-tethered alkenes of type 76 promoted by an [Ir(coe)2Cl]2/IPr catalyst (Scheme 50).96 Despite the limited scope of the process, high temperatures required, and moderate yields of products 77 (side products 78 and 79 were also obtained), this seminal publication critically showed the challenges with Ir catalysts and paved the way for further contributions in the area. Likely, the reaction involves an initial weak coordination of the iridium complex to the carbonyl moiety followed by an insertion into a C(sp3)–H bond that is adjacent to the amide nitrogen atom. The resulting iridium hydride intermediate (I) undergoes a 5-exo alkene migratory insertion. A final β-hydride elimination generates the bicyclic amide (III) coordinated to an iridium bishydride. Isomerization of III eventually provides adduct 77 (Scheme 50). The formation of 78 and 79 can be, respectively, explained by an endo cyclization of I, followed by reductive elimination or by hydrogenation of 76 by the resulting Ir bishydride species (an excess of norbornene is used as a sacrificial hydrogen acceptor to minimize this side-process).
 | ||
| Scheme 50 Ir-Catalyzed cyclization of amides initiated by C–H oxidative addition, developed by Sames and co-workers (NBE stands for norbornene). | ||
A truly remarkable hydrocarbonation reaction initiated by oxidative addition of Ir(I) to C(sp3)–H bonds was described by Shibata in 2009.97 The combination of [Ir(cod)2]OTf and BINAP in refluxing chlorobenzene allowed activating the methyl group of dimethylamides, and inserting the resulting hydride species into alkynes, to give the monoalkenylated amides 80 in moderate to good yields (Scheme 51). Deuterium labeling experiments showed a complete deuterium transfer from the methyl group of the amide to the vinylic position, as well as a fractional protonation at the methylene group. This is consistent with the participation of iridium hydride intermediate species of type I, which undergo migratory alkyne insertion and reductive elimination. Control experiments suggested that the loss of deuterium at the allylic position might be the result of a C–H bond cleavage at this position, followed by protonation using an external proton source (i.e. water).
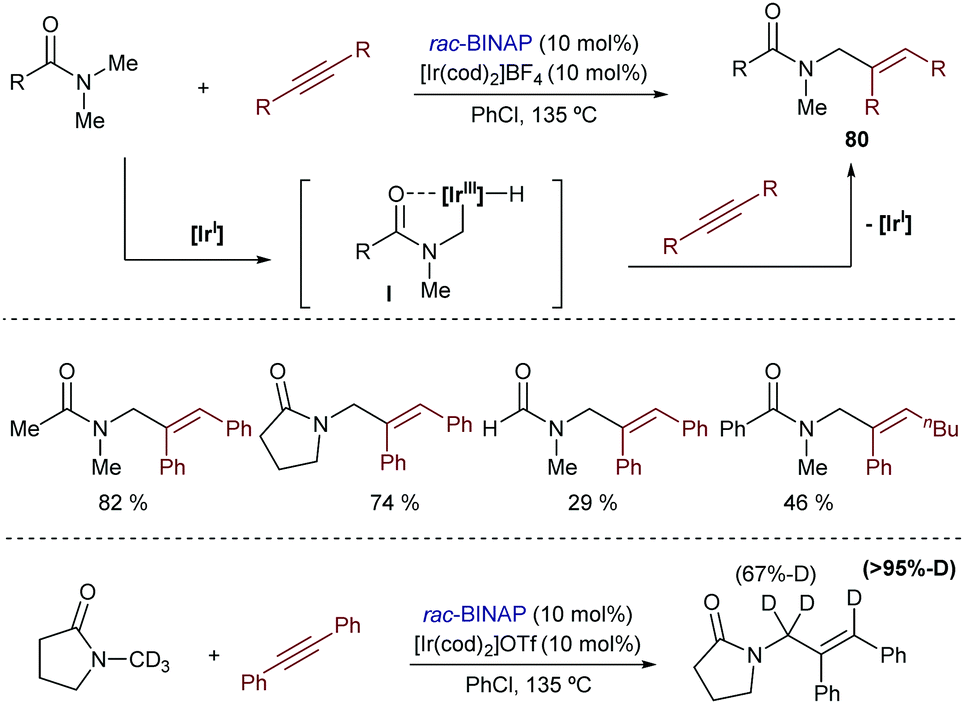 | ||
| Scheme 51 Ir-Catalyzed hydrocarbonations with alkynes developed by Shibata and co-workers. | ||
The same group extended this chemistry to 2-(alkylamino)pyridines, which in the presence of an iridium catalyst generated from [Ir(cod)2]BF4 and Tol-BINAP (L28) can add to styrenes, even in an enantioselective manner. Linear alkylation products of type 81 were selectively obtained in moderate to good yields and with enantioselectivities typically above 80% (Scheme 52).98 Deuterium labeling studies confirmed an initial cleavage of the C(sp3)–H bond adjacent to the nitrogen through an oxidative addition to generate a chiral Ir-hydride intermediate (I) that was tentatively identified by 31P-NMR. An improvement of this method was published in 2012 by the same group, demonstrating that not only styrene, but also β-unsubstituted acrylates, vinyl silanes, aliphatic alkenes and even alkynes are suitable partners. The reactions gave enantioselectivities that varied from 55 to 99% ee, depending on the C–C unsaturated partner employed.99
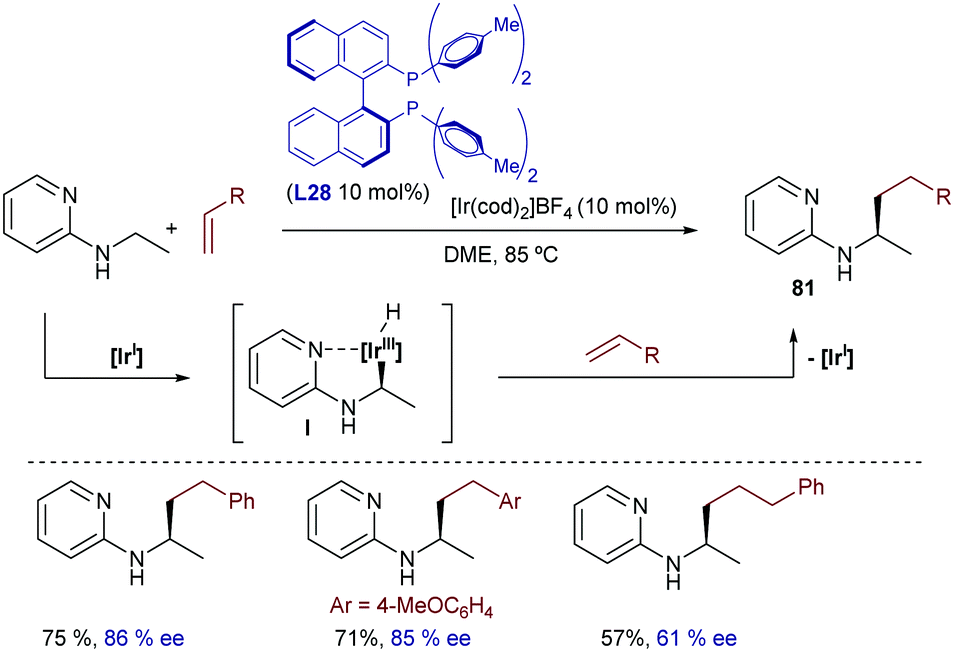 | ||
| Scheme 52 Ir-Catalyzed enantioselective alkylations of 2-(alkylamino)pyridines. | ||
Nishimura further expanded this methodology, demonstrating that the cationic iridium complex [IrCl(cod)]2/NaBArF4 is able to perform a double C–H alkylation of 2-(methylamino)pyridines, to yield α-substituted amines of type 82 (Scheme 53A).100 The reaction can also be carried out with one alkene to give an achiral product (Scheme 53A) or, alternatively, two different alkenes can be introduced in a sequential manner, provided that a vinyl silane such as triethylvinylsilane is used as the first partner. Interestingly, if BINAP is added together with the second alkene, the resulting α-substituted chiral amine (82a) can be obtained with good enantioselectivities (Scheme 53B).
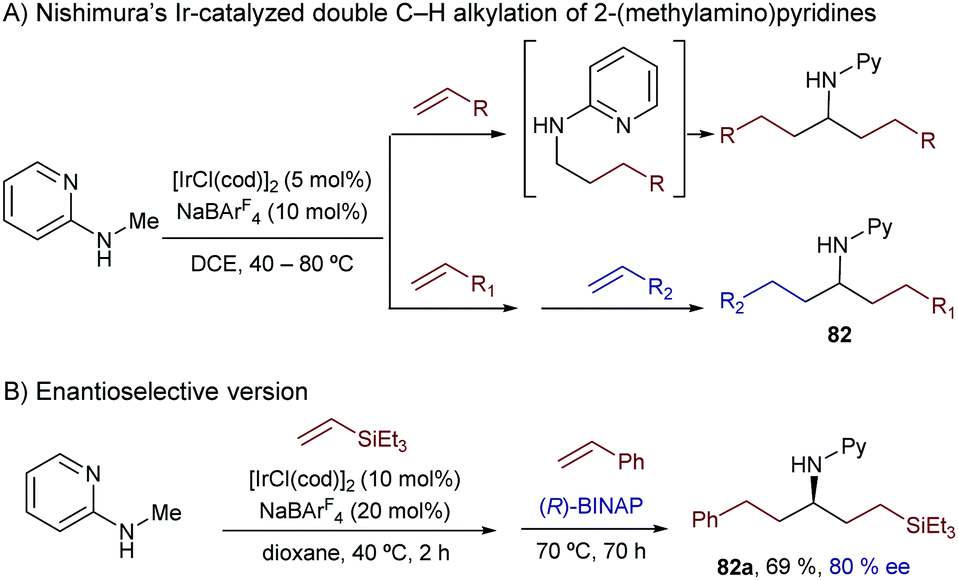 | ||
| Scheme 53 Ir-Catalyzed double alkylation of 2-(methylamino)pyridines by Nishimura and coworkers (Py stands for pyridine). | ||
The use of a nitrogen-tethered pyridine to assist the activation of C(sp3)–H bonds adjacent to a nitrogen atom was further exploited by Shibata with N-(2-pyridyl)-γ-butyrolactams of type 83 (Scheme 54).101 Remarkably, the complex prepared by [Ir(cod)2]BF4/Tol-BINAP catalyzed enantioselective alkylations with styrenes and acrylates to yield products that can be easily converted into substituted γ-amino acids by removal of the pyridine directing group. The methodology was also applied to the formal asymmetric synthesis of pyrrolam A (Scheme 54).
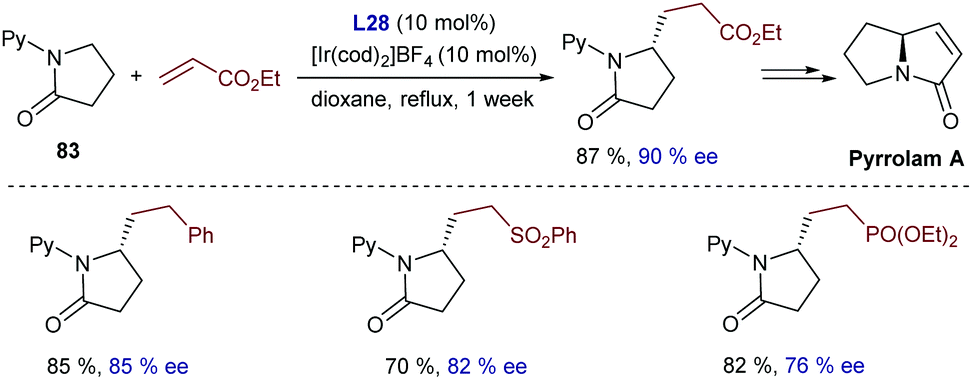 | ||
| Scheme 54 Ir-Catalyzed enantioselective alkylation of lactams developed by Shibata and co-workers. | ||
The activation of C(sp3)–H bonds in alpha to amines can also be performed using other directing groups than pyridine. Therefore, Opatz and coworkers described a hydrocarbonation of alkenes using benzoxazole scaffolds as directing groups (Scheme 55).102 In particular, using the simple cationic catalyst [Ir(cod)2]BF4 or [Ir(cod)2]BArF4, without any additional ligand, these authors demonstrated that tetrahydroquinolines like 84, as well as other related cyclic and acyclic amines, can be catalytically monoalkylated at their nitrogen-adjacent position, yielding linear products of type 85. Although harsh reductive or basic conditions are required to remove the directing group, the resulting amines are compatible with these conditions.
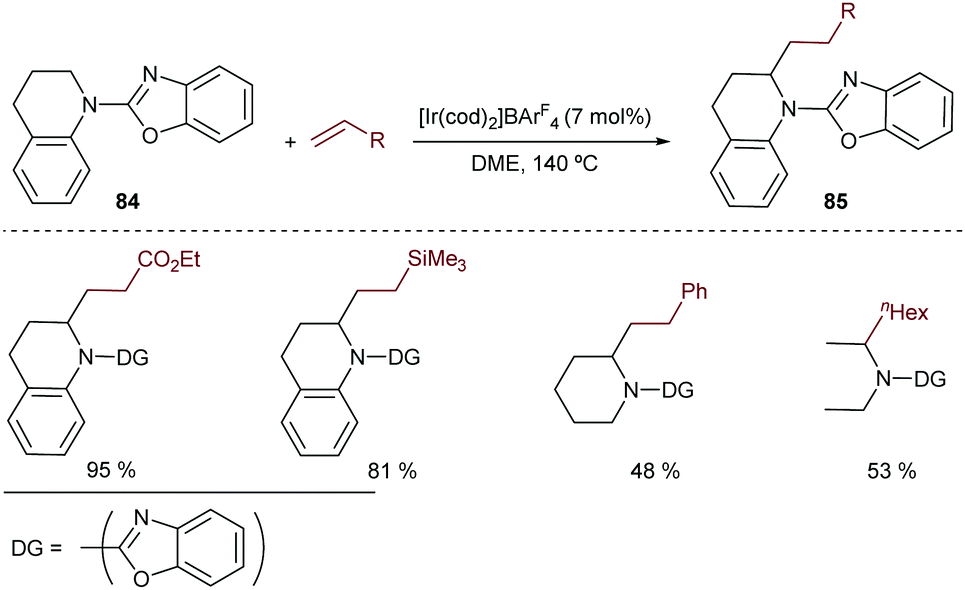 | ||
| Scheme 55 Ir-Catalyzed benzoxazole-directed alkylation developed by Opatz and co-workers. | ||
Alkoxythiocarbonyl moieties can also be successfully used as directing groups for the activation of related C(sp3)–H bonds.103 Thus, using [Ir(cod)2]OTf as a catalyst, Yu and coworkers were able to obtain α-alkylated pyrrolidines, prolines and piperidines. In many cases, mixtures of monoalkylated (86) and dialkylated (87) products were obtained (Scheme 56A). Although the mechanistic aspects of the transformation were not addressed, presumably, the process involves a C–H oxidative addition followed by a subsequent alkene migratory insertion and reductive elimination. However, it is not clear if the process involves carbo- or hydrometallation pathways.
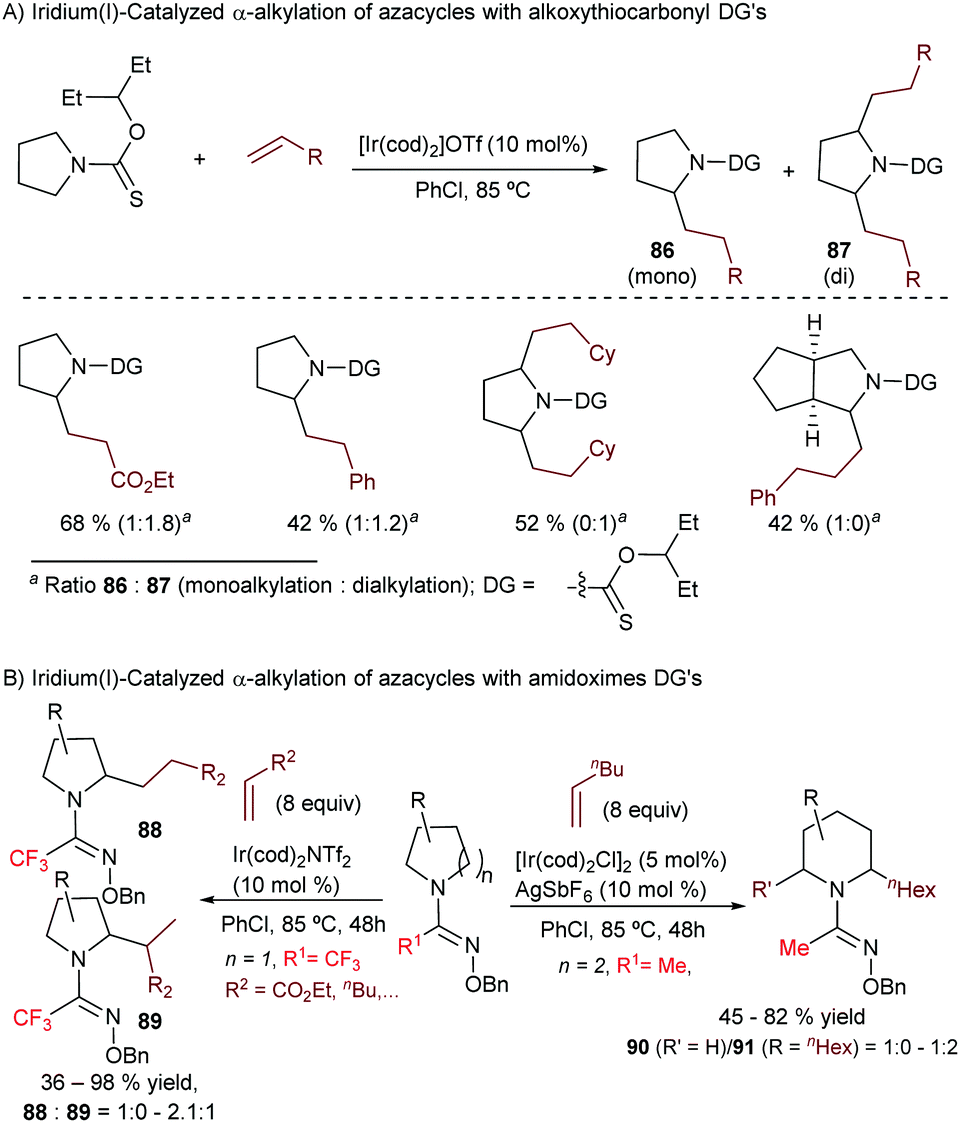 | ||
| Scheme 56 Ir-Catalyzed directed alkylation of azacycles developed by Yu and coworkers. | ||
In a more recent study, Yu and co-workers introduced the use of amidoximes as suitable directing groups for performing this type of intermolecular hydrocarbonation of alkenes with N-adjacent C(sp3)–H bonds of saturated azacycles. Depending on the type of azacycle, a different directing group and an iridium catalyst are needed.104 Thus, for the alkylation of pyrrolidines, a trifluoromethyl O-benzyl amidoxime performs best, usually providing the linear adduct (88), or mixtures of linear and branched products (Scheme 56B, left). With regard to piperidine systems, the methyl O-benzyl amidoxime provided better results, generally leading to linear mono- and di-substituted products (90 and 91) in variable yields and selectivities (Scheme 56B, right). Based on deuterium labelling experiments, the authors suggested a pathway that involves both 1,2- and 2,1-reversible migratory insertions into the Ir–H bond. Rate determining C–C reductive eliminations would provide the corresponding linear and branched isomers respectively. The viability of an alternative migratory insertion into the Ir–C bond followed by a C–H reductive elimination was not discussed.
As in the case of the hydrocarbonations based on C(sp2)–H activations, Nishimura and collaborators demonstrated that anionic amides can work as directing groups in the activation of C(sp3)–H bonds. Using hydroxoiridium catalysts, they were able to achieve the hydroalkylation of terminal alkenes with 1-methyl-1,3-diphenylurea.105 The optimal conditions involved an iridium catalyst generated in situ from [Ir(OH)(cod)]2 and the bisphosphine dippbz, at an unusually low reaction temperature of 70 °C (Scheme 57A). Although the methodology is restricted to terminal alkenes, the scope was broad, and it was further extended to the use of secondary C(sp3)–H bonds to give the indoline urea derivatives 93 (Scheme 57B).106 In the latter case, the use of [Ir(OH)(cod)]2 without any additional ancillary ligand led to the best results. The methodology tolerates a variety of terminal alkenes but is limited to very few indolines.
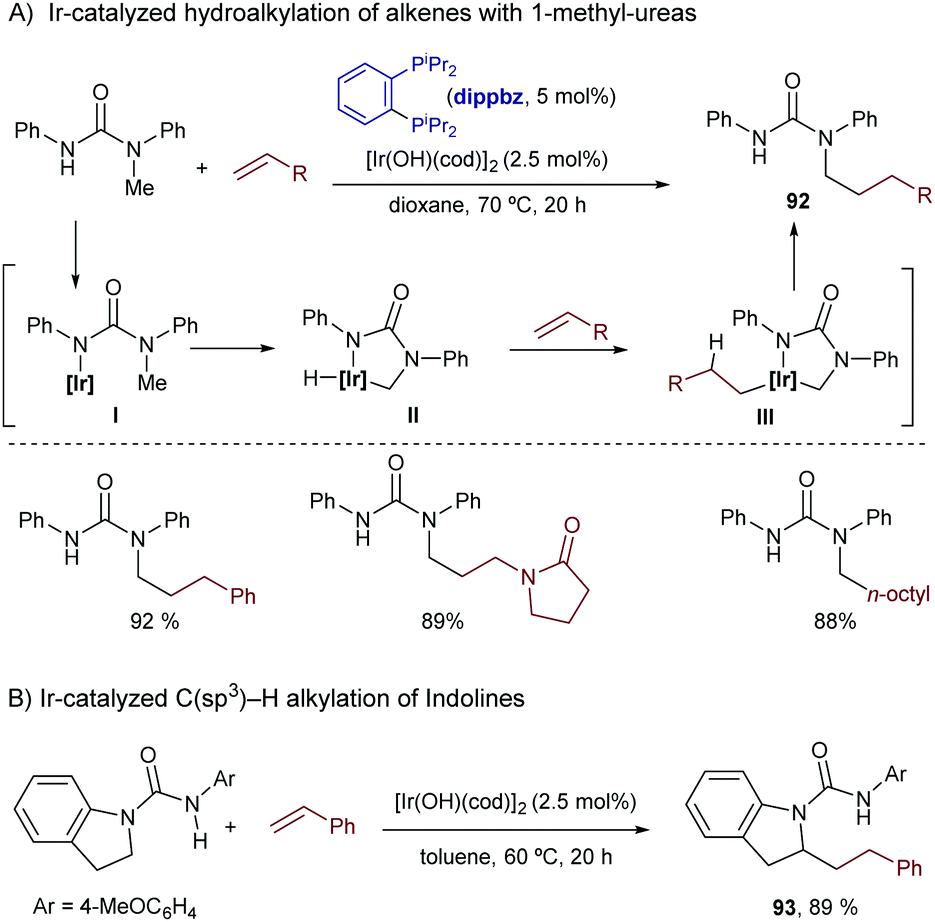 | ||
| Scheme 57 Ir-Catalyzed alkylation of ureas developed by Nishimura. | ||
These processes should proceed by the initial formation of an amidoiridium intermediate (I) that evolves through an oxidative C–H insertion to give iridacycle II. Linear-selective alkene insertion into the Ir–H bond forms III. Eventually an irreversible C(sp3)–C(sp3) reductive elimination and protonation of the amine affords the alkylated urea product (92).
Related reactions based on the C(sp3)–H bond activation adjacent to nitrogen can also be performed in an intramolecular mode. Therefore, Suginome and coworkers reported a very interesting cycloisomerization of ortho alkenyl-N-methylanilines.107 The reaction is catalyzed by [IrCl(C2H4)2]2 and DTBM-SEGPHOS, and delivers indolines of type 95 bearing a chiral carbon quaternary stereocenter, with high efficiency and excellent enantiomeric ratios. The authors proposed a mechanism involving the alkenyl-assisted insertion of the iridium complex into a C(sp3)–H bond of the dimethyl amine to generate intermediate I (Scheme 58). Subsequently, a carboiridation step, in a 5-exo fashion, delivers intermediate II which undergoes a C–H reductive elimination to yield the indoline product. According to the authors, the formation of 96 as a side product in the cyclization of 94 (R = H) can only be explained assuming a β-H elimination from the iridium-hydride intermediate II (R = H) and a subsequent isomerization. This fact allowed them to conclude that an alternative hydrometallation/C–C reductive elimination pathway is very unlikely.
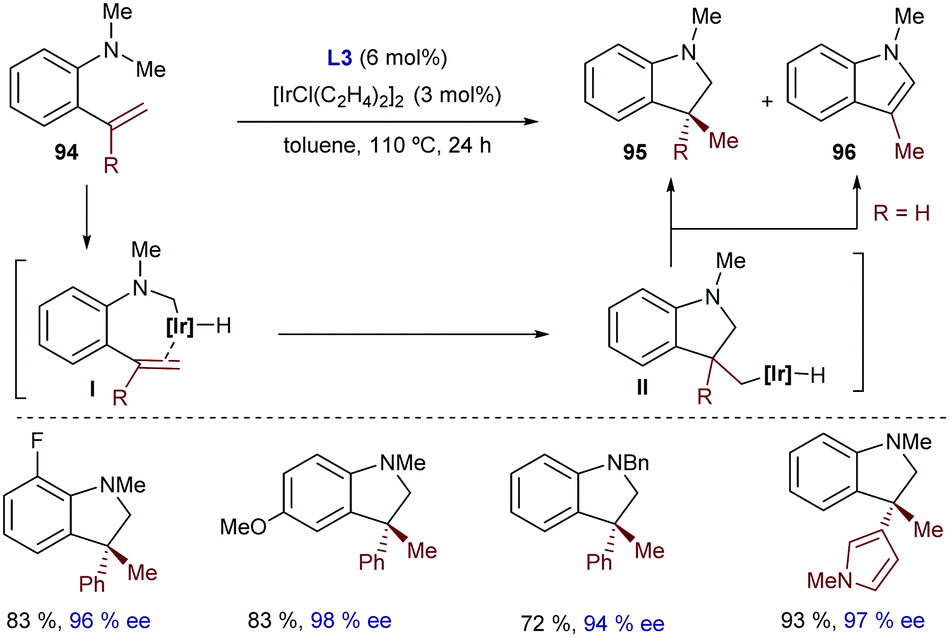 | ||
| Scheme 58 Ir-Catalyzed cycloisomerization of o-alkenyl-N-methylanilines developed by Suginome. | ||
The same authors demonstrated that it is also possible to activate a C–H bond in methoxy groups and induce related cyclizations, in this case to alkynes instead to alkenes (Scheme 59).108 Labeling experiments indicated that the initial oxidative addition is the turnover-determining step. Subsequent intramolecular syn insertion of the C–C triple bond into the Ir–C or Ir–H affords intermediate II or III. Both intermediates could evolve by reductive elimination to yield a dihydrobenzofuran product, 98, which was experimentally isolated as a side product under specific reaction conditions. An isomerization reaction of 98 through a 1,3-H shift via π-allyl iridium species IV was proposed to explain the formation of the benzofuran product 97 (Scheme 58). Although the reaction does not involve the generation of any stereocenter, the authors demonstrated that the use of DTBM-SEGPHOS is imperative to obtain not only excellent overall yields but also the complete isomerization of intermediates 97 into benzofurans 98.
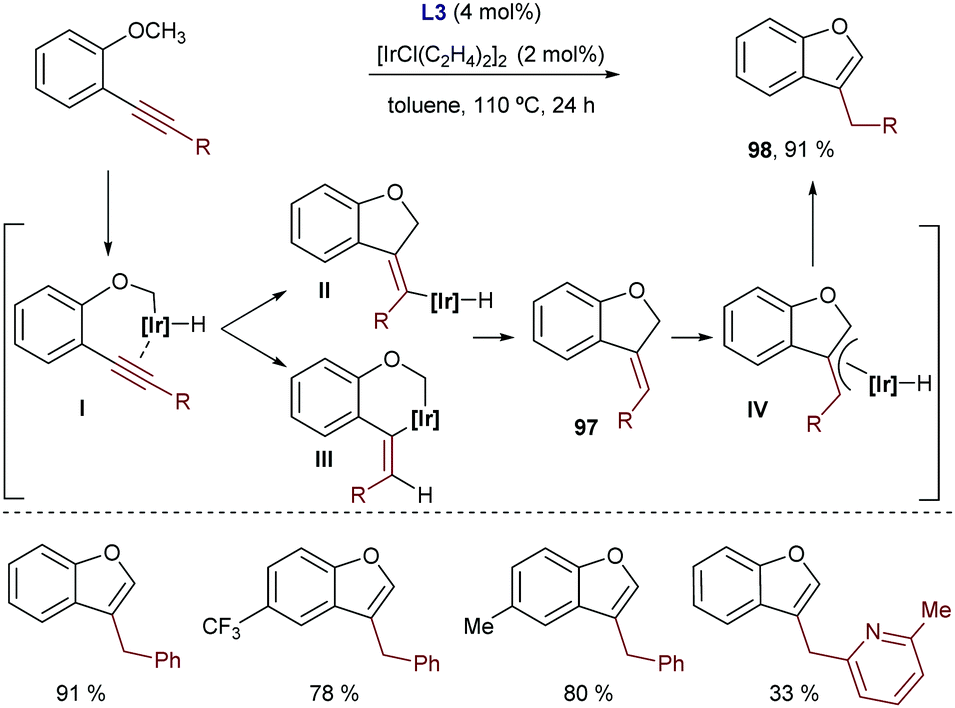 | ||
| Scheme 59 Ir-Catalyzed cycloisomerization of o-alkynyl phenols developed by Suginome. | ||
Overall, despite the above examples, this research topic is clearly underdeveloped, and there are many unknowns not only with regard to the C–H activation step, but also in terms of the substrate scope, and the influence of the elementary mechanistic steps in the reaction rate. Curiously, despite the ability of Ir catalysts to yield branched products in related hydrocarbonations of alkenes with C(sp2)–H precursors, all the examples involving C(sp3)–H bonds yield linear adducts. Thus, the development of methods to obtain branched products is still an unmet challenge.
5 Conclusions
Transition metal-catalyzed hydrocarbonation reactions represent excellent examples of transformations that fit perfectly with the requirements of sustainability and practicality of modern organic synthesis. Metals of group IX, and especially iridium complexes, are among the most effective catalysts to perform these reactions, in part because of their ability to undergo relatively easy oxidative additions to C–H bonds. Most of the examples so far described correspond to intermolecular hydroarylations, using alkynes, alkenes or allenes as unsaturated partners. Intramolecular reactions are very attractive from the constructive perspective, although the processes disclosed are still limited in terms of versatility and the substrate scope. Hydroalkenylations instead of hydroarylations have also been scarcely explored, while the addition of C(sp3)–H bonds across unsaturated partners remains to be further developed. Importantly, studies based on DFT calculations and deuterium labeling have shed light on the mechanisms of these reactions. In the case of iridium catalysts, carbometallation/C–H reductive eliminations are favored over alternative pathways involving hydrometallations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received financial support from Spanish grants (SAF2016-76689-R, CTQ2016-77047-P, CTQ2017-84767-P and ORFEO-CINQA network CTQ2016-81797-REDC), the Consellería de Cultura, Educación e Ordenación Universitaria (2015-CP082, ED431C-2017/19 and Centro Singular de Investigación de Galicia accreditation 2019–2022, ED431G 2019/03), the European Union (European Regional Development Fund-ERDF corresponding to the multiannual financial framework 2014–2020), and the European Research Council (Advanced Grant No. 340055). DFF thanks Xunta de Galicia for his postdoctoral fellowship (ED481B-2019-005).References
- (a) C. A. Busacca, D. R. Fandrick, J. J. Song and C. H. Senenayake, in Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective, ed. M. L. Crawley, B. M. Trost, John Wiley & Sons, 2012 Search PubMed; (b) J. Magano and J. R. Dunetz, in New Trends in Cross-Coupling: Theory and Applications, ed. T. J. Colacot, 2015, pp. 697–778 Search PubMed; (c) B. S. Gerstenberger, in Synthetic Methods in Drug Discovery, ed. D. Blakemore, P. Doyle and Y. Fobian, 2016, vol. 52, pp. 411–442 Search PubMed; (d) M. Picquet, Platinum Met. Rev., 2013, 57, 272–280 CrossRef.
- (a) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS; (b) R. Noyori, Nat. Chem., 2009, 1 Search PubMed; (c) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- (a) J. F. Hartwig, Catalytic C–H Functionalization in Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, South Orange, NJ, 2009 Search PubMed; (b) B.-J. Li and Z.-J. Shi in Homogeneous Catalysis for Unreactive Bond Activation, ed. Z.-J. Shi, John Wiley & Sons, 2015 Search PubMed; (c) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS; (d) R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS; (e) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS.
- For key selected reviews covering TMC hydrocarbonation reactions of C−C unsaturated bonds, see: (a) F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS; (b) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS; (c) F. Kakiuchi and T. Kochi, Synthesis, 2008, 3013–3039 CrossRef CAS; (d) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS.
- For reviews covering hydrocarbonations promoted by different transition metal catalysts, see: (a) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS; (b) N. Yoshikai, Bull. Chem. Soc. Jpn., 2014, 87, 843–857 CrossRef CAS; (c) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208–1219 CrossRef CAS; (d) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS; (e) Z. Huang, H. N. Lim, F. Mo, M. C. Young and G. Dong, Chem. Soc. Rev., 2015, 44, 7764–7786 RSC; (f) L. Wozniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef; (g) S. M. Khake and N. Chatani, Trends Chem., 2019, 1, 524–539 CrossRef; (h) K. S. Singh, Catalysts, 2019, 9, 173 CrossRef; (i) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC; (j) N. Yoshikai, Isr. J. Chem., 2017, 57, 1117–1130 CrossRef CAS; (k) M. Moselage, J. Li and L. Ackermann, ACS Catal., 2016, 6, 498–525 CrossRef CAS; (l) G. E. M. Crisenza and J. F. Bower, Chem. Lett., 2016, 45, 2–9 CrossRef CAS; (m) J. Choi and A. S. Goldman, in Iridium Catalysis, ed. P. G. Andersson, Springer-Verlag, Berlin Heidelberg, 2011, vol. 34, pp. 139–168 Search PubMed; (n) C-H Bond Activation and Catalytic Functionalization II, ed. P. H. Dixneuf, H. Doucet, Springer International Publishing, Cham, 2016, vol. 56 Search PubMed.
- For a review in the field, see: S. G. Pan and T. Shibata, ACS Catal., 2013, 3, 704–712 CrossRef CAS.
- (a) F. P. Pruchnik, Organometallic Chemistry of the Transition Elements, Modern Inorganic Chemistry, Springer, US, 1990 CrossRef; (b) J. Pospech, I. Fleischer, R. Franke, S. Buchholz and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 2852–2872 CrossRef CAS; (c) Iridium Complexes in Organic Synthesis, ed. L. A. Oro, C. Claver, Wiley-VCH, Verlag GMBH, Weinheim, 2009 Search PubMed; (d) R. H. Crabtree, in The Organometallic Chemistry of the Transition Metals, John Wiley & Sons, Hoboken, NJ, 6th edn, 2014 Search PubMed.
- Oxidative additions to Ir complexes are known since the sixties, see: (a) L. Vaska, Acc. Chem. Res., 1968, 1, 335–344 CrossRef CAS . Nonetheless, the first examples of the oxidative addition of an alkane C–H bond to a low-valent iridium center were reported in the early eighties, see: ; (b) A. H. Janowicz and R. G. Bergman, J. Am. Chem. Soc., 1982, 104, 352–354 CrossRef CAS; (c) J. K. Hoyano and W. A. G. Graham, J. Am. Chem. Soc., 1982, 104, 3723–3725 CrossRef CAS . See also: ; (d) T. H. Tulip and D. L. Thorn, J. Am. Chem. Soc., 1981, 103, 2448–2450 CrossRef CAS; (e) P. Foley, R. DiCosimo and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6713–6725 CrossRef CAS.
- Inorganic Reactions and Methods, ed. A. P. Hagen, J. J. Zuckerman, vol. 14, Wiley-VCH, 2009 Search PubMed.
- For reviews on C–H functionalization with high valent transition metal catalysts, see: (a) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (b) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS; (c) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS; (d) T. Piou and T. Rovis, Acc. Chem. Res., 2017, 51, 170–180 CrossRef; (e) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308–1318 CrossRef CAS; (f) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS; (g) J. Wencel-Delord, F. W. Patureau and F. Glorius, in C–H bond Activation and Catalytic Functionalization I, ed. P. Dixneuf and H. Doucet, Springer, 2015, pp 1–27 Search PubMed; (h) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (i) S. Wang, S.-Y. Chen and X.-Q. Yu, Chem. Commun., 2017, 53, 3165–3180 RSC; (j) S. Rej and N. Chatani, Angew. Chem., Int. Ed., 2019, 58, 8304–8329 CrossRef CAS; (k) A. Baccalini, S. Vergura, P. Dolui, G. Zanoni and D. Maiti, Org. Biomol. Chem., 2019, 17, 10119–10141 RSC; (l) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J. Q. Yu, Acc. Chem. Res., 2020, 53, 833–851 CrossRef CAS.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- F. Kakiuchi, M. Yamaguchi, N. Chatani and S. Murai, Chem. Lett., 1996, 111–112 CrossRef CAS.
- C.-H. Jun, J.-B. Hong, Y.-H. Kim and K.-Y. Chung, Angew. Chem., Int. Ed., 2000, 39, 3440–3442 CrossRef CAS.
- (a) K. Gao and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 400–402 CrossRef CAS; (b) K. Gao and N. Yoshikai, Angew. Chem., Int. Ed., 2011, 50, 6888–6892 CrossRef CAS.
- Z. Yang, H. Yu and Y. Fu, Chem. – Eur. J., 2013, 19, 12093–12103 CrossRef CAS.
- For a preliminary hydroarylation example providing high ee's but very low yields (<35%), see: (a) R. Aufdenblatten, S. Diezi and A. Togni, Monatsh. Chem., 2000, 131, 1345–1350 CrossRef CAS; (b) R. Dorta and A. Togni, Chem. Commun., 2003, 760–761 RSC.
- K. Tsuchikama, M. Kasagawa, Y.-K. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939–3942 CrossRef CAS.
- T. Shirai and Y. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9894–9897 CrossRef CAS.
- C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2116–2119 CrossRef CAS.
- S. Pan, N. Ryu and T. Shibata, J. Am. Chem. Soc., 2012, 134, 17474–17477 CrossRef CAS.
- P. S. Lee and N. Yoshikai, Org. Lett., 2015, 17, 22–25 CrossRef CAS.
- For pioneering developments, see: (a) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS; (b) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS For a review, see: ; (c) C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS . For a review based on chiral diene-iridium catalysis, see: ; (d) M. Nagamoto and T. Nishimura, ACS Catal., 2016, 7, 833–847 CrossRef.
- (a) T. Shibata and T. Shizuno, Angew. Chem., Int. Ed., 2014, 53, 5410–5413 CrossRef CAS; (b) For a review on the synthesis of planar chiral ferrocenes via TMC C–H bond activations, see: D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351–365 CrossRef CAS.
- G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258–10261 CrossRef CAS.
- G. Huang and P. Liu, ACS Catal., 2016, 6, 809–820 CrossRef CAS.
- (a) Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899–5902 CrossRef CAS; (b) M. Nagamoto and T. Nishimura, Chem. Commun., 2014, 50, 6274–6277 RSC.
- M. Zhang and G. Huang, Dalton Trans., 2016, 45, 3552–3557 RSC.
- M. Hatano, Y. Ebe, T. Nishimura and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 4010–4013 CrossRef CAS.
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Chem. Commun., 2017, 53, 2760–2763 RSC.
- M. Nagamoto, H. Yorimitsu and T. Nishimura, Org. Lett., 2018, 20, 828–831 CrossRef CAS.
- Y. Ebe, M. Onoda, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 5607–5611 CrossRef CAS.
- M. Zhang, L. Hu, Y. Lang, Y. Cao and G. Huang, J. Org. Chem., 2018, 83, 2937–2947 CrossRef CAS.
- S. Pan, N. Ryu and T. Shibata, Adv. Synth. Catal., 2014, 356, 929–933 CrossRef CAS.
- G. E. M. Crisenza, O. O. Sokolova and J. F. Bower, Angew. Chem., Int. Ed., 2015, 54, 14866–14870 CrossRef CAS.
- S. Grélaud, P. Cooper, L. J. Feron and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 9351–9356 CrossRef.
- T. Shibata, M. Michino, H. Kurita, Y. Tahara and K. S. Kanyiva, Chem. – Eur. J., 2017, 23, 88–91 CrossRef CAS.
- T. Shirai, T. Okamoto and Y. Yamamoto, Asian J. Org. Chem., 2018, 7, 1054–1056 CrossRef CAS.
- T. Shibata and H. Takano, Org. Chem. Front., 2015, 2, 383–387 RSC.
- (a) B. M. Trost, K. Imi and I. W. Davies, J. Am. Chem. Soc., 1995, 117, 5371–5372 CrossRef CAS . For a recent review on TMC alkenyl C(sp2)–H activation, see: ; (b) M. Maraswami and T.-P. Loh, Synthesis, 2019, 1049–1062 CAS.
- Y.-G. Lim, J.-S. Han, B. T. Koo and J.-B. Kang, Bull. Korean Chem. Soc., 1999, 20, 1097–1100 CAS.
- (a) F. Mo and G. Dong, Science, 2014, 345, 68–72 CrossRef CAS . For additional examples based on the stoichiometric generation of the enamide intermediate, see: ; (b) Z. Wang, B. J. Reinus and G. Dong, J. Am. Chem. Soc., 2012, 134, 13954–13957 CrossRef CAS; (c) F. Mo, H. N. Lim and G. Dong, J. Am. Chem. Soc., 2015, 137, 15518–15527 CrossRef CAS.
- Y. Dang, S. Qu, Y. Tao, X. Deng and Z. X. Wang, J. Am. Chem. Soc., 2015, 137, 6279–6291 CrossRef CAS.
- D. Xing and G. Dong, J. Am. Chem. Soc., 2017, 139, 13664–13667 CrossRef CAS.
- X. Li, H. Wu, Y. Lang and G. Huang, Catal. Sci. Technol., 2018, 8, 2417–2426 RSC.
- D. Xing, X. Qi, D. Marchant, P. Liu and G. Dong, Angew. Chem., Int. Ed., 2019, 58, 4366–4370 CrossRef CAS.
- Y. J. Zhang, E. Skucas and M. J. Krische, Org. Lett., 2009, 11, 4248–4250 CrossRef CAS.
- C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 10625–10631 CrossRef CAS.
- P. Cooper, G. E. M. Crisenza, L. J. Feron and J. F. Bower, Angew. Chem., Int. Ed., 2018, 57, 14198–14202 CrossRef CAS.
- (a) N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Chem. Lett., 1997, 425–426 CrossRef CAS; (b) N. Fujii, F. Kakiuchi, A. Yamada, N. Chatani and S. Murai, Bull. Chem. Soc. Jpn., 1998, 71, 285–298 CrossRef CAS.
- C. Aïssa and A. Fürstner, J. Am. Chem. Soc., 2007, 129, 14836–14837 CrossRef.
- (a) R. K. Thalji, K. A. Ahrendt, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 9692–9693 CrossRef CAS; (b) K. L. Tan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 2685–2686 CrossRef CAS.
- (a) R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192–7193 CrossRef CAS; (b) S. J. O’Malley, K. L. Tan, A. Watzke, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 13496–13497 CrossRef; (c) H. Harada, R. K. Thalji, R. G. Bergman and J. A. Ellman, J. Org. Chem., 2008, 73, 6772–6779 CrossRef CAS.
- Z. Ding and N. Yoshikai, Angew. Chem., Int. Ed., 2013, 52, 8574–8578 CrossRef CAS.
- T. Shibata, N. Ryu and H. Takano, Adv. Synth. Catal., 2015, 357, 1131–1135 CrossRef CAS.
- D. F. Fernández, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2017, 56, 9541–9545 CrossRef.
- Y. Lang, M. Zhang, Y. Cao and G. Huang, Chem. Commun., 2018, 54, 2678–2681 RSC.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, Mill Valley, CA, 2010 Search PubMed.
- T. Shibata, H. Kurita, S. Onoda and K. S. Kanyiva, Asian J. Org. Chem., 2018, 7, 1411–1418 CrossRef CAS.
- F. Kakiuchi, Y. Yamamoto, N. Chatani and S. Murai, Chem. Lett., 1995, 681–682 CrossRef CAS.
- D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 5604–5605 CrossRef CAS.
- S. Duttwyler, S. Chen, M. K. Takase, K. B. Wiberg, R. G. Bergman and J. A. Ellman, Science, 2013, 339, 678–682 CrossRef CAS.
- (a) Y. Shibata, Y. Otake, M. Hirano and K. Tanaka, Org. Lett., 2009, 11, 689–692 CrossRef CAS . See also: ; (b) T. Mitsudo, S.-W. Zhang, M. Nagao and Y. Watanabe, J. Chem. Soc., Chem. Commun., 1991, 598–599 RSC; (c) T. Nishimura, Y. Washitake and S. Uemura, Adv. Synth. Catal., 2007, 349, 2563–2571 CrossRef CAS; (d) N. M. Neisius and B. Plietker, Angew. Chem., Int. Ed., 2009, 48, 5752–5755 CrossRef CAS; (e) K. Meng, J. Zhang, F. Li, Z. Lin, K. Zhang and G. Zhong, Org. Lett., 2017, 19, 2498–2501 CrossRef CAS.
- (a) K. Gao, P. S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2010, 132, 12249–12251 CrossRef CAS; (b) P.-S. Lee, T. Fujita and N. Yoshikai, J. Am. Chem. Soc., 2011, 133, 17283–17295 CrossRef CAS; (c) T. Yamakawa and N. Yoshikai, Tetrahedron, 2013, 69, 4459–4465 CrossRef CAS.
- T. Yamakawa and N. Yoshikai, Org. Lett., 2013, 15, 196–199 CrossRef CAS.
- T. Satoh, Y. Nishinaka, M. Miura and M. Nomura, Chem. Lett., 1999, 615–616 CrossRef CAS.
- Y. Nishinaka, T. Satoh, M. Miura, H. Morisaka, M. Nomura, H. Matsui and C. Yamaguchi, Bull. Chem. Soc. Jpn., 2001, 74, 1727–1735 CrossRef CAS.
- K. Tsuchikama, M. Kasagawa, Y.-K. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939–3942 CrossRef CAS.
- S. Takebayashi and T. Shibata, Organometallics, 2012, 31, 4114–4117 CrossRef CAS.
- M. Nagamoto, J. Fukuda, M. Hatano, H. Yorimitsu and T. Nishimura, Org. Lett., 2017, 19, 5952–5955 CrossRef CAS.
- (a) S. Yotphan, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 2452–2453 CrossRef CAS; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645–3651 CrossRef CAS; (c) J. Ellman, Y. Matsushima, E. Phillips and R. Bergman, Synlett, 2015, 1533–1536 CrossRef.
- T. Shibata, T. Baba, H. Takano and K. S. Kanyiva, Adv. Synth. Catal., 2017, 359, 1849–1853 CrossRef CAS.
- (a) The authors indicated that an alternative mechanistic scenario based on a carbometallation and C–H reductive elimination cannot be fully excluded; (b) The use of a base such as NaOAc is essential with Ir(I) to avoid partial isomerization of the double bond of 65 to the internal position.
- D. F. Fernández, C. A. B. Rodrigues, M. Calvelo, M. Gulías, J. L. Mascareñas and F. López, ACS Catal., 2018, 8, 7397–7402 CrossRef.
- (a) M. Shirakura and M. Suginome, J. Am. Chem. Soc., 2008, 130, 5410–5411 CrossRef CAS; (b) M. Shirakura and M. Suginome, Angew. Chem., Int. Ed., 2010, 48, 3827–3829 CrossRef.
- Ni-catalyzed: (a) M. Shirakura and M. Suginome, J. Am. Chem. Soc., 2009, 131, 5060–5061 CrossRef CAS; (b) M. Shirakura and M. Suginome, Org. Lett., 2009, 11, 523–526 CrossRef CAS . Ru-catalyzed: ; (c) T. Mitsudo, Y. Nakagawa, K. Watanabe, Y. Hori, H. Misawa, H. Watanabe and Y. Watanabe, J. Org. Chem., 1985, 50, 565–571 CrossRef CAS . Pd-catalyzed: ; (d) M. Rubin, J. Markov, S. Chuprakov, D. J. Wink and V. Gevorgyan, J. Org. Chem., 2003, 68, 6251–6256 CrossRef CAS; (e) L. Villarino, R. García-Fandiño, F. López and J. L. Mascareñas, Org. Lett., 2012, 14, 2996–2999 CrossRef CAS.
- (a) B.-M. Fan, Q. Yang, J. Hu, C. Fan, S. Li, L. Yu, C. Huang, W. W. Tsang and F. Y. Kwong, Angew. Chem., Int. Ed., 2012, 51, 7821–7824 CrossRef CAS; (b) J. Hu, Q. Yang, J. Xu, C. Huang, B. Fan, J. Wang, C. Lin, Z. Bian and A. S. C. Chan, Org. Biomol. Chem., 2013, 11, 814–820 RSC; (c) Q. Yang, P. Y. Choy, B. Fan and F. Y. Kwong, Adv. Synth. Catal., 2015, 357, 2345–2350 CrossRef CAS.
- X.-Y. Bai, Z.-X. Wang and B.-J. Li, Angew. Chem., Int. Ed., 2016, 55, 9007–9011 CrossRef CAS.
- X.-Y. Bai, Z.-X. Wang, Q. Li and B.-J. Li, J. Am. Chem. Soc., 2018, 140, 506–514 CrossRef CAS.
- Z. Yu, L. Meng and Z. Lin, Organometallics, 2019, 38, 2998–3006 CrossRef CAS.
- Z.-X. Wang and B.-J. Li, J. Am. Chem. Soc., 2019, 141, 9312–9320 CrossRef.
- W. W. Zhang, S. L. Zhang and B. J. Li, Angew. Chem., Int. Ed., 2020, 59, 6874–6880 CrossRef CAS.
- For related Ir-catalyzed hydroalkynylation methods that afford γ-alkynylated products, using triisopropylsilylacetylene and α,β- or β,γ-unsaturated amides, see: Z.-X. Wang, X.-Y. Bai, H.-C. Yao and B.-J. Li, J. Am. Chem. Soc., 2016, 138, 14872–14875 CrossRef CAS.
- (a) T. Sakurada, Y.-k. Sugiyama and S. Okamoto, J. Org. Chem., 2013, 78, 3583–3591 CrossRef CAS . For a related cross-dimerization of arylacetylenes, see: ; (b) S. Ventre, E. Derat, M. Amatore, C. Aubert and M. Petit, Adv. Synth. Catal., 2013, 355, 2584–2590 CrossRef CAS.
- Y. Ueda, H. Tsurugi and K. Mashima, Angew. Chem., Int. Ed., 2020, 59, 1552–1556 CrossRef CAS.
- J.-C. Grenier-Petel and S. K. Collins, ACS Catal., 2019, 9, 3213–3218 CrossRef CAS.
- For recent reviews on Metal-Catalyzed Functionalization of C(sp3)–H see: (a) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2017, 57, 62–101 CrossRef; (b) R. R. Karimov and J. F. Hartwig, Angew. Chem., Int. Ed., 2018, 57, 4234–4241 CrossRef CAS; (c) Y. Xu and G. Dong, Chem. Sci., 2018, 9, 1424–1432 RSC; (d) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef; (e) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC.
- (a) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (b) T. A. Ramirez, B. Zhao and Y. Shi, Chem. Soc. Rev., 2012, 41, 931–942 RSC; (c) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS; (d) M. C. White and J. Zhao, J. Am. Chem. Soc., 2018, 140, 13988–14009 CrossRef CAS; (e) H. M. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS.
- C.-H. Jun, Chem. Commun., 1998, 1405–1406 RSC.
- N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935–10941 CrossRef CAS.
- A. D. Bolig and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 14544–14545 CrossRef CAS.
- T. Yoshino and S. Matsunaga, Asian J. Org. Chem., 2018, 7, 1193–1205 CrossRef CAS.
- Q. Li and Z.-X. Yu, J. Am. Chem. Soc., 2010, 132, 4542–4543 CrossRef CAS.
- Q. Li and Z.-X. Yu, Organometallics, 2012, 31, 5185–5195 CrossRef CAS.
- Q. Li and Z.-X. Yu, Angew. Chem., Int. Ed., 2011, 50, 2144–2147 CrossRef CAS.
- B. DeBoef, S. J. Pastine and D. Sames, J. Am. Chem. Soc., 2004, 126, 6556–6557 CrossRef CAS.
- K. Tsuchikama, M. Kasagawa, K. Endo and T. Shibata, Org. Lett., 2009, 11, 1821–1823 CrossRef CAS.
- S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 13, 4692–4695 CrossRef CAS.
- S. Pan, Y. Matsuo, K. Endo and T. Shibata, Tetrahedron, 2012, 68, 9009–9015 CrossRef CAS.
- H. Hattori and T. Nishimura, Adv. Synth. Catal., 2018, 360, 4827–4831 CrossRef CAS.
- Y. Tahara, M. Michino, M. Ito, K. S. Kanyiva and T. Shibata, Chem. Commun., 2015, 51, 16660–16663 RSC.
- G. Lahm and T. Opatz, Org. Lett., 2014, 16, 4201–4203 CrossRef CAS.
- A. T. Tran and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 10530–10534 CrossRef CAS.
- P. Verma, J. M. Richter, N. Chekshin, J. X. Qiao and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 5117–5125 CrossRef.
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 7200–7204 CrossRef CAS.
- I. Nakamura, D. Yamauchi and T. Nishimura, Asian J. Org. Chem., 2018, 7, 1347–1350 CrossRef CAS.
- T. Torigoe, T. Ohmura and M. Suginome, Angew. Chem., Int. Ed., 2017, 56, 14272–14276 CrossRef CAS.
- T. Torigoe, T. Ohmura and M. Suginome, Chem. – Eur. J., 2016, 22, 10415–10419 CrossRef CAS.
Footnote |
| † Current address: Merck Center for Catalysis, Princeton University, Princeton, New Jersey 08544, USA. |
| This journal is © The Royal Society of Chemistry 2020 |