
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Next-generation DNA damage sequencing
Cécile
Mingard
 a,
Junzhou
Wu
ab,
Maureen
McKeague
*cd and
Shana J.
Sturla
*a
a,
Junzhou
Wu
ab,
Maureen
McKeague
*cd and
Shana J.
Sturla
*a
aDepartment of Health Sciences and Technology, ETH Zürich, Schmelzbergstrasse 9, 8092 Zürich, Switzerland. E-mail: sturlas@ethz.ch
bDepartment of Biological Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, 02139, Massachusetts, USA
cDepartment of Pharmacology and Therapeutics, McGill University, 3655 Prom. Sir William Osler, Montreal, Quebec H3G 1Y6, Canada. E-mail: maureen.mckeague@mcgill.ca
dDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 0B8, Canada
First published on 24th September 2020
Abstract
Cellular DNA is constantly chemically altered by exogenous and endogenous agents. As all processes of life depend on the transmission of the genetic information, multiple biological processes exist to ensure genome integrity. Chemically damaged DNA has been linked to cancer and aging, therefore it is of great interest to map DNA damage formation and repair to elucidate the distribution of damage on a genome-wide scale. While the low abundance and inability to enzymatically amplify DNA damage are obstacles to genome-wide sequencing, new developments in the last few years have enabled high-resolution mapping of damaged bases. Recently, a number of DNA damage sequencing library construction strategies coupled to new data analysis pipelines allowed the mapping of specific DNA damage formation and repair at high and single nucleotide resolution. Strikingly, these advancements revealed that the distribution of DNA damage is heavily influenced by chromatin states and the binding of transcription factors. In the last seven years, these novel approaches have revealed new genomic maps of DNA damage distribution in a variety of organisms as generated by diverse chemical and physical DNA insults; oxidative stress, chemotherapeutic drugs, environmental pollutants, and sun exposure. Preferred sequences for damage formation and repair have been elucidated, thus making it possible to identify persistent weak spots in the genome as locations predicted to be vulnerable for mutation. As such, sequencing DNA damage will have an immense impact on our ability to elucidate mechanisms of disease initiation, and to evaluate and predict the efficacy of chemotherapeutic drugs.
 Cécile Mingard | Cécile Mingard received her Master's degree in Toxicology from the University of Basel in 2016. Under the supervision of Prof. Stephan Krähenbühl, she studied mechanisms of hepatotoxicity induced by tyrosine kinase inhibitors at the University Hospital of Basel. She then moved to Zürich to start a PhD at the ETH in Prof. Shana J. Sturla's lab, where she is studying genome-wide patterns of DNA damage distribution associated with mutagenesis. |
 Junzhou Wu | Junzhou Wu obtained his PhD degree (2019) from ETH Zurich under the supervision of Prof. Shana J. Sturla, concerning the development of DNA damage sequencing methods. He then received an SNF early postdoc.mobility fellowship and joined Prof. Peter Dedon's lab as a postdoctoral fellow at MIT, where he is currently developing a novel RNA sequencing technology to interrogate the epitranscriptome in human, bacterial and viral diseases. |
 Maureen McKeague | Maureen McKeague is an Assistant Professor and Canada Research Chair Tier 2 in Genomic Chemistry at McGill University. She completed a PhD (2012) in nucleic acids chemistry at Carleton University. She then received an NSERC award to perform postdoctoral work in Bioengineering at Stanford University. From 2016–2018, she carried out research in the Sturla Lab at ETH Zurich concerning the quantification and mapping of DNA damage. Researchers in the McKeague lab are dedicated to solving challenges at the interface of chemistry and the genome with projects spanning green chemistry and precision medicine. The website is https://mckeague.lab.mcgill.ca/. |
 Shana J. Sturla | Shana J. Sturla is the Professor of Toxicology at ETH Zürich. She completed her BS and PhD in Chemistry at the University of California at Berkeley and Massachusetts Institute of Technology. Her postdoctoral research focused on carcinogenesis and chemoprevention at the University of Minnesota Cancer Center. She started her independent career in 2004 at the University of Minnesota, and her team moved ETH Zürich in 2010. The ETH Laboratory of Toxicology aims to delineate relationships between chemical structure, biotransformation, and cellular response in order to understand the chemical basis of disease etiology and develop safer strategies for therapy and prevention. The website is http://www.toxicology.ethz.ch. |
1. Introduction
All physiological processes tied to cellular replication rely on the chemical integrity of DNA, and its damage is associated with a range of adverse outcomes such as accelerated aging and cancer.1–3 Endogenous cellular processes and the effects of exogenous agents, including UV irradiation and chemicals from environmental, dietary, drug, and occupational exposures, are constantly altering DNA (Fig. 1).4 These reactions create up to 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 distinct damage events in a cell each day, distorting the structure of the DNA and, if left unrepaired, potentially stalling replication or impacting gene expression.4
000 distinct damage events in a cell each day, distorting the structure of the DNA and, if left unrepaired, potentially stalling replication or impacting gene expression.4
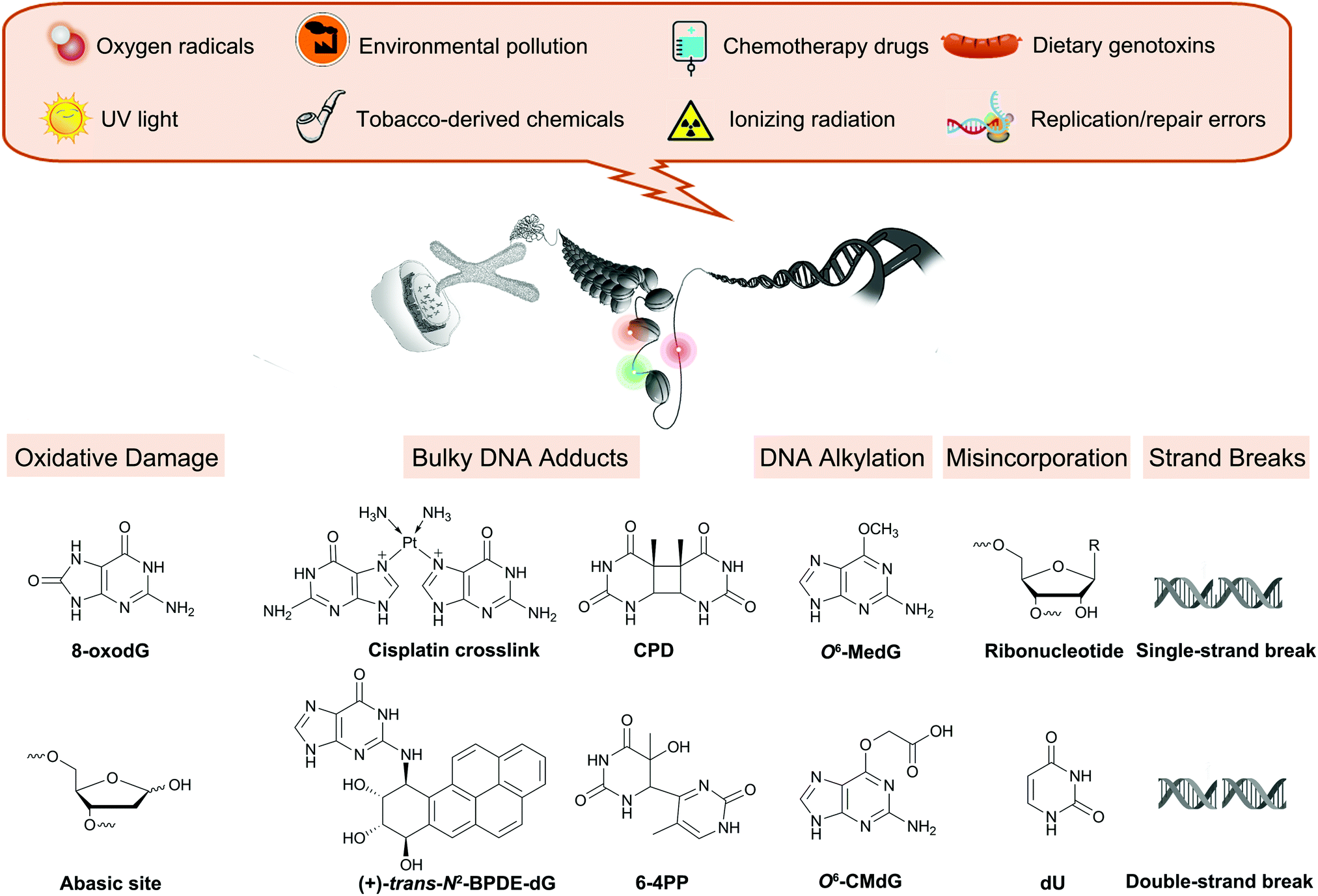 | ||
| Fig. 1 Overview of endogenous processes and exogenous exposures leading to DNA damage discussed in this review. | ||
Translesion DNA synthesis (TLS), involving specialized DNA polymerases that can bypass DNA damage, counters the cytotoxic effects of DNA replication stalling and acts in concert with DNA repair functions. In cancer therapy with DNA-binding agents, which target and stall replication, this process can contribute to drug resistance. In normal cells, TLS may be protective, but even if cytotoxicity may be avoided in the short term, DNA damage bypass can be highly mutagenic and contribute to cancer and other adverse outcomes in the long run. The biological and toxicological consequences of DNA damage, repair, and bypass depend fundamentally on not only their structure and abundance, but also their distribution in the genome, including the interplay of chemical modification and higher chromatin structures in gene expression and mutagenesis.
High-throughput sequencing has recently enabled the whole genome sequencing of numerous cancer genomes. From these large datasets, mutational signatures that describe characteristic imprints left by mutational processes, including DNA damage and repair, have been deciphered in cancer genomes.5 Because the cellular impacts of DNA damage are also the basis of the most common cancer therapy drugs, understanding the genomic distribution of DNA modification induced by anticancer drugs is a potential strategy improve the safety and efficacy of cancer therapy. While there are many techniques to study outcomes of DNA damage (i.e. mutation, cytotoxicity), there is a lag in methods available to map how DNA is initially modified, therefore limiting the ability to predict adverse or therapeutic outcomes on the basis of early measurable markers.
Defining the relationship between the distribution of chemical forms of DNA damage on a genome-wide scale with adverse or therapeutic biological outcomes is a tough nut to crack. Early models of DNA damage and mutagenesis were built around a simple direct relationship between damage formation and the acquisition of a mutation, but there is a complex interplay between genetic and epigenetic landscapes factoring into cancer evolution and progression. Indeed, cancer is driven by natural selection enabled by the evolution of mutations conferring a growth advantage.6 However, within the large mutational landscape, only some mutations are driver mutations that confer a selective growth advantage; whereas many of the other mutations are passenger mutations acquired by a cell with driver mutations.7,8
There is controversy concerning whether most mutations in cancer genomes arise from DNA replication errors or other intrinsic events (the bad luck hypothesis), which are hard to prevent9 or from extrinsic factors that, on the contrary, could be avoided.10 Indeed mutational signatures are at the core of extensive ongoing work to uncover the etiology of individual cancers, but strategies for tracking analogous precursor DNA damage signatures in the genome lag behind gene sequencing and epigenetics because of their inherent chemical complexity and variation.11
There are many well-established strategies for DNA damage quantification integrated over the whole genome as well as strategies for identifying the sequence specific locations of damage in isolated genes, but typically not both. For example, mass spectrometry12 and 32P-postlabelling13 allow high-sensitivity quantification of total DNA damage in biological samples, but do not provide sequence or location information. In contrast, ligation-mediated polymerase chain reaction (LM-PCR) is based on the principle that DNA polymerases cannot synthesize DNA past certain types of damage. Thus, LM-PCR can indicate the exact sequence and position of DNA damage on the basis of PCR termination sites; however, this method is not damage specific, meaning the chemical nature of the damage may be unknown. These strategies have various advantages, but do not allow one to relate the chemistry of damage formation with biological changes in particular genes or in the genome.
In addition to the conceptual challenge of examining the complex relationship between genome sequences, structure, regulation and potential patterns of DNA damage processes, there are two major technical challenges towards obtaining the necessary robust damage sequence data to evaluate these relationships. The first is that DNA damage events are rare on a genome-wide scale. Typically 0.1–100 endogenous DNA damage events occur per 106 nucleotides,14 and those arising from discreet interactions with particular chemicals can be of even lower magnitude such as one DNA adduct per 1011 nucleotides.15 The second challenge is that chemical damage is not typically read by DNA polymerases, and they may either stall or insert an incorrect base or combination of incorrect bases opposite the altered site. As result, the chemical identity of DNA damage is generally lost in the process of standard DNA sequencing. Nonetheless, there are several very recent examples discussed in this review of exciting and innovative approaches to address these technical challenges and yield the first insights on DNA damage distribution at the genome-wide level.
We provide here a comprehensive review of the progress in sequence-specific mapping of DNA damage. Emerging methods described in this review have addressed long-standing obstacles facing damage sequencing by including a combination of damage enrichment, damage specific recognition, and functional marking of the damage position with a sequencing-compatible adaptor (Fig. 2). A few reviews highlight specific DNA damage sequencing methods; however, no reviews exist that cover all classes of DNA damage and discuss the importance of the biological findings.16–20 For each major class of DNA damage, we first provide an overview of the occurrence and biological relevance. Next, we describe each of the novel strategies that have enabled successful DNA damage sequencing of these specific DNA damage classes (Table 1). Finally, we compare the opportunities and challenges for each of these methods, focusing on the early glimpses of biological insight enabled by each unique method. The rapid improvement and adoption of these approaches is expected to spur advances in the study and prevention of aging, cancer, and disease related to genomic instability.
| DNA damage product | Source of DNA damage | Mapping methods | Source of DNAa | Ref. |
|---|---|---|---|---|
| a When not specified the source of DNA is from immortalized cultured cell lines. | ||||
| 8-OxodG | Oxidative stress | OxiDIP-seq | Mouse, human | 55 and 56 |
| enTRAP-seq | Mouse | 57 | ||
| OG-seq | Mouse | 58 | ||
| Click-code seq | Yeast | 60 | ||
| AP-seq | Human | 59 | ||
| Platinated crosslinks | Cisplatin, oxaliplatin | (HS)-damage-seq | Human | 82, 89 and 98 |
| XR-seq | Human, mouse (in vivo) | 82 and 89–91 | ||
| Cisplatin-seq | Human | 83 | ||
| CPDs | UV light | DDIP-seq | Human | 106 |
| HS-damage-seq | Human | 98 | ||
| (t)XR-seq | Human | 84, 98 and 110 | ||
| CPD-seq | Yeast | 87 and 108 | ||
| Excision-seq | Yeast | 107 | ||
| 6-4PPs | UV light | HS-damage-seq | Human | 98 |
| XR-seq | Human | 98 | ||
| Excision-seq | Yeast | 107 | ||
| BPDE-dG | Benzo[a]pyrene | tXR-seq | Human | 110 |
| Abasic sites | Product of DNA damage, spontaneous depurination | AP-seq | Human | 59 |
| snAP-seq | Human, parasite (in vivo) | 72 | ||
| Nick-seq | Bacteria | 183 | ||
| Single-strand breaks | Product of oxidative damage, failure in DNA repair, topoisomerase activity, disintegration of sugar | SSB-Seq | Human | 187 |
| SSingLE | Human | 188 | ||
| GLOE-Seq | Human, yeast | 189 | ||
| Double-strand breaks | From SSB or fail in DNA repair | BLESS | Mouse (in vivo) | 194 |
| Break-seq | Yeast | 195 | ||
| DSBcapture | Human | 196 | ||
| END-seq | Mouse (in vivo) | 197 | ||
| GUIDE-seq | Human | 198 | ||
| BLISS | Human, mouse (in vivo) | 199 | ||
| DSB-Seq | Human | 187 | ||
| qDSB-Seq | Human, mouse (in vivo) | 200 | ||
| Ribonucleotides | Enzymatic insertion | Ribose-seq | Yeast | 214 |
| HydEN-seq | Yeast | 215 and 223 | ||
| Pu-seq | Yeast | 216 and 224 | ||
| emRiboSeq | Yeast | 217 and 218 | ||
| Uracil | Enzymatic insertion, cytosine deamination | Excision-seq | Yeast, bacteria | 107 |
| dU-seq | Human | 220 | ||
| UPD-seq | Bacteria | 221 | ||
| U-DNA-seq | Human | 222 | ||
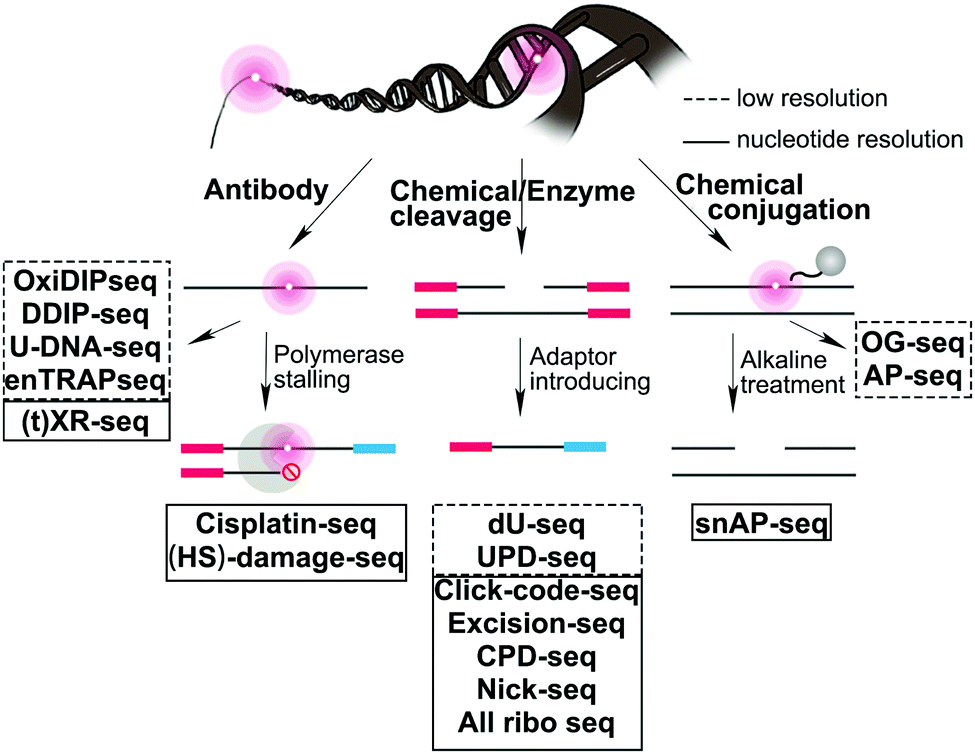 | ||
| Fig. 2 Summary of sequencing strategies used to map DNA damage formation or repair at the genome-wide level. | ||
2. Oxidative damage and 8-oxoguanine
2.1 Occurrence and relevance
Guanine has the lowest redox potential of the DNA bases, and thus can be easily oxidized to form 8-oxo-7,8-dihydrodeoxyguanine (8-oxodG) via single-electron transfer mediated by reactive oxygen species (ROS) such as superoxide, hydrogen peroxide, and hydroxyl radicals.21 ROS are generated by normal metabolic processes or as a consequence of exposure to environmental pro-oxidants, such as components of cigarette smoke, alcohol, ionizing and UV radiation, pesticides, and ozone.22 The production and scavenging of ROS are highly coordinated by cellular antioxidant networks essential for cell signaling and homeostasis.23 A mild increase of ROS production in cells and organisms has a variety of anti-aging and longevity-extending hormesis effects by stimulating endogenous defense mechanisms and stress resistance.24 However, ROS is a double-edged sword. Under typical physiological ROS levels, 8-oxodG is generated at a frequency of at least several hundred damage events per human cell per day; this rate is further increased under conditions of oxidative overload.25 Oxidative stress contributes to cancer, atherosclerosis, diabetes, aging, and pathologies of the central nervous system,26–28 making 8-oxodG an indicator of oxidative stress and a cellular biomarker of pathophysiological processes.2.2 8-OxodG repair, mutagenicity, and toxicity
Efficient search and removal of 8-oxodG is performed by the base excision repair (BER) pathway to maintain cell integrity. Three different enzymes cooperate to handle 8-oxodG in the cell, involving 8-oxo-dGTP diphosphohydrolase, 8-oxoguanine DNA glycosylase and adenine DNA glycosylase (Table 2).29–31 By monitoring the nucleotide pool, 8-oxo-dGTP diphosphohydrolase prevents the incorporation of 8-oxodG into nascent DNA. 8-Oxoguanine glycosylase acts on 8-oxodG within double stranded DNA (dsDNA), directly removing it when paired with cytosine. Additionally, persistent 8-oxodG may pair with adenine, promoted by Hoogsteen bonding during replication,32 in which case, adenine DNA glycosylase can excise the incorporated adenine. When this defense system is overwhelmed and 8-oxodG persists during replication, it is prone to G → T transversion mutation. Indeed, this type of mutation is prevalent in the MTH1/OGG1/MUTYH triple knockout mouse model (TOY-KO) and MUTYH-associated polyposis (MAP) syndrome colorectal cancer.33,34| Enzymes | Function | Genes | ||
|---|---|---|---|---|
| Human | S. cerevisiae | E. coli | ||
| 8-Oxo-dGTP diphosphohydrolase | Degrade 8-oxo-dGTP to the monophosphate | NUDT1 | PCD1 | mutT |
| Previous: MTH1 | ||||
| 8-Oxoguanine DNA glycosylase | Excise 8-oxodG when opposite dC | OGG1 | OGG1 | mutM |
| Adenine DNA glycosylase | Excise dA when opposite 8-oxodG | MUTYH | not present | mutY |
Aside from pro-mutagenic effects, 8-oxodG is also a source of toxicity when transcribed. 8-OxodG can significantly arrest transcription by direct structural interference of transcription components or the repair intermediate of 8-oxodG/OGG1.35 Furthermore, when 8-oxodG is located on the transcribed DNA strand, other consequences like erroneous bypass of the lesion by the transcribing RNA polymerase may occur. Such transcriptional mutagenesis often results in a specific C → A mutation in the RNA transcript and aberrant protein production,36 which may play a role in protein aggregation and the pathogenesis of neurodegenerative diseases, such as Alzheimer's and Parkinson's disease.37
2.3 8-OxodG and OGG1 modulate gene expression
Despite the toxicological implications of 8-oxodG, mounting evidence supports that 8-oxodG may be a cellular friend by facilitating gene activation in response to oxidative stress, countering conventional models of DNA damage effects. 8-OxodG induced gene expression involves several pathways, including direct interactions of OGG1 with transcription factors (TFs) or chromatin remodelers and allosteric transition of 8-oxodG containing G-quadruplex.When 8-oxodG is located at promoter regions, OGG1 is recruited and enhances the binding of several TFs, including hypoxia-inducible factor 1a (HIF-1α),38 signal transducer and activator of transcription 1 (STAT1),39 and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB).40,41 Reduction in OGG1 expression in rat pulmonary arterial endothelial cells strongly reduced the binding of the TF HIF-1α to the vascular endothelial growth factor gene (VEGF) promoter and reduced VEGF expression.38 OGG1 both coactivates STAT1 and induces the transcriptional activation of pro-inflammatory mediators after lipopolysaccharide (LPS) stimulation.39 In addition, the binding of OGG1 to 8-oxodG in promoter regions enhanced NF-kB/RelA binding to cis-regulatory elements and facilitated the rapid recruitment of specificity protein 1 (Sp1), transcription initiation factor II-D and phosphorylated-RNA polymerase II (Pol II), resulting in prompt gene expression upon oxidative exposure.40,41 Thus, interactions between 8-oxodG, OGG1 and relevant TFs lead to the expression of oxidative stress-induced genes.
In addition to the interactions between OGG1 and TFs, 8-oxodG in gene promoter regions regulates transcription via G-quadruplex (G4) folding. Indeed, potential G-quadruplex sequences are widely distributed in the human genome, with high enrichment in gene promoters.42,43 The formation of 8-oxodG in G-rich sequences can either impede G4–protein interactions or stall repair proteins at G4 structures which further recruit TFs. For example, the VEGF promoter contains three G-rich Sp1 binding sites, which is critical for regulating mRNA synthesis.44,45 When 8-oxodG accumulates due to hypoxia, Sp1 binding decreases in these G-rich elements, resulting in the up-regulation of VEGF transcription.38,46 These observations suggest that G4 formation activates transcription when 8-oxodG is present. Recently, Burrows et al. reported that plasmids containing 8-oxodG in G4 promoter regions produced more target protein than the same plasmid without 8-oxodG.47 The data suggest that 8-oxodG in G-rich regions of the VEGF promoter were removed by OGG1, generating abasic sites (AP sites) and destabilizing the duplex structure. This loss of stability led to the formation of a new G4 structure with an abasic-site-containing loop, which facilitated the binding and stalling of APE1 to the AP site, further stimulating TF binding and activating transcription.47–50
The emerging role of 8-oxodG as a transcriptional regulator highlights its biological and health relevance beyond classic toxicity aspects of DNA damage. However, genome-wide associations of 8-oxodG with gene expression and further with pathological processes are not understood due to the lack of precise location information of 8-oxodG in the genome. Thus, extensive efforts have been made to locate 8-oxodG with several recent high-throughput sequencing strategies providing advanced tools to understand how 8-oxodG is distributed and can modulate gene expression on a genome-wide level.
2.4 Genome-wide mapping of 8-oxodG
Initial strategies to map 8-oxodG involved enrichment by antibody pull-down of fragmented sequences containing 8-oxodG (i.e. analogous to ChIP-sequencing), where antibodies are used to selectively enrich for DNA sequences bound by a particular protein to map global protein-binding sites in cells. Following antibody binding or enrichment, 8-oxodG location could be obtained using several methods including microscopy, Sanger sequencing, high-throughput microarray assays and more recently, next-generation sequencing.The first genome-wide map of 8-oxodG was constructed nearly 14 years ago using an antibody enrichment strategy, resulting in a map of 8-oxodG in human metaphase chromosomes at a 1000 kb resolution, revealing its heterogeneous distribution in the genome.51 Specifically, immunofluorescence revealed that 8-oxodG was unevenly distributed and located primarily within regions with a high frequency of recombination and single nucleotide polymorphisms (SNPs) in cultured human lymphocytes.51 However, the relatively low resolution of optical microscopy limited the resolution of the 8-oxodG map. By Sanger sequencing, a map of 8-oxodG at a 100 base pair resolution was achieved in mouse renal cortical samples,52 allowing for 8-oxodG analysis at the gene-level. These data suggested that 8-oxodG is preferentially enriched in highly expressed genes, presenting the first clue for the potential impact of 8-oxodG on gene expression.52 However, due to the limited throughput of Sanger sequencing, the resulting map only revealed several hundred 8-oxodG sites in the mouse genome. More recently, two microarray analyses allowed for a higher throughput genome-wide mapping of 8-oxodG in kidney tissues from rats and mice (244000 probes for rat genome and 720000 probes for mouse genome).53 Both studies revealed that 8-oxodG was preferentially located in gene deserts, devoid of protein-coding genes, and correlated with lamina-associated domains.53,54
In the last 5 years, next-generation sequencing technologies have been combined with affinity enrichment strategies to achieve genome-wide high-throughput mapping of 8-oxodG. As one example, OxiDIP-seq used an 8-oxodG antibody for immunoprecipitation followed by high-throughput sequencing in human non-tumorigenic epithelial breast cells and mouse embryonic fibroblasts.55,56 The sequencing revealed that 8-oxodG sites accumulated in the transcribed regions of long genes and at DNA replication origins, overlapping with γH2AX ChIP-seq signals and double-strand breaks. Furthermore, a strong reduction of 8-oxodG was observed within promoter regions with high GC content in quiescent (G0) cells without DNA replication. As another example, an OGG1 K249Q mutant lacking glycosylase activity was used to trap a stable complex of OGG1 with the sequences containing 8-oxodG (enTRAP-seq).57 Following affinity precipitation and sequencing, enTRAP-seq revealed enrichment of 8-oxodG in transcriptionally active chromatin regions and regulatory elements such as promoters, 5′UTRs, and CpG islands in the mouse embryonic fibroblast genome. While 8-oxodG-specific binding proteins are useful tools for 8-oxodG enrichment and sequencing, further studies comparing the binding specificity of antibody clones and glycosylase mutants will help to understand apparently conflicting results.
Besides protein-based enrichment, two chemical enrichment methods have also been developed for high-throughput sequencing of 8-oxodG. The first approach was based on the selective oxidation of 8-oxodG to form an electrophilic intermediate that can be specifically recognized and labelled with amine-terminated biotin for affinity enrichment (OG-seq).58 In mouse embryonic fibroblast cells, 8-oxodG levels were elevated in promoters, 5′-UTRs, 3′-UTRs and G4 structures in comparison with the baseline random distribution throughout the genome. The second chemical-based enrichment approach was based on the reaction between an AP site released from 8-oxodG and an aldehyde reactive probe (AP-seq), enabling both the specific recognition and enrichment of 8-oxodG sequences.59 In HepG2 cells, a reduction of 8-oxodG was found in functional elements such as promoters, exons, TF binding sites, and termination sites in a seemingly GC content-dependent manner. However, AP-seq has been used to sequence other aldehyde-containing nucleotides, to be discussed in the abasic site section. Depending on the biological questions addressed, a potential drawback of these protein- and chemical-based enrichment methods is lack of nucleotide resolution, preventing determination of sequence-specific 8-oxodG occurrence and distribution at the resolution level, for example of mutational signatures.
A nucleotide resolution map of 8-oxodG is of interest to better understand sequence context effects of 8-oxodG formation and repair, and origins of mutational signatures. Recently, we reported a nucleotide resolution sequencing method, click-code-seq, to map 8-oxodG.60 In this approach, 8-oxodG sites are specifically recognized and removed by an 8-oxodG glycosylase, generating a gap with a free 3′-hydroxyl at the damage site. Next, a synthetic O-3′-propargyl modified nucleotide (prop-dGTP) is incorporated into the resulting gap by DNA polymerase, giving rise to a 3′-alkynyl-modified end. The 3′-alkynyl DNA is then ligated to a 5′-azido-modified code sequence via a copper(I)-catalyzed click reaction, resulting in triazole-linked DNA that can be amplified by DNA polymerases.61Via this process, 8-oxodG sites are stably labelled with a code sequence that serves as a tag for affinity enrichment, an adaptor for PCR amplification, and a sequencing-compatible marker of the damage locations.60
Using click-code-seq, a single-base resolution whole genome map of DNA oxidation was obtained for S. cerevisiae.60 On a genome level, the first G in a 5′-GG-3′ dimer was more frequently oxidized than in other contexts. By analyzing 8-oxodG within discrete genomic features, especially transcription start sites (TSS), transcription terminator sites, DNase I hypersensitive sites, and autonomously replicating sequences, less 8-oxodG could be observed relative to the average coverage over the entire genome. On the other hand, telomeres, nucleosomes, and positions of low RNA Pol II occupancy had higher 8-oxodG frequency. Meanwhile, nucleosomes with post-translational modifications that accelerate nucleosome unwrapping had less 8-oxodG compared to nucleosomes without these modifications. These data suggest that chromatin accessibility may shape 8-oxodG distribution, with an accumulation of 8-oxodG in regions of reduced chromatin accessibility where repair proteins cannot penetrate.
All of the genome-wide sequencing methods for 8-oxodG reported to date rely on damaged sample enrichment (Fig. 3). However, 8-oxodG can also be sequenced directly at nucleotide resolution without enrichment using third generation sequencing technologies, such as single-molecule real-time sequencing62 and nanopore sequencing.63–65
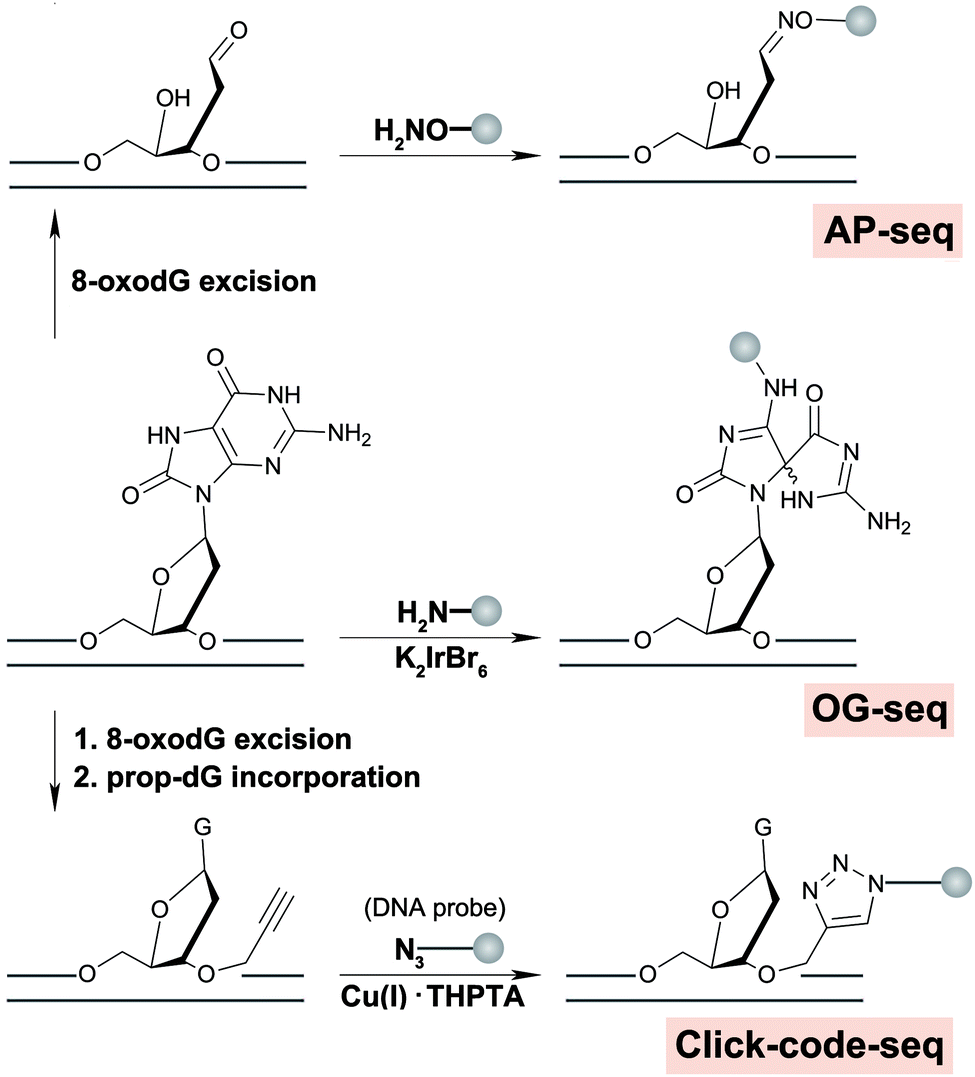 | ||
| Fig. 3 Chemical reactions involved in strategies for genome-wide mapping for 8-oxodG or subsequently formed AP sites. | ||
Finally, a number of methods have the potential to detect 8-oxodG at nucleotide resolution, but were designed for one gene or one position, such as DNA hybridization probes containing a non-natural nucleoside specific for 8-oxodG,66 LM-PCR,67 third base pair based amplification,68 BER-mediated deletion mutation69 and Hoogsteen base pairing-mediated PCR-sequencing.70 These technologies are faster and cheaper than whole genome sequencing and may be used as diagnostic tools to detect 8-oxodG hotspots within the genome.
From the 10 currently available genomic maps of 8-oxodG in biological contexts ranging from yeast and rodents to cultured human cells, and with resolutions varying from thousands of kb to a single nucleotide, a consistently emerging observation is that there is a non-uniform genomic distribution of 8-oxodG. In particular, DNA oxidation depends on the heterogeneous structure of a chromosome, consisting of protein-bound regions, open regulatory regions, and actively transcribed genes. However, it is too early to make strong biological conclusions from these data due to the differing species, conditions, library preparation protocols, and processing. Further methodological improvements are needed to understand, eliminate, or correct for embedded biases, as well as to control for artefactual DNA oxidation during sample preparation, a notorious problem in DNA oxidation analysis. Potential biases may arise from the binding specificity of different antibody clones/glycosylases, reaction selectivity of chemical probes, adaptor ligation, and PCR amplification.71–73 Meanwhile, artefactual 8-oxodG may arise from genomic DNA extraction and DNA shearing, leading to false positive reads during sequencing.74 Additionally, further work is anticipated to improve data reliability and the sensitivity of 8-oxodG sequencing methods. In the future, systematic sequencing studies of DNA oxidation are expected with a complement of robust methods to reveal a genomic basis of cellular oxidative stress responses.
3. Cisplatin
3.1 Relevance and repair
Cisplatin (cis-diamminedichloroplatinum II, [Pt(NH3)2Cl2]), and related platinum compounds such as oxaliplatin (C8H14N2O4Pt), are drugs used to treat a variety of solid tumors including breast, ovarian, head and neck, testicular, bladder, lung, brain, and esophageal cancers.75–77 Platinum-based drugs bind to DNA, forming interstrand or intrastrand DNA crosslinks, especially Pt-d(GpG), followed by Pt-d(ApG) and Pt-d(GpNpG).75,78 Once platinated, replicative DNA synthesis is stalled and, if left unrepaired, can lead to DNA double-strand breaks and cell death.79 Cisplatin–DNA crosslinks are recognized and removed by nucleotide excision repair (NER) machinery, and upregulation of NER function is often observed in resistant tumors, along with increased cisplatin glutathione conjugation, increased cellular efflux, or proficient bypass of Pt-DNA damage by translesion DNA synthesis.80,81 Because cisplatin resistance deteriorates the prognosis for cancer patient survival, recent DNA damage repair mapping techniques aim to define the genomic distribution of cisplatin crosslinks and the influence of altered NER function in cancer cells.3.2 Genome-wide mapping of cisplatin–DNA damage
Two methods, damage-seq and cisplatin-seq, have been used to map cisplatin damage in human cells at single nucleotide resolution. Due to the low abundance of damage, both methods require an initial enrichment step. Damage-seq involves using an antibody specific either for cisplatin or oxaliplatin damage,82 and cisplatin-seq involves using the HMGB1 domain A protein, which binds to distorted helices induced by cisplatin–DNA damage (Fig. 4).83 In developing the cisplatin-seq approach, different constructs of HMGB1 protein were tested for their affinity towards cisplatin-induced distorted DNA structures. HMGB1 domain A was found to be the most specific for binding cisplatin-induced damage. Interestingly, in the damage-seq study, enrichment for cis-GG and cis-AG damage products, which was previously claimed to be impossible using any commercially available antibody for Pt-DNA damage,83 were successful. In both studies, only intrastrand crosslinks could be mapped due to the specificity of these antibodies and proteins, but this selectivity is biologically relevant since intrastrand crosslinks are the most potent in killing cancer cells.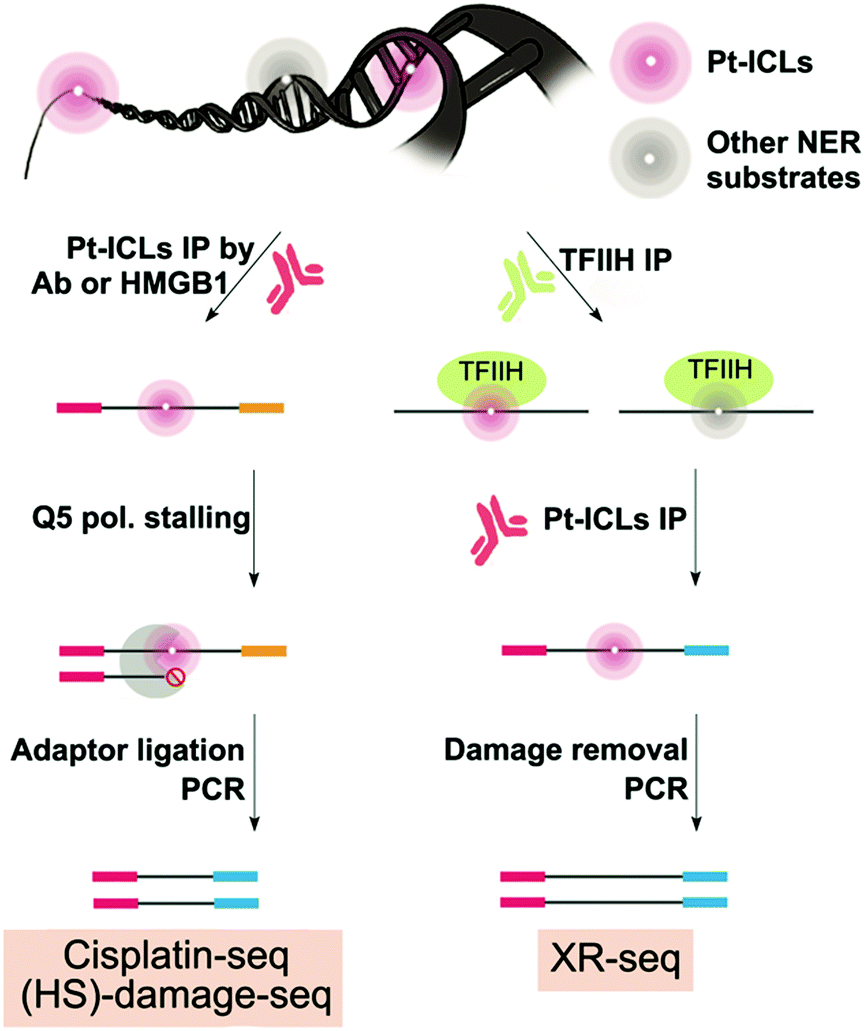 | ||
| Fig. 4 Genome-wide mapping methods for cisplatin–DNA damage formation and NER repair events at single-nucleotide resolution. XR-seq and damage-seq were used to map oxaliplatin DNA damage formation and NER repair events as well. | ||
Following enrichment, both methods then take advantage of the fact that cisplatin–DNA damage stalls polymerases to mark the specific location of damage during PCR (Fig. 4). Specifically, a biotinylated primer is used in damage seq for amplification of the enriched DNA from human lymphocytes with the high-fidelity Q5 polymerase. Q5 polymerase stalls upon encountering Pt-DNA damage such that DNA synthesis termination sites mark the site of the damage. Next, different sized DNA fragments yielded from the biotinylated primer were purified using streptavidin beads. Finally, a second adapter is ligated, and the resulting DNA library is sequenced by next-generation sequencing. Alignment of the sequencing reads with a human reference genome then allows for the identification of damage sites. Cisplatin-seq follows a very similar protocol as damage-seq for DNA damage location site marking. Damage-seq and cisplatin-seq methods reported the first genome-wide maps of cisplatin damage distribution in the human genome at single nucleotide resolution.
In addition to formation of DNA damage, DNA repair is expected to have a major role in shaping DNA damage distribution in the genome. Therefore, Sancar and co-workers examined damage distribution with damage-seq, but also used another method called XR-seq to map NER repair events in order to relate damage formation and repair patterns on a genome-wide level.82 The XR-seq method was previously described to map UV damage, but was adapted to map NER repair of cisplatin (Fig. 4).84 Therefore, the methodological details of XR-seq will be discussed below in the UV section. XR-seq requires DNA damage reversion for proper strand amplification containing cisplatin damage. Reversion was achieved by using sodium cyanide which can remove platinated DNA damage. While XR-seq was already a known technique, the strength of this study resided in the establishment of the damage-seq method which involved mapping any cisplatin damage present in genomic DNA and not only repair events.82
3.3 Genome-wide profiles of cisplatin damage in human cells
Damage-seq and cisplatin-seq studies led to the first genomic maps of cisplatin-induced DNA damage at single nucleotide resolution, opening a new possibility towards understanding anti-cancer agent efficacy and strategies to overcome resistance in cancer patients on the basis of genome-wide damage profiles. Both approaches mapped cisplatin damage in the human genome, though with different cell lines and cisplatin doses. Nonetheless, in both cases, G-G dinucleotides were highly enriched at damage sites in accordance with previous observations that the major cisplatin intrastrand crosslink is Pt-d(GpG),75 and to a lesser extent, A–G dinucleotides were enriched at the damage sites, confirming Pt-d(ApG) to be the second most prevalent intrastrand crosslink. This latter observation refutes that the pull-down is specific to only one type of intrastrand crosslink. Finally, in both studies an increase in damage at the TSS was also noted.An important strength of the damage-seq study82 was the coupling of damage formation data derived from damage-seq with damage-specific NER events derived from XR-seq. The coupling of these types of data permitted several key findings. First, sequence context analysis revealed a preference for cisplatin damage formation at G–G dinucleotides downstream of A, but in damage repair data the preference switched to a T upstream and a G downstream, meaning that the first was more prevalently formed but the latter was more resistant to repair. These sequence context findings are in conflict with previous biochemical studies testing DNA damage recognition for NER where the preference was for an A both up- and down- stream.85 Therefore, further studies are needed to determine whether the differences are due to the cellular context or due to biases introduced in the library preparation. Second, comparing the damage distribution maps over time as well as the XR-seq maps indicated that NER repair is the main driver in shaping the distribution of platinum-DNA damage. In particular, overall damage formation was fairly uniform in genomic regions with the exception of only a slightly higher damage abundance at the TSS and a slightly lower one at the TES. Interestingly, less damage was found on the transcribed strand (TS) which is consistent with a key repair process dictating the distribution of damage is TC-NER.86
Damage-seq and XR-seq were compared to nucleosome occupancy data for the same lymphocyte GM12878 cell line annotated in the ENCODE database. This analysis suggested how chromatin folding may impact damage distribution. A 5% reduction in damage formation was observed within the nucleosome center, whereas repair was substantially inhibited due to the inaccessibility of the nucleosome center. As such, the overall damage load was higher in the nucleosome center, aligned with observations made for UV-induced photodimers.87
There are several important unique findings worth noting from the cisplatin-seq study.83 The combined cisplatin damage-seq, XR-seq study mainly focused on chromosome 17 which contains TP53, whereas the cisplatin-seq study provided a more detailed investigation of all chromosomes and mitochondrial DNA. Interestingly, in comparison to the fairly uniform distribution of cisplatin damage load observed using damage-seq, results from cisplatin-seq differed up to 3-fold amongst chromosomes. In addition, mitochondrial DNA (mtDNA) carried the largest amount of cisplatin damage, likely because NER does not take place in the mitochondria.88 This finding was supported in later studies in mice (described below).89 Furthermore, short-duration cisplatin exposure led to less damage on the mtDNA light strand, which carries more genes than the heavy strand, suggesting protection or repair proficiency especially for the mtDNA light strand by an as yet undefined mechanism. Unlike damage-seq, which benefited from the previously annotated nucleosome occupancy, the cisplatin-seq study additionally performed ChIP-seq on HeLa cells to determine the influence of chromatin states on damage distribution. Here, an increase in cisplatin damage was observed to coincide with nucleosome signals, suggesting that there is preferred crosslinking of cisplatin on nucleosomes.
These last results83 contradict the damage-seq study82 and may be due to the lack of repair data. Specifically, the higher cisplatin damage load could be the result of a lack of repair rather than a preference for damage formation. Subsequent studies mapping NER events in mice support this possibility, having demonstrated a very rapid peak of transcription-coupled NER (TC-NER) activity 2 hours after cisplatin exposure.90 Given that cisplatin-seq was performed following cell exposure after 3–24 h, it is likely that TC-NER already took place. Finally, cisplatin-seq data was compared with ChIP-seq data, indicating that the occupancy of DNA binding proteins Pol II, EZH2, and CTCF coincide with cisplatin damage. The conclusion of this comparison is that there is an increase in DNA damage formation at sites where NER accessibility is reduced. As repair seems to play a major role in cisplatin crosslink distribution, further efforts should characterize the influence of genomic architecture on repair accessibility. Additionally, because XR-seq data represent a snapshot of repair at a given moment, these sequencing techniques should be applied in a time-recovery course to investigate how damage distribution changes over time. Finally, cisplatin doses investigated were substantially higher than therapeutic levels; therefore, future studies should address relevant doses.
Damage-seq and XR-seq have been applied to investigate the effect of cisplatin chronochemotherapy on genome-wide cisplatin damage distribution and repair across different organs in mice, the first time DNA damage mapping has been performed in vivo.89–91 In addition to addressing a basis for cisplatin resistance, a second motivation to map cisplatin damage concerns potential off-target effects, for example, in chronochemotherapy, to find the optimal time of the day where the drugs will most efficiently kill cancer cells while reducing toxicity in other organs. Thus, NER of cisplatin–DNA damage was characterized using XR-seq and applied to analyze mouse liver and kidney due to the common side effects of nephrotoxicity and hepatotoxicity.91 Damage in TSs was repaired up to 10-fold more efficiently than in non-transcribed strands (NTSs), an observation potentially explained by TC-NER being more active than global genome NER (GG-NER). Indeed TC-NER is active all the time because it depends on transcription, whereas a peak was observed at a specific time within the circadian rhythm (here it was Zeitgeber time ZT08) for GG-NER. This study showed when each gene strand will be repaired giving the first circadian DNA damage repair map in mice and is now being extended by the same group to obtain individual circadian map in different human tumor cell lines.
In a second study, this time mimicking clinical dosing (70 days), the same approach combining damage-seq and XR-seq was used, to characterize damage maps in mouse liver.90 Results indicated that up to 5 weeks were need to completely remove platinated DNA crosslinks from the mouse genome. Again, 90% of TSs were repaired after only 2 days, whereas damage persisted for NTS, which might have a detrimental effect on healthy cells by causing replication fork arrest, leading to cell death. Therefore, TC-NER should be considered as being the dominant form of cisplatin damage repair following drug administration, and therefore could be an important pathway for additional targeted therapeutic strategies.
Finally, the most recent study of the genome-wide distribution of cisplatin–DNA damage coupled XR-seq, damage-seq, and RNA-seq data for mouse kidney, liver, lung, and spleen.89 The study revealed that the rate of NER on the TS and NTS of active genes is positively correlated with gene expression. Specifically, repair in the TS and NTS increases with gene expression and plateaus in the TS among highly expressed genes. The data further suggest that cellular transcription stimulates the repair of damage in the NTS due to the fact that transcription is associated with an open chromatin conformation and increased accessibility to repair machinery. Interestingly, the spleen carried the least cisplatin damage, which could be explained by the fact that genes thought to be associated with cisplatin transport were downregulated (atp7B & Steap3). Finally, consistent with cell-based results, patterns of damage distribution appear to mainly be driven by repair activity.
4. UV damage (CPD and 6-4PP)
4.1 Occurrence, relevance, and repair
Ultraviolet light (UV) is associated with the occurrence of at least 95% of all skin cancer cases. UVC (254 nm) and UVB (290–320 nm) wavelength ranges photoexcite pyrimidines in DNA to form cyclic dimers,92 the most frequent of which are cyclobutane pyrimidine dimers (CPDs) and 6-4 pyrimidine-pyrimidine photoproducts (6-4PPs).87 UVC light is less of concern as it is mainly blocked by the atmosphere.93 UVA light (320–400 nm) can also lead to CPD DNA damage to a lesser extent, but can penetrate the skin layers more deeply, reaching the dermis.94 Additionally, UVB and UVA can lead to the formation of oxidative damage (8-oxodG) in DNA.92 It is well-known that formation of pyrimidine dimers occur predominantly at TT sites, with much lower amounts of the corresponding TC, CT, and CC dimers.95 Furthermore, CPDs are more abundant and likely more cytotoxic, but 6-4PPs are more mutagenic.96 All of these bulky DNA damage events are the basis of human skin cancer because they can mispair during DNA replication giving rise to mutations.The pathological role of DNA photodimerization was compellingly revealed through the characterization of human genetic deficiencies in XP proteins, contributing to the rare human disease xeroderma pigmentosum. It was later understood to be part of the NER machinery, which effectively removes these dimers from cells and protects against mutagenicity.97 Similar to cisplatin crosslinks, UV-induced DNA damage is removed by NER and therefore UV damage repair can also be investigated using mapping methods specific to NER repair events.98 If cellular repair is overwhelmed, there are deleterious implications to the cell including cell death95 due to the stalling of replicative polymerases during DNA synthesis.99 Finally, as specialized TLS polymerases bypass UV lesions,96,100,101 there is a characteristic mutation signature comprised mainly of C →T and CC → TT mutations102,103 that correspond to signatures found in skin cancer.5 To test whether damage distribution is predictive of mutations, there is a need for more insight into the genome wide location of UV damage and identification of regions which are recalcitrant to NER repair.
4.2 Genome-wide mapping of UV damage
Given the strong association of skin cancer with the UV-induced mutational signature,102 methods to map UV damage in the genome are essential to resolve the gap between DNA damage formation and mutagenesis. Two precursor studies obtained CPD maps with lower resolution by using antibodies for enrichment followed by microarrays hybridization in yeast and human cells.104,105 Another study used a similar strategy of damaged DNA immunoprecipitation (DDIP-seq) but followed by next-generation sequencing to map CPD damage at a resolution of 100–300 base pair in nuclear and mitochondrial DNA in human cells.106 At least four methods have been developed to map genome-wide UV damage formation or repair with single-nucleotide resolution: excision-seq, CPD-seq, XR-seq, and HS-damage-seq (Fig. 5). Excision-seq and CPD-seq rely on enzymes that cleave upstream or downstream of UV damage. XR-seq is unique in that it does not directly map UV-specific damage formation, but rather it maps the occurrence of NER events occurring at UV damage sites. Finally, HS-damage-seq is an improvement of the damage-seq method which was previously used and described in this review to map cisplatin and oxaliplatin-induced DNA damage. As discussed above, damage first requires specific enrichment via an antibody against the DNA damage followed by stalling of a high-fidelity polymerase used for DNA replication to mark the damage location during library preparation. The first two methods (i.e. excision-seq and CPD-seq) have been used to map damage in yeast, and XR-seq and HS-damage-seq has been effectively extended to gain insight on human genome from cultured cells.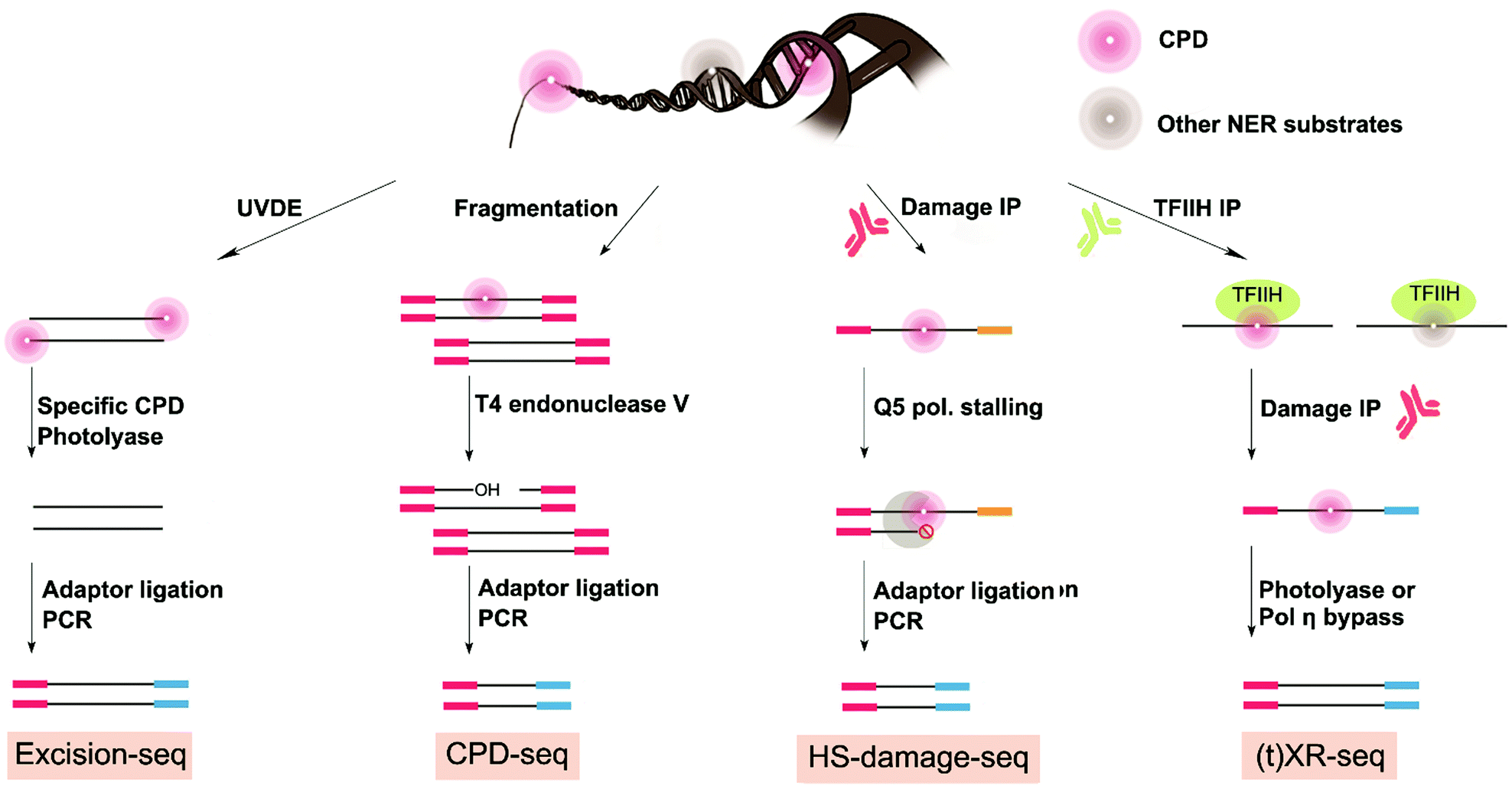 | ||
| Fig. 5 Genome-wide mapping methods for CPDs damage formation and NER repair events at single-nucleotide resolution. XR-seq, HS-damage-seq and excision-seq were used to map 6-4PPs damage formation and NER repair events as well. | ||
Of the four strategies reported to date addressing the genome-wide mapping distribution of UV damage at single-nucleotide resolution, excision-seq was the first to map UV damage at the genome wide level.107 Genomic yeast DNA was selectively digested by Ultraviolet Endonuclease Damage (UVDE) enzyme, which cleaves upstream CPD and 6-4PPs (Fig. 5). This enzymatic digestion releases short damaged dsDNA fragments that need to be repaired before amplification. Next, specificity for either CPD or 6-4PPs mapping was achieved by repairing the fragments with specific photolyases. Specifically, Vibrio cholera CPD photolyase or X. laevis 6-4 photolyase was used to repair the pyrimidine dimers into mono pyrimidines, thus allowing for end-repair of the damage of interest and enabling adapter ligation for NGS library preparation. DNA fragment ends read in the sequencing data thus correspond to the location of previous UV damage.
With a very similar approach to excision-seq, CPD-seq also achieved precise genome-wide mapping of CPD damage, while also adding a new dimension to previous information on the distribution of photodimers in the yeast genome by integrating insight on the impact of repair and chromatin structure.87 Genomic yeast DNA treated with UV was fragmented, end-repaired, dA-tailed, and ligated to adapters prior to digestion by T4 endonuclease V that specifically cleaves downstream CPD damage generating single-strand DNA breaks with AP sites at the 3′ end. Next, the APE1 enzyme was used to remove the AP sites, releasing 3′-OHs at the end of the short ssDNA breaks to allow ligation and sequencing (Fig. 5).108 CPD-seq specifically permitted mapping of UV CPD damage at single nucleotide resolution in yeast.
Application of excision-seq and CPD-seq to map CPD photodimers in UV-exposed yeast, despite the use of UV doses 100 times higher for excision-seq, revealed similar sequence-associated preferences for photodimerization. As expected, CPD dimers primarily occurred between two Ts. The next most prevalent CPD sequence pairings were T–C, C–T, and C–C. Excision-seq additionally mapped 6-4PPs, indicating T–C is the most abundant followed by, T–T, C–C, and C–T. These results confirm older chromatography data.109 A key benefit of the sequencing approach is the ability to examine the sequence context surrounding the dimer positions. Specifically, excision-seq data indicated a preference for an A downstream of 6-4PPs in yeast and this same preference was also observed in later experiments in human cells.107 As the downstream A preference was not observed in the CPD dataset, it was concluded that the UVDE enzymes did not introduce this sequence bias; however, it is possible that this is an artifact related to the sequence preference of X. laevis 6-4 photolyase. While both studies revealed similar sequence preference, excision-seq revealed a uniform distribution of UV damage (CPD and 6-4PPs) in the yeast genome, whereas CPD-seq indicated that CPD damage distribution was not uniform. These differences may be a result of either the methodology or repair.
One strength of the CPD-seq study is that NER repair, chromatin structure, and their influence on CPD damage distribution were measured. Notably, UV damage formation and repair were reduced at strong nucleosome positions. Furthermore, NER was inhibited at translational positions near the strongly positioned nucleosome dyad and CPD formation within the nucleosome was lower at inward rotational DNA and higher in outward rotational settings. The interpretation of these data is that inward-rotated DNA is protected from UV damage because of DNA bending and flexibility imposed by the nucleosome structure (i.e. due to the principle that two pyrimidines need to be close and correctly aligned to form a dimer). Interestingly, cells might use the inward setting to protect A-T rich regions, which are more prone to be damaged by UV. Another finding was that there was significantly less CPD at TF-binding sites, suggesting TFs may act as guardians of important DNA sequences. While more studies are required, the CPD-seq study suggests UV-induced damage distribution is strongly influenced by nucleosomes and TF-binding sites.
Both excision-seq and CPD-seq methods are effective at mapping UV damage, however the use of digestion enzymes brings potential liabilities with regards to damage specificity and the potential for introducing artifacts during library preparation. For instance, UVDE enzymes can cleave after other bulky DNA adducts, or there could be certain sequence contexts in which the excision/repair enzymes are more efficient. In both studies, results were presented from experiments involving high levels of UV exposure (10000 J m−2 for excision-seq and 125 J m−2 for CPD-seq) with no indication of a dose–response relationship or threshold for effective mapping. Additionally, a common limitation is that these methods may not be entirely specific and cannot be generally extended to every bulky DNA adduct because damage specific digestion/repair enzymes are required. Finally, these observations contrast subsequent findings in the human genome, highlighting that DNA damage distribution might be unique to certain species, or even to certain cancers.
The distribution of UV damage in the genome has also been characterized using more generally-applicable methods for bulky adducts: specifically XR-seq and HS-damage-seq (Fig. 5).84,98 Rather than measuring damage itself, XR-seq uses NER's unique characteristic of releasing excised 30-mer damaged fragments during repair. These small fragments are isolated from genomic DNA based on their low-molecular-weight and subjectivity to specific NER repair protein immunoprecipitation (TFIIHα). Fragments that are pulled down can then be subjected to a second damage-specific immunoprecipitation (in this case for CPD or 6-4PPs). Finally, either CPD or 6-4PPs photolyases are used to repair the damaged fragments to allow PCR amplification and sequencing. XR-seq was applied to map UV damage repair in human cells. However, given that XR-seq captures a map of NER repair of bulky adducts, it is a general approach and has also been used to map cisplatin and BPDE damage as well as NER events in various model organism including bacteria, plant, yeast, mouse, and human.110–115
HS-damage-seq can also be considered a general approach for mapping bulky DNA damage. In fact, HS-damage-seq is based on the previously published damage-seq method (used to map for cisplatin and oxaliplatin damage82) but includes an extra antibody enrichment for UV damage. As such, HS-damage-seq results in an increase in sensitivity needed to map more physiologically relevant exposure conditions and only requires 1 μg of input DNA. Briefly, HS-damage-seq requires initial immunoprecipitation of the damage of interest. This enrichment process is followed by a primer extension using the pull-down fragments as templates and a high-fidelity polymerase to perform DNA synthesis. The high-fidelity polymerase, like in cisplatin-seq and damage-seq described above, stalls at the site of the bulky damage leading to a termination site and production of a shorter DNA fragment attached to biotin. The resulting synthesized strands are purified by a biotin–streptavidin system and then undergo a subtractive hybridization step to further remove undamaged strands prior to amplification. As such, more than 95% of the reads generated through sequencing are specific to the damage of interest, also increasing sensitivity. To compare HS-damage-seq, a new cisplatin map was generated using only 1 μg of input DNA. Importantly, this new map compared favorably with the original cisplatin map generated using damage-seq,98 confirming that the new HS-damage seq method enabled more sensitive mapping of damage. HS-damage-seq and XR-seq are excellent methods to understand UV damage distribution dynamics and can be further combined to understand the basis of damage-induced cancer in human cells.
4.3 Genome-wide profiles of UV damage and repair in human cells
XR-seq and HS-damage-seq were combined to map UV damage in human skin fibroblast cells with deficiencies in GG or TC-NER pathways. Specifically, cells were exposed to environmentally relevant UV levels (20 J m−2) and DNA was collected at different time points (ranging from 20 to 240 min). The overall sequence context preference for UV damage was similar to what was reported in yeast using CPD-seq. However, when focusing on repair preferences and the influence of chromatin, several interesting patterns emerged.First, global genome repair of CPD was slightly higher around the TSSs of genes. While the reason is not clear, it may be due to the higher levels of CPD formation in this area. However, it is more likely that the higher CPD repair in the TSS regions is a direct consequence of the higher levels of the TF TFIIHα binding, which ultimately initiates global NER. Additionally, the correlation between 16 different TF-binding sites and damage occurrence was investigated. However, no general pattern relevant to all TF-binding sites emerged. Rather, the relevance of TF-binding sites in damage formation is dependent on the TF and type of damage. As one example, the BHLEH40 TF position correlates with higher 6-4PPs but lower cisplatin damage load.
This study also confirmed that CPD damage is mainly repaired by TC-NER, while 6-4PPs are mainly repaired by GG-NER.98 However, CPD distribution was the same regardless of chromatin states whereas the occurrence of 6-4PPs varied by chromatin state. In particular, 6-4PPs were most abundant in poised and active promoters as well as in repetitive regions, but had a lower frequency in heterochromatin. Therefore, similar to the findings of 8-oxodG and cisplatin, UV damage distribution appears to be the result of fairly uniform damage formation, but heterogeneous repair. In summary, the combination of XR-seq and HS-damage-seq helps to understand the importance of damage formation and damage repair in providing the overall map of damage and may be useful to more closely link damage patterns with mutagenesis and mutational signatures.
5. Benzo[a]pyrene (BPDE-dG)
5.1 Relevance and repair
Benzo[a]pyrene (BaP) is a known human carcinogen, particularly associated with lung cancer in patients who smoke.116 BaP is a polycyclic aromatic carbon environmental pollutant resulting from incomplete combustion.117 While BaP is found naturally in fossil fuels, shale and crude oils, and coal tars,117 human exposure to BaP is mainly from diet or cigarette smoke.118 In cells, BaP can be metabolized by CYP1A1 and CYP1B1 to form the ultimate carcinogen diol epoxide (BPDE), which forms BPDE-DNA adducts, mainly by addition to the N2 position of guanine.119 BPDE-dG DNA damage is also repaired by NER120 and, if unrepaired, BPDE-dG induces apoptosis and necrosis associated with mitochondrial signalling, and DNA double-strand breaks.121,122 As such, different TLS polymerases are capable of bypassing BPDE-dG; Pol η is highly error prone when bypassing BPDE-dG by inserting an A or T more efficiently than C,123 on the other hand, Pol κ can perform error-free TLS.124 Pol ι is unable to bypass BPDE-dG whereas in combination with Rev 1, pol ζ is error-prone.125 Thus, BDPE-dG DNA damage typically gives rise to G → T transversions, preferentially at CpG dinucleotides.126 Using cleavage enzymes on genomic DNA, followed by LM-PCR with specific TP53 exons primers, preferential formation of BPDE-dG in hotspot codons 273 and 248 in the human TP53 gene was observed, indicating a causal link between BPDE-dG damage and cancer development.127,128 Unfortunately, it is not yet feasible to extend this strategy to an entire genome, and modern methods for whole genome identification of BPDE-dG are highly desirable.5.2 Genome-wide mapping of benzo[a]pyrene
Since BPDE-dG DNA damage is repaired by NER, XR-seq mapping at the genome-wide level is possible (Fig. 6).110 XR-seq, described above in the context of UV and cisplatin, involves capturing the short oligomers that are released during NER of bulky adducts. The fragments that are positive for damage are enriched for with the use of specific antibodies for the damage of interest. The captured fragments are then repaired to reverse the damage and are used for NGS library preparation and sequencing.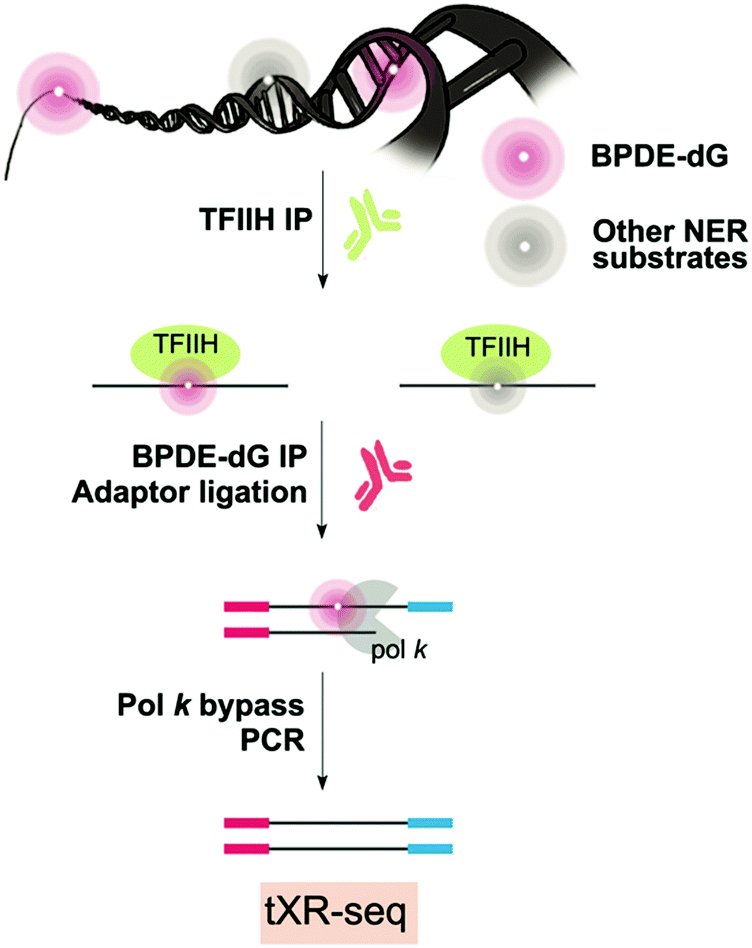 | ||
| Fig. 6 Genome-wide mapping method for BPDE-dG NER repair events. | ||
In the case of applying XR-seq to map BaP damage, there is one major challenge: no enzymes to reverse BPDE-dG damage. Therefore, XR-seq was modified by incorporating a translesion DNA synthesis step during PCR amplification using pol κ for N2-BPDE-dG and pol η for CPD, permitting bypass of the damage. This variation on XR-seq, termed tXR-seq expands its capacity to map damage repaired by NER. Nonetheless, there is still a limitation related to availability of an error-free TLS polymerase or combination of polymerases that bypass the damage of interest. After validating the tXR-seq method comparing the previous XR-seq CPD data with new tXR-seq CPD data, tXR-seq was applied to provide the first human repair events map of BPDE-dG DNA damage associated with a wide range of exposure in humans.
5.3 Genome-wide profiles of BPDE-dG repair in human cells
To map NER repair events of BPDE-dG using tXR-seq, DNA was extracted from a human lymphocyte cell line well characterized by the ENCODE project concerning chromatin states and regulatory region information (GM12878). Cells were treated with 2 μM BPDE and repair was allowed to take place for 1 h before DNA extraction. Preferred sequence contexts, such as a high frequency of CG dinucleotides for BPDE-dG were found in the NER excised fragments. The higher frequency of CG is likely related to the increased formation of BPDE-dG at CpG islands;129 however, is it not clear whether these preferred sequence contexts are related to damage formation or repair.Damage formation data, for instance from a damage-seq experiment, would be essential to discriminate between these two processes. Furthermore, the sequence coverage in this experiment was insufficient to make any claims regarding the repair of relevant genes involved in cancer development, such as TP53 hotpots. However, in comparing the repair of BPDE-dG to previous repair data (i.e. UV damage), the authors observed that the rate of the NER machinery repair of BPDE-dG damage ranged between fast repair for 6-4PPs and slower repair for CPD. Furthermore, the data revealed that BPDE-dG damage was only slightly more prevalent on the NTS, suggesting a minor role of TC-NER. This contradicts mutational signature data, specifically signature 4 associated with tobacco exposure, which shows the G → T mutations exhibit strong transcriptional strand bias.130 This difference could be explained by a tissue type difference, given that lymphocytes, which were used in the tXR-seq study are not the most biologically relevant model for tobacco exposure. Regardless, the novel method tXR-seq is an improvement of the previous XR-seq which could not have been applied to map BPDE-dG DNA damage. The main limitation of tXR-seq is the requirement of identifying TLS polymerases which are known to be more efficient in bypassing BPDE-dG DNA damage in certain sequence contexts, therefore introducing a small bias during the one round PCR amplification. Next studies on benzo[a]pyrene damage distribution should aim at treating cells with meaningful doses of BaP or BPDE, use higher sequencing depths, improved bioinformatics and statistical analysis, and couple repair data to new damage formation datasets.
6. Methylating agents (O6-methylguanine)
6.1 Relevance and occurrence
O 6-methyl-2′-deoxyguanosine (O6-MedG) only contributes ∼5% of total damage induced by alkylating agents, but it has dramatic biological effects compared with more abundant N-methyl adducts.131 Endogenous methylation results mainly from S-adenosyl methionine132 which forms 10–30 residues of O6-MedG per cell each day.133 Exogenous alkylating agents also contribute to the occurrence of O6-MedG in the genome, including natural and anthropogenic components of air, water, and food, as well as several chemotherapeutic agents.134 For example, the oral methylating agent temozolomide (TMZ) is widely used to treat brain cancers, including glioblastoma multiforme and astrocytomas.135 While there is evidence that TMZ extends the survival of patients, resistance often occurs due to increased expression of the repair enzyme alkylguanine alkyltransferase (AGT).136The primary repair of O6-MedG is through direct reversion by AGT. Biochemical assays,137 crystal structure data,138 cellular studies,139 and transgenic mouse studies140 all confirm that AGT repairs O6-MedG and other O6-alkylguanine DNA damage, and that its expression greatly reduces the incidence of mutations caused by exposure to methylating agents. On the other hand, in the case of TMZ therapy, AGT overexpression renders TMZ less effective. To mitigate this problem, the methylation status of the AGT gene promoter, MGMT, is used as a diagnostic strategy for stratifying treatment regimes, and its epigenetic silencing in tumor cells is associated with glioma sensitivity to TMZ.141,142 AGT removes alkyl groups located on the O6-position of guanine in one direct transfer step, regenerating the undamaged guanine residue. The current model of AGT-mediated repair involves cooperative binding of the AGT protein to the minor groove of DNA. AGT then scans the genomic DNA and flips the O6-MedG residue into its active site, permitting the transfer of the alkyl group and releasing the dealkylated DNA.143 Several biochemical studies have revealed that sequence context, including the opposing base, impacts the repair of O6-MedG by AGT.137,144–146 In contrast, recent work from Essigmann and coworkers showed no specific mutational patterns arising from AGT repair in mouse cells treated with the alkylation agent N-methyl-N-nitrosourea (MNU).147 However, there is little work on AGT accessibility to different chromatin states in mammalian cells. Thus, developing a mapping method for methylation formation and repair processes would help fill this gap.148
O 6-MedG is highly mutagenic as a result of mispairing during DNA replication.149 Replicative polymerases and TLS polymerases including Pol η, Pol κ and Pol ζ can bypass O6-MedG, causing misincorporation of T with up to a 10-fold misinsertion.150–153 The O6-MedG:T mismatch results in a G → A transition mutation upon the second round of synthesis. In addition to being mutagenic, O6-MedG is cytotoxic via a unique indirect process resulting from the recognition of the O6-MedG:T mispairing by the mismatch repair pathway.154–156 This mispairing gives rise to the mutational spectra mainly composed of G → A transitions.157 In particular, signature 11 is observed in malignant melanomas and glioblastoma multiforme treated with the alkylating agent temozolomide.5 Likewise, in vitro immortalized primary murine embryonic fibroblasts treated with the methylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) showed signature 11.158 A key defining feature of signature 11 is the prevalence of these G → A transition mutations;159 and thus suggests that signature 11 is relevant to O6-MedG damage derived from exposure to methylating agents. However, there are no methods to map O6-MedG at a single nucleotide resolution. Their development would allow for the extrapolation of a damage spectrum that could directly be compared with mutational signature, highlighting the contribution of specific DNA damage into the final mutational load.
6.2 Sequence-based DNA damage analysis
Despite the high volume of sequencing methods and studies addressing methylated cytosine (5-methylcytosine, 5-mC)160–163 and methylated adenosine (6′-methyladenosine, 6-mA),164–167 there are no genome-wide sequencing methods for mapping O6-MedG or other chemically-induced alkyl DNA damage. The lack of methods is likely a consequence of both the base pair misincorporation by and stalling of polymerases opposite O6-MedG168 – whereas natural modifications such as 5-mC and 6-mA are more readily amplified. Despite this major challenge, there has been recent progress in using unnatural nucleobases that specifically pair with this damage as an approach to identify or amplify it within particular sequence contexts by hybridization of polymerase-based amplification strategies.169,170A nanoparticle-based-hybridization strategy permitted sensitive detection of O6-MedG in a sequence-specific manner.169 In this approach, elongated hydrophobic nucleobase analogues were designed to base pair to O6-MedG. As such, short oligonucleotides that contained the nucleobase analogue formed a more stable duplex with a complementary sequence that contained O6-MedG vs. G. To develop a biosensor from this system, the nucleobase-containing oligonucleotides were conjugated to gold nanoparticles. In this way, the gold nanoparticles served as a quantitative dose-responsive optical readout, where dispersed nanoparticles displayed a bright red color and aggregated nanoparticles resulted in a measurable blue color. In particular, the abundance of O6-MedG located at a mutational hot spot could be quantified within the human KRAS gene in mixtures with competing unmodified DNA. While many challenges remain before this approach can be implemented for biological studies, the approach is suited for massively parallel sequence probes for analysis of multiple genes, but not whole genome analysis. Moreover, high levels of input DNA are needed,171 thus, additional enrichment of samples would be needed. In spite of these hurdles, this proof-of-concept suggests hybridization-based assays that incorporate nucleotide modifications as a strategy for detecting DNA damage within defined genomic contexts.
A second chemistry-oriented approach to the selective amplification of O6-MedG in defined sequence contexts and a potential basis for mapping DNA alkylation damage involves the use of artificial nucleotides incorporated opposite the damage site by a DNA polymerase. In this way, DNA adducts can be marked by a non-natural nucleotide rather than by inserting a mismatched T which causes a loss of the damage identity. Furthermore, if a polymerase can efficiently incorporate the artificial nucleotide, exponential amplification of the marked damage may also be possible, thus enabling the detection of extremely low abundance DNA damage. Polymerase-mediated incorporation of synthetic nucleotides opposite a DNA adduct has been reported for AP sites,172 8-oxodG,173cis-platinated guanine,174 as well as for O6-alkyldG.170,175 The detection of damaged bases was achieved thanks to the development of new artificial nucleotides that were designed and tested for the base pairing and that are reviewed here.176 Recently, we have used this strategy to quantify and localize the related mutagenic O6-alkyl-G adduct O6-carboxymethyldG by amplification with artificial nucleotides.177
7. DNA sugar and backbone damage
We have so far reviewed methods for mapping various DNA damage types including oxidative DNA damage (i.e. 8-oxodG), cisplatin-derived damage (i.e. crosslinks), UV damage (i.e. 6-4PPs and CPD), and alkylation damage including BPDE-dG and O6-MedG, focusing on how the chemistry of the damage drives strategies to map them. However, from these various chemically-induced adducts and their repair, several downstream forms of DNA damage also occur. For example, AP sites are a common product of DNA damage and are generated as an intermediate in base excision repair (BER). Single-strand breaks (SSBs) also arise due to oxidative DNA damage or a failure in DNA repair and thus are the most common form of DNA damage. SSBs may also lead to double-strand breaks (DSBs) which, if unrepaired, lead to cell death and, if mis-repaired, cause chromosomal translocations, an early step in carcinogenesis. Finally, due to the high levels of ribonucleotide precursors present as potential substrates during DNA synthesis and repair, many are incorporated into DNA. In particular, studies have addressed the distribution of uracil in DNA. Here, we introduce recent technologies to map these general forms of DNA damage and summarize the most important biological insights resulting.7.1 Abasic sites
AP sites commonly arise as a by-product of DNA damage, either because damage formation typically accelerates depurination rates,178 or because they are generated as an intermediate in base excision repair, wherein small DNA base modifications such as 8-oxodG or uracil are removed by DNA glycosylases.179 Another source of AP-sites is in the maintenance of epigenetic DNA base modifications by thymine DNA glycosylase when removing 5-formylcytosine (5-fC) or 5-carboxylcytosine.180 Typically, AP sites are recognized by AP-endonuclease and repaired with reinsertion of the correct base. However, if not repaired, their persistence can lead to mutations, stalling of polymerases and genomic instability.181 It has been shown that AP sites can cluster in the genome during DNA replication, but little is known about their precise location in the genome and in which sequence context they preferentially form or persist.182Three methods were recently developed to sequence AP sites; AP-seq,59 snAP-seq72 (Fig. 7) and Nick-seq,183 with the latter two having single-nucleotide resolution. AP-seq and snAP-seq rely on the reactivity of the aldehyde group exposed in the acyclic form of the 2′-deoxyribose ring to tag the AP site with biotin. Following tagging, the DNA is enriched using streptavidin, recovered, and prepared for NGS, where AP sites are called and thus mapped to the genome. Since the epigenetic base modifications 5-fC and 5-formyluracil (5-fU) contain reactive aldehydes, sometimes occurring at a higher abundance than AP sites, snAP-seq includes an additional step involving alkaline cleavage in which only the AP sites (i.e. not the formylated bases) lead to DNA strand scission. This selection increases the specificity of the capture.184 As such, the DNA fragments containing AP sites are released from the biotin pull down and are recovered for NGS, whereas DNA fragments containing 5-fC and 5-fU remain immobilized.72
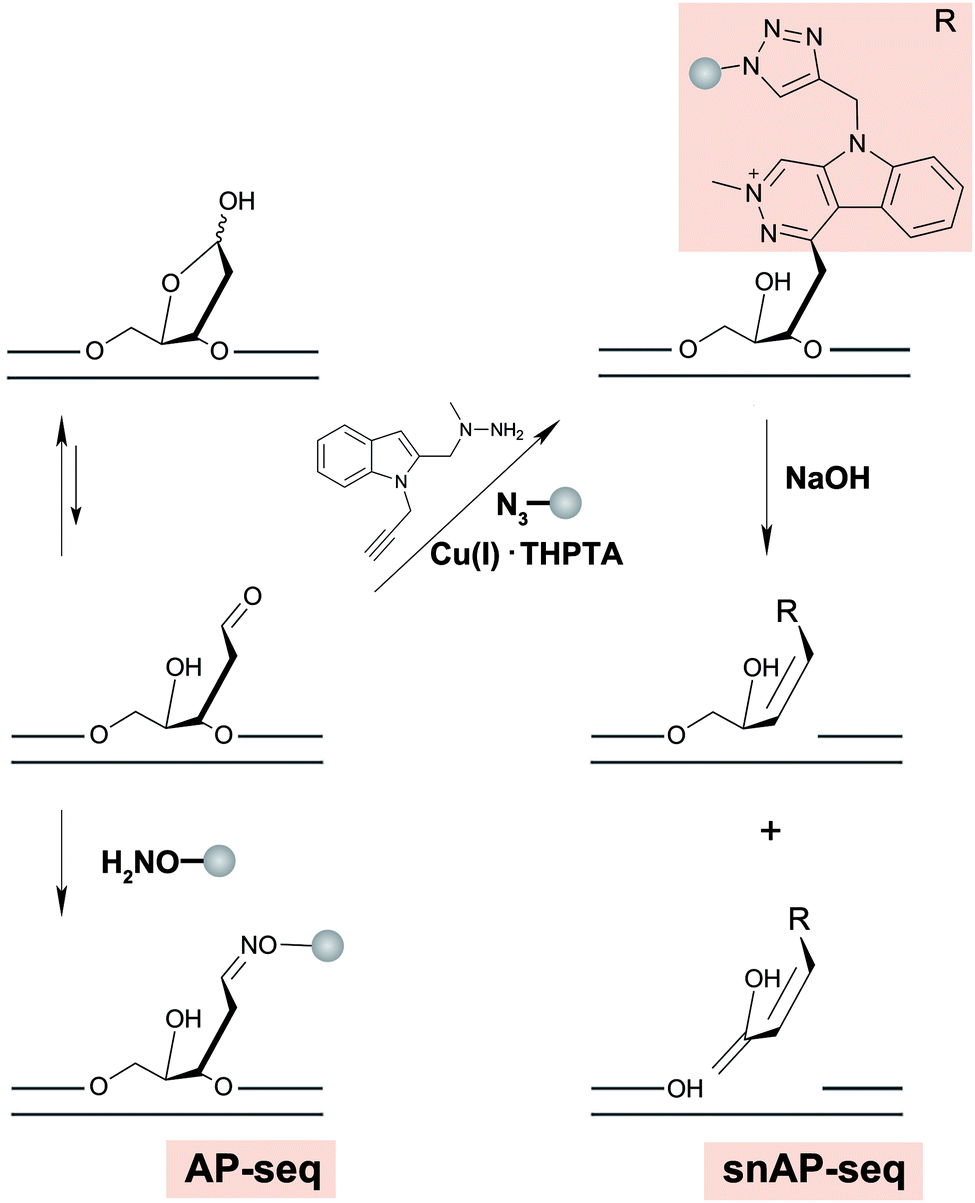 | ||
| Fig. 7 Chemical reactions involved in strategies for genome-wide mapping of AP sites. | ||
More recently, Nick-seq was reported to use endonuclease IV (Endo IV), an AP endonuclease to create new strand breaks at AP sites after blocking pre-existing breaks. Nick-seq is a versatile method that could generally be used for any DNA damage that can be converted into a single-strand break. The strand breaks originating from AP sites were captured at the 3′- and 5′-ends using two complementary strategies: nick translation with α-thio-dNTPs for 5′-end sequencing and terminal transferase tailing for 3′-end sequencing. The co-location of AP sites at both ends increases the sensitivity and specificity of the resulting map. As such, these AP-specific sequencing methods provide efficient approaches for further exploring the genome-wide biological impacts of AP sites.
AP sites have been so far mapped in parasitic worms (Leishmania m.), bacteria and human cell lines (HepG2 and HeLa). In the snAP-seq study, AP site distribution at single nucleotide resolution in human cells competent or not for BER (APE1) was investigated. No specific locations where AP sites might accumulate were identified, so it was concluded that AP site accumulation is stochastic in a cell population. On the other hand, when data was binned to characterize genomic regions more broadly, APE1 was identified as especially proficient in repairing AP sites in regulatory and genic regions. This latter observation highlights the paradox that with single-nucleotide resolution it might be harder to make claims due to the heterogeneity present between each cell and that often, more informative information may be derived based on lower resolution analysis.
7.2 Single-strand breaks
SSBs are one of the most common types of DNA damage. They can arise from oxidative stress, DNA repair failure, activity of topoisomerase, or destruction of the sugar backbone.185 SSBs can cause the replication fork collapse, leading to even worse damage such as DSBs.186 Thus three methods have been developed to map SSBs including SSB-Seq,187 SSiNGLe (single-strand break mapping at nucleotide genome level)188 and GLOE-Seq.189In the SSB-Seq study, SSBs were tagged with nucleotides attached to digoxigenin during a nick translation step with DNA polymerase I followed by an immunoprecipitation with an antibody anti-digoxigenin.187 Results suggested that SSBs induced by topoisomerase II were primarily located in the promoter regions of genes in human cell lines.
The SSiNGLe method involves tagging the 3′-OH terminus of a DNA strand, which represents the position of a SSB, by adding a poly A tail with a terminal transferase. Helicos Single Molecule Sequencing (SMS) and Illumina platforms (ILM) were used to map SSBs in genomic DNA. Extracted DNA is first fragmented by MNase leaving 3′ primer-phosphate ends which are not recognized by the terminal transferase. The polydA tail is then either captured on a flow cell harboring chain of dT oligonucleotides for SMS or amplified with oligo-dT primers before adapter ligation for Illumina sequencing. The distribution of SSBs was characterized in a variety of human and mouse cell lines and called the breakome. The patterns of distribution of SSBs changed across cell types as well as within the same cell type in response to anti-cancer drugs. Thus, the breakome of peripheral blood mononuclear cells from patients correlated with age, which shows the close association between SSBs and aging-related disease states.
Similarly, GLOE-Seq takes advantage of the presence of the 3′-OH terminus but introduced a new strategy to eliminate the polydA and the possible repetitive sequence limitation inherent to the Illumina platform. Thus, a ligation strategy on the 3′-OH terminus of SSB with a biotinylated adaptor followed by a biotin pull down was carried out. The strength of this method is its applicability to double-strand breaks, Okazaki fragments and to any type of DNA damage that that can be converted into a nick or a gap with a free 3′OH terminus. The distribution of SSBs was mainly located in the leading strand due to polymerase ε activity. The authors suggest that since polymerase ε incorporates ribonucleotides, the main cause of SSBs are repair intermediates of ribonucleotides misincorporations.
7.3 Double-strand breaks
DNA DSBs are an ultimate fate of diverse forms of DNA damage. For example, DSBs arise indirectly from two closely located SSBs or during the repair of other DNA damages. DSBs are also formed due to replication fork collapse, which occurs during the replication of SSBs or damaged bases.190 However, DSBs also arise during transcription, meiosis, and replication stress.191,192 Regardless of the etiology, unresolved DSBs occur at an estimated rate of 50 breaks per cell per day and may lead to cell death or mutagenesis, including translocations, deletions and amplifications.193 As such, there have been numerous methods developed to sequence the location of DSBs in whole genomes.First developed in 2013, the BLESS (breaks labeling and enrichment on streptavidin and sequencing) method involves the ligation of DSBs to a biotinylated linker. Following streptavidin enrichment, an additional linker is added which allows PCR amplification and sequencing of DNA fragments containing the DSBs.194 Follow-up methods to this basic ligation and enrichment strategy include Break-seq,195 DSBCapture,196 END-seq,197 GUIDE-seq,198 BLISS,199 DSB-Seq,187 and qDSB-Seq.200 Notable improvements on the original method include use for mapping DSBs in mice in vivo (i.e. END-seq) and increased sensitivity (i.e. DSBCapture,196 END-seq,197 and BLISS199). For example, the authors impressively demonstrated that END-seq is sensitive enough to detect a single DSB within a sample of 10000 cells.
The most recently reported method, qDSB-Seq, uses site-specific endonucleases to create a DSB in a controlled manner, using this material for quantitative normalization of the subsequent analysis. As such, the key innovation of qDSB-Seq is combining nucleotide-resolution mapping for localization and simultaneous quantification of DSBs. The coupling of damage localization with direct quantification is a very new concept which might play a central role when investigating dose–response relationships for DNA damaging agents.
7.4 Ribonucleotides
Ribonucleotides, the building blocks for RNA synthesis, are also incorporated into nuclear DNA by several polymerases, including DNA polymerases,201–203 RNA primase,204 and PrimPol.205 As a result, ribonucleotides are by far the most abundant noncanonical nucleotides present in eukaryotic cells. As an example, an excess of 1.3 million ribonucleotide sites are introduced into the nuclear genome each cell division in mouse embryonic fibroblasts.206 To maintain genome stability, ribonucleotides are quickly removed in multiple ways, including Okazaki fragment (OF) maturation, ribonucleotide excision repair (RER), topoisomerase 1 (TOP1) cleavage, and prokaryotic nucleotide excision repair.207 Unrepaired ribonucleotides represent a major threat to genome integrity due to the reactive hydroxyl group at the 2′ position of the ribose sugar,208 leading to strand breaks,209 deletion,210 cell cycle checkpoint activation,211 abortive DNA ligation,212 aberrant recombination,206 and the formation of protein-DNA crosslinks.213Strategies to map ribonucleotides at single-nucleotide resolution are based on specific alkaline cleavage or enzymatic cleavage of ribonucleotides, because of the highly reactive 2′-hydroxyl groups (Fig. 8). One strategy is to use Arabidopsis thaliana tRNA ligase (AtRNL) to ligate the 2′-phosphate termini of DNA derived from alkaline cleavage to the 5′-phosphate terminus of the same DNA strand to produce an ssDNA circle containing the embedded ribonucleotide (Ribose-seq).214 Sequencing results using Ribose-seq in yeast show a higher rNMP incorporation on the newly synthesized leading strand than lagging strand. This is consistent with results showing that leading strand DNA Pol ε shows lower fidelity to ribonucleotides than the lagging strand Pol δ.214 Other sequencing methods include HydEn-seq215 that involves ligation of an adaptor directly to the free 5′-OH end and Pu-seq216 that uses random hexamer primer extension to synthesize a flush end adjacent to the initial ribose. Another strategy, emRiboSeq,217,218 uses the RNase H2 to recognize and cleave the ribonucleotides site, generating 3′-OH and 5′-phosphate groups. All these four methods have been applied to budding yeast, including strains with mutant replication polymerases that introduce excess ribonucleotides into DNA.
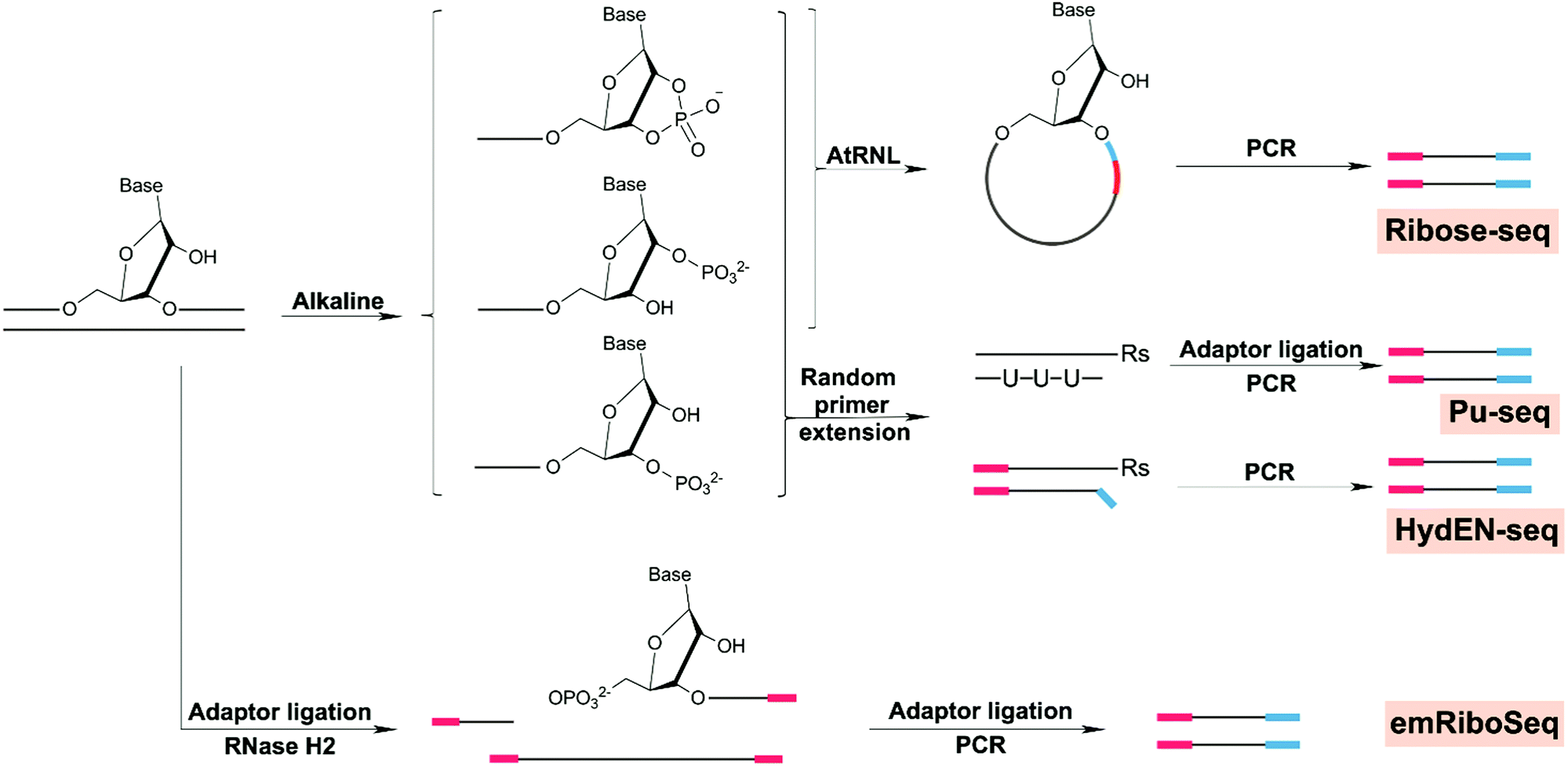 | ||
| Fig. 8 Chemical reactions involved in strategies for genome-wide mapping of ribonucleotides. | ||
To date, all applications of ribonucleotide sequencing have focused on leveraging the distribution of ribonucleotides as markers of replication enzymology. Error-prone synthesis by Pol α is retained in yeast and incorporates ribonucleotides into 1.5% of the mature genome. The different methods used all obtained similar results concerning replication polymerases, and it appears that these methods are well suited to address broader biologically relevant or toxicology studies. For example, there are several open questions regarding the impact of ribonucleotide misincorporation and the influence of chromatin state, transcriptional activity and local sequence context on ribonucleotides distribution. Furthermore, future studies may be useful for understanding the genomic connections between embedded ribonucleotides and diseases related to RNase H2 mutation.
7.5 Uracil
Uracil in genomic DNA (dU) results from cytosine deamination or direct incorporation during DNA synthesis. Cytosine deamination leads to a U:G mispairing, eventually leading to C > T mutations whereas dUTP incorporation instead of dTTP leads to A:U mispairing, which is not directly mutagenic.219 Uracil also can be repaired by uracil glycosylases (UDG enzymes) involved in BER, resulting in abasic sites and mutation. Therefore, there are several methods aimed at locating uracils.Excision-seq was first used to map uracil in genomic DNA.107 Specifically, cleavage at the site of the uracil, using UDG and T4 endonuclease IV coupled to Excision-Seq, generated a single-nucleotide resolution map of uracil distribution. The authors observed that distribution of uracil in the genome is tightly correlated with replication timing and hypothesized that it arises from changes in nucleotide pool composition during replication. This observation could be important to relate uracil distribution to mutational signatures where a replication timing factor is also observed.
Surprisingly, since its report in 2014, excision-seq was no longer used, but lower resolution methods were developed such as dU-seq,220 UPD-seq221and U-DNA-seq.222 Alternatively a gap-ligation approach developed by Burrows and et al. could in theory be used to map any lesions repaired by BER but has not yet been used for whole genomes.69
dU-seq uses UDG to remove uracil bases and replace them with biotinylated nucleotides to yield pull-down fragments for sequencing. The authors found that uracil content was high at centromeres in human genome. UPD-seq also uses UDG to remove uracil and then tag the abasic sites, forming a very easy disulfide link and a biotin-containing chemical (ssARP). With the UPD-seq strategy, the so-called uracilome was defined in a bacterial strain active in human apolipoprotein B mRNA-editing enzyme catalytic subunit (APOBEC), allowing for correlation of the mutational footprint left by cytosine deamination enzymes.
Finally U-DNA-seq involves the use of a uracil sensor (a mutant of human protein UNG2 one of the BER glycosylase of uracil) to locate uracil damage followed by immunoprecipitation and purification before sequencing of the uracil-enriched DNA. Furthermore, uracil distribution in human tumors cells upon chemotherapeutic treatment with raltitrexed and 5-fluoro-2′-deoxyuridine moves from heterochromatin regions to euchromatin (active transcriptional regions).
8. Conclusion
The field of DNA damage sequencing is new and advancing rapidly. There is massive data available concerning landscapes of mutation in disease, and relative to this little on how the chemistry-driven process of DNA damage and its patterns of distribution in the genome shape them. The more we gain insight into damage distribution both in regards to local sequence context and higher-scale genome arrangement, the better we will be able to link mutational signatures observed in human cancer to their etiological basis in a potentially prognostic manner. Thus, this review is useful to consider a broad spectrum of DNA damage sources, both derived from specific chemical modifications, such as carcinogen-DNA adducts, as well as general forms of damage, such as strand breaks, in regards to the state of the art on how to sequence them and emerging biological implications.In the future, DNA damage maps are expected to be an important tool in the sequencing arsenal for studying mutagenesis, carcinogenesis, aging processes, and responsiveness to DNA damaging drugs. However, sequencing DNA damage in a reliable and robust manner still requires significant work. Notably, most of the methods discussed here rely often on a specificity step and therefore cannot be extended to different forms of DNA damage. This specificity is required because damage products are present at such low frequencies and DNA samples need to be enriched for analysis. For 8-oxodG, platinated DNA, UV damage, benzo[a]pyrene DNA damage and uracil, enrichment has been achieved using antibodies. Apart from such antibodies, which could be devised de novo, albeit with significant time and expense, other enrichment strategies such as specific excision/cleavage or chemical conjugation are exclusively applicable to particular forms of damage that they naturally target. Thus, specific direct excision of the damage or cleavage next to the damage site to insert a probe/adapter have also been possible for 8-oxodG and UV damage, in addition to ribonucleotides and abasic sites. Finally damage-specific chemical conjugation has been used to enrich 8-oxodG and abasic sites.
Enriched samples of damaged DNA fragments, obtained i.e. by antibody pull-down, could be sequenced directly after damage removal, or by using TLS polymerases to bypass the DNA damage. The sequencing resolution of this general approach depends on the basis of fragmentation and resulting size of the fragments sequenced. In the case of tXR-seq and XR-seq, the fragments were released by NER enzymes, and they were short (20 mer). Thus, a single damaged nucleotide position could be identified because the damage was present exactly in the middle of the resulting oligonucleotides. With OxiDIP-seq and enTRAPseq, however, the resolution depended on the size of the sonicated or digested DNA fragments, around several hundred base-pair resolution. A significant advance was introduced by the cisplatin-seq, damage-seq and HS-damage-seq methods in order to obtain single-nucleotide resolution data, namely a high-fidelity polymerase that stalls at the site of DNA damage during a single PCR round. While this high-fidelity polymerase strategy was effective for large adducts including platinated crosslinks, UV damage and BPDE-dG which reliably block synthesis, such an approach is unlikely to work for smaller DNA damage such as 8-oxodG or O6-MedG. Third-generation sequencing technologies could be in this case potentially useful as (single-molecular real-time sequencing and nanopore sequencing) that do not require amplification and work on small epigenetic DNA modifications or DNA damage.64,225–231
The second strategy involving specific DNA strand cleavage or enzyme excision of the damage was also suited to map DNA damage independent of their size. This strategy was based on the recognition of DNA damage by nature's own recognition systems, such as Fpg for 8-oxodG, UVDE for UV damage, T4 endonuclease V for CPD damage, RNase H2 for ribonucleotides, or UDG for uracil. These methods created a nick or gap at the damage location, enabling the introduction of a sequencing adaptor/enrichment probe by ligation, as in excision-seq, CPD-seq, all ribonucleotide seq, dU-seq or UDP-seq, or by click chemistry, as in click-code-seq. One of the largest shortcomings of these strategies was false positive reads from background gap or nick sites generated during DNA sonication for instance. Thus, excision-seq, Pu-seq, and HydEN-seq corrected for this by fragmenting genomic DNA directly with cleavage treatment therefore removing the sonication step. CPD-seq, emRiboseq, ribose-seq, click-code-seq and nick-seq corrected the false positive signals by blocking the already present nick or gap with an adapter before excision/cleavage, and only then a second adapter/ddNTP was introduced for specific amplification. Finally, as in the case of antibodies, enzymatic cleavage enrichment methods also are potentially limited in regards to specificity. Namely, the excision enzymes might not be only specific to one substrate and therefore the mapping is the result of the enzyme substrate scope rather than a single DNA damage. Nonetheless, such approaches are valuable to understand the biological implication of an enzyme in a controlled experiment with controlled exposures that generate DNA damage.
A third key strategy for damage sequencing has involved direct chemical conjugation of DNA damage with affinity probes, such as OG-seq for 8-oxodG, AP-seq and snAP-seq for abasic sites. Damage-enriched DNA could be used directly for sequencing with ∼100 bp resolution (OG-seq, AP-seq) or fragmented at damage sites by alkaline treatment, in order mark the single-nucleotide position (snAP-seq). Chemical conjugation can also pose the problem of specificity as the probe might react with other aldehydes group present on DNA, such as 2-deoxyribose oxidation.
Despite the impressive obstacles that have been overcome with damage sequencing, there remain significant limitations. One of these is the possibility to determine both quantities and locations of DNA damage, which are critically needed to evaluate dose–response relationships to DNA-damaging agents. So far only qDSB-Seq was able to accurately quantify the number of DSBs simultaneously with their location by using calibrated samples that have a known amount of DSBs. Additionally, when working with large genomes such as human or mouse genomes, billions of reads need to be sequenced to draw robust biological conclusions in specific genes. Finally, bioinformatics pipelines have been developed for each of the strategies, integrating key aspects such as data normalization, however there are no standardized pipelines as for more common sequencing analysis such as for variant calling. Despite these aspects that are undergoing development, damage sequencing has already offered the opportunity to understand dynamics of damage formation and repair at the genome-wide level in a variety of organisms.
Given the difficulties of uncovering discreet chemical reactions on the scale of the genome, it is astounding that there are finally several possibilities to use sequencing methods to address chemically induced DNA damage distribution from the level of local sequence context, to the influence of transcription factors and higher chromatin structures. Amongst these data, almost all have illuminated that there is a heterogeneous distribution of DNA damage in the genome, resulting from combined dynamics of damage formation and repair activity both being influenced to varying extents by sequence context, DNA protein binding sites and nucleosome positioning. For instance, coupling of damage sequencing with nucleosome location data have revealed how the rotational setting of wrapped DNA around histones influence damage formation and repair. Additionally, DNA damage such as 8-oxodG in gene promoters have also been found to have a regulatory role in gene expression. As DNA damage distribution may be an early event dictating potentially adverse effects, anticipated future development of diverse DNA damage maps is expected to help understand and better predict etiologies of mutational landscapes in human cancer genomes and other biological processes driven by genome instability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Jasmina Büchel for helping to review the oxidative damage literature, Lucia Wang for helping to edit the manuscript, and Mary O’Reilly for work on figure design. This work was supported by the Swiss National Foundation Grants 185020 and 186332, and Krebsliga Schweiz Grant KFS-4443-02-2018.Notes and references
- T. Ishikawa, S. S. Zhang, X. Qin, Y. Takahashi, H. Oda, Y. Nakatsuru and F. Ide, Cancer Sci., 2004, 95, 112–117 Search PubMed.
- J. L. Barnes, M. Zubair, K. John, M. C. Poirier and F. L. Martin, Biochem. Soc. Trans., 2018, 46, 1213–1224 Search PubMed.
- H. A. Waldron, Br. J. Ind. Med., 1983, 40, 390–401 Search PubMed.
- A. Tubbs and A. Nussenzweig, Cell, 2017, 168, 644–656 Search PubMed.
- L. B. Alexandrov, S. Nik-Zainal, D. C. Wedge, S. A. Aparicio, S. Behjati, A. V. Biankin, G. R. Bignell, N. Bolli, A. Borg, A. L. Borresen-Dale, S. Boyault, B. Burkhardt, A. P. Butler, C. Caldas, H. R. Davies, C. Desmedt, R. Eils, J. E. Eyfjord, J. A. Foekens, M. Greaves, F. Hosoda, B. Hutter, T. Ilicic, S. Imbeaud, M. Imielinski, N. Jager, D. T. Jones, D. Jones, S. Knappskog, M. Kool, S. R. Lakhani, C. Lopez-Otin, S. Martin, N. C. Munshi, H. Nakamura, P. A. Northcott, M. Pajic, E. Papaemmanuil, A. Paradiso, J. V. Pearson, X. S. Puente, K. Raine, M. Ramakrishna, A. L. Richardson, J. Richter, P. Rosenstiel, M. Schlesner, T. N. Schumacher, P. N. Span, J. W. Teague, Y. Totoki, A. N. Tutt, R. Valdes-Mas, M. M. van Buuren, L. van 't Veer, A. Vincent-Salomon, N. Waddell, L. R. Yates, I. Australian Pancreatic Cancer Genome, I. B. C. Consortium, I. M.-S. Consortium, I. PedBrain, J. Zucman-Rossi, P. A. Futreal, U. McDermott, P. Lichter, M. Meyerson, S. M. Grimmond, R. Siebert, E. Campo, T. Shibata, S. M. Pfister, P. J. Campbell and M. R. Stratton, Nature, 2013, 500, 415–421 Search PubMed.
- H. Easwaran, H. C. Tsai and S. B. Baylin, Mol. Cell, 2014, 54, 716–727 Search PubMed.
- T. Sakoparnig, P. Fried and N. Beerenwinkel, PLoS Comput. Biol., 2015, 11, e1004027 Search PubMed.
- I. Bozic, T. Antal, H. Ohtsuki, H. Carter, D. Kim, S. Chen, R. Karchin, K. W. Kinzler, B. Vogelstein and M. A. Nowak, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18545–18550 Search PubMed.
- C. Tomasetti and B. Vogelstein, Science, 2015, 347, 78–81 Search PubMed.
- International Agency for Research on Cancer, 2015 January 13, Press release, Retrieved from https://www.iarc.fr/wpcontent/uploads/2018/07/pr231_E.pdf.
- X. Huang, D. Wojtowicz and T. M. Przytycka, Bioinformatics, 2018, 34, 330–337 Search PubMed.
- B. Hwa Yun, J. Guo, M. Bellamri and R. J. Turesky, Mass Spectrom. Rev., 2020, 39, 55–82 Search PubMed.
- D. H. Phillips and V. M. Arlt, Methods Mol. Biol., 2020, 2102, 291–302 Search PubMed.
- R. De Bont and N. van Larebeke, Mutagenesis, 2004, 19, 169–185 Search PubMed.
- B. Ma, I. Stepanov and S. S. Hecht, Toxics, 2019, 7, 16 Search PubMed.
- D. B. Sloan, A. K. Broz, J. Sharbrough and Z. Wu, Trends Biotechnol., 2018, 36, 729–740 Search PubMed.
- Y. Yu, P. Wang, Y. Cui and Y. Wang, Anal. Chem., 2018, 90, 556–576 Search PubMed.
- W. Li and A. Sancar, Environ. Mol. Mutagen., 2020, 1–16, DOI:10.1002/em.22365.
- P. Mao and J. J. Wyrick, DNA Repair, 2019, 81, 102645 Search PubMed.
- A. R. Poetsch, Comput. Struct. Biotechnol. J., 2020, 18, 207–219 Search PubMed.
- J. Cadet and J. R. Wagner, Cold Spring Harbor Perspect. Biol., 2013, 5, 2 Search PubMed.
- A. Phaniendra, D. B. Jestadi and L. Periyasamy, Indian J. Clin. Biochem., 2015, 30, 11–26 Search PubMed.
- C. Espinosa-Diez, V. Miguel, D. Mennerich, T. Kietzmann, P. Sanchez-Perez, S. Cadenas and S. Lamas, Redox Biol., 2015, 6, 183–197 Search PubMed.
- S. I. Rattan, Ageing Res. Rev., 2008, 7, 63–78 Search PubMed.
- J. Allgayer, N. Kitsera, C. von der Lippen, B. Epe and A. Khobta, Nucleic Acids Res., 2013, 41, 8559–8571 Search PubMed.
- G. H. Kim, J. E. Kim, S. J. Rhie and S. Yoon, Exp. Neurobiol., 2015, 24, 325–340 Search PubMed.
- S. Prasad, S. C. Gupta, M. K. Pandey, A. K. Tyagi and L. Deb, Oxid. Med. Cell. Longevity, 2016, 2016, 5010423 Search PubMed.
- T. Hussain, B. Tan, Y. Yin, F. Blachier, M. C. Tossou and N. Rahu, Oxid. Med. Cell. Longevity, 2016, 2016, 7432797 Search PubMed.
- S. S. David, V. L. O'Shea and S. Kundu, Nature, 2007, 447, 941–950 Search PubMed.
- N. C. Bauer, A. H. Corbett and P. W. Doetsch, Nucleic Acids Res., 2015, 43, 10083–10101 Search PubMed.
- T. Nunoshiba, R. Ishida, M. Sasaki, S. Iwai, Y. Nakabeppu and K. Yamamoto, Nucleic Acids Res., 2004, 32, 5339–5348 Search PubMed.
- S. Shibutani, M. Takeshita and A. P. Grollman, Nature, 1991, 349, 431–434 Search PubMed.
- M. Ohno, K. Sakumi, R. Fukumura, M. Furuichi, Y. Iwasaki, M. Hokama, T. Ikemura, T. Tsuzuki, Y. Gondo and Y. Nakabeppu, Sci. Rep., 2014, 4, 4689 Search PubMed.
- A. Viel, A. Bruselles, E. Meccia, M. Fornasarig, M. Quaia, V. Canzonieri, E. Policicchio, E. D. Urso, M. Agostini, M. Genuardi, E. Lucci-Cordisco, T. Venesio, A. Martayan, M. G. Diodoro, L. Sanchez-Mete, V. Stigliano, F. Mazzei, F. Grasso, A. Giuliani, M. Baiocchi, R. Maestro, G. Giannini, M. Tartaglia, L. B. Alexandrov and M. Bignami, EBioMedicine, 2017, 20, 39–49 Search PubMed.
- J. Allgayer, N. Kitsera, S. Bartelt, B. Epe and A. Khobta, Nucleic Acids Res., 2016, 44, 7267–7280 Search PubMed.
- T. T. Saxowsky, K. L. Meadows, A. Klungland and P. W. Doetsch, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18877–18882 Search PubMed.
- S. Basu, G. Je and Y. S. Kim, Exp. Mol. Med., 2015, 47, e179 Search PubMed.
- V. Pastukh, J. T. Roberts, D. W. Clark, G. C. Bardwell, M. Patel, A. B. Al-Mehdi, G. M. Borchert and M. N. Gillespie, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2015, 309, L1367–1375 Search PubMed.
- H. S. Kim, B. H. Kim, J. E. Jung, C. S. Lee, H. G. Lee, J. W. Lee, K. H. Lee, H. J. You, M. H. Chung and S. K. Ye, Free Radical Biol. Med., 2016, 93, 12–22 Search PubMed.
- X. Ba, A. Bacsi, J. Luo, L. Aguilera-Aguirre, X. Zeng, Z. Radak, A. R. Brasier and I. Boldogh, J. Immunol., 2014, 192, 2384–2394 Search PubMed.
- L. Pan, B. Zhu, W. Hao, X. Zeng, S. A. Vlahopoulos, T. K. Hazra, M. L. Hegde, Z. Radak, A. Bacsi, A. R. Brasier, X. Ba and I. Boldogh, J. Biol. Chem., 2016, 291, 25553–25566 Search PubMed.
- V. S. Chambers, G. Marsico, J. M. Boutell, M. Di Antonio, G. P. Smith and S. Balasubramanian, Nat. Biotechnol., 2015, 33, 877–881 Search PubMed.
- A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2020, 142, 1115–1136 Search PubMed.
- G. Schafer, T. Cramer, G. Suske, W. Kemmner, B. Wiedenmann and M. Hocker, J. Biol. Chem., 2003, 278, 8190–8198 Search PubMed.
- P. Agrawal, E. Hatzakis, K. Guo, M. Carver and D. Yang, Nucleic Acids Res., 2013, 41, 10584–10592 Search PubMed.
- D. W. Clark, T. Phang, M. G. Edwards, M. W. Geraci and M. N. Gillespie, Free Radical Biol. Med., 2012, 53, 51–59 Search PubMed.
- A. M. Fleming, Y. Ding and C. J. Burrows, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2604–2609 Search PubMed.
- A. M. Fleming, J. Zhu, S. A. Howpay Manage and C. J. Burrows, J. Am. Chem. Soc., 2019, 141, 11036–11049 Search PubMed.
- A. M. Fleming and C. J. Burrows, DNA Repair, 2017, 56, 75–83 Search PubMed.
- A. M. Fleming, J. Zhu, Y. Ding and C. J. Burrows, Nucleic Acids Res., 2019, 47, 5049–5060 Search PubMed.
- M. Ohno, T. Miura, M. Furuichi, Y. Tominaga, D. Tsuchimoto, K. Sakumi and Y. Nakabeppu, Genome Res., 2006, 16, 567–575 Search PubMed.
- S. Akatsuka, T. T. Aung, K. K. Dutta, L. Jiang, W. H. Lee, Y. T. Liu, J. Onuki, T. Shirase, K. Yamasaki, H. Ochi, Y. Naito, T. Yoshikawa, H. Kasai, Y. Tominaga, K. Sakumi, Y. Nakabeppu, Y. Kawai, K. Uchida, A. Yamasaki, T. Tsuruyama, Y. Yamada and S. Toyokuni, Am. J. Pathol., 2006, 169, 1328–1342 Search PubMed.
- M. Yoshihara, L. Jiang, S. Akatsuka, M. Suyama and S. Toyokuni, DNA Res., 2014, 21, 603–612 Search PubMed.
- S. Akatsuka, G. H. Li, S. Kawaguchi, T. Takahashi, M. Yoshihara, M. Suyama and S. Toyokuni, Free Radical Res., 2020, 1–11, DOI:10.1080/10715762.2020.1733548.
- F. Gorini, G. Scala, G. Di Palo, G. I. Dellino, S. Cocozza, P. G. Pelicci, L. Lania, B. Majello and S. Amente, Nucleic Acids Res., 2020, 48, 4309–4324 Search PubMed.
- S. Amente, G. Di Palo, G. Scala, T. Castrignano, F. Gorini, S. Cocozza, A. Moresano, P. Pucci, B. Ma, I. Stepanov, L. Lania, P. G. Pelicci, G. I. Dellino and B. Majello, Nucleic Acids Res., 2019, 47, 221–236 Search PubMed.
- Y. Fang and P. Zou, Biochemistry, 2020, 59, 85–89 Search PubMed.
- Y. Ding, A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2017, 139, 2569–2572 Search PubMed.
- A. R. Poetsch, S. J. Boulton and N. M. Luscombe, Genome Biol., 2018, 19, 215 Search PubMed.
- J. Wu, M. McKeague and S. J. Sturla, J. Am. Chem. Soc., 2018, 140, 9783–9787 Search PubMed.
- A. H. El-Sagheer, A. P. Sanzone, R. Gao, A. Tavassoli and T. Brown, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11338–11343 Search PubMed.
- A. B. Clark, S. A. Lujan, G. E. Kissling and T. A. Kunkel, DNA Repair, 2011, 10, 476–482 Search PubMed.
- A. E. Schibel, N. An, Q. Jin, A. M. Fleming, C. J. Burrows and H. S. White, J. Am. Chem. Soc., 2010, 132, 17992–17995 Search PubMed.
- N. An, A. M. Fleming, H. S. White and C. J. Burrows, ACS Nano, 2015, 9, 4296–4307 Search PubMed.
- L. H. H. Aung, Y. Wang, Z.-Q. Liu, Z. Li, Z. Yu, X. Chen, J. Gao, P. Shan, Z. Zhou and P. Li, bioRxiv, 2020 DOI:10.1101/2020.02.17.951582.
- Y. Taniguchi, R. Kawaguchi and S. Sasaki, J. Am. Chem. Soc., 2011, 133, 10322 Search PubMed.
- M. Nomoto, R. Yamaguchi, K. Kohno and H. Kasai, Oncogene, 2002, 21, 1649–1657 Search PubMed.
- J. Riedl, Y. Ding, A. M. Fleming and C. J. Burrows, Nat. Commun., 2015, 6, 8807 Search PubMed.
- J. Riedl, A. M. Fleming and C. J. Burrows, J. Am. Chem. Soc., 2016, 138, 491–494 Search PubMed.
- J. Park, J. W. Park, H. Oh, F. S. Maria, J. Kang and X. Tian, PLoS One, 2016, 11, e0155792 Search PubMed.
- P. Rossner, Jr. and R. J. Sram, Free Radical Res., 2012, 46, 492–522 Search PubMed.
- Z. J. Liu, S. Martinez Cuesta, P. van Delft and S. Balasubramanian, Nat. Chem., 2019, 11, 629–637 Search PubMed.
- M. G. Ross, C. Russ, M. Costello, A. Hollinger, N. J. Lennon, R. Hegarty, C. Nusbaum and D. B. Jaffe, Genome Biol., 2013, 14, R51 Search PubMed.
- M. Costello, T. J. Pugh, T. J. Fennell, C. Stewart, L. Lichtenstein, J. C. Meldrim, J. L. Fostel, D. C. Friedrich, D. Perrin, D. Dionne, S. Kim, S. B. Gabriel, E. S. Lander, S. Fisher and G. Getz, Nucleic Acids Res., 2013, 41, e67 Search PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 Search PubMed.
- R. K. Mehmood, Oncol. Rev., 2014, 8, 256 Search PubMed.
- J. Graham, M. Mushin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2004, 3, 11–12 Search PubMed.
- A. L. Pinto and S. J. Lippard, Biochim. Biophys. Acta, 1985, 780, 167–180 Search PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 Search PubMed.
- J. T. Reardon, A. Vaisman, S. G. Chaney and A. Sancar, Cancer Res., 1999, 59, 3968–3971 Search PubMed.
- K. V. Ferry, T. C. Hamilton and S. W. Johnson, Biochem. Pharmacol., 2000, 60, 1305–1313 Search PubMed.
- J. Hu, J. D. Lieb, A. Sancar and S. Adar, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11507–11512 Search PubMed.
- X. Shu, X. Xiong, J. Song, C. He and C. Yi, Angew. Chem., Int. Ed., 2016, 55, 14246–14249 Search PubMed.
- J. Hu, S. Adar, C. P. Selby, J. D. Lieb and A. Sancar, Genes Dev., 2015, 29, 948–960 Search PubMed.
- S. U. Dunham and S. J. Lippard, Biochemistry, 1997, 36, 11428–11436 Search PubMed.
- T. Furuta, T. Ueda, G. Aune, A. Sarasin, K. H. Kraemer and Y. Pommier, Cancer Res., 2002, 62, 4899–4902 Search PubMed.
- P. Mao, M. J. Smerdon, S. A. Roberts and J. J. Wyrick, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 9057–9062 Search PubMed.
- Z. Yang, L. M. Schumaker, M. J. Egorin, E. G. Zuhowski, Z. Guo and K. J. Cullen, Clin. Cancer Res., 2006, 12, 5817–5825 Search PubMed.
- A. Yimit, O. Adebali, A. Sancar and Y. Jiang, Nat. Commun., 2019, 10, 309 Search PubMed.
- Y. Yang, Z. Liu, C. P. Selby and A. Sancar, J. Biol. Chem., 2019, 294, 11960–11968 Search PubMed.
- Y. Yang, O. Adebali, G. Wu, C. P. Selby, Y. Y. Chiou, N. Rashid, J. Hu, J. B. Hogenesch and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4777–E4785 Search PubMed.
- G. P. Pfeifer and A. Besaratinia, Photochem. Photobiol. Sci., 2012, 11, 90–97 Search PubMed.
- S. Roy, Adv. Exp. Med. Biol., 2017, 996, 207–219 Search PubMed.
- M. Watson, D. M. Holman and M. Maguire-Eisen, Semin. Oncol. Nurs., 2016, 32, 241–254 Search PubMed.
- A. C. Kneuttinger, G. Kashiwazaki, S. Prill, K. Heil, M. Muller and T. Carell, Photochem. Photobiol., 2014, 90, 1–14 Search PubMed.
- R. P. Sinha and D. P. Hader, Photochem. Photobiol. Sci., 2002, 1, 225–236 Search PubMed.
- A. R. Lehmann, S. Kirk-Bell, C. F. Arlett, M. C. Paterson, P. H. Lohman, E. A. de Weerd-Kastelein and D. Bootsma, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 219–223 Search PubMed.
- J. Hu, O. Adebali, S. Adar and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6758–6763 Search PubMed.
- L. K. Lerner, G. Francisco, D. T. Soltys, C. R. Rocha, A. Quinet, A. T. Vessoni, L. P. Castro, T. I. David, S. O. Bustos, B. E. Strauss, V. Gottifredi, A. Stary, A. Sarasin, R. Chammas and C. F. Menck, Nucleic Acids Res., 2017, 45, 1270–1280 Search PubMed.
- A. R. Lehmann, FEBS Lett., 2005, 579, 873–876 Search PubMed.
- Z. Li, H. Zhang, T. P. McManus, J. J. McCormick, C. W. Lawrence and V. M. Maher, Mutat. Res., 2002, 510, 71–80 Search PubMed.
- S. Nik-Zainal, J. E. Kucab, S. Morganella, D. Glodzik, L. B. Alexandrov, V. M. Arlt, A. Weninger, M. Hollstein, M. R. Stratton and D. H. Phillips, Mutagenesis, 2015, 30, 763–770 Search PubMed.
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. L. Lin, G. R. Ordonez, G. R. Bignell, K. Ye, J. Alipaz, M. J. Bauer, D. Beare, A. Butler, R. J. Carter, L. Chen, A. J. Cox, S. Edkins, P. I. Kokko-Gonzales, N. A. Gormley, R. J. Grocock, C. D. Haudenschild, M. M. Hims, T. James, M. Jia, Z. Kingsbury, C. Leroy, J. Marshall, A. Menzies, L. J. Mudie, Z. Ning, T. Royce, O. B. Schulz-Trieglaff, A. Spiridou, L. A. Stebbings, L. Szajkowski, J. Teague, D. Williamson, L. Chin, M. T. Ross, P. J. Campbell, D. R. Bentley, P. A. Futreal and M. R. Stratton, Nature, 2010, 463, 191–196 Search PubMed.
- Y. Teng, M. Bennett, K. E. Evans, H. Zhuang-Jackson, A. Higgs, S. H. Reed and R. Waters, Nucleic Acids Res., 2011, 39, e10 Search PubMed.
- A. G. Zavala, R. T. Morris, J. J. Wyrick and M. J. Smerdon, Nucleic Acids Res., 2014, 42, 893–905 Search PubMed.
- A. S. Alhegaili, Y. Ji, N. Sylvius, M. J. Blades, M. Karbaschi, H. G. Tempest, G. D. D. Jones and M. S. Cooke, Int. J. Mol. Sci., 2019, 20, 20 Search PubMed.
- D. S. Bryan, M. Ransom, B. Adane, K. York and J. R. Hesselberth, Genome Res., 2014, 24, 1534–1542 Search PubMed.
- P. Mao and J. J. Wyrick, Methods Mol. Biol., 2020, 2175, 79–94 Search PubMed.
- T. Douki and J. Cadet, Biochemistry, 2001, 40, 2495–2501 Search PubMed.
- W. Li, J. Hu, O. Adebali, S. Adar, Y. Yang, Y. Y. Chiou and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6752–6757 Search PubMed.
- O. Adebali, Y. Y. Chiou, J. Hu, A. Sancar and C. P. Selby, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E2116–E2125 Search PubMed.
- J. Hu, C. P. Selby, S. Adar, O. Adebali and A. Sancar, J. Biol. Chem., 2017, 292, 15588–15597 Search PubMed.
- J. Hu, W. Li, O. Adebali, Y. Yang, O. Oztas, C. P. Selby and A. Sancar, Nat. Protoc., 2019, 14, 248–282 Search PubMed.
- W. Li, W. Liu, A. Kakoki, R. Wang, O. Adebali, Y. Jiang and A. Sancar, J. Biol. Chem., 2019, 294, 5914–5922 Search PubMed.
- Y. Yang, J. Hu, C. P. Selby, W. Li, A. Yimit, Y. Jiang and A. Sancar, J. Biol. Chem., 2019, 294, 210–217 Search PubMed.
- N. Tretyakova, B. Matter, R. Jones and A. Shallop, Biochemistry, 2002, 41, 9535–9544 Search PubMed.
- A. I. Roig, U. Eskiocak, S. K. Hight, S. B. Kim, O. Delgado, R. F. Souza, S. J. Spechler, W. E. Wright and J. W. Shay, Gastroenterology, 2010, 138, 1012–1021.e1015 Search PubMed.
- D. J. Madureira, F. T. Weiss, P. Van Midwoud, D. E. Helbling, S. J. Sturla and K. Schirmer, Chem. Res. Toxicol., 2014, 27, 443–453 Search PubMed.
- C. Genies, A. Maitre, E. Lefebvre, A. Jullien, M. Chopard-Lallier and T. Douki, PLoS One, 2013, 8, e78356 Search PubMed.
- H. Mu, N. E. Geacintov, J. H. Min, Y. K. Zhang and S. Broyde, Chem. Res. Toxicol., 2017, 30, 1344–1354 Search PubMed.
- Y. Jiang, X. Zhou, X. Chen, G. Yang, Q. Wang, K. Rao, W. Xiong and J. Yuan, Mutat. Res., 2011, 726, 75–83 Search PubMed.
- E. W. Tung, N. A. Philbrook, C. L. Belanger, S. Ansari and L. M. Winn, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2014, 760, 64–69 Search PubMed.
- D. Chiapperino, H. Kroth, I. H. Kramarczuk, J. M. Sayer, C. Masutani, F. Hanaoka, D. M. Jerina and A. M. Cheh, J. Biol. Chem., 2002, 277, 11765–11771 Search PubMed.
- O. Rechkoblit, Y. Zhang, D. Guo, Z. Wang, S. Amin, J. Krzeminsky, N. Louneva and N. E. Geacintov, J. Biol. Chem., 2002, 277, 30488–30494 Search PubMed.
- K. Hashimoto, Y. Cho, I. Y. Yang, J. Akagi, E. Ohashi, S. Tateishi, N. de Wind, F. Hanaoka, H. Ohmori and M. Moriya, J. Biol. Chem., 2012, 287, 9613–9622 Search PubMed.
- J. H. Yoon, L. E. Smith, Z. Feng, M. Tang, C. S. Lee and G. P. Pfeifer, Cancer Res., 2001, 61, 7110–7117 Search PubMed.
- M. F. Denissenko, A. Pao, M. Tang and G. P. Pfeifer, Science, 1996, 274, 430–432 Search PubMed.
- G. P. Pfeifer, M. F. Denissenko, M. Olivier, N. Tretyakova, S. S. Hecht and P. Hainaut, Oncogene, 2002, 21, 7435–7451 Search PubMed.
- D. J. Weisenberger and L. J. Romano, J. Biol. Chem., 1999, 274, 23948–23955 Search PubMed.
- D. H. Phillips, DNA Repair, 2018, 71, 6–11 Search PubMed.
- D. Fu, J. A. Calvo and L. D. Samson, Nat. Rev. Cancer, 2012, 12, 104–120 Search PubMed.
- B. Rydberg and T. Lindahl, EMBO J., 1982, 1, 211–216 Search PubMed.
- A. V. Ignatov, K. A. Bondarenko and A. V. Makarova, Acta Nat., 2017, 9, 12–26 Search PubMed.
- J. Klapacz, L. H. Pottenger, B. P. Engelward, C. D. Heinen, G. E. Johnson, R. A. Clewell, P. L. Carmichael, Y. Adeleye and M. E. Andersen, Mutat. Res., Rev. Mutat. Res., 2016, 767, 77–91 Search PubMed.
- H. S. Friedman, T. Kerby and H. Calvert, Clin. Cancer Res., 2000, 6, 2585–2597 Search PubMed.
- G. Jiang, A. J. Jiang, Y. Xin, L. T. Li, Q. Cheng and J. N. Zheng, Mol. Biol. Rep., 2014, 41, 6659–6665 Search PubMed.
- R. Coulter, M. Blandino, J. M. Tomlinson, G. T. Pauly, M. Krajewska, R. C. Moschel, L. A. Peterson, A. E. Pegg and T. E. Spratt, Chem. Res. Toxicol., 2007, 20, 1966–1971 Search PubMed.
- E. M. Duguid, P. A. Rice and C. He, J. Mol. Biol., 2005, 350, 657–666 Search PubMed.
- A. Goder, G. Nagel, A. Kraus, B. Dorsam, N. Seiwert, B. Kaina and J. Fahrer, Carcinogenesis, 2015, 36, 817–831 Search PubMed.
- J. Fahrer, J. Frisch, G. Nagel, A. Kraus, B. Dorsam, A. D. Thomas, S. Reissig, A. Waisman and B. Kaina, Carcinogenesis, 2015, 36, 1235–1244 Search PubMed.
- J. R. Perry, N. Laperriere, C. J. O'Callaghan, A. A. Brandes, J. Menten, C. Phillips, M. Fay, R. Nishikawa, J. G. Cairncross, W. Roa, D. Osoba, J. P. Rossiter, A. Sahgal, H. Hirte, F. Laigle-Donadey, E. Franceschi, O. Chinot, V. Golfinopoulos, L. Fariselli, A. Wick, L. Feuvret, M. Back, M. Tills, C. Winch, B. G. Baumert, W. Wick, K. Ding, W. P. Mason and I. Trial, N. Engl. J. Med., 2017, 376, 1027–1037 Search PubMed.
- C. Happold, P. Roth, W. Wick, N. Schmidt, A. M. Florea, M. Silginer, G. Reifenberger and M. Weller, J. Neurochem., 2012, 122, 444–455 Search PubMed.
- A. E. Pegg, Chem. Res. Toxicol., 2011, 24, 618–639 Search PubMed.
- M. McKeague, C. Otto, M. H. Raz, T. Angelov and S. J. Sturla, ACS Chem. Biol., 2018, 13, 2534–2541 Search PubMed.
- T. E. Spratt, J. D. Wu, D. E. Levy, S. Kanugula and A. E. Pegg, Biochemistry, 1999, 38, 6801–6806 Search PubMed.
- A. S. Meyer, M. D. McCain, Q. Fang, A. E. Pegg and T. E. Spratt, Chem. Res. Toxicol., 2003, 16, 1405–1409 Search PubMed.
- P. Thongararm, B. I. Fedeles, S. Khumsubdee, A. L. Armijo, L. Kim, A. Thiantanawat, J. Promvijit, P. Navasumrit, M. Ruchirawat, R. G. Croy and J. M. Essigmann, Chem. Res. Toxicol., 2020, 33, 625–633 Search PubMed.
- A. T. Gordon, F. C. Manning, D. P. Cooper, G. P. Margison, P. J. O'Connor and M. A. Billett, Biochem. Soc. Trans., 1993, 21, 374S Search PubMed.
- J. J. Warren, L. J. Forsberg and L. S. Beese, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19701–19706 Search PubMed.
- Z. Li, A. H. Pearlman and P. Hsieh, DNA Repair, 2016, 38, 94–101 Search PubMed.
- L. D. Williams and B. R. Shaw, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1779–1783 Search PubMed.
- L. Haracska, S. Prakash and L. Prakash, Mol. Cell. Biol., 2003, 23, 1453–1459 Search PubMed.
- A. M. Woodside and F. P. Guengerich, Biochemistry, 2002, 41, 1027–1038 Search PubMed.
- S. Quiros, W. P. Roos and B. Kaina, Cell Cycle, 2010, 9, 168–178 Search PubMed.
- K. Yoshioka, Y. Yoshioka and P. Hsieh, Mol. Cell, 2006, 22, 501–510 Search PubMed.
- J. M. Soll, R. W. Sobol and N. Mosammaparast, Trends Biochem. Sci., 2017, 42, 206–218 Search PubMed.
- R. Benigni, F. Palombo and E. Dogliotti, Mutat. Res., 1992, 267, 77–88 Search PubMed.
- M. Olivier, A. Weninger, M. Ardin, H. Huskova, X. Castells, M. P. Vallee, J. McKay, T. Nedelko, K. R. Muehlbauer, H. Marusawa, J. Alexander, L. Hazelwood, G. Byrnes, M. Hollstein and J. Zavadil, Sci. Rep., 2014, 4, 4482 Search PubMed.
- S. L. Poon, J. R. McPherson, P. Tan, B. T. Teh and S. G. Rozen, Genome Med., 2014, 6, 24 Search PubMed.
- M. Weber, J. J. Davies, D. Wittig, E. J. Oakeley, M. Haase, W. L. Lam and D. Schubeler, Nat. Genet., 2005, 37, 853–862 Search PubMed.
- L. Shen, H. Wu, D. Diep, S. Yamaguchi, A. C. D'Alessio, H. L. Fung, K. Zhang and Y. Zhang, Cell, 2013, 153, 692–706 Search PubMed.
- M. J. Booth, T. W. Ost, D. Beraldi, N. M. Bell, M. R. Branco, W. Reik and S. Balasubramanian, Nat. Protoc., 2013, 8, 1841–1851 Search PubMed.
- T. A. Clark, X. Lu, K. Luong, Q. Dai, M. Boitano, S. W. Turner, C. He and J. Korlach, BMC Biol., 2013, 11, 4 Search PubMed.
- Y. Fu, G. Z. Luo, K. Chen, X. Deng, M. Yu, D. Han, Z. Hao, J. Liu, X. Lu, L. C. Dore, X. Weng, Q. Ji, L. Mets and C. He, Cell, 2015, 161, 879–892 Search PubMed.
- G. Zhang, H. Huang, D. Liu, Y. Cheng, X. Liu, W. Zhang, R. Yin, D. Zhang, P. Zhang, J. Liu, C. Li, B. Liu, Y. Luo, Y. Zhu, N. Zhang, S. He, C. He, H. Wang and D. Chen, Cell, 2015, 161, 893–906 Search PubMed.
- E. L. Greer, M. A. Blanco, L. Gu, E. Sendinc, J. Liu, D. Aristizabal-Corrales, C. H. Hsu, L. Aravind, C. He and Y. Shi, Cell, 2015, 161, 868–878 Search PubMed.
- T. P. Wu, T. Wang, M. G. Seetin, Y. Lai, S. Zhu, K. Lin, Y. Liu, S. D. Byrum, S. G. Mackintosh, M. Zhong, A. Tackett, G. Wang, L. S. Hon, G. Fang, J. A. Swenberg and A. Z. Xiao, Nature, 2016, 532, 329–333 Search PubMed.
- J. M. Voigt and M. D. Topal, Carcinogenesis, 1995, 16, 1775–1782 Search PubMed.
- I. A. Trantakis, A. Nilforoushan, H. A. Dahlmann, C. K. Stauble and S. J. Sturla, J. Am. Chem. Soc., 2016, 138, 8497–8504 Search PubMed.
- L. A. Wyss, A. Nilforoushan, D. M. Williams, A. Marx and S. J. Sturla, Nucleic Acids Res., 2016, 44, 6564–6573 Search PubMed.
- B. Dorsam, N. Seiwert, S. Foersch, S. Stroh, G. Nagel, D. Begaliew, E. Diehl, A. Kraus, M. McKeague, V. Minneker, V. Roukos, S. Reissig, A. Waisman, M. Moehler, A. Stier, A. Mangerich, F. Dantzer, B. Kaina and J. Fahrer, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4061–E4070 Search PubMed.
- T. J. Matray and E. T. Kool, Nature, 1999, 399, 704–708 Search PubMed.
- Y. Taniguchi, Y. Kikukawa and S. Sasaki, Angew. Chem., Int. Ed., 2015, 54, 5147–5151 Search PubMed.
- A. Nilforoushan, A. Furrer, L. A. Wyss, B. van Loon and S. J. Sturla, J. Am. Chem. Soc., 2015, 137, 4728–4734 Search PubMed.
- L. A. Wyss, A. Nilforoushan, F. Eichenseher, U. Suter, N. Blatter, A. Marx and S. J. Sturla, J. Am. Chem. Soc., 2015, 137, 30–33 Search PubMed.
- M. H. Raz, C. M. N. Aloisi, H. L. Gahlon and S. J. Sturla, Acc. Chem. Res., 2019, 52, 1391–1399 Search PubMed.
- C. M. N. Aloisi, A. Nilforoushan, N. Ziegler and S. J. Sturla, J. Am. Chem. Soc., 2020, 142, 6962–6969 Search PubMed.
- T. Lindahl and B. Nyberg, Biochemistry, 1972, 11, 3610–3618 Search PubMed.
- O. D. Scharer and J. Jiricny, BioEssays, 2001, 23, 270–281 Search PubMed.
- R. M. Kohli and Y. Zhang, Nature, 2013, 502, 472–479 Search PubMed.
- S. Boiteux and M. Guillet, DNA Repair, 2004, 3, 1–12 Search PubMed.
- P. D. Chastain, 2nd, J. Nakamura, S. Rao, H. Chu, J. G. Ibrahim, J. A. Swenberg and D. G. Kaufman, FASEB J., 2010, 24, 3674–3680 Search PubMed.
- B. Cao, X. Wu, J. Zhou, H. Wu, L. Liu, Q. Zhang, M. S. DeMott, C. Gu, L. Wang, D. You and P. C. Dedon, Nucleic Acids Res., 2020, 48, 6715–6725 Search PubMed.
- T. Pfaffeneder, B. Hackner, M. Truss, M. Munzel, M. Muller, C. A. Deiml, C. Hagemeier and T. Carell, Angew. Chem., Int. Ed., 2011, 50, 7008–7012 Search PubMed.
- K. W. Caldecott, Nat. Rev. Genet., 2008, 9, 619–631 Search PubMed.
- A. Kuzminov, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8241–8246 Search PubMed.
- L. Baranello, F. Kouzine, D. Wojtowicz, K. Cui, T. M. Przytycka, K. Zhao and D. Levens, Int. J. Mol. Sci., 2014, 15, 13111–13122 Search PubMed.
- H. Cao, L. Salazar-Garcia, F. Gao, T. Wahlestedt, C. L. Wu, X. Han, Y. Cai, D. Xu, F. Wang, L. Tang, N. Ricciardi, D. Cai, H. Wang, M. P. S. Chin, J. A. Timmons, C. Wahlestedt and P. Kapranov, Nat. Commun., 2019, 10, 5799 Search PubMed.
- A. M. Sriramachandran, G. Petrosino, M. Méndez-Lago, A. J. Schäfer, L. S. Batista-Nascimento, N. Zilio and H. D. Ulrich, Mol. Cell, 2020, 78, 975–985.e977 Search PubMed.
- P. A. Jeggo and M. Lobrich, Oncogene, 2007, 26, 7717–7719 Search PubMed.
- B. Schwer, P. C. Wei, A. N. Chang, J. Kao, Z. Du, R. M. Meyers and F. W. Alt, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2258–2263 Search PubMed.
- F. Baudat, Y. Imai and B. de Massy, Nat. Rev. Genet., 2013, 14, 794–806 Search PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 Search PubMed.
- N. Crosetto, A. Mitra, M. J. Silva, M. Bienko, N. Dojer, Q. Wang, E. Karaca, R. Chiarle, M. Skrzypczak, K. Ginalski, P. Pasero, M. Rowicka and I. Dikic, Nat. Methods, 2013, 10, 361–365 Search PubMed.
- E. A. Hoffman, A. McCulley, B. Haarer, R. Arnak and W. Feng, Genome Res., 2015, 25, 402–412 Search PubMed.
- S. V. Lensing, G. Marsico, R. Hansel-Hertsch, E. Y. Lam, D. Tannahill and S. Balasubramanian, Nat. Methods, 2016, 13, 855–857 Search PubMed.
- A. Canela, S. Sridharan, N. Sciascia, A. Tubbs, P. Meltzer, B. P. Sleckman and A. Nussenzweig, Mol. Cell, 2016, 63, 898–911 Search PubMed.
- S. Q. Tsai, Z. Zheng, N. T. Nguyen, M. Liebers, V. V. Topkar, V. Thapar, N. Wyvekens, C. Khayter, A. J. Iafrate, L. P. Le, M. J. Aryee and J. K. Joung, Nat. Biotechnol., 2015, 33, 187–197 Search PubMed.
- W. X. Yan, R. Mirzazadeh, S. Garnerone, D. Scott, M. W. Schneider, T. Kallas, J. Custodio, E. Wernersson, Y. Li, L. Gao, Y. Federova, B. Zetsche, F. Zhang, M. Bienko and N. Crosetto, Nat. Commun., 2017, 8, 15058 Search PubMed.
- Y. Zhu, A. Biernacka, B. Pardo, N. Dojer, R. Forey, M. Skrzypczak, B. Fongang, J. Nde, R. Yousefi, P. Pasero, K. Ginalski and M. Rowicka, Nat. Commun., 2019, 10, 2313 Search PubMed.
- M. P. Roettger, K. A. Fiala, S. Sompalli, Y. Dong and Z. Suo, Biochemistry, 2004, 43, 13827–13838 Search PubMed.
- L. M. Dieckman, R. E. Johnson, S. Prakash and M. T. Washington, Biochemistry, 2010, 49, 7344–7350 Search PubMed.
- E. Crespan, A. Furrer, M. Rösinger, F. Bertoletti, E. Mentegari, G. Chiapparini, R. Imhof, N. Ziegler, S. J. Sturla, U. Hübscher, B. van Loon and G. Maga, Nat. Commun., 2016, 7, 10805 Search PubMed.
- S. A. Nick McElhinny, D. Kumar, A. B. Clark, D. L. Watt, B. E. Watts, E. B. Lundstrom, E. Johansson, A. Chabes and T. A. Kunkel, Nat. Chem. Biol., 2010, 6, 774–781 Search PubMed.
- A. Diaz-Talavera, P. A. Calvo, D. Gonzalez-Acosta, M. Diaz, G. Sastre-Moreno, L. Blanco-Franco, S. Guerra, M. I. Martinez-Jimenez, J. Mendez and L. Blanco, Sci. Rep., 2019, 9, 1121 Search PubMed.
- M. A. Reijns, B. Rabe, R. E. Rigby, P. Mill, K. R. Astell, L. A. Lettice, S. Boyle, A. Leitch, M. Keighren, F. Kilanowski, P. S. Devenney, D. Sexton, G. Grimes, I. J. Holt, R. E. Hill, M. S. Taylor, K. A. Lawson, J. R. Dorin and A. P. Jackson, Cell, 2012, 149, 1008–1022 Search PubMed.
- A. Vaisman and R. Woodgate, DNA Repair, 2015, 29, 74–82 Search PubMed.
- Y. Li and R. R. Breaker, J. Am. Chem. Soc., 1999, 121, 5364–5372 Search PubMed.
- S. N. Huang, J. S. Williams, M. E. Arana, T. A. Kunkel and Y. Pommier, EMBO J., 2017, 36, 361–373 Search PubMed.
- A. R. Clausen, M. S. Murray, A. R. Passer, L. C. Pedersen and T. A. Kunkel, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 16802–16807 Search PubMed.
- S. P. Linke, K. C. Clarkin, A. Di Leonardo, A. Tsou and G. M. Wahl, Genes Dev., 1996, 10, 934–947 Search PubMed.
- P. Tumbale, J. S. Williams, M. J. Schellenberg, T. A. Kunkel and R. S. Williams, Nature, 2014, 506, 111–115 Search PubMed.
- S. N. Andres, M. J. Schellenberg, B. D. Wallace, P. Tumbale and R. S. Williams, Environ. Mol. Mutagen., 2015, 56, 1–21 Search PubMed.
- K. D. Koh, S. Balachander, J. R. Hesselberth and F. Storici, Nat. Methods, 2015, 12, 251–257 Search PubMed.
- A. R. Clausen, S. A. Lujan, A. B. Burkholder, C. D. Orebaugh, J. S. Williams, M. F. Clausen, E. P. Malc, P. A. Mieczkowski, D. C. Fargo, D. J. Smith and T. A. Kunkel, Nat. Struct. Mol. Biol., 2015, 22, 185–191 Search PubMed.
- Y. Daigaku, A. Keszthelyi, C. A. Muller, I. Miyabe, T. Brooks, R. Retkute, M. Hubank, C. A. Nieduszynski and A. M. Carr, Nat. Struct. Mol. Biol., 2015, 22, 192–198 Search PubMed.
- J. Ding, M. S. Taylor, A. P. Jackson and M. A. Reijns, Nat. Protoc., 2015, 10, 1433–1444 Search PubMed.
- M. A. M. Reijns, H. Kemp, J. Ding, S. M. de Proce, A. P. Jackson and M. S. Taylor, Nature, 2015, 518, 502–506 Search PubMed.
- H. E. Krokan, F. Drabløs and G. Slupphaug, Oncogene, 2002, 21, 8935–8948 Search PubMed.
- X. Shu, M. Liu, Z. Lu, C. Zhu, H. Meng, S. Huang, X. Zhang and C. Yi, Nat. Chem. Biol., 2018, 14, 680–687 Search PubMed.
- R. Sakhtemani, V. Senevirathne, J. Stewart, M. L. W. Perera, R. Pique-Regi, M. S. Lawrence and A. S. Bhagwat, J. Biol. Chem., 2019, 294, 15037–15051 Search PubMed.
- H. L. Pálinkás, A. Békési, G. Róna, L. Pongor, G. Tihanyi, E. Holub, Á. Póti, C. Gemma, S. Ali, M. J. Morten, E. Rothenberg, M. Pagano, D. Szüts, B. Győrffy and B. G. Vértessy, bioRxiv, 2020 DOI:10.1101/2020.03.04.976977.
- C. D. Orebaugh, S. A. Lujan, A. B. Burkholder, A. R. Clausen and T. A. Kunkel, Methods Mol. Biol., 2018, 1672, 329–345 Search PubMed.
- A. Keszthelyi, Y. Daigaku, K. Ptasinska, I. Miyabe and A. M. Carr, Nat. Protoc., 2015, 10, 1786–1801 Search PubMed.
- T. A. Clark, K. E. Spittle, S. W. Turner and J. Korlach, Genome Integr., 2011, 2, 10 Search PubMed.
- J. Wang, M. Y. Li, J. Yang, Y. Q. Wang, X. Y. Wu, J. Huang, Y. L. Ying and Y. T. Long, ACS Cent. Sci., 2020, 6, 76–82 Search PubMed.
- Z. Feng, G. Fang, J. Korlach, T. Clark, K. Luong, X. Zhang, W. Wong and E. Schadt, PLoS Comput. Biol., 2013, 9, e1002935 Search PubMed.
- X. Zhang, N. E. Price, X. Fang, Z. Yang, L. Q. Gu and K. S. Gates, ACS Nano, 2015, 9, 11812–11819 Search PubMed.
- E. E. Schadt, O. Banerjee, G. Fang, Z. Feng, W. H. Wong, X. Zhang, A. Kislyuk, T. A. Clark, K. Luong, A. Keren-Paz, A. Chess, V. Kumar, A. Chen-Plotkin, N. Sondheimer, J. Korlach and A. Kasarskis, Genome Res., 2013, 23, 129–141 Search PubMed.
- K. M. Zatopek, V. Potapov, L. L. Maduzia, E. Alpaslan, L. Chen, T. C. Evans, Jr., J. L. Ong, L. M. Ettwiller and A. F. Gardner, DNA Repair, 2019, 80, 36–44 Search PubMed.
- Y. Wang, K. M. Patil, S. Yan, P. Zhang, W. Guo, Y. Wang, H. Y. Chen, D. Gillingham and S. Huang, Angew. Chem., Int. Ed., 2019, 58, 8432–8436 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |