
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fundamental studies of functional nucleic acids: aptamers, riboswitches, ribozymes and DNAzymes
Ronald
Micura
 *a and
Claudia
Höbartner
*b
*a and
Claudia
Höbartner
*b
aInstitute of Organic Chemistry and Center for Molecular Biosciences Innsbruck CMBI, Leopold-Franzens University Innsbruck, Innsbruck, Austria. E-mail: ronald.micura@uibk.ac.at
bInstitute of Organic Chemistry and Center for Nanosystems Chemistry CNC, Julius-Maximilians University Würzburg, Würzburg, Germany. E-mail: claudia.hoebartner@uni-wuerzburg.de
First published on 18th September 2020
Abstract
This review aims at juxtaposing common versus distinct structural and functional strategies that are applied by aptamers, riboswitches, and ribozymes/DNAzymes. Focusing on recently discovered systems, we begin our analysis with small-molecule binding aptamers, with emphasis on in vitro-selected fluorogenic RNA aptamers and their different modes of ligand binding and fluorescence activation. Fundamental insights are much needed to advance RNA imaging probes for detection of exo- and endogenous RNA and for RNA process tracking. Secondly, we discuss the latest gene expression–regulating mRNA riboswitches that respond to the alarmone ppGpp, to PRPP, to NAD+, to adenosine and cytidine diphosphates, and to precursors of thiamine biosynthesis (HMP-PP), and we outline new subclasses of SAM and tetrahydrofolate-binding RNA regulators. Many riboswitches bind protein enzyme cofactors that, in principle, can catalyse a chemical reaction. For RNA, however, only one system (glmS ribozyme) has been identified in Nature thus far that utilizes a small molecule – glucosamine-6-phosphate – to participate directly in reaction catalysis (phosphodiester cleavage). We wonder why that is the case and what is to be done to reveal such likely existing cellular activities that could be more diverse than currently imagined. Thirdly, this brings us to the four latest small nucleolytic ribozymes termed twister, twister-sister, pistol, and hatchet as well as to in vitro selected DNA and RNA enzymes that promote new chemistry, mainly by exploiting their ability for RNA labelling and nucleoside modification recognition. Enormous progress in understanding the strategies of nucleic acids catalysts has been made by providing thorough structural fundaments (e.g. first structure of a DNAzyme, structures of ribozyme transition state mimics) in combination with functional assays and atomic mutagenesis.
 Ronald Micura | Ronald Micura studied chemistry at the University of Linz. He received his PhD in the field of phycobilin pigments under the supervision of Karl Grubmayr in 1995. Immediately thereafter he joined the laboratory of Albert Eschenmoser at ETH Zürich, and then moved to The Scripps Research Institute, to perform postdoctoral research on alternative nucleic acids. He started independent research funded by an APART-fellowship (Austrian Academy of Science) at the University of Linz. In 2004, he was appointed Professor of Organic Chemistry at the University of Innsbruck. His research focuses on the chemistry, chemical biology, and biophysics of RNA. |
 Claudia Höbartner | Claudia Höbartner studied at TU Wien and ETH Zürich, and received her PhD under the supervision of Ronald Micura at the University of Innsbruck in 2004. After postdoctoral work with Scott Silverman at the University of Illinois at Urbana Champaign, she joined the Max Planck Institute for Biophysical Chemistry in Göttingen as a research group leader. In 2014 she was appointed at the University of Göttingen, and since 2017 she holds a Chair of Organic Chemistry at the University of Würzburg. Her main research interests concern the chemistry and chemical biology of functional nucleic acids and RNA modifications. |
Introduction
Nature has endowed nucleic acids, both DNA and RNA, with fascinating properties that allow them to fold into complex three-dimensional structures forming the basis for their functional diversity and reactivity.1–3 RNA and DNA cannot only mould pockets to recognize small molecules with high specificity and selectivity,4 they also possess the inherent propensity for structural ambiguities and alterations in shape which are frequently applied as the molecular basis for distinct signalling pathways in the cell.5,6 Additionally, nucleic acids are dynamic in structure on timescales that vary from milliseconds to minutes and even hours which is crucial for ligand recognition and discrimination on the one hand (fast events),7–9 and on the other hand, for folding/refolding of alternative secondary structures (slower events), such as terminator/antiterminator stem formation to regulate gene expression.10 On top, these structural and dynamic properties are key to triggering reactivity, culminating in the occurrence of nucleic acids catalysts.11,12With regard to structural diversity and reactivity, RNA is often deemed the more powerful sibling of DNA. This impression originates from the many naturally occurring representatives in form of non-coding RNAs with important cellular functions (e.g. riboswitches).4 Nevertheless, many functional nucleic acids, RNA and DNA, have been generated artificially in the test tube and are not limited to the functions found in Nature. The activities of ligand binding (sensors) and catalysis (ribozymes, DNAzymes) have been combined in aptazymes,13,14 although the currently known combinations are mostly limited to triggering “simple” reactions, namely phosphodiester cleavage or ligation. Only few artificial nucleic acids have been identified, which employ small molecules such as nicotinamide adenine dinucleotide (NAD+/NADH) or thiamine as cofactors. However, these ribozymes use synthetic substrates that were fine-tuned for the in vitro selection process.15,16 Thus far, nucleic acids that are able to conduct reactions equivalent to modern cellular transformations, for instance, selective methylation in trans, using cofactors such as cobalamine or S-adenosyl methionine (SAM),17 or any other natural methyl group donor, have not yet been identified. Finding such reactivity would have a major impact on the (protein-free) RNA world hypothesis and prebiotic chemistry.18,19
In the laboratory, functional nucleic acids are identified by in vitro selection (also known as SELEX) starting from random RNA libraries. The concept was introduced by the Szostak20 and Gold21 research groups about 30 years ago, and consists of repeated cycles of selection and amplification for the enrichment of RNA sequences with desired functions. The functions range from specific non-covalent interactions with small and large molecules to catalysing various chemical reactions of diverse substrates. Although there are numerous successful examples known today, and the procedures are constantly refined and expanded by modern technologies,22–24 the outcome of in vitro selection (also known as SELEX) is still highly unpredictable. Moreover, it is currently impossible to design functional nucleic acids from first principles. Fundamental biochemical and biophysical studies of the NAs provide much needed data on the thermodynamic and kinetic levels. High throughput methods reveal insights into folding and activity landscapes at an impressive pace.25,26 Combined with machine learning approaches, these methods may form the basis for new algorithms to generate aptamer and ribozyme candidates in silico, and to derive guidelines for the evolution of high selectivity, high rate acceleration and multiple turnover. In the protein world, important steps in this direction have been successfully demonstrated, with the de novo design of protein folds, enzymes, logic gates and molecular switches.27–30 Similar exciting advances are to be expected in the field of nucleic acids research.31,32
This review summarizes recent developments in the fields of in vitro selected fluorogenic RNA aptamers and riboswitch discovery, and discusses more intensively the structures and mechanistic elucidations of recently discovered natural ribozymes, which we contrast with in vitro selected DNAzymes catalysing phosphodiester cleavage and ligation chemistry (Fig. 1).
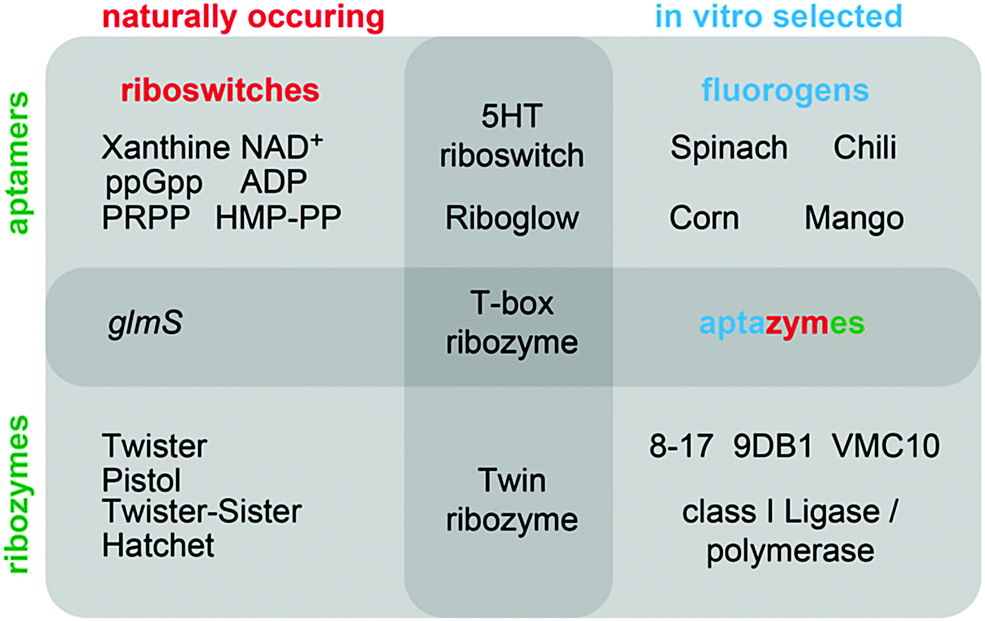 | ||
| Fig. 1 Overview of classes of functional nucleic acids (with selected examples) covered in this review and their interconnections. | ||
Fluorogen-activating RNA aptamers
Organic chromophores have been early targets for the in vitro selection of RNA aptamers.20,33 The successful discrimination of closely related structural analogues of anthraquinone dyes was demonstrated by large differences in binding affinity. The Reactive Blue A and sulforhodamine-binding aptamers constituted early examples of specific recognition of anionic ligands despite the polyanionic nature of RNA. About 10 years later, based on the crystal structure of the Malachite green aptamer,34 it was recognized that ligand binding resulted in restricted conformational freedom of the ligand and this observation culminated in the discovery of strong fluorescence enhancement by the malachite green and sulforhodamine-binding aptamers.35In the last decade, fluorescence turn-on aptamers have been an emerging class of synthetic functional nucleic acids for RNA tracking and visualization in cellular and molecular biology. Also termed fluorescent light-up probes, or fluorogen-activating aptamers (FLAPs), an increasing number of in vitro evolved 30 to 100 nt long RNAs are now known to activate the fluorescence of various classes of conditional fluorophores. Most of the recent examples and their applications have been summarized in excellent reviews.3,36–39 Here, we focus on different mechanisms and structural principles of fluorescence activation and discuss open challenges to be addressed by fundamental biophysical studies in combination with further evolution and engineering.
HBI-binding aptamers – Spinach, Broccoli, Corn, Chili
The first aptamer in the family of “RNA mimics of green fluorescent protein (GFP)” was named spinach.40 It was selected to bind the difluorinated analogue of the GFP chromophore, named DFHBI (Fig. 2A). The Spinach RNA binds the deprotonated ligand, and the emission wavelength and quantum yield resemble enhanced GFP. Broccoli is a second generation aptamer that was selected with a modified DFHBI analogue, DFHBI-1T, and was optimized to function in cellular conditions at low Mg2+ concentration.41 Spinach and Broccoli RNAs share similar primary and secondary structures, and use the same structure-based mechanism of fluorescence activation that relies on reducing non-radiative deactivation of the excited state. The ligand binding site involves a guanine quadruplex of complex topology.42,43 The ligand stacks on top of one G-quartet, and, in spinach, it is sandwiched between the G-quartet and a base-triple (Fig. 2B). The extended π-stacking interactions reduce the conformational flexibility of the ligand, resulting in rigidification and reduced conformational flexibility, thus promoting fluorescence emission. Although the 3D structure of Broccoli has not been explicitly solved, it is reported to be highly related to Spinach, and single point mutations are known to tune fluorescence emission wavelengths and intensities, as well as RNA folding kinetics.44–46In vitro evolution of Spinach based on microfluidic screening of water-in-oil droplets resulted in a variant named iSpinach,47 with reduced salt-sensitivity, higher thermal stability and increased brightness, despite containing an almost identical core structure of the quadruplex in the ligand binding site.48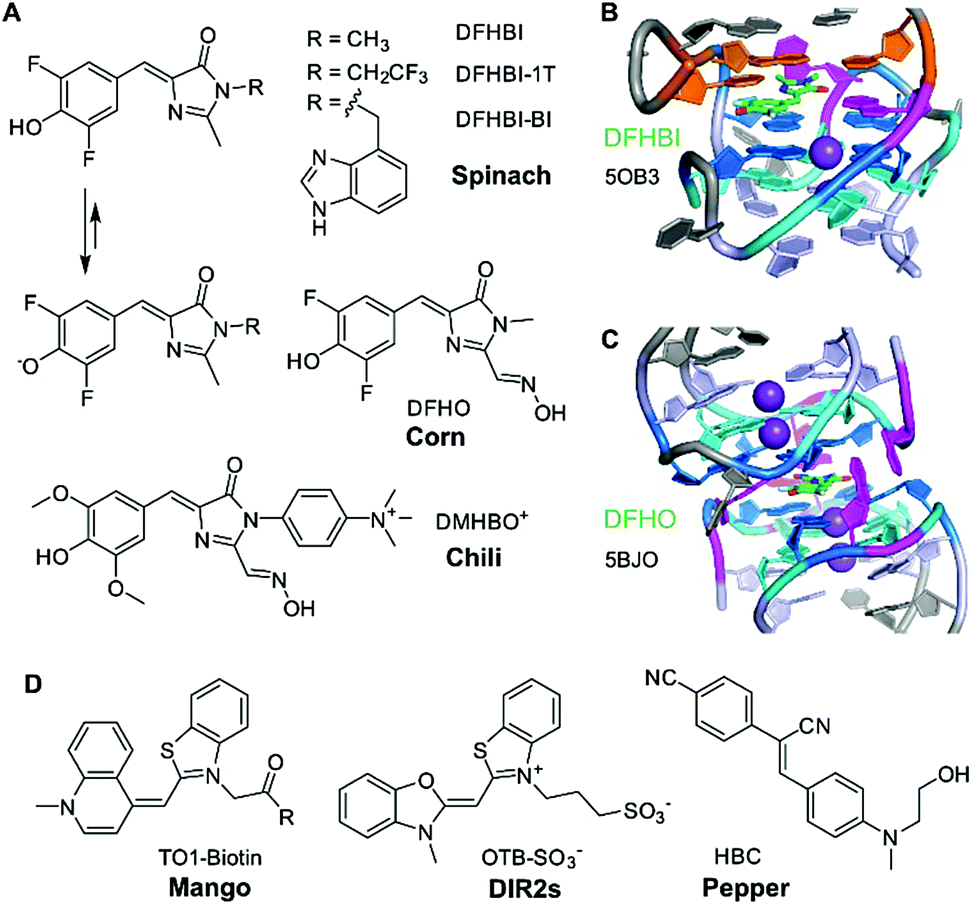 | ||
| Fig. 2 Fluorogen-activating RNA aptamers. (A) HBI ligands of Spinach, Broccoli, Corn and Chili. (B) Core of iSpinach with DFHBI sandwiched between G-quartet (blue, cyan) and UAU base triple (orange), held in place by H-bonding with guanine and methyl π interaction with adenine (magenta). (C) Dimeric quadruplex of Corn with H-bonding contacts to the oxime side chain and phenolate oxygen. (D) Ligand structures for Mango, DIR2s, and Pepper aptamers. | ||
Another quadruplex-containing aptamer named corn was obtained in an in vitro selection experiment with an extended derivative of DFHBI that carries an oxime sidechain, named DFHO.49 Corn differs from Spinach and Broccoli not only in the redshifted emission wavelength and enhanced photostability, but also in the architecture of the ligand binding site.50 Spinach and Broccoli are monomeric aptamers, while Corn forms a dimer, and the ligand is bound at the interface of the two interacting RNA molecules (Fig. 2C).50 Interestingly, the quadruplex dimer is also formed in the apo state (i.e., in the absence of the ligand), with a collapsed, symmetric conformation.51 Moreover, structures of Corn in complex with the quadruplex-binders thioflavin T (ThT) and thiazol orange (TO) helped to rationalize their strong fluorescence activation by binding to the Corn quadruplex dimer.51
Spinach, Broccoli and Corn bind fluorinated HBI analogues with low pKa values that are easily deprotonated and exist predominantly as phenolates under physiological conditions. In contrast, the Chili aptamer,52 which is derived from the earlier reported 13-2 aptamer,40 binds and activates the dimethoxy-substituted analogue DMHBI exclusively in the protonated state. The phenolic proton is lost upon excitation of the bound ligand, resulting in fluorescence emission from the phenolate form. The excited state proton transfer (ESPT) leads to the large Stokes shifts of >130 nm observed in Chili. Moreover, Chili is able to activate the emission of several ligands with different substitution patterns, resulting in fluorescence emission in the green, yellow and red wavelength range.53 The introduction of a permanent positive charge in the ligand sidechain in DMHBI+ and DMHBO+ led to strongly enhanced affinities, and maintained high selectivity for the Chili aptamer. The structure of any Chili RNA–ligand complex has not yet been reported. However, the high guanine content (48% G), and preliminary NMR spectra suggest the presence of a quadruplex core structure.42,53 Initial hints into the organization of the aptamer and the relative location of the ligand were obtained by supramolecular FRET.54 The fluorescent nucleobase analogue 4-cyanoindol was covalently incorporated into the RNA aptamer and served as donor for energy transfer to the non-covalently bound DMHBI+/DMHBO+ ligands that served as acceptor.54 Additional fundamental studies of ESPT photophysics in combination with structural studies may reveal the identity and function of the proton acceptor. Preliminary data regarding the involvement of a crucial nucleobase nitrogen have been obtained by mutagenesis experiments, but more detailed investigations are needed to surface the mechanism of how Chili mimics the large Stokes shift (LSS) fluorescent proteins, such as LSSmOrange and LSSmKate.55
TO-binding aptamers – Mango
The second heavily investigated and structurally well-characterised class of fluorogenic aptamers is the Mango family, which binds to derivatives of thiazole orange (TO). Originally selected to bind biotinylated TO1 with a PEG linker,56 four generations of the Mango aptamer were evolved by advanced methods, including fluorescence-activated droplet sorting.47,57,58 The structures of Mango variants from generations I–IV are known, and have recently been discussed in detail.3,58–61 The core of all Mango aptamers contains a G-quadruplex, primarily responsible for stacking and rigidification of the bound ligand. While the first generation included the PEG linker and the biotin in packing onto the G-quartet next to TO1,61 later generations exhibited improved fluorophore planarity resulting in higher brightness. Mango-III and iMango-III exhibited additional sophisticated structural elements, such as an unusual pseudoknot structure containing non-canonical trans base pairs in parallel strand orientation.58,62Additional recent light-up aptamers
Besides HBI- and TO-binding aptamers described above, several other aptamers have recently been reported to enhance the brightness of conditional fluorophores. These include the DIR2s aptamer that binds asymmetric cyanine dyes such as dimethylindole red (DIR) and oxazole thiazole blue (OTB).63 Interestingly, this aptamer does not form a G-quadruplex core for planarization of the fluorophore, but instead uses a mixed-sequence tetrad to promote rigidification, supported by a single adenine stacking on top of the thiazole ring.64The most recent “spicy” addition to the family of fluorogenic vegetable and fruit aptamers for RNA imaging is the Pepper aptamer.65 The small ca. 45 nt long RNA binds and activates a family of eight related synthetic dyes, derived from substituted aminobenzylidene cyanophenyl acetonitriles (HBC, Fig. 2D) that exhibit a structurally rigid electron acceptor and a strong electron donor to produce emission colours in the range from cyan to red. Structural analysis of the predicted bulged stem-loop core will likely reveal the molecular basis of this colourful diversity. To avoid confusion, it should be noted that a second strategy for RNA tracking was recently introduced under the name Pepper RNA, which follows a very different mechanism based on RNA-mediated stability of a fluorescent protein.66
A different fluorogenic mechanism is employed by the recently reported silicon rhodamine-binding RNA aptamer (SiRA).67 Specific binding of silicon rhodamine to the RNA shifts the equilibrium between the closed non-colored spirolactone and the open fluorescent zwitterion, resulting in turn-on of NIR fluorescence emission.
Besides fluorophore rigidification, another common mechanism of fluorescence enhancement is the RNA-based disruption of chromophore–quencher conjugates. In this case, binding of RNA aptamers either to the fluorophore or to the quencher results in enhanced emission. Early examples include non-emissive fluorescein derivatives with 4-methoxyphenyl-piperazinyl side chains that served as donors by photoinduced electron transfer (PET).68 Aptamers have been evolved for selective binding of black-hole-quenchers to eliminate contact-mediated quenching of fluorescence.69,70 Other rationally designed fluoromodules used rhodamine dyes with appended dimethylaniline or dinitroaniline quenchers.71 The SRB-2 aptamer, termed rainbow, then allowed for dual colour fluorogenic RNA imaging with low background in live cells.72,73 Another recently introduced concept takes advantage of self-quenching upon fluorophore dimerization. A cell-permeable fluorogenic dimer of sulforhodamine B (Gemini-561) was used for the in vitro selection and resulted in the dimeric o-Coral aptamer with enhanced performance for RNA imaging.74
The first example of a natural aptamer that was repurposed for the development of a fluorogenic RNA imaging platform was the cobalamin riboswitch.75 Cobalamin served as fluorescence quencher when appended to different fluorescent dyes, and binding to the RNA elicited fluorescence enhancement due to reduction of contact quenching. This strategy holds promise for engineering of differently coloured fluoromodules with high affinity. However, the relatively high background of the Riboglow platform is an inherent difficulty encountered in systems that rely on contact quenching in solution.
Riboswitch RNAs
Riboswitches are utilized by bacteria to sense metabolites and ions in order to regulate gene expression. To date, riboswitches that respond to nearly fourty distinct small ligands have been discovered and experimentally validated.76,77 For most of them, X-ray crystallography has revealed the three-dimensional structures of their ligand-binding aptamer domains, shedding light on the enormous diversity of RNA folds and RNA recognition patterns.78–80 The first riboswitch validation reports date back to 200281–84 and covered riboswitches that are very common and are present in bacteria from nearly all lineages. They respond to thiamine pyrophosphate, cobalamine, adenine, guanine, S-adenosylmethionine, glycine, lysine, cyclic diguanylate, and many more. Other riboswitches are exceedingly rare and appear in only a few species. Nearly all of the currently known riboswitches have been identified by a single laboratory (Ronald R. Breaker). They predict that there are potentially many thousands of distinct bacterial riboswitches remaining to be discovered.76 Recent articles on riboswitch diversity and distribution cover biocomputational aspects,76,77 several reviews summarize their structural concepts,78–80 and some focus on latest developments to use riboswitches as sensor tools in biotechnological and biomedical applications.10,14,85–90Here, we concisely focus on the most recent additions to riboswitches (Fig. 3) and on a few reports that aim at utilizing riboswitch scaffolds to engineer ligand specificities and chemical reactivity towards systems that function as riboswitch–ribozymes.
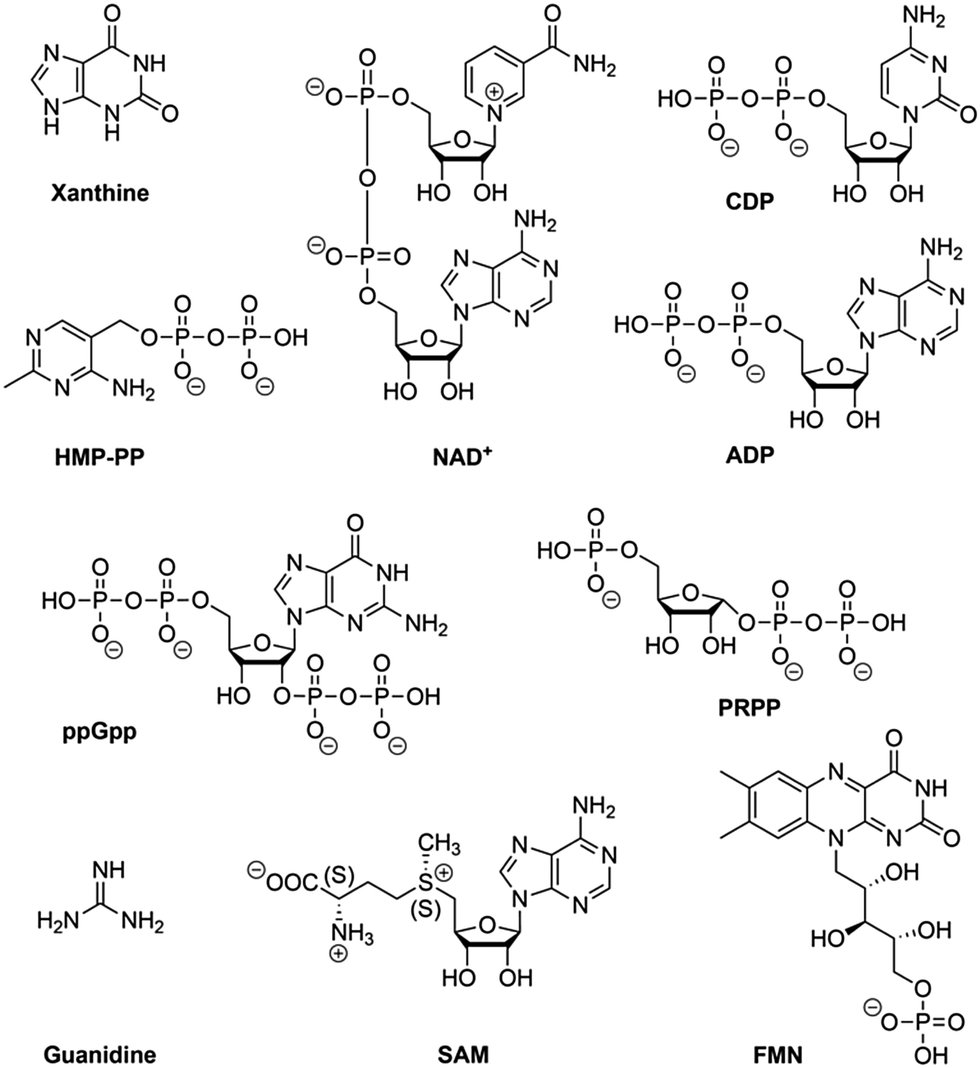 | ||
| Fig. 3 Ligands of recently discovered bacterial mRNA riboswitch classes. | ||
Recent additions to natural riboswitch systems
In this context, we stress that the NAD+ aptamer discriminates NAD+ over NADH by a factor of about two, while NADP+ is not binding to this RNA. The assignment to NAD+ as cognate ligand was based on the higher cellular concentration of NAD+ (∼2.6 mM) compared to NADH (∼120 μM) in bacteria.92 Furthermore, it cannot be excluded that a riboswitch exists for NADP as well, however, such an RNA motif is likely distinct from the NAD+ motif because the 2′-OH is tightly recognized in the pocket by H-bonding93 and a 2′-OPO32− group (as present in NADP+) would cause steric interference.
Riboswitch goes ribozyme – and an outlook on non-protein RNA methyltransferases
Many riboswitches bind protein enzyme cofactors that can catalyse or mediate chemical reactions.115,116 Thus far, however, only one RNA system (glmS riboswitch–ribozyme) has been identified in nature that utilizes a small molecule – namely glucosamine-6-phosphate – to participate directly in reaction catalysis (phosphodiester cleavage).117 We wonder why not more such examples have been found and what we have to undertake to disclose these likely existing cellular activities that could be more widespread than currently imagined. Moreover, in the hypothetical RNA world,118–122 ribozymes with the capability to conduct more diverse reactions in a primitive cell might have taken the role of today's protein enzymes. Such ribozymes could have evolved from riboswitches, thereby utilizing the aptamer binding pockets for proper positioning of substrates and cofactors. A recent study delineates such a pathway for the evolution of an aminoacylation ribozyme which could be part of a primitive translation system.123Building selection strategies on natural riboswitch scaffolds could also impact the field of ‘aptazymes’,14,126,127 namely the development of allosteric RNA devices that currently predominantly rely on de novo selected small molecule aptamers (e.g. theophylline, tetracycline, etc.) with adjoining self-cleaving small ribozymes of natural origin (hammerhead, HDV, twister, pistol, etc.).128–130 Such allosteric RNA devices are increasingly being regarded as effective tools for monitoring enzyme evolution, optimizing engineered metabolic pathways, modulating splicing, facilitating gene discovery, and as regulators of nucleic acid–based therapeutics, including next-generation gene therapy.
Due to the limited understanding of context-dependent structure–function relationships, the identification of functional riboswitch devices can be significantly advanced by large-scale-screening of aptamer-effector-domain designs. The latter is often impeded by the lack of appropriate cellular high-throughput methods.
RNA harbours all structural and functional requirements to bind small molecules and to catalyse more complex chemical reactions beyond phosphodiester cleavage. In particular, the identification of RNA methylation tools seems to be at hand, however, this has not yet been demonstrated, neither by in vitro selection, nor by identification of an existing cellular system (Fig. 4). Finding evidence for both would have a tremendous impact on the RNA world hypothesis and prebiotic chemistry, on RNA epitranscriptomics, and on biotechnological RNA tool development.
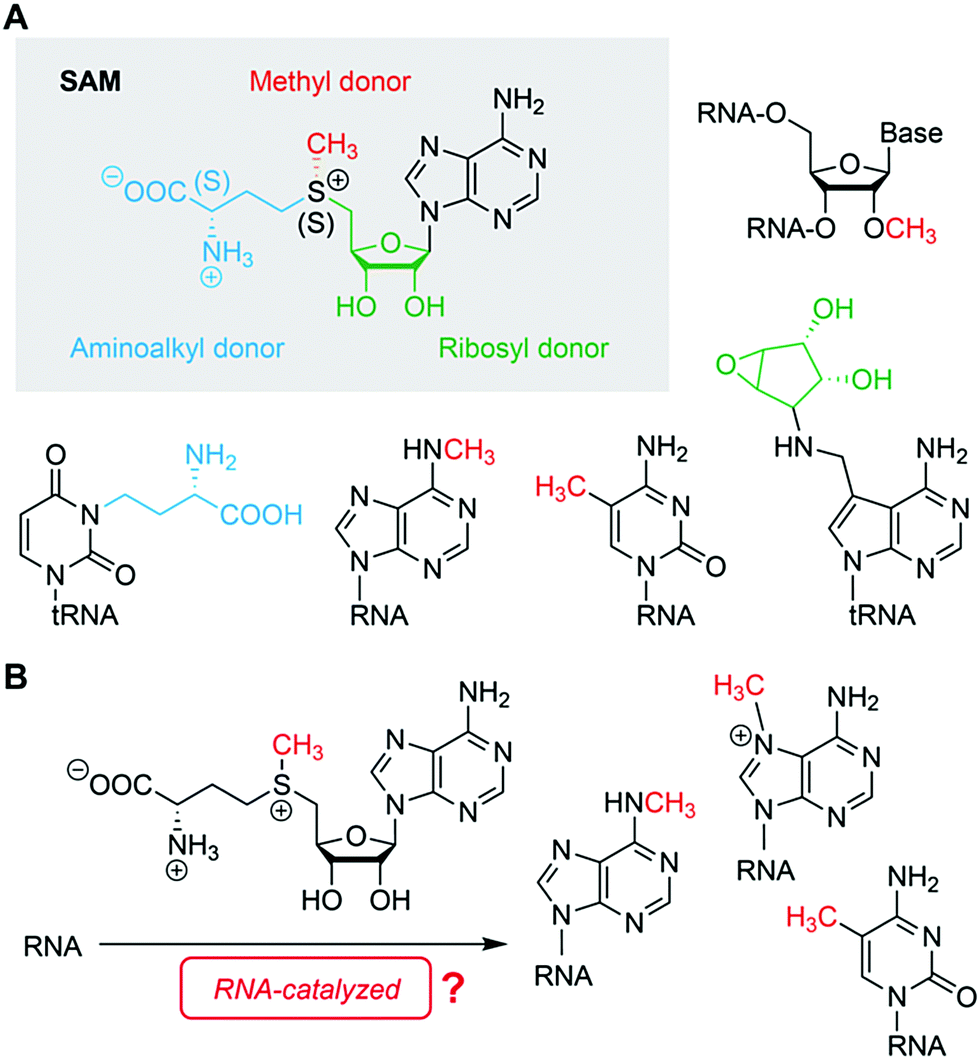 | ||
| Fig. 4 S-Adenosylmethionine (SAM). (A) Chemical structure of the cofactor and known reactivities (grey shadow) and protein-enzyme mediated reaction products of SAM with RNA targets. (B) Hypothesis for RNA-catalysed methylation reactions using SAM as cofactor in a prebiotic RNA world. | ||
Catalytic nucleic acids – ribozymes & DNAzymes
Both RNA and DNA can fold into structures that catalyse chemical reactions. For naturally occurring systems, these reactions mostly concern RNA phosphodiester cleavage and ligation, besides peptide bond formation in the peptidyl transferase center of the ribosome,136 while no examples for naturally occurring catalytic DNAs are known thus far.137,138 With respect to in vitro selected RNA and DNA, the scope of reactions that can be catalysed is much broader, including N7 alkylations (mentioned above),132,135 aminoacylations,26,139,140 Michael additions,141 Diels–Alder reactions,142,143 and many more.137,144Below we will focus on recent developments in the field of phosphodiester-cleaving and -forming ribozymes and DNAzymes only.
Small self-cleaving ribozymes
The first ribozymes were discovered by Thomas Cech and Sidney Altman in the eighties and this finding changed the paradigm that proteins were the sole catalytic molecules in living organism. Earlier, RNA was considered to be information carrier (messenger RNA, mRNA), structural scaffold of the ribosome (ribosomal RNA, rRNA), and interface between them as amino acid transporter and decoder (transfer RNA, tRNA). RNA's functions, however, go well beyond these tasks, and ribozymes represent one important class of naturally occurring functional RNAs. Most of them catalyse RNA backbone cleavage or the converse, RNA ligation. Ribozymes can be grouped into splicing ribozymes and cleaving ribozymes,145 with the latter divided further into trans-cleaving ribonuclease P,146 and small self-cleaving (or ‘nucleolytic’) ribozymes. To date, nine distinct small self-cleaving ribozyme classes have been described, including hairpin,147–150 hammerhead,151–155 hepatitis delta virus (HDV) and HDV-like motifs,156–159 glucosoamine-6-phosphate synthase (glmS),117,160,161 and Neurospora Varkud satellite (VS).162,163 More recently, twister,164 twister sister,165 hatchet,165 and pistol165 motifs have been added.Concerning the biological function of small self-cleaving ribozymes in living systems, rather little is known.166 For instance, HDV ribozymes promote their own cleavage from the transcript during rolling circle replication of the hepatitis delta virus. Also hairpin and VS ribozymes are connected to viral genome replication.167 The function of hammerhead ribozymes with thousands of representatives in diverse organisms is less clear. A role during pre-mRNA biosynthesis is conceivable but other putative biological functions are mainly linked to the genomic context and hence remain speculative.166
Numerous chemical, biochemical, and biophysical studies have been performed, that can be compared and contrasted with structural studies of most ribozymes, usually based on X-ray crystallography.166–173 Small self-cleaving ribozymes conduct site-specific internal transesterification which involves the ribose 2′-hydroxy group adjacent to the scissile phosphate. Through a SN2-type mechanism, a penta-coordinated phosphorane transition state is passed,174 and finally results in two products. The product RNA 5′ of the scissile phosphate retains this phosphate in form of a 2′,3′-cyclic phosphate while the 3′ RNA product obtains free hydroxyl termini. To support the reaction, the ribozyme can engage up to four potential strategies (Fig. 5).175–177 First, the 2′-oxygen, phosphor, and 5′-oxygen atoms should ideally lie on a straight line for nucleophilic attack (α catalysis);178 second, the enhanced negative charge on the nonbridging phosphate oxygens in the transition state should be electrostatically compensated (β catalysis); third, the proton from the attacking 2′-OH nucleophile should be removed (γ catalysis – general base catalysis); and fourth, the developing negative charge on the 5′ oxygen leaving group should be neutralized by donating a proton (δ catalysis, general acid catalysis) (Fig. 5). Recently an advanced ontology was developed for discussion of these strategies that refines this established framework into primary, secondary, and tertiary contributions to enable a more precise description of the reaction mechanism with respect to structure and bonding.179
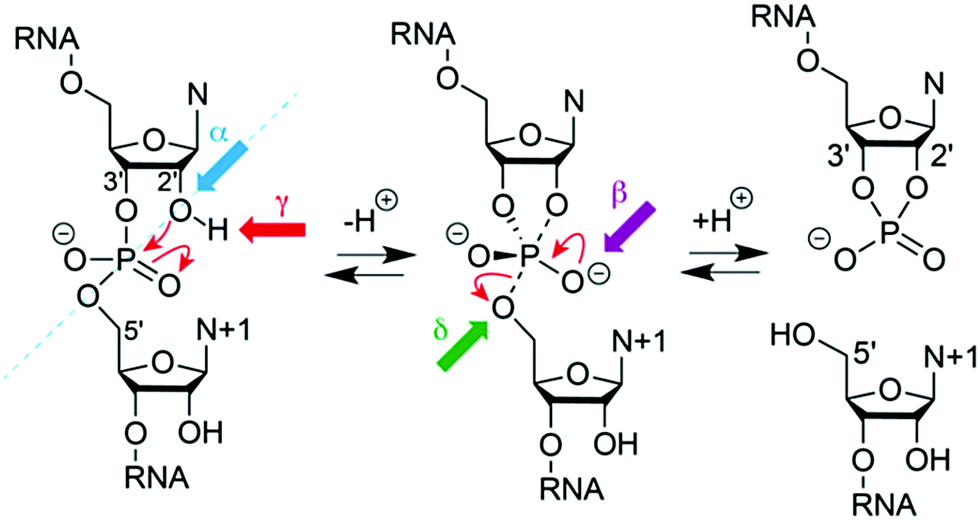 | ||
| Fig. 5 Model for phosphodiester cleavage in small self-cleaving ribozymes.180 The to-be-cleaved (“scissile”) phosphate is proposed to form a pentacoordinate transition state by SN2-like in-line attack of the nucleophilic 2′-hydroxy group (α catalysis, blue); thereby, neutralization of the (developing) negative charge on nonbridging phosphate oxygen atoms (β catalysis, purple) may support, as well as deprotonation of the 2′-hydroxy group (γ catalysis, red) and/or neutralization of negative charge on the 5′-oxygen atom by protonation (δ catalysis, green). The 5′ cleavage product carries the original phosphate in form or a 2′,3′-cyclic phosphate. | ||
Moreover, the cleavage of the phosphodiester bond is not necessarily concerted. The transesterification process can proceed stepwise involving transition states that are ‘tight’ and asynchronous.174,181 The individual architectures of the active sites encountered in distinct ribozyme classes determine the reaction pathway and transition states and these features are responsible for the broad range of cleavage rates, spanning several orders of magnitude (<0.1 h−1 to >1 s−1).
For protein-based enzymes, the structurally distinct side chains of 21 amino acids provide sufficient diversity to contribute to reaction catalysis.176 In contrast, the four nucleosides in ribozymes offer a limited scope of functionalities that affects electrostatic catalysis and general acid–base catalysis at neutral pH. Due to the fact that ionization of the nucleobases and the 2′-OH of the ribose requires rather harsh acidic or basic conditions, the first reports thus suggested ribozymes to rather be metalloenzymes with the RNA serving as scaffold to place hydrated Mg2+ ions as effectors.182,183 Later, it was found that many self-cleaving ribozymes are functional in the absence of divalent metal cations. Experimental evidence was collected for pKa-shifted active site nucleobases that participate directly in catalysis in some ribozymes, and nowadays, the general acid–base catalysis is a widely accepted concept for rate enhancement in small self-cleaving ribozymes.167,184 Nevertheless, direct effects of pH and metal ions on ribozyme catalysis are usually difficult to distinguish from indirect effects on folding (and fold stabilization) of a ribozyme and require advanced and careful experimentation.176
In the years 2014 and 2015, the ribozyme field was reinvigorated by two studies describing the identification of four novel ribozymes, termed twister,164 twister sister,165 pistol,165 and hatchet.165 These discoveries initiated comprehensive investigations applying modern experimental and computational approaches to further increase our understanding of ribozyme-mediated catalysis.
Twister ribozyme
Among the four most recently discovered ribozymes, twister has been investigated most intensively by biochemical,164,165,185 structural,186–189 and chemical189 approaches providing insights into the topological constraints contributing to catalysis. The clear distinctions observed in the active site and the P1 segment of the available crystal structures left room for miscellaneous interpretations and speculations. In particular, the observation of Mg2+ ions in inner-sphere coordination to the scissile phosphate in two of the structures caused debates on their relevance for catalysis (Fig. 6).190–192 Analysis of thio effects and metal ion rescues for phosphorothioate substrates assigned them a minor role.189,193 Key functions, however, can be attributed to conserved nucleosides in the active site, namely a guanine and the adenine directly at the cleavage site, both involved in general acid–base catalysis. Interestingly, for the adenine, the crucial site of protonation in its role of δ catalysis was narrowed down to the N3 position in the architectural framework of the ribozyme, distinct to the usually preferred N1 protonation site of adenosine.189,193,194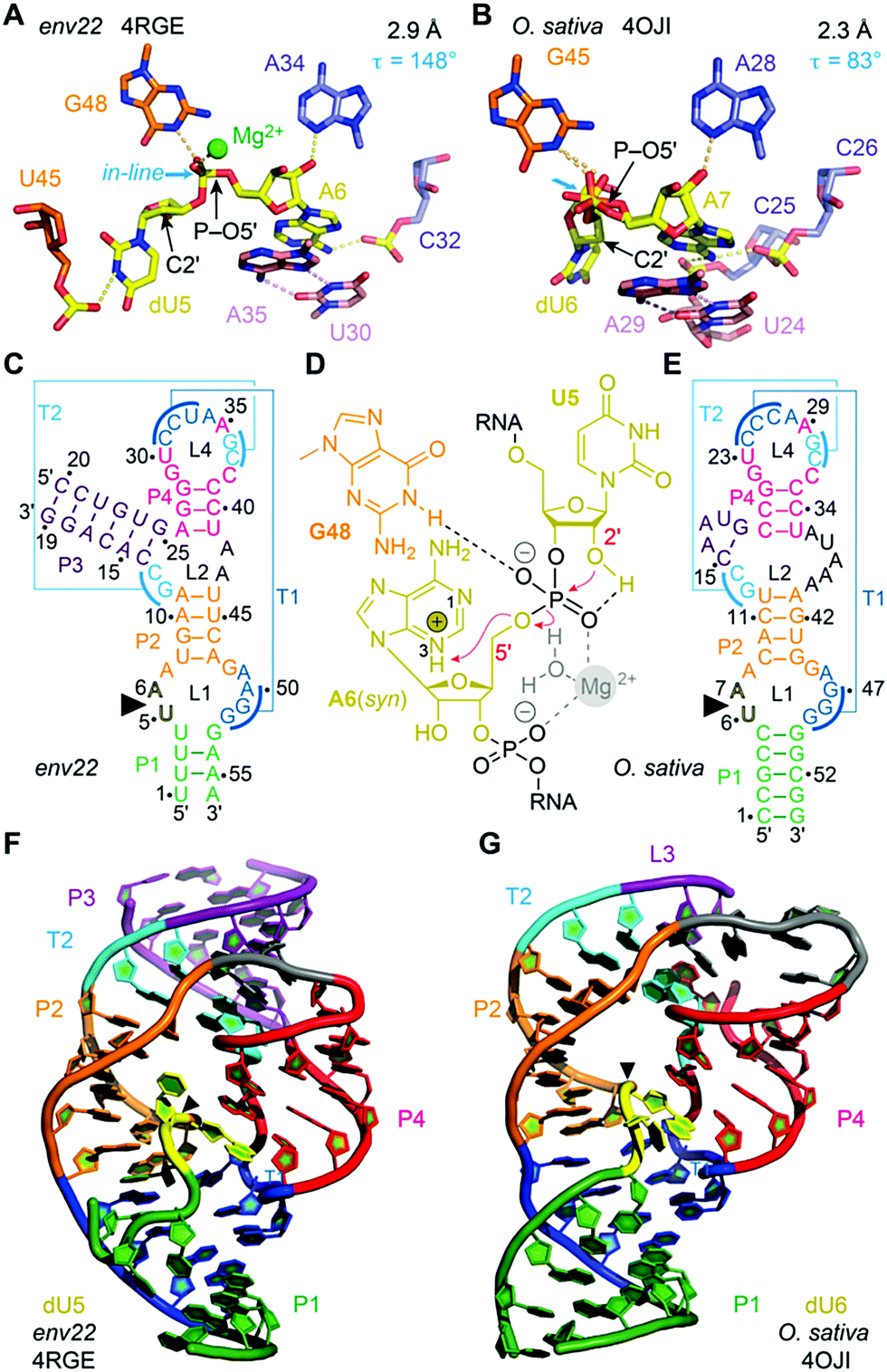 | ||
| Fig. 6 Twister ribozyme. (A) Pocket for dU–A active site in the 2.9 Å resolution structure of the env22 twister ribozyme with emphasis on the position of the C2′ of dU relative to the P–O5′ bond. (B) Pocket for dU–A active site pocket in the 2.3 Å resolution structure of the O. sativa twister ribozyme. (C) Secondary structure model of env22 ribozyme. (D) Current model for the twister cleavage mechanism; major roles G48 (γ catalysis) and A6 (δ catalysis via N3), minor role: hydrated Mg2+. (E) Secondary structure model of O. sativa ribozyme. (F) Three-dimensional structure in cartoon presentation of the dU modified env22 ribozyme and (G) the dU modified O. sativa ribozyme. Note the differences in segment P1 (green): ‘back-folding’ of nucleosides (U1 and U4) of segment P1 for env22 to form base triplets, and fully Watson–Crick base-paired stem P1 for O. sativa, respectively. Black triangles indicate the cleavage positions. | ||
Another insightful perspective was provided by recent studies that analysed the δ catalysis role of this adenine via chemical rescue.195,196 Using inhibited twister ribozyme variants with 1- and 3-deazaadenosine modifications, the authors observed significant chemical rescue effects in the presence of small molecules such as imidazole and histidine. Brønsted plots for the twister variants supported a model in which small molecules rescue catalytic activity via a proton transfer mechanism, suggesting that the conserved adenosine directly at the cleavage site in the wild type is involved in proton transfer, most likely general acid catalysis. Additionally, it was demonstrated that an 8-bromoadenosine modified twister ribozyme is faster than a native counterpart, supporting crystallographic data that show that this adenosine is in syn conformation when conducting proton transfer.
A further peculiarity of twister is that it retains wild-type activity when the phylogenetically conserved stem P1 is deleted, able to cleave a single nucleotide only.189 This feature was already insinuated by the crystal structures of twister, with two of them showing a fully base paired P1 stem (Fig. 6G) whereas two others showed disrupted base pairs instead, with two of the nucleobases participating through fold back in formation of stacked base triples (Fig. 6F).190,192 A single-molecule FRET study on twister ribozyme folding rationalizes this behaviour by revealing that the active site-embracing pseudoknot fold is preserved in the P1-lacking ribozyme and in the cleaved 3′-product RNA where P1 cannot form (Fig. 7).197 The rigid fold of the cleaved 3′-fragment that retains its compacted pseudoknot fold despite the absence of stem P1 is furthermore consistent with the poor turnover efficiency of the twister ribozyme. Taken together, the phylogenetically conserved stem P1 is not only dispensable for cleavage activity but also for folding into the active structure. The reason for P1 conservation remains unclear and very likely concerns features other than chemical mechanism and formation of the cleavage-competent ribozyme fold.
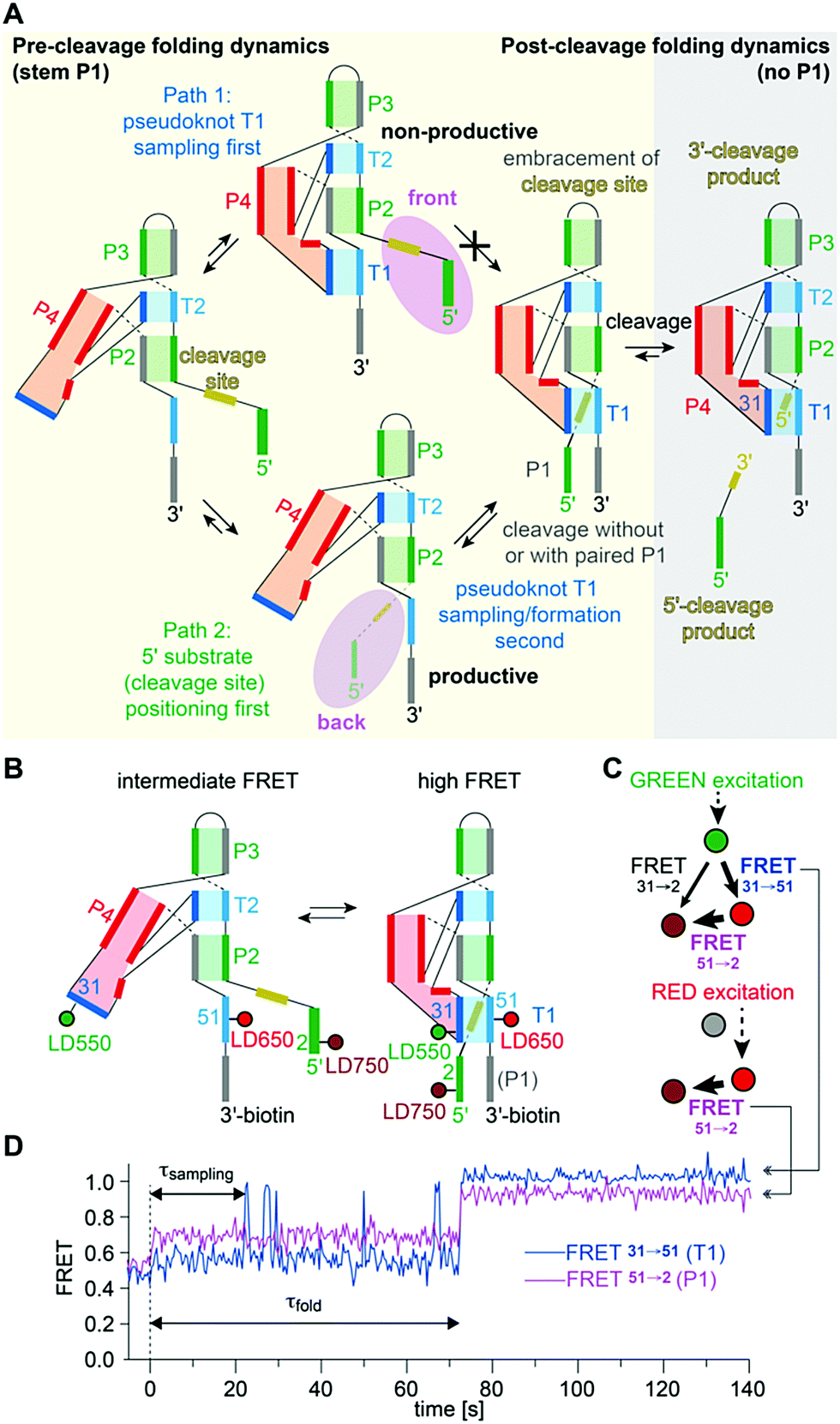 | ||
| Fig. 7 Twister ribozyme. (A) Alternative folding pathways to achieve the catalytically active pseudoknot fold as derived from a comprehensive 2- and 3-color FRET study based on a series of probes with distinct label positions.197 (B) Labelling pattern and cartoon of anticipated dynamics. (C) Schematics of alternating laser excitation (ALEX) experiments. (D) Representative FRET time course after stopped-flow Mg2+ addition (dotted line indicates 2 mM final concentration). | ||
Finally, a series of computational studies have followed and further described this ribozyme, essentially supporting the above mentioned experimental findings and interpretations.198–201
Pistol ribozyme
For the pistol ribozyme, our understanding of the catalytic mechanism of phosphodiester cleavage has grown significantly over the past years (Fig. 8A). Initial structural and biochemical data hinted primarily at a general acid–base mechanism involving purine nucleobases in the active site.165,202–204 Later, this view was expanded because intensive atomic mutagenesis-based assays shed light on the mechanistic role of divalent metal ions.205,206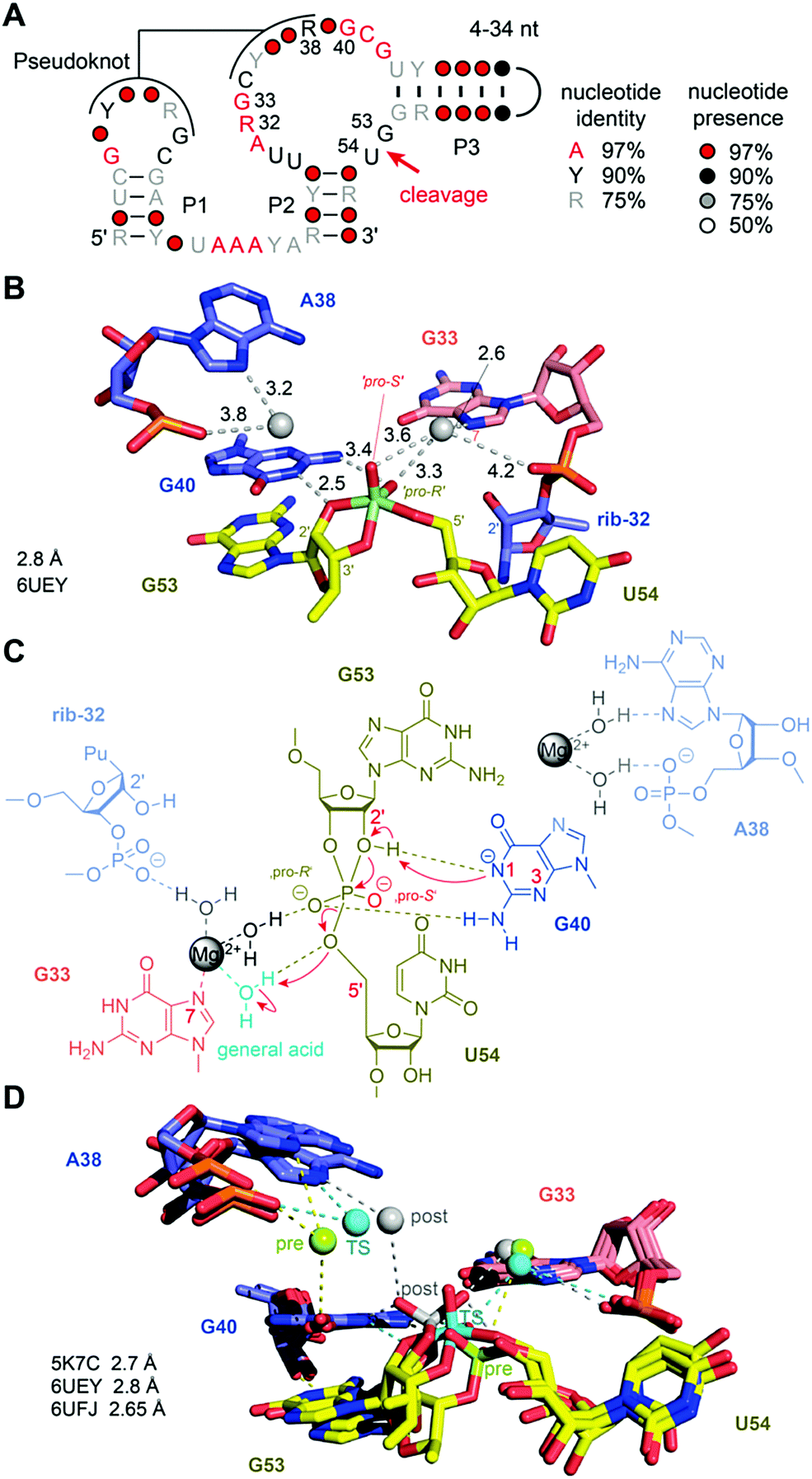 | ||
| Fig. 8 Pistol ribozyme. (A) Secondary structure model of the consensus sequence. (B) Pocket for active site G53–U54 observed in the crystal structure of a vanadate transition state mimic. Crucial atom distances (below 4.5 Å) that indicate direct or water-mediated hydrogen bonds and/or metal ion interactions are shown by dashed lines. The values in black are distances in Å. Note that the number of distances can exceed the number of possible hydrogen bonds for an atom. (C) Current model for pistol cleavage mechanism; major roles G40 (γ catalysis) and hydrated Mg2+ coordinated to N7–G33 (δ catalysis), minor role: hydrated Mg2+ coordinated to A38. (D) Superpositions of the pre-cleavage state (M2+ and scissile phosphate in green color), TS analogue vanadate (cyan), and post-cleavage state (grey), revealing the repositioning of the Mg2+ ion close to O6 of G40 in the pre-cleavage state towards the pro-S oxygen atom of the scissile phosphate in the cyclophosphate product. | ||
The first three-dimensional structures of the pistol ribozyme were available in 2016, solved by X-ray crystallographic methods with a resolution of 2.7 Å (env25 pistol)203 and 3.0 Å (env27 pistol),204 respectively. Both structures represent a precatalytic conformation that has been trapped based on mutants carrying a hydrogen atom instead of the native 2′ OH nucleophile at the cleavage site. Moreover, the two structures are in very good agreement with respect to the overall topology and the cleavage site alignments and implied nucleobase-mediated acid–base catalysis of the cleavage reaction,192 involving a guanine (G40) and seemingly, an adenine (A32) in the active site. Shortly after, a chemical study based on atomic mutagenesis demonstrated that a Mg2+ ion that is innersphere-coordinated to the N7 atom of another guanine in the active site (G33) plays a key role in catalysis (rather than A32).205 The disruption of this coordination site by N7-to-C7H mutation caused a 1000-fold decrease in cleavage rate. This was reconfirmed by an independent study.207
Very recently, additional crystal structures have been published, providing three-dimensional snapshots that now cover further states along the reaction coordinate of pistol phosphodiester cleavage, representing the transition state (in form of a vanadate mimic) (Fig. 8B) and the product (2′,3′ cyclophosphate).206 This broad structural foundation resulted in a profound proposal of the underlying chemical mechanism. Essentially, a hydrated Mg2+ ion remains innersphere-coordinated to the N7 nitrogen of G33 in all three states, which supports its proposed role as acid in general acid base chemistry (δ and β catalysis).206 Interestingly, a second hydrated Mg2+ ion approaches the to-be-cleaved phosphate from its binding site in the pre-cleavage state to stretch out for water-mediated hydrogen bonding to one of the non bridging oxygen atoms of the cyclophosphate product.206 The major role of the second Mg2+ ion appears to be stabilization of the product conformation.
Not long ago, the three-dimensional structure of a hammerhead ribozyme (HHRz) TS analogue was described.155 This vanadate complex revealed important rearrangements compared to the earlier obtained pre-cleavage HHRz structures. The active site contracted, placing a guanine (G10.1) closer to the cleavage site (Fig. 9). This guanine appears similar to G33 in the pistol ribozyme because it coordinates a divalent ion through N7 and a backbone phosphate (A9). Although the distance to the scissile phosphate is farther compared to the arrangement in pistol, the HHRz vanadate structure also suggested a stabilization of the transition state by water-mediated H-bonding with the scissile phosphate. A second divalent ion was found in innersphere coordination to O6 of a guanine (G12). This guanine fulfills the role of a general base in the hammerhead ribozyme active site with the metal ion tuning the pKa value to support deprotonation of the attacking 2′-OH of C17. Importantly, the deletion of O6 in a G12Ap mutant of the hammerhead ribozyme was responsible for a significant rate reduction (by a factor of 103),209 and this finding underlines the importance of innersphere coordination of the metal ion to this atom. Distinct to HHRz, the role of the second Mg2+ with respect to pistol cleavage acceleration is minor, however, it appears to stabilize the conformation of the cyclophosphate product in the complex.
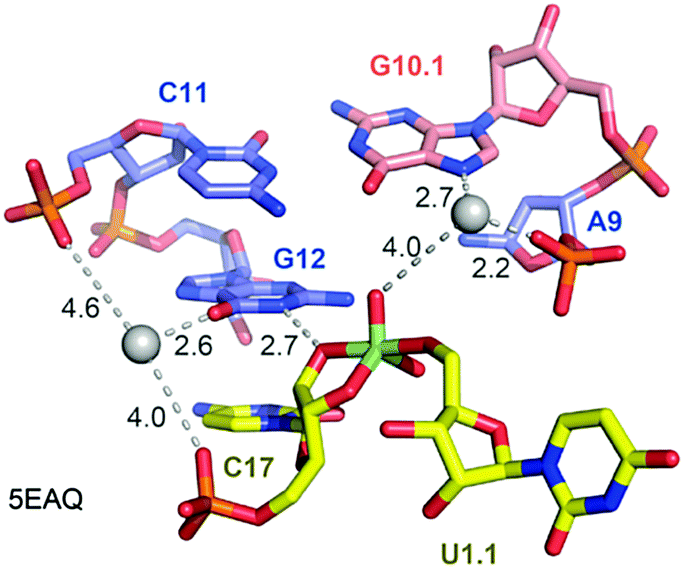 | ||
| Fig. 9 Hammerhead ribozyme. Pocket for active site C17-U1.1 (transition state mimic; vanadate) for purposes of comparison with the pistol ribozyme pocket. Crucial atom distances indicating possible interactions are shown by dashed lines. The values in black represent distances in Å. | ||
Twister-sister ribozyme
The twister-sister ribozyme adopts either a three-way or four-way junctional fold comprising internal and terminal loops with conserved residues resembling those in the twister ribozyme (Fig. 10A).165 Self-cleavage occurs between cytidine (C62) and an adenosine (A63) of an internal loop that is on the opposite side when compared to the twister ribozyme secondary structure model.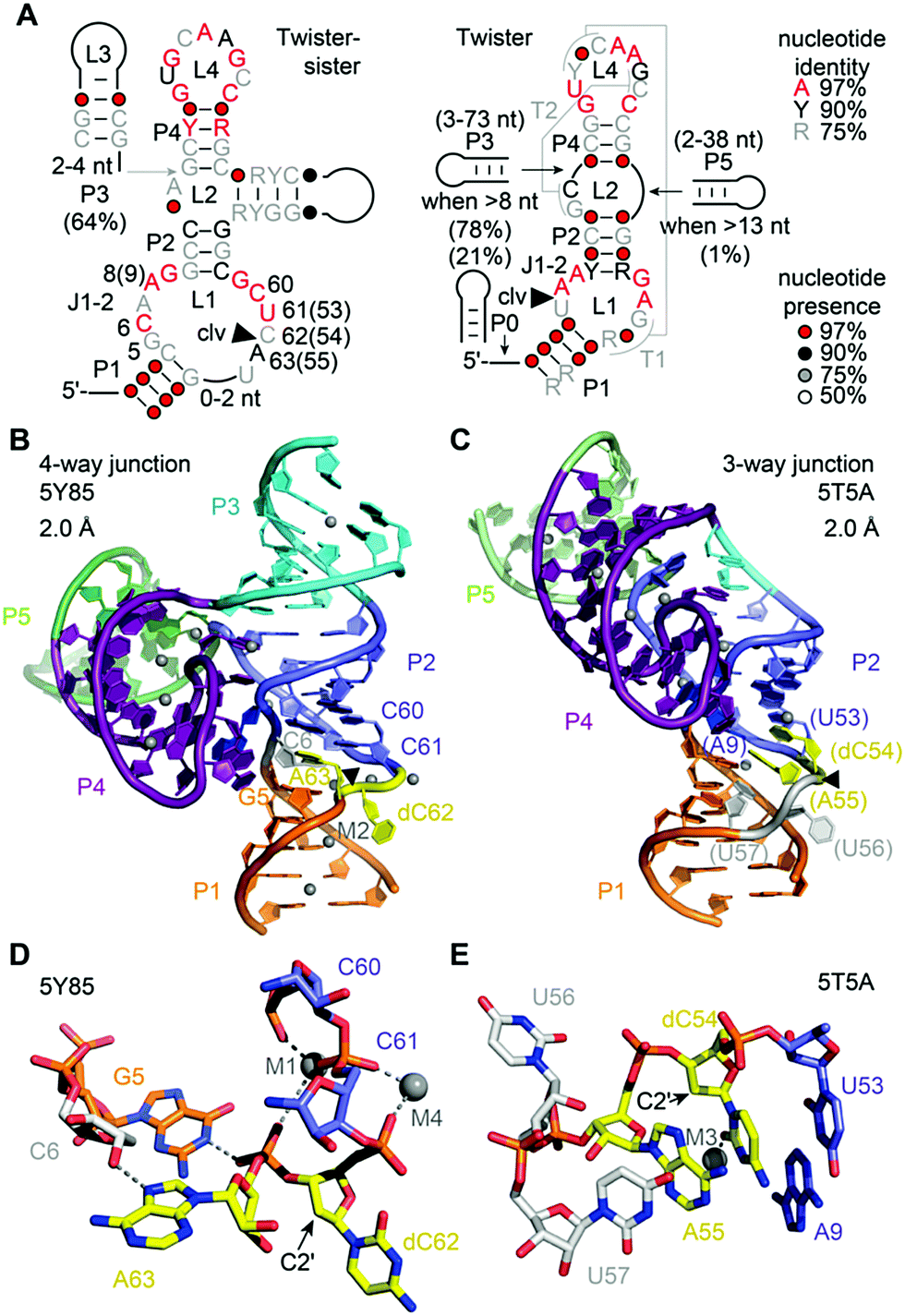 | ||
| Fig. 10 Twister-sister ribozyme. (A) Secondary structure model of the consensus sequence of twister-sister (left) and comparison to twister (right). (B) Three-dimensional structure in cartoon presentation of the dC modified 4-way junctional (4WJ) ribozyme and (C) the dC modified 3-way junctional (3WJ) ribozyme. Note the differences at the cleavage site (yellow): splayed apart (dC62 and A63) for 4WJ, and stacked in double helix (dC54 and A55) for 3WJ. Black triangles indicate the cleavage positions. (D) Pocket for dC–A active site in the 2 structure of the 4WJ ribozyme with emphasis on the G5–scissile phosphate interaction. (E) Pocket for dC–A active site pocket in the structure of the 3WJ ribozyme. | ||
To date only two structures exist of this ribozyme, representing the precatalytic state (Fig. 10B and C).210,211 Both structures display the terminal loop with a high number of conserved nucleotides projecting from a large continuous helical fold onto the minor groove of the partially zippered-up internal L1 loop segment associated with a second continuous helix (Fig. 10B and C). The two structures, however, show significant distinctions in the active site arrangement (Fig. 10D and E).
For the four-way junctional fold, the bases C and A at the cleavage site are splayed apart. The C is directed outwards and A directed inwards and held tightly within the ribozyme core (Fig. 10D).210 Remarkably, the modeled 2′-OH is far from adopting an ideal angle for in-line attack at the scissile phosphate. Importantly, the N1H of an active pocket guanine (G5) and an innersphere water of a hydrated Mg2+ cation (labeled M1 in Fig. 10D) form hydrogen bonds to the non bridging phosphate oxygens, fixating the scissile phosphate. In addition, a second hydrated Mg2+ (labeled M2 in Fig. 10B) is located in a way that an innersphere water is in hydrogen bond distance of the (modeled) 2′-O nucleophile of C. Mutations of the active pocket guanine and inwardly-oriented and anchored active site adenine abolish cleavage, while mutations of the flexible and outwardly-oriented cytosine have no influence on activity.
In the three-way junctional fold, the in-line conformation required for attack of the 2′-OH at the scissile phosphate is also not adopted (Fig. 10E).211 Interestingly, the 2′-OH nucleophile is within hydrogen bonding distance of an innersphere water of a hydrated Mg2+ cation (labeled M3 in Fig. 10E). Furthermore, the C and A are stacked, and C adopts a fixated and A a flexible arrangement in the three-way junctional structure,211 unlike to the alignments of C and A in the four-way junctional structure.210 Moreover, the oxygens of the scissile phosphate do not interact with a G and also not with a hydrated Mg2+ in the three-way junctional structure,211 as it is in the four-way junctional structure.210 These stark differences observed for the catalytic pocket of the pre-catalytic states attest the necessity for further studies that reveal the architecture of transition state mimics of the twister-sister ribozyme at high resolution to shed light on the chemical mechanism.
Based on the structure of the 3WJ twister-sister ribozyme, a model for the functional active state of the ribozyme from molecular simulation proposed either direct or Mg2+ mediated involvement of a cytosine (C7) in the active site pocket for the cleavage reaction.212
Hatchet ribozyme
The hatchet motif is unusual because its cleavage site is located at the very 5′ end of its secondary structure model.165,185,212 The only other ribozymes that are wholly downstream of their cleavage sites are the HDV family of ribozymes213 and the glmS riboswitch–ribozyme, keeping in mind that the latter is dependent on the cofactor GlcN6P for its activity.160,161The hatchet ribozyme sequence contains thirteen highly conserved residues interspersed amongst bulge elements linking stem segments but no obvious tertiary interactions could be deduced from the biochemical data alone.185,212 RNA strand scission in the hatchet ribozyme requires divalent Mg2+ cations. The maximum cleavage rate kobs was determined to approx. 4 min−1, reaching a plateau on increasing the pH to 7.5. Rate measurements on scissile phosphorothioate analogues suggested a crucial contact between the scissile phosphate and a functional group in the active site of the ribozyme.214 Moreover, the pH reactivity profile was determined and implied that the attacking 2′-OH is activated by a functional moiety that has a pKa value of about 7.214
To date, only one crystal structure of a hatchet ribozyme is available, representing the post-cleavage state (Fig. 11).215 The ribozyme crystallised as dimer with strand swap in the 3′-end region. Mutations in the 4 nt dimerisation site revealed that the monomeric form is fully active. The hatchet ribozyme comprises a pair of two parallel-aligned long helices P2–P1 (H12) and P3–L3–P4 (H34) (Fig. 11A). The long-distance interaction between terminal loop L1 and internal loop L3 identified in the tertiary fold, together with several bridged nucleotides in internal L2, appear to anchor the relative alignments of H12 and H34.
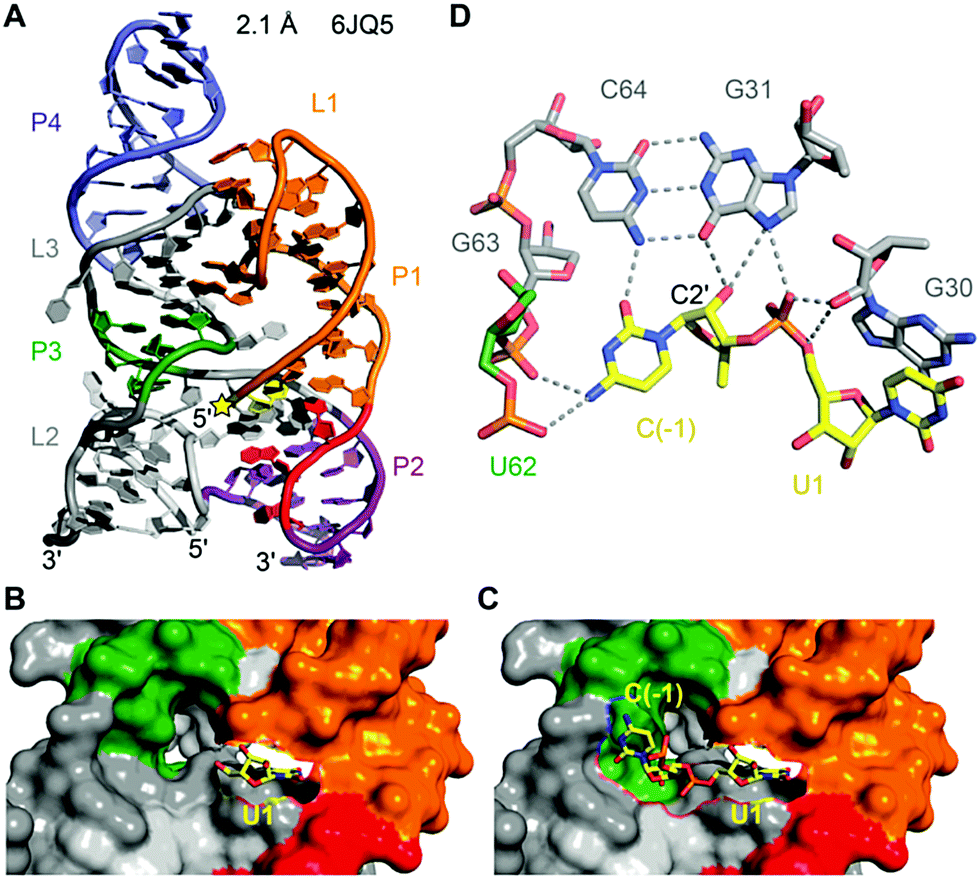 | ||
| Fig. 11 Hatchet ribozyme. (A) Three-dimensional fold in cartoon presentation of the ribozyme product (post-cleavage state). The site of cleavage is labeled with a yellow star. (B) The ribozyme product is shown in surface representation with U1 in sticks (yellow). A cavity is visible adjacent to the leaving group 5′-OH of U1 which seems of sufficient size to accommodate the scissile phosphate and a nucleoside 5′ of it. (C) Model of the hatchet ribozyme comprising the C(−1)–U1 cleavage site shown in sticks (yellow) and otherwise in surface representation. C(−1) was modeled based on the shape of the cavity on the hatchet ribozyme surface. (D) The proposed model for the precatalytic state of the ribozyme, in which C(−1) forms extensive hydrogen bonds with nucleosides and the phosphate backbone in the pocket. The modelled conformation is in-line and indicates functional groups of nucleotides that may contribute to catalysis. In particular, N7 of G31 is a candidate based on the finding that a 7-deaza-G31 mutant was inactive. | ||
Despite the cleavage site being located at the 5′ end of the sequence, it is positioned in the center of the tertiary structure of the hatchet ribozyme (Fig. 11A).215 Moreover, the dispersed conserved nucleotides in the secondary structure of the hatchet ribozyme are all brought into proximity and aligned around the cleavage site. Their alignments result in the generation of a pocket with the leaving group 5′-OH of U1 pointing toward the cavity (Fig. 11B). The dimensions of the pocket are capable of accommodating both the (modeled) C(−1) and the scissile phosphate linked to U1 (Fig. 11C). The C(−1) and U1 are splayed apart and the 2′-OH is ideally positioned for an in-line attack at the scissile phosphate (Fig. 11D). The model further highlights likely interactions of C(−1) and the to-be-cleaved phosphate with functional groups of nucleosides in the pocket which are potential candidates for general base and general acid catalysis. In particular, this concerns a G of a conserved G–C base pair (G31–C64) whose mutation to 7-deazaguanine renders the ribozyme inactive. To gain deeper insights into hatchet catalysis, structure determination of pre-cleavage and transition state mimics are needed.
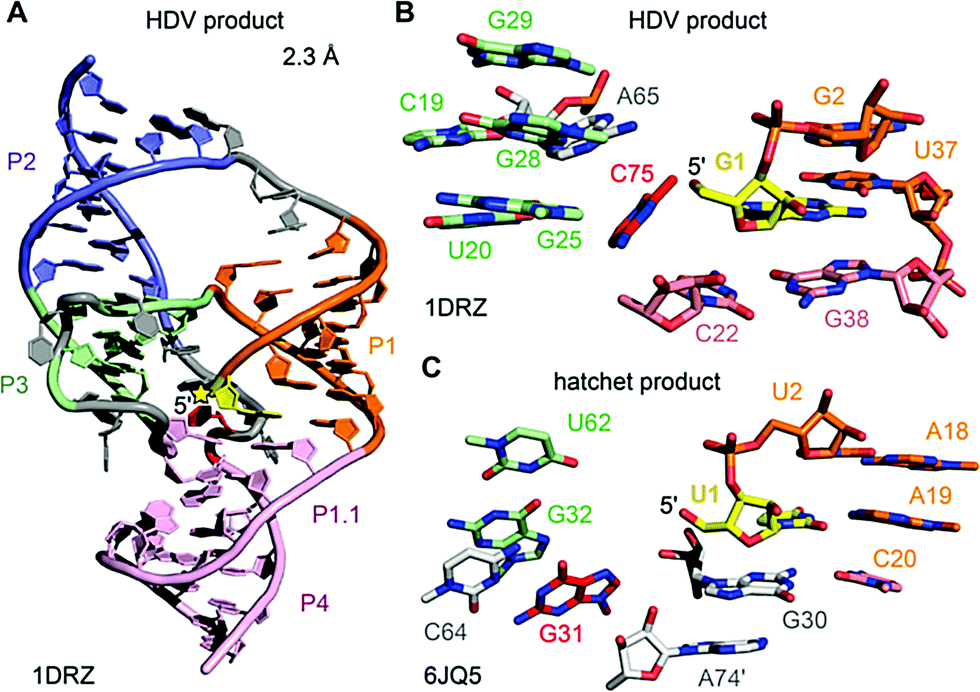 | ||
| Fig. 12 HDV ribozyme. (A) Three-dimensional fold in cartoon presentation of the HDV ribozyme product (post-cleavage state). The site of cleavage is labeled with a yellow star. (B) Pocket of the HDV cleavage site with G1 shown in sticks (yellow). C75 (red) acts as general acid in δ catalysis. (C) Pocket of the hatchet cleavage site with U1 shown in sticks (yellow). G31 (red) has been proposed to play a crucial role in catalysis. Its precise role remains to be determined. | ||
The first HDV ribozyme structure pointed at a nucleoside (C75) in its active site pocket that was later ascertained to play a key role in catalysis. The hatchet product structure now points at a specific guanine (G31) in its pocket. Whether this nucleoside participates in general acid–base catalysis has yet to be determined. In the product structures of both ribozymes, no Mg2+ ions were identified in the vicinity of the cleavage site. However, the crystal structures of the pre-cleavage state that followed for HDV RNA later on, revealed a Mg2+ ion in the pocket.216,217 Today the general view is that the protonated C75 of HDV is directly interacting with the scissile phosphate during the cleavage process,218 which is additionally supported by electrostatic interactions with the metal ion bound in the active site.216–220
RNA-cleaving deoxyribozymes
The first DNA enzyme with endoribonuclease activity was identified by Breaker and Joyce using in vitro selection in 1994.221 Since then, numerous examples of RNA-cleaving DNA enzymes have been reported, all of them derived from synthetic DNA libraries.222,223 Up to now, no natural catalytically active DNA has been found. RNA-cleaving DNA enzymes are practically useful tools for fundamental biochemical research,224 and deoxyribozymes have been explored as mRNA-targeting agents for the downregulation of disease-relevant genes (before the advent of other potent gene regulation approaches such as RNAi and CRISPR/Cas strategies).225 The largest and most diverse field of applications is found in bio- and nanotechnology, where DNAzymes are used as components of analytical devices for biosensors and logic gates.226–228 Recent reviews summarize diverse metal ion-dependent transesterification DNA enzymes and their applications.228–230 In many analytical applications, the substrate is a DNA strand with a single ribonucleotide incorporated, linked to a FRET pair, with donor and acceptor placed before and after the cleavage site.231 A DNA sequence with a single ribonucleotide was also used as a substrate for in vitro selections that aimed at generalizing the substrate sequence tolerance at the cleavage site.232,233 Nearly all 16 dinucleotide junctions can now be cleaved, but only few DNA enzymes were tested on all-RNA substrates. Compared to the single-ribonucleotide-containing substrate, significantly reduced cleavage rates and yields were observed.224 Nevertheless, interesting analytical applications have been reported, including quantitative analysis of Na+ ion concentrations in cells.234 Below, we summarize structural and mechanistic aspects of RNA-cleaving DNA enzymes, and discuss recently reported DNA enzymes for the detection of posttranscriptional RNA modifications.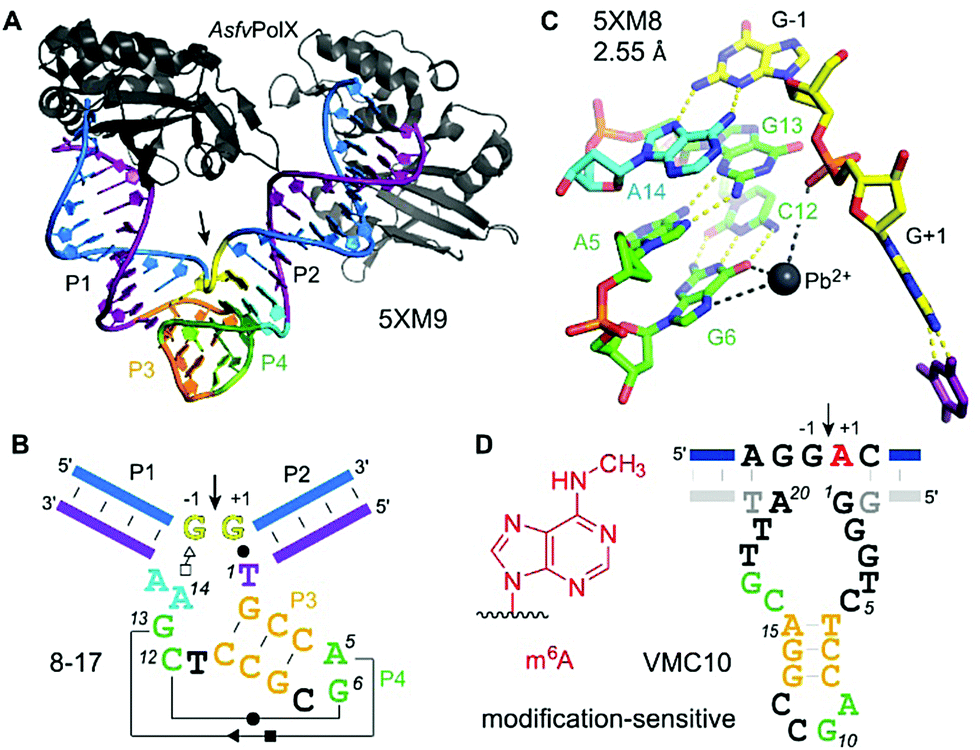 | ||
| Fig. 13 8–17 and VMC10 DNAzymes. (A) Tertiary structure and (B) secondary structure overview of RNA-cleaving 8–17 DNAzyme (crystallized with AsfvPolX protein, grey), GG kink at the cleavage site in yellow. (C) Excerpt of the catalytic core showing the G13 acting as a base for deprotonation of 2′-OH (missing in the structure) and Pb2+ coordinating to G6. (D) Secondary structure of m6A-sensitive VMC10 DNAzyme.240 Features suggested to resemble similar functions compared to 8–17 are shown in matching colors to (B). | ||
The structure revealed a surprising twisted pseudoknot in the catalytic core, with long-range base-pairing interactions that could not be predicted from a large set of biochemical data.222,236 The pseudoknot contains very short base-paired regions, of 3 and 2 bp respectively. The 2 bp interaction consists of the Watson–Crick base pair G6–C12 and a non-canonical Hoogsten-sugar edge base pair A5–G13. The pseudoknot architecture positions the guanine G13 to serve as a general base to activate the 2′-OH by proton abstraction (not present in the structure), consistent with biochemical data.238 Divalent metal ions are essential for activity of 8–17, with Mg2+ and Mn2+ as the most common cofactors for practical use.224 Interestingly, Pb2+ has been shown as the most active metal ion for 8–17 activation.239
The structure of 8–17 was solved with Pb2+, and a partially occupied metal ion binding site was found that suggests interaction with O6 and/or N7 of G6 and with the non-bridging phosphate oxygen atoms, likely via water-mediated contacts in the hydration shell. Evidence for general acid–base catalysis was also established by (bio)chemical atomic mutagenesis data,238 and analysed in detail in combination with molecular dynamics (MD) simulations.241,242 Such investigations may reveal the degree of shared mechanistic features between artificial RNA-cleaving DNA catalysts and naturally evolved ribozyme counterparts. However, the MD simulations were carried out with Pb2+, and it is currently unclear if the results are transferable to the more biologically relevant Mg2+. This question is of special interest, since FRET analyses revealed different folding landscapes with different metal ions.243,244 Thus, future structures of the 8–17 DNAzyme in complex with an RNA substrate strand carrying a 2′-OMe or 2′-F modified nucleotide at the cleavage site, and in the presence of Mg2+ are highly desirable. Along the same lines, it will be interesting to study the structural features of Na+-specific DNAzymes, for which a single point mutation has recently been described to result in switching the catalytic mechanism from general base to general acid catalysis.245
The 2′-O-methylation of rRNA was the first posttranscriptional RNA modification studied by RNA-cleaving deoxyribozymes.246 For mechanistic reasons, methylation at the cleavage site inhibits the DNA enzyme, and the methylated RNA remains intact. In contrast, nucleobase modifications 5-methylcytidine (m5C), 5-methyluridine (m5U), and pseudouridine (ψ) in tRNA did not interfere with DNA-catalysed RNA cleavage. Both modifications barely influenced cleavage rate and yield of 10–23 DNAzymes, and were detected at the 5′-terminus of the downstream fragment by TLC and LC-MS methods.247,248 However, 10–23 is limited to cleaving purine|pyrimidine (R|Y) junctions, and thus is not useful for the analysis of modified purine nucleotides. Selective DNA enzymes that result in distinct cleavage events for modified RNA and unmodified RNA would simplify the analysis. The first RNA-cleaving DNA enzyme that showed accelerated cleavage of modified RNA was shown to distinguish N6-methyladenosine (m6A) from unmodified adenosine at the cleavage site.240 Surprisingly, better selectivity was obtained in the opposite direction, i.e. it was easier to find DNA enzymes that are strongly inhibited by m6A. The VMC10 DNA enzyme (Fig. 13D) efficiently cleaved unmodified RNA and left methylated RNA intact. Such enzymes are useful for the enrichment of sparsely methylated RNA, and can be used to validate predicted m6A sites in isolated cellular RNA.145,249
Besides the development of tools for epitranscriptomic research, it is of fundamental interest to understand if and how DNA enzymes distinguish different methylation/modification states of the target RNA. In the absence of any crystal structure, the responsible supramolecular contacts remain speculative. While m6A-specific reader proteins recognize m6A via a methyl–π interaction with heteroaromatic amino acid side chains (such as the tryptophan cage in YTH proteins),250 no analogous interactions are known in RNA/DNA-only structures. However, preferred structural motifs are known in RNA, in which m6A together with Mg2+ has a stabilizing effect,251 and others in which m6A modulates base-pairing patterns,252–254 or RNA annealing kinetics.255 In the DNA active site of VMC10, it is thus conceivable, that the methyl group of m6A interferes with an essential H-bonding interaction within the core of the deoxyribozyme and thus inhibits catalytic activity. Interestingly, the predicted VMC10 secondary structure shares features reminiscent of 8–17 (Fig. 13). However, no interference of m6A can be predicted from the orientation of the nucleotides around the cleavage site.237 Likely, additional contacts are formed between the RNA substrate and the guanine-rich bulge between the predicted P2 and P3 stems.
Compared to m6A, much stronger activating effects on DNA enzymes were found with a larger nucleobase modification, N6-isopentenyl adenosine (i6A).256 The prenylated adenosine is a natural tRNA modification that is conserved in certain tRNAs in bacteria and eukaryotes,257 and the hydrophobic nature of i6A was suggested to facilitate its association to membranes.258 It remains to be determined which type of non-covalent interactions are most influential to explain the pronounced preference of 2500-fold faster cleavage of i6A RNA compared to unmodified RNA.256
The findings on m6A and i6A-selective DNAzymes spur further interest in the development of DNA catalysts targeting other RNA modifications. For example, it would be quite spectacular to evolve DNAzymes that can discriminate the three natural singly methylated cytidine isomers m3C, m4C and m5C from unmodified cytidine. Ideally, individual DNAzymes should selectively cleave only one out of the four targets. Since none of the previously reported 8–17 analogues is particularly good at cleaving N|C junctions in all-RNA substrates,232,259 advanced in vitro evolution strategies need to be developed to overcome the tyranny of the 8–17 motif.236 Deep sequencing analyses of enriched selection libraries with in situ analyses of cleavage activity and selectivity26,260 may lead to the identification of new catalytic DNA motifs for the site-specific interrogation of difficult-to-cleave RNA substrates and isomers of posttranscriptional RNA modifications.
DNA-cleaving DNA enzymes
In contrast to RNA transesterification, the site-specific cleavage of DNA is more challenging to achieve, because the internal nucleophile for attack at the scissile phosphate is missing. Several different cleavage mechanism were employed by DNA-cleaving DNA enzymes, including Cu2+-mediated oxidative cleavage, and strand breaks at apurinic sites after DNA-catalysed deglycosylation.261 A serendipitous discovery of a bimetallic deoxyribozyme that required Zn2+ and Mn2+ for hydrolytic phosphodiester cleavage activity, generated well-defined DNA fragments with impressive rate enhancements and multiple turnover.262 Further selections in the presence of different metals produced DNA-cleaving sequences that required only Zn2+,263 or were reactive with low concentrations of lanthanides.264 An elegant selection strategy involving circular DNA revealed generally applicable Zn2+-dependent DNA-cleaving DNAzymes,265,266 that were engineered for mass production of DNA origami.267 Deoxyribozyme pairs were constructed into self-cleaving cassettes that enabled the generation of all necessary single strands for several DNA origami structures from large-scale bacteriophage cultures.Nucleic acid-catalysed formation of phosphodiester bonds in RNA
Formally, the reverse of phosphodiester cleavage, is the formation of a new 3′–5′ linkage between two RNA fragments. This connection can be furnished by different mechanisms. The microscopic reversibility of the cleavage by transesterification, initiated by the 2′-OH attack on the scissile phosphodiester, suggests that regioselective opening of the cyclic phosphate by attack from the 5′-OH results in regiospecific ligation. In the laboratory, this reaction is employed by the twin ribozyme for RNA labelling and mutagenesis.268 The twin ribozyme is an engineered version of the hairpin ribozyme found in the satellite RNA of tobacco ringspot virus, and thus it is a variant of the small nucleolytic ribozymes described above.In contrast to opening of 2′,3′-cyclophosphates, an alternative practically useful ligation reaction involves the 3′-OH as the nucleophile and an activated 5′-phosphate as the electrophile. Most commonly, 5′-triphosphates are used, but 5′-adenylates, nicotinamides, or substituted 5′-phosphorimidazolides have also been employed.269–271 Fundamentally, this reaction resembles the elongation step catalysed by RNA polymerases, and is thus also of great interest in the context of origin of life research and the RNA world, in which an RNA polymerase ribozyme would have been able to copy functional RNA molecules.118,272
RNA-ligating ribozymes & polymerase ribozymes
In contrast to the detailed knowledge available on RNA/DNA-catalysed RNA cleavage reactions summarized above, very little is known on structures and mechanisms of RNA ligase and RNA polymerase ribozymes. The crystal structures for two types of RNA ligases were reported between 2007 and 2011,273–275 but since then no additional structures have become available of any other ligase or polymerase ribozymes.The class I ligase276 and the L1 ligase277 share the arrangement of the ligation substrates in a discontinuous double helix, firmly hybridized to a complementary template strand (Fig. 14). An engineered version of the class I ligase is still the fastest ribozyme described today with a kcat of 100 min−1.274 The crystal structures of the pre- and post-catalytic states reveal complex catalytic strategies. In the pre-catalytic state of the class I ligase, the 5′-triphosphate takes an unexpected orientation in the major groove of the primer–template duplex.273 The 3′-OH nucleophile is positioned for in-line attack with the help of a Mg2+ ion, and interactions with nucleobase and ribose functional groups also play important roles.273
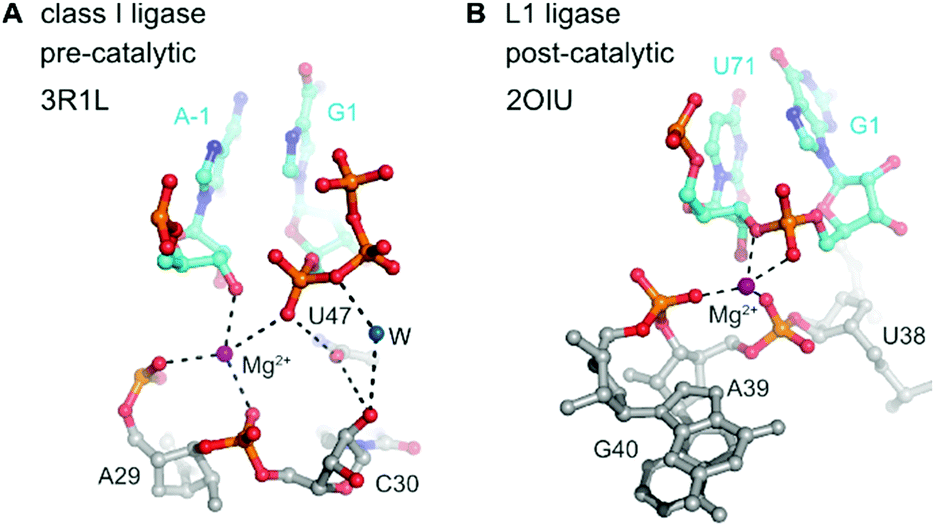 | ||
| Fig. 14 Class I ligase and L1 ligase ribozymes. Positioning of the reactive nucleotides in the active centre of class I ligase274 (A) and L1 ligase275 (B) ribozymes. Nucleotides at ligation junction are coloured in cyan, and pre- and postcatalytic structures are compared. | ||
Structural analogues of the L1 ligase, were derived from independent selections, originally in the absence of cytidine (R3 ligase), and then re-selected with all four nucleotides (R3C ligase) and further evolved for enhanced catalytic efficiencies (F1 ligase).278,279 Recently, large-scale mutagenesis and the evaluation of more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sequence variants resulted in the systematic minimization of the catalytic core to 18 nucleotides (minF1).280 The class I and R3C ligases were the starting points for the evolution of RNA polymerase ribozymes, and their evolutionary history was recently summarized.272 Some of the most exciting and creative strategies towards the synthesis of long more complex RNAs involved the evolution in ice,281 the evolution of a cross-chiral RNA ligase ribozyme,282 and the templated polymerization of RNA trinucleotide triphosphates (triplet building blocks).283 A highly optimized ribozyme was able to synthesize DNA polymers on an RNA template.284 The most recent ribozyme generation was shown to synthesise its own evolutionary ancestor in three pieces, which self-assembled into an active class I RNA ligase.285
000 sequence variants resulted in the systematic minimization of the catalytic core to 18 nucleotides (minF1).280 The class I and R3C ligases were the starting points for the evolution of RNA polymerase ribozymes, and their evolutionary history was recently summarized.272 Some of the most exciting and creative strategies towards the synthesis of long more complex RNAs involved the evolution in ice,281 the evolution of a cross-chiral RNA ligase ribozyme,282 and the templated polymerization of RNA trinucleotide triphosphates (triplet building blocks).283 A highly optimized ribozyme was able to synthesize DNA polymers on an RNA template.284 The most recent ribozyme generation was shown to synthesise its own evolutionary ancestor in three pieces, which self-assembled into an active class I RNA ligase.285
RNA-ligation catalysed by DNA (deoxyribozymes)
The generally useful RNA ligase deoxyribozyme named 9DB1 was reported by Silverman in 2005.286 It was derived from an in vitro selection experiment, in which a selection pressure demanded the regioselective formation of only 3′,5′ linkages.286 The crystal structure of 9DB1 was reported in 2016 as the first three-dimensional structure of a DNA enzyme in a catalytically relevant conformation.287 The structure provided unprecedented insights into the architecture of an active site made of DNA. The 9DB1 DNA complexed with its RNA ligation product forms a double pseudoknot in which two thymidine loop nucleotides interact with the A–G RNA nucleotides directly at the ligation junction (Fig. 15). These interactions could not be predicted from the available biochemical data.288–290 The insights from the crystal structure enabled engineering of the deoxyribozyme to broaden the substrate scope of RNA ligation.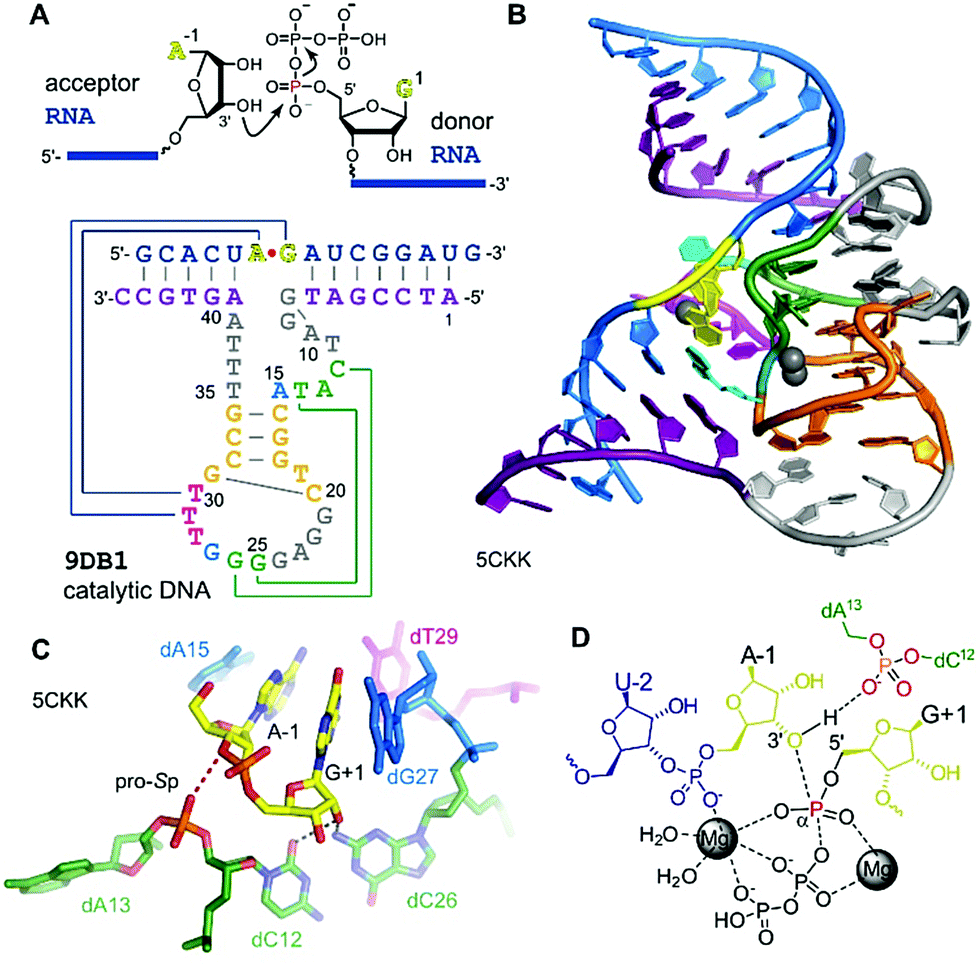 | ||
| Fig. 15 9DB1 DNAzyme. (A) Reaction catalysed by 9DB1 RNA ligase deoxyribozyme and secondary structure of DNA enzyme in complex with ligated RNA product. (B) Global architecture of the DNA catalyst. (C) Active site of 9DB1. The ligation junction, the base-pairing (magenta) and stacking (blue) nucleotides, the putatively catalytic dA13 phosphate, oriented by the long-range base pair (green) brings the pro-Sp non bridging oxygen in proximity to the 3′-oxygen of A-1. (D) Schematic illustration of the transition state model from QM/MM,291 suggesting a concerted phosphoryl and proton transfer involving the coordination of two metal ions. | ||
The crystal structure revealed a static picture of the post-catalytic product-bound state of the DNA enzyme and was unable to explain the involvement of magnesium ions as cofactors for accelerating RNA ligation. An internucleotide phosphodiester oxygen was suggested to make a critical contact,287 and this was supported by computational analyses and molecular dynamics simulations that revealed a plateau in the energy landscape, suggesting a concerted asynchronous transition state.291 The concerted phosphoryl and proton transfer pathway was found to be endergonic, and the release of the protonated pyrophosphate in complex with Mg2+ ions was suggested to drive the reaction towards ligation. The direct protonation of the phosphate needs further experimental confirmation, since slight rearrangements in the active site could support an alternative explanation, as recently suggested by density functional theory (DFT) calculations of an alternative active-site model of 9DB1.292 However, this earlier model did not consider the involvement of metal ions, which are required for catalysis.286,290
On the other hand, the proposed contacts of the metal ions in the active site with the triphosphate and RNA substrate were experimentally validated by phosphorothioate substitution and analysis of reaction rates.291 Nevertheless, structural models in the pre-catalytic state with bound metal ions and the pyrophosphate leaving group, with a transition state analogue or with an α,β-non-hydrolysable triphosphate analogue are highly desired. The structure revealed a broader range of sugar–phosphate backbone conformations in the DNA core than in natural and synthetic ribozymes.287 This suggests that the missing of the 2′-OH groups of DNA expands the conformational flexibility of DNA relative to RNA, to allow additional modes of interaction. This concept is likely to be employed by other deoxyribozymes, for which the structure is not yet known.
(Deoxy)ribozymes for covalent RNA labelling
RNA-ligating ribozymes and deoxyribozymes can not only activate the 3′-terminal hydroxy group, but the active site can be directed to a specific internal nucleotide and activate the 2′-OH for nucleophilic attack on a triphosphate, resulting in the formation of 2′,5′-branched RNA.293 Surprisingly, such branching reactions were identified when in vitro selections for RNA-ligating deoxyribozymes were carried out in the absence of a selection pressure for formation of the linear native connection.294 These unexpected DNA enzymes were turned into useful tools for generating artificial lariat RNAs (that contained the native linkage at the branch site),295–297 and for the attachment of oligonucleotides that modulated the folding and function of larger RNAs and ribozymes.298,299 Moreover, the branch-forming deoxyribozyme 10DM24 was engineered for RNA labelling.300,301 Together with the finding, that the ligation reaction was significantly accelerated by lanthanide ions (e.g. Tb3+),302 fast and efficient RNA labelling was achieved by the attachment of functionalized or fluorescently labelled guanines via the 2′,5′-branched linkage.303 Sine lanthanide ions cannot completely replace divalent Mg2+ or Mn2+, they are more likely to play a supporting structural role rather than being directly involved in the catalytic mechanism.The rather complex metal ion requirements of 10DM24 may be overcome by alternative nucleic acid catalysts. In this respect, ribozymes for a comparable labelling strategy of RNA were recently described.304,305 These RNA catalysts resulted from a targeted in vitro selection strategy that used a structured RNA library to guide the ribozyme to the predetermined labelling site, and were directly selected with ATP derivatives as small-molecule labelling reagents.305 To enhance the bioorthogonality of this RNA labelling strategy, a new generation of ribozymes was developed that used antiviral nucleotide analogues as substrates.304 In particular, tenofovir-transferase ribozymes were obtained that are orthogonal to the adenylyl-transferase ribozymes, and generate a branched phosphonate linkage instead of the phosphate linkage (Fig. 16). Site-specific installation of FRET reporters was demonstrated on small and large RNAs (5S, 16S and 23S rRNA) in the context of total cellular RNA.304,305 These studies pave the way for future evolution of catalytic RNA labelling tools in cells.
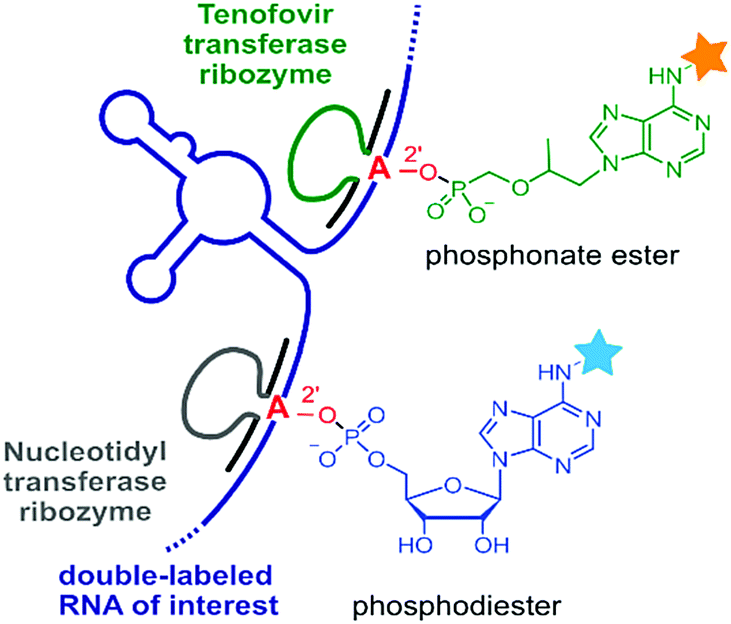 | ||
| Fig. 16 RNA-catalysed RNA labelling reactions produce 2′-linked phosphodiester or phosphonate esters with fluorescent nucleotide analogues as substrates. Orthogonal ribozymes allow site-specific dual labelling of RNA.304,305 | ||
Outlook on further fundamental studies on ribozymes and deoxyribozymes
Besides important insights into the mechanism of ribozymes and DNAzymes that are anticipated from chemical and biophysical studies, as well as from crystal structures of ribozymes/DNAzymes (pre- and post-catalytic states, transition state analogues), exploring their conformational dynamics by modern single molecule FRET and NMR spectroscopic methods would contribute to a deeper understanding of their catalytic activity. The expected micro- to milliseconds timescale of nucleoside dynamics in the active site, and of nucleosides at tertiary interactions should be accessible by using selective 13C labelling patterns and relaxation dispersion NMR experiments.306–310 In this way, populations of nucleoside conformations (concerning e.g. ribose pucker, glycosidic bond angles, etc.) that are crucial for approaching the transition state of phosphodiester cleavage could be revealed. Finally, using selective 15N labelling patterns, nucleobase tautomers and/or ionic forms potentially involved in catalysis could be made visible and this would represent a first direct spectroscopic verification in ribozymes/DNAzymes.Additional exciting future directions target the evolution of nucleic acid catalysts for reactions not yet known to be catalysed by RNA and DNA. A particular challenge is to find ribozymes and deoxyribozymes that modulate the modification state of RNA, directly by installing RNA modifications or by removing them, rather than degrading modified RNA or the unmodified counterpart. Fundamental research will then advance nucleic acid enzymes beyond analytical tools, and will allow entrance to the synthetic biology regime as an active modulator.
Summary
This review summarised common versus distinct structural and functional strategies that are applied by fluorogen-activating RNA aptamers, recently discovered bacterial riboswitches, and ribozymes/DNAzymes that catalyse phosphodiester chemistry. Fundamental studies on their structure and folding, their dynamics and reactivity, and their modification and labelling are continuously performed because the understanding of nucleic acids inherent molecular properties is needed to harness these modules for advanced applications in biotechnology and biomedicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Olga Krasheninina (Innsbruck) and Christian Steinmetzger (Würzburg) for critical reading of the manuscript. We thank all members of the Micura and Höbartner groups for their enthusiasm in making our projects happen and for valuable discussions during the course of writing this review. This work was supported by the Austrian Science Fund FWF (P27947, P31691, F8011-B), the Austrian Research Promotion Agency FFG [West Austrian BioNMR 858017], and the Wiener Wissenschafts-, Forschungs- und Technologiefonds (WWTF LS17-003), the German Science Foundation DFG (SPP 1784) and the European Research Council [ERC consolidator grant 682586 to C. H.].Notes and references
- Z. Miao and E. Westhof, Annu. Rev. Biophys., 2017, 46, 483–503 Search PubMed.
- D. J. Patel, Cold Spring Harbor Perspect. Biol., 2016, 8, a018754 Search PubMed.
- R. J. Trachman and A. R. Ferre-D'Amare, Q. Rev. Biophys., 2019, 52, e8 Search PubMed.
- A. Roth and R. R. Breaker, Annu. Rev. Biochem., 2009, 78, 305–334 Search PubMed.
- R. Micura and C. Höbartner, ChemBioChem, 2003, 4, 984–990 Search PubMed.
- B. Fürtig, S. Nozinovic, A. Reining and H. Schwalbe, Curr. Opin. Struct. Biol., 2015, 30, 112–124 Search PubMed.
- S. Ray, J. R. Widom and N. G. Walter, Chem. Rev., 2018, 118, 4120–4155 Search PubMed.
- M. F. Juette, D. S. Terry, M. R. Wasserman, Z. Zhou, R. B. Altman, Q. Zheng and S. C. Blanchard, Curr. Opin. Chem. Biol., 2014, 20, 103–111 Search PubMed.
- L. R. Ganser, M. L. Kelly, D. Herschlag and H. M. Al-Hashimi, Nat. Rev. Mol. Cell Biol., 2019, 20, 474–489 Search PubMed.
- A. V. Bedard, E. D. M. Hien and D. A. Lafontaine, Biochim. Biophys. Acta, Gene Regul. Mech., 2020, 1863, 194501 Search PubMed.
- A. I. Taylor, G. Houlihan and P. Holliger, Cold Spring Harbor Perspect. Biol., 2019, 11, a032490 Search PubMed.
- D. Gillingham, S. Geigle and O. Anatole von Lilienfeld, Chem. Soc. Rev., 2016, 45, 2637–2655 Search PubMed.
- D. Balke, R. Hieronymus and S. Müller, Adv. Biochem. Eng./Biotechnol., 2020, 170, 21–35 Search PubMed.
- Y. Yokobayashi, Curr. Opin. Chem. Biol., 2019, 52, 72–78 Search PubMed.
- S. Tsukiji, S. B. Pattnaik and H. Suga, Nat. Struct. Biol., 2003, 10, 713–717 Search PubMed.
- P. Cernak and D. Sen, Nat. Chem., 2013, 5, 971–977 Search PubMed.
- N. Muthmann, K. Hartstock and A. Rentmeister, Wiley Interdiscip. Rev.: RNA, 2020, 11, e1561 Search PubMed.
- M. Neveu, H. J. Kim and S. A. Benner, Astrobiology, 2013, 13, 391–403 Search PubMed.
- S. K. Wolk, W. S. Mayfield, A. D. Gelinas, D. Astling, J. Guillot, E. N. Brody, N. Janjic and L. Gold, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8236–8242 Search PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 Search PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 Search PubMed.
- S. Breuers, L. L. Bryant, T. Legen and G. Mayer, Methods, 2019, 161, 3–9 Search PubMed.
- M. R. Gotrik, T. A. Feagin, A. T. Csordas, M. A. Nakamoto and H. T. Soh, Acc. Chem. Res., 2016, 49, 1903–1910 Search PubMed.
- K. H. Cole and A. Luptak, Methods Enzymol., 2019, 621, 329–346 Search PubMed.
- R. Nutiu, R. C. Friedman, S. Luo, I. Khrebtukova, D. Silva, R. Li, L. Zhang, G. P. Schroth and C. B. Burge, Nat. Biotechnol., 2011, 29, 659–664 Search PubMed.
- A. D. Pressman, Z. Liu, E. Janzen, C. Blanco, U. F. Müller, G. F. Joyce, R. Pascal and I. A. Chen, J. Am. Chem. Soc., 2019, 141, 6213–6223 Search PubMed.
- Z. Chen, R. D. Kibler, A. Hunt, F. Busch, J. Pearl, M. Jia, Z. L. VanAernum, B. I. M. Wicky, G. Dods, H. Liao, M. S. Wilken, C. Ciarlo, S. Green, H. El-Samad, J. Stamatoyannopoulos, V. H. Wysocki, M. C. Jewett, S. E. Boyken and D. Baker, Science, 2020, 368, 78–84 Search PubMed.
- J. B. Siegel, A. Zanghellini, H. M. Lovick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael and D. Baker, Science, 2010, 329, 309–313 Search PubMed.
- E. Marcos, B. Basanta, T. M. Chidyausiku, Y. Tang, G. Oberdorfer, G. Liu, G. V. Swapna, R. Guan, D. A. Silva, J. Dou, J. H. Pereira, R. Xiao, B. Sankaran, P. H. Zwart, G. T. Montelione and D. Baker, Science, 2017, 355, 201–206 Search PubMed.
- F. Lapenta, J. Aupic, Z. Strmsek and R. Jerala, Chem. Soc. Rev., 2018, 47, 3530–3542 Search PubMed.
- J. D. Yesselman, D. Eiler, E. D. Carlson, M. R. Gotrik, A. E. d'Aquino, A. N. Ooms, W. Kladwang, P. D. Carlson, X. Shi, D. A. Costantino, D. Herschlag, J. B. Lucks, M. C. Jewett, J. S. Kieft and R. Das, Nat. Nanotechnol., 2019, 14, 866–873 Search PubMed.
- M. J. Wu, J. O. L. Andreasson, W. Kladwang, W. Greenleaf and R. Das, ACS Synth. Biol., 2019, 8, 1838–1846 Search PubMed.
- L. A. Holeman, S. L. Robinson, J. W. Szostak and C. Wilson, Folding Des., 1998, 3, 423–431 Search PubMed.
- C. Baugh, D. Grate and C. Wilson, J. Mol. Biol., 2000, 301, 117–128 Search PubMed.
- J. R. Babendure, S. R. Adams and R. Y. Tsien, J. Am. Chem. Soc., 2003, 125, 14716–14717 Search PubMed.
- S. Neubacher and S. Hennig, Angew. Chem., Int. Ed., 2019, 58, 1266–1279 Search PubMed.
- F. Bouhedda, A. Autour and M. Ryckelynck, Int. J. Mol. Sci., 2018, 19, 44 Search PubMed.
- M. You and S. R. Jaffrey, Annu. Rev. Biophys., 2015, 44, 187–206 Search PubMed.
- Y. Xia, R. Zhang, Z. Wang, J. Tian and X. Chen, Chem. Soc. Rev., 2017, 46, 2824–2843 Search PubMed.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 Search PubMed.
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 Search PubMed.
- K. D. Warner, M. C. Chen, W. Song, R. L. Strack, A. Thorn, S. R. Jaffrey and A. R. Ferre-D'Amare, Nat. Struct. Mol. Biol., 2014, 21, 658–663 Search PubMed.
- H. Huang, N. B. Suslov, N. S. Li, S. A. Shelke, M. E. Evans, Y. Koldobskaya, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2014, 10, 686–691 Search PubMed.
- G. S. Filonov, W. Song and S. R. Jaffrey, Biochemistry, 2019, 58, 1560–1564 Search PubMed.
- E. A. Ageely, Z. J. Kartje, K. J. Rohilla, C. L. Barkau and K. T. Gagnon, ACS Chem. Biol., 2016, 11, 2398–2406 Search PubMed.
- J. C. Savage, M. A. Davare and U. Shinde, Chem. Commun., 2020, 56, 2634–2637 Search PubMed.
- A. Autour, E. Westhof and M. Ryckelynck, Nucleic Acids Res., 2016, 44, 2491–2500 Search PubMed.
- P. Fernandez-Millan, A. Autour, E. Ennifar, E. Westhof and M. Ryckelynck, RNA, 2017, 23, 1788–1795 Search PubMed.
- W. Song, G. S. Filonov, H. Kim, M. Hirsch, X. Li, J. D. Moon and S. R. Jaffrey, Nat. Chem. Biol., 2017, 13, 1187–1194 Search PubMed.
- K. D. Warner, L. Sjekloca, W. Song, G. S. Filonov, S. R. Jaffrey and A. R. Ferre-D'Amare, Nat. Chem. Biol., 2017, 13, 1195–1201 Search PubMed.
- L. Sjekloca and A. R. Ferre-D'Amare, Cell Chem. Biol., 2019, 26, 1159–1168 Search PubMed . e1154.
- C. Steinmetzger, N. Palanisamy, K. R. Gore and C. Höbartner, Chem. – Eur. J., 2019, 25, 1931–1935 Search PubMed.
- C. Steinmetzger, I. Bessi, A. K. Lenz and C. Höbartner, Nucleic Acids Res., 2019, 47, 11538–11550 Search PubMed.
- C. Steinmetzger, C. Bäuerlein and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 6760–6764 Search PubMed.
- D. M. Shcherbakova, M. A. Hink, L. Joosen, T. W. Gadella and V. V. Verkhusha, J. Am. Chem. Soc., 2012, 134, 7913–7923 Search PubMed.
- E. V. Dolgosheina, S. C. Jeng, S. S. Panchapakesan, R. Cojocaru, P. S. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 Search PubMed.
- A. Autour, S. C. Y. Jeng, A. D. Cawte, A. Abdolahzadeh, A. Galli, S. S. S. Panchapakesan, D. Rueda, M. Ryckelynck and P. J. Unrau, Nat. Commun., 2018, 9, 656 Search PubMed.
- R. J. Trachman, 3rd, A. Autour, S. C. Y. Jeng, A. Abdolahzadeh, A. Andreoni, R. Cojocaru, R. Garipov, E. V. Dolgosheina, J. R. Knutson, M. Ryckelynck, P. J. Unrau and A. R. Ferre-D'Amare, Nat. Chem. Biol., 2019, 15, 472–479 Search PubMed.
- R. J. Trachman, 3rd, A. Abdolahzadeh, A. Andreoni, R. Cojocaru, J. R. Knutson, M. Ryckelynck, P. J. Unrau and A. R. Ferre-D'Amare, Biochemistry, 2018, 57, 3544–3548 Search PubMed.
- R. J. Trachman, 3rd, R. Cojocaru, D. Wu, G. Piszczek, M. Ryckelynck, P. J. Unrau and A. R. Ferre-D'Amare, Structure, 2020, 28, 776–785 Search PubMed.
- R. J. Trachman, 3rd, N. A. Demeshkina, M. W. L. Lau, S. S. S. Panchapakesan, S. C. Y. Jeng, P. J. Unrau and A. R. Ferre-D'Amare, Nat. Chem. Biol., 2017, 13, 807–813 Search PubMed.
- R. J. Trachman, 3rd, J. R. Stagno, C. Conrad, C. P. Jones, P. Fischer, A. Meents, Y. X. Wang and A. R. Ferre-D'Amare, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2019, 75, 547–551 Search PubMed.
- X. Tan, T. P. Constantin, K. L. Sloane, A. S. Waggoner, M. P. Bruchez and B. A. Armitage, J. Am. Chem. Soc., 2017, 139, 9001–9009 Search PubMed.
- S. A. Shelke, Y. Shao, A. Laski, D. Koirala, B. P. Weissman, J. R. Fuller, X. Tan, T. P. Constantin, A. S. Waggoner, M. P. Bruchez, B. A. Armitage and J. A. Piccirilli, Nat. Commun., 2018, 9, 4542 Search PubMed.
- X. Chen, D. Zhang, N. Su, B. Bao, X. Xie, F. Zuo, L. Yang, H. Wang, L. Jiang, Q. Lin, M. Fang, N. Li, X. Hua, Z. Chen, C. Bao, J. Xu, W. Du, L. Zhang, Y. Zhao, L. Zhu, J. Loscalzo and Y. Yang, Nat. Biotechnol., 2019, 37, 1287–1293 Search PubMed.
- J. Wu, S. Zaccara, D. Khuperkar, H. Kim, M. E. Tanenbaum and S. R. Jaffrey, Nat. Methods, 2019, 16, 862–865 Search PubMed.
- R. Wirth, P. Gao, G. U. Nienhaus, M. Sunbul and A. Jäschke, J. Am. Chem. Soc., 2019, 141, 7562–7571 Search PubMed.
- B. A. Sparano and K. Koide, J. Am. Chem. Soc., 2007, 129, 4785–4794 Search PubMed.
- S. Sato, M. Watanabe, Y. Katsuda, A. Murata, D. O. Wang and M. Uesugi, Angew. Chem., Int. Ed., 2015, 54, 1855–1858 Search PubMed.
- A. Murata, S. Sato, Y. Kawazoe and M. Uesugi, Chem. Commun., 2011, 47, 4712–4714 Search PubMed.
- A. Arora, M. Sunbul and A. Jäschke, Nucleic Acids Res., 2015, 43, e144 Search PubMed.
- M. Sunbul and A. Jäschke, Nucleic Acids Res., 2018, 46, e110 Search PubMed.
- M. Sunbul and A. Jäschke, Angew. Chem., Int. Ed., 2013, 52, 13401–13404 Search PubMed.
- F. Bouhedda, K. T. Fam, M. Collot, A. Autour, S. Marzi, A. Klymchenko and M. Ryckelynck, Nat. Chem. Biol., 2020, 16, 69–76 Search PubMed.
- E. Braselmann, A. J. Wierzba, J. T. Polaski, M. Chrominski, Z. E. Holmes, S. T. Hung, D. Batan, J. R. Wheeler, R. Parker, R. Jimenez, D. Gryko, R. T. Batey and A. E. Palmer, Nat. Chem. Biol., 2018, 14, 964–971 Search PubMed.
- P. J. McCown, K. A. Corbino, S. Stav, M. E. Sherlock and R. R. Breaker, RNA, 2017, 23, 995–1011 Search PubMed.
- N. Pavlova, D. Kaloudas and R. Penchovsky, Gene, 2019, 708, 38–48 Search PubMed.
- A. Peselis, A. Gao and A. Serganov, Biochimie, 2015, 117, 100–109 Search PubMed.
- A. Serganov and D. J. Patel, Curr. Opin. Struct. Biol., 2012, 22, 279–286 Search PubMed.
- A. Serganov and D. J. Patel, Annu. Rev. Biophys., 2012, 41, 343–370 Search PubMed.
- A. Nahvi, N. Sudarsan, M. S. Ebert, X. Zou, K. L. Brown and R. R. Breaker, Chem. Biol., 2002, 9, 1043 Search PubMed.
- A. S. Mironov, I. Gusarov, R. Rafikov, L. E. Lopez, K. Shatalin, R. A. Kreneva, D. A. Perumov and E. Nudler, Cell, 2002, 111, 747–756 Search PubMed.
- W. Winkler, A. Nahvi and R. R. Breaker, Nature, 2002, 419, 952–956 Search PubMed.
- X. Nou and R. J. Kadner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7190–7195 Search PubMed.
- Z. F. Hallberg, Y. Su, R. Z. Kitto and M. C. Hammond, Annu. Rev. Biochem., 2017, 86, 515–539 Search PubMed.
- T. H. T. Chau, D. H. A. Mai, D. N. Pham, H. T. Q. Le and E. Y. Lee, Int. J. Mol. Sci., 2020, 21, 3192 Search PubMed.
- K. B. Arnvig, Biochem. Soc. Trans., 2019, 47, 1091–1099 Search PubMed.
- J. D. Munzar, A. Ng and D. Juncker, Chem. Soc. Rev., 2019, 48, 1390–1419 Search PubMed.
- R. Galizi and A. Jaramillo, Curr. Opin. Biotechnol., 2019, 55, 103–113 Search PubMed.
- T. S. Lotz and B. Suess, Microbiol. Spectrum, 2018, 6, RWR-0025-2018 Search PubMed.
- D. Yu and R. R. Breaker, RNA, 2020, 26, 960–968 Search PubMed.
- S. N. Malkowski, T. C. J. Spencer and R. R. Breaker, RNA, 2019, 25, 1616–1627 Search PubMed.
- L. Huang, J. Wang and D. Lilley, RNA, 2020, 26, 878–887 Search PubMed.
- R. M. Atilho, G. Mirihana Arachchilage, E. B. Greenlee, K. M. Knecht and R. R. Breaker, eLife, 2019, 8, e45210 Search PubMed.
- M. E. Sherlock, N. Sudarsan, S. Stav and R. R. Breaker, eLife, 2018, 7, e33908 Search PubMed.
- J. W. Nelson, R. M. Atilho, M. E. Sherlock, R. B. Stockbridge and R. R. Breaker, Mol. Cell, 2017, 65, 220–230 Search PubMed.
- M. E. Sherlock, S. N. Malkowski and R. R. Breaker, Biochemistry, 2017, 56, 352–358 Search PubMed.
- M. E. Sherlock and R. R. Breaker, Biochemistry, 2017, 56, 359–363 Search PubMed.
- C. W. Reiss, Y. Xiong and S. A. Strobel, Structure, 2017, 25, 195–202 Search PubMed.
- R. A. Battaglia, I. R. Price and A. Ke, RNA, 2017, 23, 578–585 Search PubMed.
- L. Huang, J. Wang, T. J. Wilson and D. M. J. Lilley, Cell Chem. Biol., 2017, 24, 1407–1415 Search PubMed . e1402.
- R. A. Battaglia and A. Ke, Wiley Interdiscip. Rev.: RNA, 2018, e1482, DOI:10.1002/wrna.1482.
- M. E. Sherlock, N. Sudarsan and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6052–6057 Search PubMed.
- A. J. Knappenberger, C. W. Reiss and S. A. Strobel, eLife, 2018, 7, e36381 Search PubMed.
- A. Peselis and A. Serganov, Nat. Chem. Biol., 2018, 14, 887–894 Search PubMed.
- M. E. Sherlock, H. Sadeeshkumar and R. R. Breaker, Biochemistry, 2019, 58, 401–410 Search PubMed.
- P. L. Sazani, R. Larralde and J. W. Szostak, J. Am. Chem. Soc., 2004, 126, 8370–8371 Search PubMed.
- A. C. Wolter, A. K. Weickhmann, A. H. Nasiri, K. Hantke, O. Ohlenschlager, C. H. Wunderlich, C. Kreutz, E. Duchardt-Ferner and J. Wöhnert, Angew. Chem., Int. Ed., 2017, 56, 401–404 Search PubMed.
- W. C. Winkler, A. Nahvi, N. Sudarsan, J. E. Barrick and R. R. Breaker, Nat. Struct. Biol., 2003, 10, 701–707 Search PubMed.
- R. T. Fuchs, F. J. Grundy and T. M. Henkin, Nat. Struct. Mol. Biol., 2006, 13, 226–233 Search PubMed.
- G. Mirihana Arachchilage, M. E. Sherlock, Z. Weinberg and R. R. Breaker, RNA Biol., 2018, 15, 371–378 Search PubMed.
- A. Sun, C. Gasser, F. Li, H. Chen, S. Mair, O. Krasheninina, R. Micura and A. Ren, Nat. Commun., 2019, 10, 5728 Search PubMed.
- X. Chen, G. Mirihana Arachchilage and R. R. Breaker, RNA, 2019, 25, 1091–1097 Search PubMed.
- T. D. Ames, D. A. Rodionov, Z. Weinberg and R. R. Breaker, Chem. Biol., 2010, 17, 681–685 Search PubMed.
- J. C. Cochrane and S. A. Strobel, RNA, 2008, 14, 993–1002 Search PubMed.
- A. Kirschning, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.201914786.
- W. C. Winkler, A. Nahvi, A. Roth, J. A. Collins and R. R. Breaker, Nature, 2004, 428, 281–286 Search PubMed.
- G. F. Joyce and J. W. Szostak, Cold Spring Harbor Perspect. Biol., 2018, 10, a034801 Search PubMed.
- C. Anastasi, F. F. Buchet, M. A. Crowe, A. L. Parkes, M. W. Powner, J. M. Smith and J. D. Sutherland, Chem. Biodiversity, 2007, 4, 721–739 Search PubMed.
- H. Okamura, A. Crisp, S. Hübner, S. Becker, P. Rovo and T. Carell, Angew. Chem., Int. Ed., 2019, 58, 18691–18696 Search PubMed.
- J. E. Hein and D. G. Blackmond, Acc. Chem. Res., 2012, 45, 2045–2054 Search PubMed.
- M. Bolli, R. Micura and A. Eschenmoser, Chem. Biol., 1997, 4, 309–320 Search PubMed.
- S. Ishida, N. Terasaka, T. Katoh and H. Suga, Nat. Chem. Biol., 2020, 16, 702–709 Search PubMed.
- J. W. Weaver and A. Serganov, Nat. Struct. Mol. Biol., 2019, 26, 1081–1083 Search PubMed.
- E. B. Porter, J. T. Polaski, M. M. Morck and R. T. Batey, Nat. Chem. Biol., 2017, 13, 295–301 Search PubMed.
- J. L. Vinkenborg, N. Karnowski and M. Famulok, Nat. Chem. Biol., 2011, 7, 519–527 Search PubMed.
- M. Famulok, J. S. Hartig and G. Mayer, Chem. Rev., 2007, 107, 3715–3743 Search PubMed.
- M. Spöring, M. Finke and J. S. Hartig, Curr. Opin. Biotechnol., 2019, 63, 34–40 Search PubMed.
- M. Wieland and J. S. Hartig, Angew. Chem., Int. Ed., 2008, 47, 2604–2607 Search PubMed.
- C. Berens, F. Groher and B. Suess, Biotechnol. J., 2015, 10, 246–257 Search PubMed.
- B. Strobel, M. Spöring, H. Klein, D. Blazevic, W. Rust, S. Sayols, J. S. Hartig and S. Kreuz, Nat. Commun., 2020, 11, 714 Search PubMed.
- C. Wilson and J. W. Szostak, Nature, 1995, 374, 777–782 Search PubMed.
- S. Ameta and A. Jäschke, Chem. Sci., 2013, 4, 957–964 Search PubMed.
- A. K. Sharma, J. J. Plant, A. E. Rangel, K. N. Meek, A. J. Anamisis, J. Hollien and J. M. Heemstra, ACS Chem. Biol., 2014, 9, 1680–1684 Search PubMed.
- R. I. McDonald, J. P. Guilinger, S. Mukherji, E. A. Curtis, W. I. Lee and D. R. Liu, Nat. Chem. Biol., 2014, 10, 1049–1054 Search PubMed.
- T. R. Cech, Science, 2000, 289, 878–879 Search PubMed.
- S. K. Silverman, Trends Biochem. Sci., 2016, 41, 595–609 Search PubMed.
- K. Schlosser and Y. Li, Chem. Biol., 2009, 16, 311–322 Search PubMed.
- M. Illangasekare, G. Sanchez, T. Nickles and M. Yarus, Science, 1995, 267, 643–647 Search PubMed.
- H. Murakami, H. Saito and H. Suga, Chem. Biol., 2003, 10, 655–662 Search PubMed.
- G. Sengle, A. Eisenführ, P. S. Arora, J. S. Nowick and M. Famulok, Chem. Biol., 2001, 8, 459–473 Search PubMed.
- B. Seelig and A. Jäschke, Chem. Biol., 1999, 6, 167–176 Search PubMed.
- M. Chandra and S. K. Silverman, J. Am. Chem. Soc., 2008, 130, 2936–2937 Search PubMed.
- L. L. Martin, P. J. Unrau and U. F. Müller, Life, 2015, 5, 247–268 Search PubMed.
- A. Serganov and D. J. Patel, Nat. Rev. Genet., 2007, 8, 776–790 Search PubMed.
- A. V. Kazantsev, A. A. Krivenko, D. J. Harrington, S. R. Holbrook, P. D. Adams and N. R. Pace, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13392–13397 Search PubMed.
- J. M. Buzayan, W. L. Gerlach and G. Bruening, Nature, 1986, 323, 349–353 Search PubMed.
- M. J. Fedor, J. Mol. Biol., 2000, 297, 269–291 Search PubMed.
- P. B. Rupert and A. R. Ferre-D'Amare, Nature, 2001, 410, 780–786 Search PubMed.
- P. B. Rupert, A. P. Massey, S. T. Sigurdsson and A. R. Ferre-D'Amare, Science, 2002, 298, 1421–1424 Search PubMed.
- M. Martick, L. H. Horan, H. F. Noller and W. G. Scott, Nature, 2008, 454, 899–902 Search PubMed.
- A. Mir, J. Chen, K. Robinson, E. Lendy, J. Goodman, D. Neau and B. L. Golden, Biochemistry, 2015, 54, 6369–6381 Search PubMed.
- G. A. Prody, J. T. Bakos, J. M. Buzayan, I. R. Schneider and G. Bruening, Science, 1986, 231, 1577–1580 Search PubMed.
- M. Martick and W. G. Scott, Cell, 2006, 126, 309–320 Search PubMed.
- A. Mir and B. L. Golden, Biochemistry, 2016, 55, 633–636 Search PubMed.
- L. Sharmeen, M. Y. Kuo, G. Dinter-Gottlieb and J. Taylor, J. Virol., 1988, 62, 2674–2679 Search PubMed.
- A. R. Ferre-D'Amare, K. Zhou and J. A. Doudna, Nature, 1998, 395, 567–574 Search PubMed.
- A. Ke, K. Zhou, F. Ding, J. H. Cate and J. A. Doudna, Nature, 2004, 429, 201–205 Search PubMed.
- S. R. Das and J. A. Piccirilli, Nat. Chem. Biol., 2005, 1, 45–52 Search PubMed.
- D. J. Klein and A. R. Ferre-D'Amare, Science, 2006, 313, 1752–1756 Search PubMed.
- J. C. Cochrane, S. V. Lipchock and S. A. Strobel, Chem. Biol., 2007, 14, 97–105 Search PubMed.
- D. M. Lilley, RNA, 2004, 10, 151–158 Search PubMed.
- N. B. Suslov, S. DasGupta, H. Huang, J. R. Fuller, D. M. Lilley, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2015, 11, 840–846 Search PubMed.
- A. Roth, Z. Weinberg, A. G. Chen, P. B. Kim, T. D. Ames and R. R. Breaker, Nat. Chem. Biol., 2014, 10, 56–60 Search PubMed.
- Z. Weinberg, P. B. Kim, T. H. Chen, S. Li, K. A. Harris, C. E. Lunse and R. R. Breaker, Nat. Chem. Biol., 2015, 11, 606–610 Search PubMed.
- A. R. Ferre-D'Amare and W. G. Scott, Cold Spring Harbor Perspect. Biol., 2010, 2, a003574 Search PubMed.
- Z. Zhong and G. Chen, Nat. Chem. Biol., 2015, 11, 830–831 Search PubMed.
- R. M. Jimenez, J. A. Polanco and A. Luptak, Trends Biochem. Sci., 2015, 40, 648–661 Search PubMed.
- E. A. Doherty and J. A. Doudna, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 457–475 Search PubMed.
- J. A. Doudna and T. R. Cech, Nature, 2002, 418, 222–228 Search PubMed.
- E. Westhof, J. Mol. Recognit., 2007, 20, 1–3 Search PubMed.
- W. G. Scott, L. H. Horan and M. Martick, Prog. Mol. Biol. Transl. Sci., 2013, 120, 1–23 Search PubMed.
- V. Singh, B. I. Fedeles and J. M. Essigmann, RNA, 2015, 21, 1–13 Search PubMed.
- D. L. Kellerman, D. M. York, J. A. Piccirilli and M. E. Harris, Curr. Opin. Chem. Biol., 2014, 21, 96–102 Search PubMed.
- J. C. Cochrane and S. A. Strobel, Acc. Chem. Res., 2008, 41, 1027–1035 Search PubMed.
- M. J. Fedor and J. R. Williamson, Nat. Rev. Mol. Cell Biol., 2005, 6, 399–412 Search PubMed.
- G. M. Emilsson, S. Nakamura, A. Roth and R. R. Breaker, RNA, 2003, 9, 907–918 Search PubMed.
- G. A. Soukup and R. R. Breaker, RNA, 1999, 5, 1308–1325 Search PubMed.
- P. C. Bevilacqua, M. E. Harris, J. A. Piccirilli, C. Gaines, A. Ganguly, K. Kostenbader, S. Ekesan and D. M. York, ACS Chem. Biol., 2019, 14, 1068–1076 Search PubMed.
- R. R. Breaker, G. M. Emilsson, D. Lazarev, S. Nakamura, I. J. Puskarz, A. Roth and N. Sudarsan, RNA, 2003, 9, 949–957 Search PubMed.
- B. P. Weissman, N. S. Li, D. York, M. Harris and J. A. Piccirilli, Biochim. Biophys. Acta, 2015, 1854, 1737–1745 Search PubMed.
- A. M. Pyle, Science, 1993, 261, 709–714 Search PubMed.
- M. Yarus, FASEB J., 1993, 7, 31–39 Search PubMed.
- J. A. Doudna and J. R. Lorsch, Nat. Struct. Mol. Biol., 2005, 12, 395–402 Search PubMed.
- C. Gasser, J. Gebetsberger, M. Gebetsberger and R. Micura, Nucleic Acids Res., 2018, 46, 6983–6995 Search PubMed.
- Y. Liu, T. J. Wilson, S. A. McPhee and D. M. Lilley, Nat. Chem. Biol., 2014, 10, 739–744 Search PubMed.
- D. Eiler, J. Wang and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13028–13033 Search PubMed.
- A. Ren, M. Kosutic, K. R. Rajashankar, M. Frener, T. Santner, E. Westhof, R. Micura and D. J. Patel, Nat. Commun., 2014, 5, 5534 Search PubMed.
- M. Kosutic, S. Neuner, A. Ren, S. Flur, C. Wunderlich, E. Mairhofer, N. Vusurovic, J. Seikowski, K. Breuker, C. Höbartner, D. J. Patel, C. Kreutz and R. Micura, Angew. Chem., Int. Ed., 2015, 54, 15128–15133 Search PubMed.
- J. Gebetsberger and R. Micura, Wiley Interdiscip. Rev.: RNA, 2017, 8, e1402 Search PubMed.
- R. R. Breaker, ACS Chem. Biol., 2017, 12, 886–891 Search PubMed.
- A. Ren, R. Micura and D. J. Patel, Curr. Opin. Chem. Biol., 2017, 41, 71–83 Search PubMed.
- T. J. Wilson, Y. Liu, C. Domnick, S. Kath-Schorr and D. M. Lilley, J. Am. Chem. Soc., 2016, 138, 6151–6162 Search PubMed.
- E. Fuchs, C. Falschlunger, R. Micura and K. Breuker, Nucleic Acids Res., 2019, 47, 7223–7234 Search PubMed.
- K. J. Messina and P. C. Bevilacqua, J. Am. Chem. Soc., 2018, 140, 10578–10582 Search PubMed.
- K. J. Messina, R. Kierzek, M. A. Tracey and P. C. Bevilacqua, Biochemistry, 2019, 58, 4857–4868 Search PubMed.
- N. Vusurovic, R. B. Altman, D. S. Terry, R. Micura and S. C. Blanchard, J. Am. Chem. Soc., 2017, 139, 8186–8193 Search PubMed.
- C. S. Gaines and D. M. York, J. Am. Chem. Soc., 2016, 138, 3058–3065 Search PubMed.
- M. N. Ucisik, P. C. Bevilacqua and S. Hammes-Schiffer, Biochemistry, 2016, 55, 3834–3846 Search PubMed.
- C. S. Gaines, T. J. Giese and D. M. York, ACS Catal., 2019, 9, 5803–5815 Search PubMed.
- A. A. Kognole and A. D. MacKerell, Jr., Biophys. J., 2020, 118, 1424–1437 Search PubMed.
- K. A. Harris, C. E. Lunse, S. Li, K. I. Brewer and R. R. Breaker, RNA, 2015, 21, 1852–1858 Search PubMed.
- A. Ren, N. Vusurovic, J. Gebetsberger, P. Gao, M. Juen, C. Kreutz, R. Micura and D. J. Patel, Nat. Chem. Biol., 2016, 12, 702–708 Search PubMed.
- L. A. Nguyen, J. Wang and T. A. Steitz, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1021–1026 Search PubMed.
- S. Neuner, C. Falschlunger, E. Fuchs, M. Himmelstoss, A. Ren, D. J. Patel and R. Micura, Angew. Chem., Int. Ed., 2017, 56, 15954–15958 Search PubMed.
- M. Teplova, C. Falschlunger, O. Krasheninina, M. Egger, A. Ren, D. J. Patel and R. Micura, Angew. Chem., Int. Ed., 2020, 59, 2837–2843 Search PubMed.
- T. J. Wilson, Y. Liu, N. S. Li, Q. Dai, J. A. Piccirilli and D. M. J. Lilley, J. Am. Chem. Soc., 2019, 141, 7865–7875 Search PubMed.
- A. T. Torelli, J. Krucinska and J. E. Wedekind, RNA, 2007, 13, 1052–1070 Search PubMed.
- J. Han and J. M. Burke, Biochemistry, 2005, 44, 7864–7870 Search PubMed.
- L. Zheng, E. Mairhofer, M. Teplova, Y. Zhang, J. Ma, D. J. Patel, R. Micura and A. Ren, Nat. Commun., 2017, 8, 1180 Search PubMed.
- Y. Liu, T. J. Wilson and D. M. J. Lilley, Nat. Chem. Biol., 2017, 13, 508–513 Search PubMed.
- C. S. Gaines and D. M. York, Angew. Chem., Int. Ed., 2017, 56, 13392–13395 Search PubMed.
- N. Riccitelli and A. Luptak, Prog. Mol. Biol. Transl. Sci., 2013, 120, 123–171 Search PubMed.
- S. Li, C. E. Lünse, K. A. Harris and R. R. Breaker, RNA, 2015, 21, 1845–1851 Search PubMed.
- L. Zheng, C. Falschlunger, K. Huang, E. Mairhofer, S. Yuan, J. Wang, D. J. Patel, R. Micura and A. Ren, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10783–10791 Search PubMed.
- J. H. Chen, R. Yajima, D. M. Chadalavada, E. Chase, P. C. Bevilacqua and B. L. Golden, Biochemistry, 2010, 49, 6508–6518 Search PubMed.
- B. L. Golden, Biochemistry, 2011, 50, 9424–9433 Search PubMed.
- B. Gong, J. H. Chen, P. C. Bevilacqua, B. L. Golden and P. R. Carey, Biochemistry, 2009, 48, 11961–11970 Search PubMed.
- P. Thaplyal, A. Ganguly, B. L. Golden, S. Hammes-Schiffer and P. C. Bevilacqua, Biochemistry, 2013, 52, 6499–6514 Search PubMed.
- G. J. Kapral, S. Jain, J. Noeske, J. A. Doudna, D. C. Richardson and J. S. Richardson, Nucleic Acids Res., 2014, 42, 12833–12846 Search PubMed.
- R. R. Breaker and G. F. Joyce, Chem. Biol., 1994, 1, 223–229 Search PubMed.
- S. K. Silverman, Nucleic Acids Res., 2005, 33, 6151–6163 Search PubMed.
- G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791–836 Search PubMed.
- S. K. Silverman and D. A. Baum, Methods Enzymol., 2009, 469, 95–117 Search PubMed.
- A. A. Fokina, D. A. Stetsenko and J. C. Francois, Expert Opin. Biol. Ther., 2015, 15, 689–711 Search PubMed.
- L. Gong, Z. Zhao, Y. F. Lv, S. Y. Huan, T. Fu, X. B. Zhang, G. L. Shen and R. Q. Yu, Chem. Commun., 2015, 51, 979–995 Search PubMed.
- K. Hwang, Q. Mou, R. J. Lake, M. Xiong, B. Holland and Y. Lu, Inorg. Chem., 2019, 58, 13696–13708 Search PubMed.
- L. Ma and J. Liu, iScience, 2020, 23, 100815 Search PubMed.
- X. B. Zhang, R. M. Kong and Y. Lu, Annu. Rev. Anal. Chem., 2011, 4, 105–128 Search PubMed.
- Y. Lin, Z. Yang, R. J. Lake, C. Zheng and Y. Lu, Angew. Chem., Int. Ed., 2019, 58, 17061–17067 Search PubMed.
- S. H. Mei, Z. Liu, J. D. Brennan and Y. Li, J. Am. Chem. Soc., 2003, 125, 412–420 Search PubMed.
- R. P. Cruz, J. B. Withers and Y. Li, Chem. Biol., 2004, 11, 57–67 Search PubMed.
- K. Schlosser, J. Gu, L. Sule and Y. Li, Nucleic Acids Res., 2008, 36, 1472–1481 Search PubMed.
- S. F. Torabi, P. Wu, C. E. McGhee, L. Chen, K. Hwang, N. Zheng, J. Cheng and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5903–5908 Search PubMed.
- S. W. Santoro and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 Search PubMed.
- K. Schlosser and Y. Li, ChemBioChem, 2010, 11, 866–879 Search PubMed.
- H. Liu, X. Yu, Y. Chen, J. Zhang, B. Wu, L. Zheng, P. Haruehanroengra, R. Wang, S. Li, J. Lin, J. Li, J. Sheng, Z. Huang, J. Ma and J. Gan, Nat. Commun., 2017, 8, 2006 Search PubMed.
- M. Cepeda-Plaza, C. E. McGhee and Y. Lu, Biochemistry, 2018, 57, 1517–1522 Search PubMed.
- A. K. Brown, J. Li, C. M. Pavot and Y. Lu, Biochemistry, 2003, 42, 7152–7161 Search PubMed.
- M. V. Sednev, V. Mykhailiuk, P. Choudhury, J. Halang, K. E. Sloan, M. T. Bohnsack and C. Höbartner, Angew. Chem., Int. Ed., 2018, 57, 15117–15121 Search PubMed.
- A. Ganguly, B. P. Weissman, J. A. Piccirilli and D. M. York, ACS Catal., 2019, 9, 10612–10617 Search PubMed.
- S. Ekesan and D. M. York, Nucleic Acids Res., 2019, 47, 10282–10295 Search PubMed.
- H. K. Kim, J. Liu, J. Li, N. Nagraj, M. Li, C. M. Pavot and Y. Lu, J. Am. Chem. Soc., 2007, 129, 6896–6902 Search PubMed.
- H. K. Kim, I. Rasnik, J. Liu, T. Ha and Y. Lu, Nat. Chem. Biol., 2007, 3, 763–768 Search PubMed.
- L. Ma, S. Kartik, B. Liu and J. Liu, Nucleic Acids Res., 2019, 47, 8154–8162 Search PubMed.
- M. Buchhaupt, C. Peifer and K.-D. Entian, Anal. Biochem., 2007, 361, 102–108 Search PubMed.
- M. Hengesbach, M. Meusburger, F. Lyko and M. Helm, RNA, 2008, 14, 180–187 Search PubMed.
- D. Graber, H. Moroder, J. Steger, K. Trappl, N. Polacek and R. Micura, Nucleic Acids Res., 2010, 38, 6796–6802 Search PubMed.
- M. Bujnowska, J. Zhang, Q. Dai, E. M. Heideman and J. Fei, J. Biol. Chem., 2020, 295, 6992–7000 Search PubMed.
- D. P. Patil, B. F. Pickering and S. R. Jaffrey, Trends Cell Biol., 2018, 28, 113–127 Search PubMed.
- B. Liu, D. K. Merriman, S. H. Choi, M. A. Schumacher, R. Plangger, C. Kreutz, S. M. Horner, K. D. Meyer and H. M. Al-Hashimi, Nat. Commun., 2018, 9, 2761 Search PubMed.
- N. Liu, Q. Dai, G. Zheng, C. He, M. Parisien and T. Pan, Nature, 2015, 518, 560–564 Search PubMed.
- C. Roost, S. R. Lynch, P. J. Batista, K. Qu, H. Y. Chang and E. T. Kool, J. Am. Chem. Soc., 2015, 137, 2107–2115 Search PubMed.
- R. Micura, W. Pils, C. Höbartner, K. Grubmayr, M. O. Ebert and B. Jaun, Nucleic Acids Res., 2001, 29, 3997–4005 Search PubMed.
- H. Shi, B. Liu, F. Nussbaumer, A. Rangadurai, C. Kreutz and H. M. Al-Hashimi, J. Am. Chem. Soc., 2019, 141, 19988–19993 Search PubMed.
- A. Liaqat, C. Stiller, M. Michel, M. V. Sednev and C. Höbartner, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202006218.
- U. Schweizer, S. Bohleber and N. Fradejas-Villar, RNA Biol., 2017, 14, 1197–1208 Search PubMed.
- T. Janas, T. Janas and M. Yarus, RNA, 2012, 18, 2260–2268 Search PubMed.
- J. C. Lam, J. B. Withers and Y. Li, J. Mol. Biol., 2010, 400, 689–701 Search PubMed.
- Y. Yokobayashi, Methods, 2019, 161, 41–45 Search PubMed.
- T. L. Sheppard, P. Ordoukhanian and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7802–7807 Search PubMed.
- M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 Search PubMed.
- Y. Xiao, E. C. Allen and S. K. Silverman, Chem. Commun., 2011, 47, 1749–1751 Search PubMed.
- V. Dokukin and S. K. Silverman, Chem. Sci., 2012, 3, 1707–1714 Search PubMed.
- H. Gu, K. Furukawa, Z. Weinberg, D. F. Berenson and R. R. Breaker, J. Am. Chem. Soc., 2013, 135, 9121–9129 Search PubMed.
- V. Dhamodharan, S. Kobori and Y. Yokobayashi, ACS Chem. Biol., 2017, 12, 2940–2945 Search PubMed.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 Search PubMed.
- R. Welz, K. Bossmann, C. Klug, C. Schmidt, H. J. Fritz and S. Müller, Angew. Chem., Int. Ed., 2003, 42, 2424–2427 Search PubMed.
- T. Walton, S. DasGupta, D. Duzdevich, S. S. Oh and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 5741–5748 Search PubMed.
- F. Wachowius and P. Holliger, ChemSystemsChem, 2019, 1, 1–4 Search PubMed.
- Y. Fujita, H. Furuta and Y. Ikawa, RNA, 2009, 15, 877–888 Search PubMed.
- F. Wachowius, J. Attwater and P. Holliger, Q. Rev. Biophys., 2017, 50, e4 Search PubMed.
- D. M. Shechner and D. P. Bartel, Nat. Struct. Mol. Biol., 2011, 18, 1036–1042 Search PubMed.
- D. M. Shechner, R. A. Grant, S. C. Bagby, Y. Koldobskaya, J. A. Piccirilli and D. P. Bartel, Science, 2009, 326, 1271–1275 Search PubMed.
- M. P. Robertson and W. G. Scott, Science, 2007, 315, 1549–1553 Search PubMed.
- D. P. Bartel and J. W. Szostak, Science, 1993, 261, 1411–1418 Search PubMed.
- M. P. Robertson and A. D. Ellington, Nat. Biotechnol., 1999, 17, 62–66 Search PubMed.
- J. Rogers and G. F. Joyce, RNA, 2001, 7, 395–404 Search PubMed.
- M. P. Robertson and G. F. Joyce, Chem. Biol., 2014, 21, 238–245 Search PubMed.
- Y. Nomura and Y. Yokobayashi, Nucleic Acids Res., 2019, 47, 8950–8960 Search PubMed.
- J. Attwater, A. Wochner and P. Holliger, Nat. Chem., 2013, 5, 1011–1018 Search PubMed.
- J. T. Sczepanski and G. F. Joyce, Nature, 2014, 515, 440–442 Search PubMed.
- J. Attwater, A. Raguram, A. S. Morgunov, E. Gianni and P. Holliger, eLife, 2018, 7, e35255 Search PubMed.
- B. Samanta and G. F. Joyce, eLife, 2017, 6, e31153 Search PubMed.
- K. F. Tjhung, M. N. Shokhirev, D. P. Horning and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2906–2913 Search PubMed.
- W. E. Purtha, R. L. Coppins, M. K. Smalley and S. K. Silverman, J. Am. Chem. Soc., 2005, 127, 13124–13125 Search PubMed.
- A. Ponce-Salvatierra, K. Wawrzyniak-Turek, U. Steuerwald, C. Höbartner and V. Pena, Nature, 2016, 529, 231–234 Search PubMed.
- F. Wachowius, F. Javadi-Zarnaghi and C. Höbartner, Angew. Chem., Int. Ed., 2010, 49, 8504–8508 Search PubMed.
- F. Wachowius and C. Höbartner, J. Am. Chem. Soc., 2011, 133, 14888–14891 Search PubMed.
- L. Büttner, J. Seikowski, K. Wawrzyniak, A. Ochmann and C. Höbartner, Bioorg. Med. Chem., 2013, 21, 6171–6180 Search PubMed.
- J. Aranda, M. Terrazas, H. Gomez, N. Villegas and M. Orozco, Nat. Catal., 2019, 2, 544–552 Search PubMed.
- E. J. Mattioli, A. Bottoni and M. Calvaresi, J. Chem. Inf. Model., 2019, 59, 1547–1553 Search PubMed.
- E. Zelin, Y. Wang and S. K. Silverman, Biochemistry, 2006, 45, 2767–2771 Search PubMed.
- R. L. Coppins and S. K. Silverman, Nat. Struct. Mol. Biol., 2004, 11, 270–274 Search PubMed.
- Y. Wang and S. K. Silverman, J. Am. Chem. Soc., 2003, 125, 6880–6881 Search PubMed.
- Y. Wang and S. K. Silverman, Angew. Chem., Int. Ed., 2005, 44, 5863–5866 Search PubMed.
- F. Javadi-Zarnaghi and C. Höbartner, Chem. – Eur. J., 2016, 22, 3720–3728 Search PubMed.
- E. Zelin and S. K. Silverman, ChemBioChem, 2007, 8, 1907–1911 Search PubMed.
- E. Zelin and S. K. Silverman, Chem. Commun., 2009, 767–769 Search PubMed.
- D. A. Baum and S. K. Silverman, Angew. Chem., Int. Ed., 2007, 46, 3502–3504 Search PubMed.
- C. Höbartner and S. K. Silverman, Angew. Chem., Int. Ed., 2007, 46, 7420–7424 Search PubMed.
- F. Javadi-Zarnaghi and C. Höbartner, J. Am. Chem. Soc., 2013, 135, 12839–12848 Search PubMed.
- L. Büttner, F. Javadi-Zarnaghi and C. Höbartner, J. Am. Chem. Soc., 2014, 136, 8131–8137 Search PubMed.
- M. Ghaem Maghami, S. Dey, A. K. Lenz and C. Höbartner, Angew. Chem., Int. Ed., 2020, 59, 9335–9339 Search PubMed.
- M. Ghaem Maghami, C. P. M. Scheitl and C. Höbartner, J. Am. Chem. Soc., 2019, 141, 19546–19549 Search PubMed.
- M. A. Juen, C. H. Wunderlich, F. Nussbaumer, M. Tollinger, G. Kontaxis, R. Konrat, D. F. Hansen and C. Kreutz, Angew. Chem., Int. Ed., 2016, 55, 12008–12012 Search PubMed.
- E. Strebitzer, A. Rangadurai, R. Plangger, J. Kremser, M. A. Juen, M. Tollinger, H. M. Al-Hashimi and C. Kreutz, Chemistry, 2018, 24, 18903–18906 Search PubMed.
- B. Chen, A. P. Longhini, F. Nussbaumer, C. Kreutz, J. D. Dinman and T. K. Dayie, Chem. – Eur. J., 2018, 24, 5462–5468 Search PubMed.
- Y. Xue, B. Gracia, D. Herschlag, R. Russell and H. M. Al-Hashimi, Nat. Commun., 2016, 7, ncomms11768 Search PubMed.
- I. J. Kimsey, K. Petzold, B. Sathyamoorthy, Z. W. Stein and H. M. Al-Hashimi, Nature, 2015, 519, 315–320 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |