
Selective, bifunctional Cu–WOx/Al2O3 catalyst for hydrodeoxygenation of fatty acids†
Sagar
Janampelli
a,
Govind
Sethia
b and
Srinivas
Darbha
 *a
*a
aCatalysis and Inorganic Chemistry Division, CSIR-National Chemical Laboratory, Pune-411008, India. E-mail: d.srinivas@ncl.res.in; Fax: +91 20 25902633; Tel: +91 20 25902018
bInorganic Materials and Catalysis Division, CSIR-Central Salt & Marine Chemicals Research Institute, Gijubhai Bhadheka Marg, Bhavnagar-364002, India
First published on 27th November 2019
Abstract
Selective hydrodeoxygenation of oleic acid (OA; in a batch reactor, at 300 °C, 30 bar H2 pressure, reaction time of 1 h and reactant-to-catalyst weight ratio of 2 g OA/0.2 g) forming n-octadecane in yields as high as 93% over a non-noble metal, sulfur-free, bimetallic Cu–WOx/Al2O3 catalyst is reported for the first time. Several Cu–WOx/Al2O3 compositions were prepared by a sequential wet-impregnation method and evaluated. A catalyst with 10 wt% Cu and 4 wt% W enabled the highest activity and selectivity. Upon adding WOx, the amount of moderate and strong acid sites increased and more Cu in the catalyst was in reduced electron-rich metallic (Cu0) state. The crystallite size and dispersion of Cu were little affected. WOx promoted the fatty acid hydrodeoxygenation activity of Cu. While a monometallic Cu catalyst (10Cu/Al2O3) yielded n-octadecane and n-heptadecane along with high amounts of intermediate octadecanol and octadecanal products, the bimetallic catalyst (10Cu–4WOx/Al2O3) gave mainly n-octadecane. Acidity and the high amount of reduced Cu0 species are responsible for the high catalytic hydrodeoxygenation performance of this bimetallic catalyst.
Introduction
Fuel-grade hydrocarbons (HCs) produced from inedible vegetable oils, animal fats and fatty acids are a sustainable alternative to fossil-derived transport fuels.1–4 Their deployment leads to energy independence, reduced greenhouse gas (GHG) emissions, savings on oil import bill and rural empowerment. Their exploitation in transportation aligns with the mandate of several nations towards the use of renewable fuels.5 Deoxygenation is the pathway by which these HCs are produced.6,7 It is achieved by two means: (1) hydrodeoxygenation (HDO), wherein oxygen in fatty acid is taken out as water under a hydrogen environment, and (2) decarboxylation/decarbonylation (DCO), wherein oxygen is removed as CO2/CO. In the former route (HDO), a HC with carbon chain length the same as that of the reactant fatty compound is produced (for example, oleic acid (OA) is converted into octadecane, C18), while in the latter route (DCO), a HC with carbon chain length shorter by one unit than the original fatty compound is produced (i.e., OA is converted into heptadecane, C17). The renewable HCs thus formed outperform the fossil-derived fuels with cetane number (CN) ranging from 85 to 99 compared to 45 to 55 for the conventional diesel. They can be used as stand-alone fuel in conventional diesel engines after mild hydroisomerization.Neste Oil, UOP/Eni (Ecofining™), Honeywell (Green Jet Fuel Technology) and ConocoPhillips–Tyson Foods announced commercial processes of renewable HCs using conventional hydrotreating catalysts such as sulfided CoMo and sulfided NiMo supported on Al2O3.8–12 These processes operate at high temperature (325–380 °C) and hydrogen pressure (50–80 bar). Continuous sulfidation of the catalyst by passing a sulfur-containing compound in the reactor is essential for its stable performance.13 While the starting fatty compound does not contain sulfur in its composition, the product HC contains it due to sulfur leaching. The DCO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HDO product ratio in this process is often in the range of 69:21 to 20:80.14,15
:HDO product ratio in this process is often in the range of 69:21 to 20:80.14,15
Supported noble metals (Pd and Pt) are the other class of deoxygenation catalysts. They operate at moderate temperature (250–300 °C) and in the absence of hydrogen.16–18 However, a hydrogen supply (5–10 bar) extends the life of the catalyst, avoiding coke formation on the catalyst surface.19 The absence of hydrogen leads to side reactions such as cracking and heavier compound formation, especially when unsaturated feeds are employed. Unlike that for the conventional hydrotreating catalysts, DCO is the main reaction pathway (>95%) over the noble metal catalysts.16–18 Acidic metal oxide-promoted noble metals are more efficient catalysts operating at lower temperatures (180–260 °C).20–24 Adsorption and activation of the fatty compound on the acidic metal oxide and hydrogenation/hydrogenolysis of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on the noble metal selectively lead to the HDO product over these bifunctional catalysts. Limited stability, high cost and restricted availability are some drawbacks of the supported noble metal catalysts that impede their industrial application. Thus, development of an affordable, highly active and stable deoxygenation catalyst is still a challenge.
O bond on the noble metal selectively lead to the HDO product over these bifunctional catalysts. Limited stability, high cost and restricted availability are some drawbacks of the supported noble metal catalysts that impede their industrial application. Thus, development of an affordable, highly active and stable deoxygenation catalyst is still a challenge.
In view of the above challenges, there were several efforts to develop non-sulfided non-noble metal catalysts including mono-elemental and bi-elemental Cu, Ni and Co.14,15,25–29 In general, the reduced (non-sulfided) NiMo catalysts showed inferior activity and HC yields compared to the conventional sulfided NiMo catalysts. By optimizing the synergistic atomic ratio of 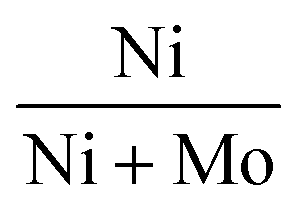 to 0.8 in the reduced NiMo catalysts (instead of 0.3 in the sulfided NiMo catalysts), vegetable oil conversion close to that of the sulfided NiMo catalyst was achieved.30,31 But then, the sulfided catalyst (1Ni3Mo–S) gave higher HC yield (sunflower oil conversion = 94%, HC yield = 21 wt% with the distribution of C18 = 13 wt%, C17 = 7.0%, C16 = 0.8%, C15 = 0.4 wt%) than the reduced catalyst (3.5Ni0.5Mo) (sunflower oil conversion = 81%, HC yield = 14.2 wt% with the distribution of C18 = 5.5 wt%, C17 = 8.0%, C16 = 0.3%, C15 = 0.4 wt%). DCO (as compared to HDO) is the predominant deoxygenation pathway over the reduced, non-sulfided catalysts.30,31 Wu et al.32 compared the performance of ZrO2-supported Cu and Ni catalysts in decarboxylation of stearic acid at 330 °C after 5 h of run under an inert atmosphere. The yield of C17 was only 8% over 20 wt% Cu/ZrO2, while it was 30% over 20 wt% Ni/ZrO2. Berenblyum et al.33 compared the performance of 5 wt% Ni and Cu catalysts supported on γ-Al2O3 and WO3/ZrO2 in the deoxygenation of stearic acid under 14 bar hydrogen at 350 °C after 6 h of reaction. 1-Heptadecene (formed through decarbonylation of stearic acid) was the main product over 5 wt% Cu/Al2O3, whereas n-heptadecane (formed through decarboxylation) was the major product on 5 wt% Ni/Al2O3 catalyst. Unlike that for the ZrO2 support,33 the deoxygenation performance of 5 wt% Cu/Al2O3 was higher than that of 5 wt% Ni/Al2O3 (yield of total C17 compounds was 78% over the Cu and 36% over the Ni catalyst). 5 wt% Cu/WO3/ZrO2 and 5 wt% Ni/WO3/ZrO2 exhibited complete conversion of stearic acid. However, cracking was the major reaction over these catalysts. Total C17 compound yield was only 7% over 5 wt% Cu/WO3/ZrO2 and 11% over 5 wt% Ni/WO3/ZrO2. Denk et al.34 found that Cu increases the activity of Ni for decarbonylation of 1-octadecanal by increasing the electron density of Ni in the bimetallic NixCu1−x/ZrO2 catalyst. Thus, the nature of the metal and support and reaction conditions (temperature, pressure and gas environment) influence the catalyst performance and DCO selectivity over non-noble metal catalysts. Highly performing and selective HDO catalysts are still a challenge.
to 0.8 in the reduced NiMo catalysts (instead of 0.3 in the sulfided NiMo catalysts), vegetable oil conversion close to that of the sulfided NiMo catalyst was achieved.30,31 But then, the sulfided catalyst (1Ni3Mo–S) gave higher HC yield (sunflower oil conversion = 94%, HC yield = 21 wt% with the distribution of C18 = 13 wt%, C17 = 7.0%, C16 = 0.8%, C15 = 0.4 wt%) than the reduced catalyst (3.5Ni0.5Mo) (sunflower oil conversion = 81%, HC yield = 14.2 wt% with the distribution of C18 = 5.5 wt%, C17 = 8.0%, C16 = 0.3%, C15 = 0.4 wt%). DCO (as compared to HDO) is the predominant deoxygenation pathway over the reduced, non-sulfided catalysts.30,31 Wu et al.32 compared the performance of ZrO2-supported Cu and Ni catalysts in decarboxylation of stearic acid at 330 °C after 5 h of run under an inert atmosphere. The yield of C17 was only 8% over 20 wt% Cu/ZrO2, while it was 30% over 20 wt% Ni/ZrO2. Berenblyum et al.33 compared the performance of 5 wt% Ni and Cu catalysts supported on γ-Al2O3 and WO3/ZrO2 in the deoxygenation of stearic acid under 14 bar hydrogen at 350 °C after 6 h of reaction. 1-Heptadecene (formed through decarbonylation of stearic acid) was the main product over 5 wt% Cu/Al2O3, whereas n-heptadecane (formed through decarboxylation) was the major product on 5 wt% Ni/Al2O3 catalyst. Unlike that for the ZrO2 support,33 the deoxygenation performance of 5 wt% Cu/Al2O3 was higher than that of 5 wt% Ni/Al2O3 (yield of total C17 compounds was 78% over the Cu and 36% over the Ni catalyst). 5 wt% Cu/WO3/ZrO2 and 5 wt% Ni/WO3/ZrO2 exhibited complete conversion of stearic acid. However, cracking was the major reaction over these catalysts. Total C17 compound yield was only 7% over 5 wt% Cu/WO3/ZrO2 and 11% over 5 wt% Ni/WO3/ZrO2. Denk et al.34 found that Cu increases the activity of Ni for decarbonylation of 1-octadecanal by increasing the electron density of Ni in the bimetallic NixCu1−x/ZrO2 catalyst. Thus, the nature of the metal and support and reaction conditions (temperature, pressure and gas environment) influence the catalyst performance and DCO selectivity over non-noble metal catalysts. Highly performing and selective HDO catalysts are still a challenge.
We report here for the first time a highly active and HDO selective, bimetallic Cu–WOx/Al2O3 catalyst for the deoxygenation of oleic acid (OA, a representative fatty acid). The amounts of Cu and W in the catalyst were varied. WOx promoted the HDO activity of Cu/Al2O3. The HDO/DCO product ratio over the catalyst of this study (Cu–WOx/Al2O3) is higher than that known for reduced/sulfided NiMo catalysts. The high HC yield and low carbon loss over the Cu–WOx/Al2O3 catalyst can lead to an improved fatty acid deoxygenation process.
Experimental
Catalyst preparation
Monometallic Cu/Al2O3 catalysts with Cu loading in the range of 5–20 wt% and bimetallic Cu–WOx/Al2O3 catalysts with Cu loading at 10 wt% and W in the range of 2–16 wt% were prepared by a wet-impregnation method. γ-Al2O3 supplied by Süd-Chemie India Pvt. Ltd., New Delhi, was used as a catalyst support in the preparations.Initially, a known quantity of ammonium metatungstate ((NH4)6W12O39·xH2O; 99.9%, Alfa Aesar) was dissolved in 10 ml of Milli-Q water. To that, 1 g of γ-Al2O3 powder was added. The suspension was agitated at 40 °C for 16 h. Water was removed over a rotary evaporator. The solid formed was dried at 110 °C for 12 h and calcined at 450 °C (ramp rate = 2 °C min−1) for 4 h. The material thus formed was designated as yWOx/Al2O3.
In the second step, Cu (10 wt%) was deposited on yWOx/Al2O3. The required amount of Cu(NO3)2·3H2O was dissolved in 10 ml of Milli-Q water. To it, 1 g of yWOx/Al2O3 (prepared as above) was added. It was stirred at 40 °C for 4 h. Water was removed over a rotary evaporator (maintained at 65 °C). The solid formed was collected, dried at 110 ° C for 12 h, and calcined at 450 °C for 4 h. It was reduced at 350 °C for 2.5 h in a flow of hydrogen (20 ml min−1). The catalyst thus obtained was labeled as 10Cu–yWOx/Al2O3.
Catalyst characterization procedure
Metal dispersion (DCu), active metal surface area (SCu; per gram metal) and average crystallite size (CSCu) of Cu were determined using a CO-chemisorption technique performed on a Quantachrome Autosorb iQ instrument. In a typical experiment, about 0.2 g of the catalyst was charged into a U-shaped quartz tube. It was degassed at 200 °C (heating rate = 20 °C min−1) in a helium flow (20 ml min−1) for 2 h and then reduced with hydrogen (20 ml min−1) at 350 °C (heating rate = 20 °C min−1) for 2.5 h. Subsequently, it was evacuated (at 350 °C) for 2 h and the sample temperature was lowered to 40 °C (in 1 min) under vacuum. Then, CO gas was introduced and the chemisorption measurements were performed under the following conditions: equilibrium tolerance = 0, equilibrium time = 3 min, adsorption points = 80, 160, 240, 320, 400, 480 and 560 mm Hg, analysis temperature = 40 °C, thermal equilibrium time = 10 min and leak test = 1 min. The following formulae were used to determine the textural properties of supported copper metal.35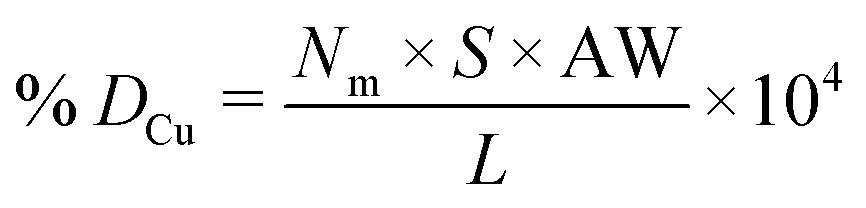
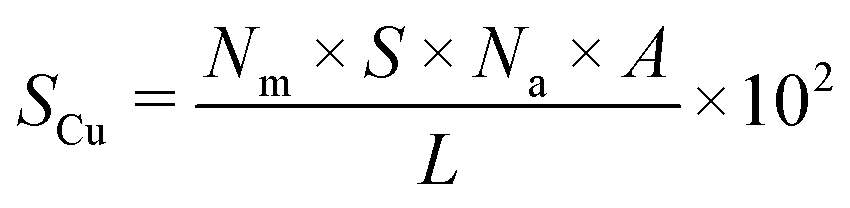
Here, Nm is the monolayer CO uptake in units of mol g−1, S is the stoichiometric ratio i.e., number of surface metal atoms covered by each chemisorbed CO molecule, AW is the atomic weight of copper (63.546 g mol−1), L is percent loading of copper, Na is the Avogadro number (6.023 × 1023 atoms per mol), A is the cross-sectional area occupied by each active surface Cu atom (6.803 × 10−20 m2 per atom), f is a particle shape correction factor (considering a spherical/cubic structure, its value is taken as 6) and Z is the density of copper (8.92 × 106 g m−3). SCu is expressed in units of m2 gCu−1 and CSCu is expressed in nm. The value of the stoichiometric ratio (S) is considered as two (2Cu + CO → Cu(CO)Cu).36
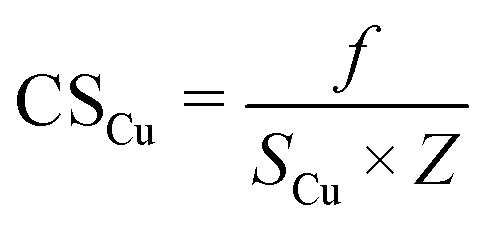
X-ray diffraction (XRD) patterns of reduced catalyst powders were recorded in the 2θ range of 5–90° and with a scan rate of 2° min−1 on an X'Pert Pro Philips diffractometer equipped with a Cu Kα radiation source (λ = 0.15406 nm) and a proportional counter detector. The textural properties of the catalysts were determined at −196 °C using a Micromeritics ASAP 2020 instrument. Nitrogen adsorption–desorption isotherms were collected in the relative pressure (P/Po) range of 0.01 to 0.99. Prior to nitrogen adsorption, the catalyst samples were outgassed in vacuum at 200 °C for 6 h to achieve clean surfaces. The specific surface area (SBET) was determined by the Brunauer–Emmett–Teller (BET) method (P/P0 = 0.01–0.2) and the total pore volume (PV) and pore diameter (PD) were determined using the Barret–Joyner–Halenda (BJH) model.
The acidity of the reduced catalyst was determined by temperature-programmed desorption of ammonia (NH3-TPD) performed on a Micromeritics Auto Chem 2910 instrument. In a typical experiment, 0.1 g of reduced catalyst was taken in a U-shaped quartz tube. Prior to analysis, pre-treatment was performed in a helium flow (30 ml min−1) at 200 °C for 1 h. Then, the temperature of the sample was brought down to 100 °C and ammonia in helium (10 vol%) was fed for 1 h (at a flow rate of 30 ml min−1). Later, it was flushed with helium (30 ml min−1) for 1 h at the same temperature. Baseline stability was checked prior to the analysis. TPD measurements were recorded in the temperature range of 75 to 550 °C. The plot obtained was deconvoluted (using Origin 6.1 or Pro 8.5) into different peaks corresponding to NH3 desorption from acid sites of different strengths. The area of the peak and prior calibration enabled quantification of the acid sites.
Transmission electron microscopy (TEM) images of reduced catalyst samples were recorded on a Technai-G2 T20 super twin instrument fitted with a 200 kV field emission gun. The catalyst powder was dispersed in isopropanol, sonicated, drop-casted on a TEM copper grid and dried at 25 °C. Digital micrograph (Gatan) software was used to calculate the size of particles. Around 100–150 particles were considered in particle size estimation.
Hydrogen-temperature-programmed reduction (H2-TPR) measurements were performed on a Micromeritics Auto Chem 2910 instrument. In a typical procedure, 0.1 g of unreduced catalyst sample was charged in a U-shaped quartz tube. It was heated at 200 °C for 1 h and cooled to 50 °C. The TPR pattern was recorded in the temperature range of 50 to 800 °C while flowing 5 vol% H2 in Ar (30 ml min−1).
X-ray photoelectron spectroscopy (XPS) studies of the reduced catalysts were conducted on a Thermo Fisher Scientific Instrument, UK (model: K-Alpha+) equipped with an Al Kα anode (1486.6 eV) in a transmission lens mode and a multi-channel plate (MCP) detector. The lines of XPS were referenced to C1s appearing at 284.8 eV. The XP spectra were deconvoluted using XPSPEAK41 software.
Reaction procedure and product analysis
Catalytic tests were performed using a Parr stainless steel batch reactor (300 ml) equipped with a temperature controller. Prior to the reaction, the catalyst was reduced in a flow of hydrogen (20 ml min−1) at 350 °C for 2.5 h. In a typical reaction, known quantities of OA (Sigma Aldrich), n-heptane (solvent, 99.9%, Merck) and the reduced catalyst were placed in an autoclave which was then flushed and pressurized with hydrogen gas (5–20 bar). The temperature of the reactor was raised to 240 to 300 °C and the reaction was conducted for 1 to 5 h. After the reaction, the autoclave was cooled to room temperature (25 °C) and depressurized. The catalyst was separated by centrifugation/filtration. The solvent in the liquid portion was removed over a rotary evaporator and the product isolated was subjected to analysis by titration with NaOH solution (for acid value estimation and OA conversion) and to gas chromatography (GC, for product selectivity) as reported by us earlier.23,24Results and discussion
Representative TEM images of 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 are shown in Fig. 1. Particle size distribution curves (ESI,† Fig. S1) pointed out a narrow size distribution of Cu particles in the range of 1–5 nm. The average particle size of Cu in 10Cu/A2O3 was 2.5 nm and that in 10Cu–4WOx/Al2O3 was 3 nm. Thus, tungsten addition (4 wt%) affected only marginally the Cu particle size. XRD studies revealed that Cu and WO3 crystallites in the reduced catalysts are below the X-ray detection limit of 3 nm. No separate peaks other than the XRD peaks of γ-Al2O3 were detected for 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 (Fig. 2). | ||
| Fig. 1 TEM images of reduced 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 catalysts. | ||
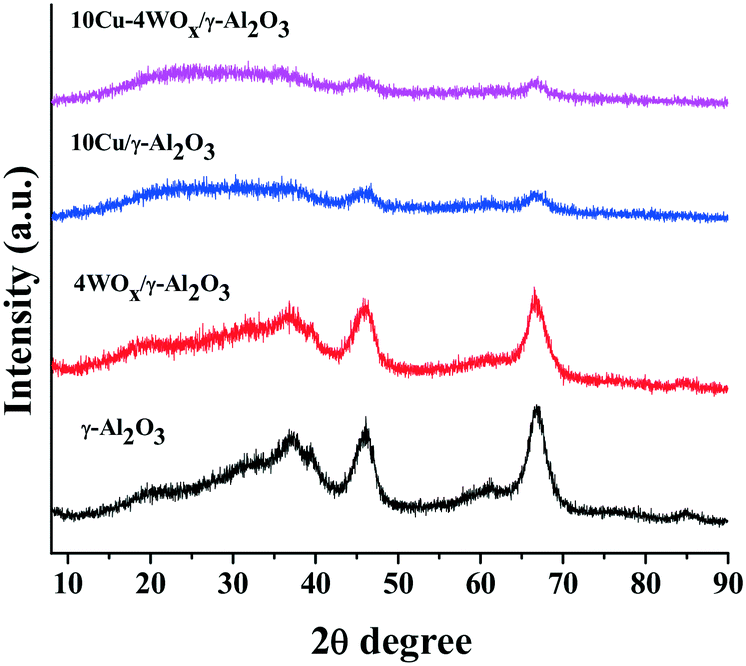 | ||
| Fig. 2 XRD patterns of γ-Al2O3 and reduced 4WOx/Al2O3, 10Cu/Al2O3 and 10Cu–4WOx/Al2O3. | ||
CO-chemisorption data of xCu/Al2O3 and 10Cu–yWOx/Al2O3 catalysts are presented in Table 1. The percentage of metal dispersion (DCu), active metal surface area (SCu) and average crystallite size (CSCu) of Cu were determined from the monolayer CO uptake values. WOx/Al2O3 did not adsorb CO on its surface. Hence, CO-chemisorption was chosen to measure the dispersion and crystallite size of Cu in copper-loaded samples. The monolayer CO uptake value of different catalysts decreased with increasing Cu and W contents (Table 1). The decrease for a monometallic catalyst (xCu/Al2O3) was from 90 to 54 μmol g−1 and for a bimetallic catalyst (10Cu–yWOx/Al2O3) was from 87 to 59 μmol g−1. This variation is mainly attributed to an increase in the crystallite size of Cu (from 5 to 31 nm for xCu/Al2O3 and from 10 to 14 nm for 10Cu–yWOx/Al2O3) with increasing metal content. The active metal surface area (per gram of Cu) decreased with increasing metal content from 148 to 22 m2 g−1 for xCu/Al2O3 and from 71 to 48 m2 g−1 for 10Cu–yWOx/Al2O3. Metal dispersion decreased from 23% to 3% for xCu/Al2O3 and from 11% to 7% for 10Cu–yWOx/Al2O3. It may be noted that tungsten addition by 4 wt% did not alter the CO-chemisorption data of the Cu catalyst (10Cu/Al2O3 and 10Cu–4WOx/Al2O3 had nearly the same metal textural property values; Table 1). But with higher amounts of W (8–16 wt%), the crystallite size of Cu in 10Cu–yWOx/Al2O3 had increased and metal dispersion had decreased (Table 1). A similar observation in variation of CO uptake values and metal crystallite sizes was also reported by Regalbuto et al.37 for Pt/WOx/SiO2 catalysts with different amounts of Pt and W. The average crystallite size of Pt increased with increasing Pt and W contents. Zhou et al.38 reported that the dispersion of Pt (measured by CO chemisorption) drops on adding WOx. The low dispersion in the Pt/WOx system was attributed to a partial coverage of the Pt surface by the WOx species.39 They confirmed this by diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy of adsorbed CO. The band at 1837 cm−1 due to bridging CO on the Pt surface (present in Pt/ZrO2) had disappeared in the case of Pt/WOx/ZrO2, inferring a decrease in neighboring Pt atoms on the surface due to partial coverage by the WOx species.39 Thus, WOx addition reduced the exposed surface metal sites as a consequence of increased metal crystallite size and/or by surface coverage of metal with WOx species.
| Catalyst | S BET (m2 g−1) | CO-chemisorption | NH3-TPD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Monolayer CO uptake (μmol g−1) | S Cu (m2 gCu−1) | CSCu (nm) | D Cu (%) | Total acidity (mmol g−1) | Acid site distribution (mmol g−1) | ||||
| Weak (75–200 °C) | Medium (200–350 °C) | Strong (>350 °C) | |||||||
| γ-Al2O3 | 352 | — | — | — | — | 0.99 | 0.256 | 0.551 | 0.183 |
| 5Cu/Al2O3 | 244 | 90 | 148 | 5 | 23 | 1.00 | 0.344 | 0.556 | 0.100 |
| 10Cu/Al2O3 | 183 | 89 | 73 | 9 | 11 | 1.12 | 0.314 | 0.691 | 0.115 |
| 15Cu/Al2O3 | 171 | 68 | 37 | 18 | 6 | 0.93 | 0.279 | 0.608 | 0.043 |
| 20Cu/Al2O3 | 134 | 54 | 22 | 31 | 3 | 0.88 | 0.269 | 0.571 | 0.040 |
| 10Cu–4WOx/Al2O3 | 179 | 87 | 71 | 10 | 11 | 1.38 | 0.417 | 0.638 | 0.325 |
| 10Cu–8WOx/Al2O3 | 168 | 79 | 65 | 10 | 10 | 1.56 | 0.422 | 0.919 | 0.219 |
| 10Cu–12WOx/Al2O3 | 165 | 63 | 52 | 13 | 8 | 1.65 | 0.438 | 0.996 | 0.216 |
| 10Cu–16WOx/Al2O3 | 159 | 59 | 48 | 14 | 7 | 1.67 | 0.462 | 1.077 | 0.130 |
γ-Al2O3 used in this study had a specific surface area (SBET) of 352 m2 g−1. With increasing Cu (5 to 20 wt%) and W (4 to 16 wt%) loading, a marked decrease in SBET of Al2O3 (to 134 m2 g−1) was observed (Table 1). The total pore volume (PV) had also decreased (ESI,† Fig. S2 and Table S1), inferring that the pore mouth of γ-Al2O3 is blocked partially with Cu and WOx particles, affecting the textural property values of alumina. The surface density of tungsten (ρW; atoms per nm2) was estimated using the following equation:
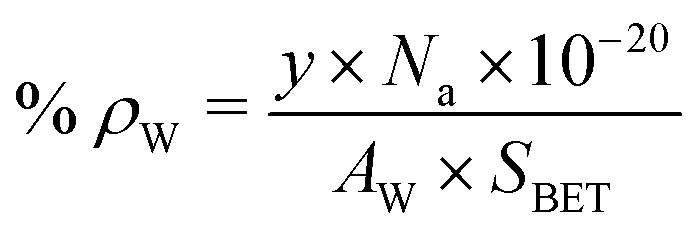
The acidity of the catalysts was determined from NH3-TPD measurements. All the catalysts showed a broad NH3 desorption curve in the temperature range of 75 to 550 °C, which was deconvoluted into desorption peaks arising from acid sites of different strengths. Representative NH3-TPD profiles (continuous lines) and deconvoluted plots (dotted lines) of 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 are shown in Fig. 3. Desorption in the temperature range of 75–200 °C is attributed to that arising from the weak acidic sites. Desorption in the temperature range of 200 to 350 °C is attributed to that arising from the acid sites of medium strength, and that above 350 °C is attributed to that of strong acid sites. The overall acidity of the catalyst increased (from 0.99 to 1.12 mmol g−1) with increasing Cu content up to a value of 10 wt% and above that it decreased to 0.88 mmol g−1. A composition with 10 wt% Cu led to a highly acidic catalyst. WOx addition affected the acidity of the Cu catalyst. It increased the acidity of the catalyst from 1.12 to 1.67 mmol g−1. The weak, medium and strong acid site distribution of 10Cu/Al2O3 was 0.31, 0.69 and 0.12 mmol g−1 and that of 10Cu–4WOx/Al2O3 was 0.42, 0.64 and 0.33 mmol g−1, respectively. In other words, a significant increase in weak and strong acid sites and a decrease in medium acid sites was noted on tungsten addition. With a higher amount of tungsten (>4 wt%), the strong acid sites decreased, while the weak and medium acid sites increased (Table 1). A catalyst composition of 10 wt% Cu and 4 wt% W had the highest amount of strong acid sites. At lower concentrations, W is known to be in a highly dispersed state and at higher concentrations, oligomeric tungsten and WO3 type species are known to be formed.38 While the dispersed tungsten species strongly interacts with the support (Al2O3) leading to strong acid sites, the oligomeric and crystalline WO3 species lead to weak and medium strength acid sites. Si et al.42 found by Fourier transform infrared (FT-IR) spectroscopy of adsorbed NH3 that WOx–ZrO2 samples contain Lewis and Brønsted acid sites. NH3 coordinated to the Lewis acid sites showed characteristic IR bands of NH3 at 1600 and 1200 cm−1 due to σas and σs modes. NH3 coordinated to the Brønsted acid sites (as NH4+) showed bands at 1680 and 1470 cm−1 due to σs and σas modes. When CuO was deposited (CuO/WOx–ZrO2), the intensity of the IR bands due to NH4+ ions decreased, indicating reduction in the concentration of Brønsted acid sites, and those of NH3 coordinated to Lewis acid sites increased. The higher concentration of weak and medium acid sites and lower concentration of strong acid sites observed in the present study for the copper catalysts with >4 wt% W agrees well with the report of Si et al.42
 | ||
| Fig. 3 NH3-TPD profiles (continuous black curve) and deconvoluted plots (dotted blue curves) of reduced 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 catalysts. | ||
10CuO/Al2O3 and 10CuO–4WOx/Al2O3 showed two overlapping reduction peaks in H2-TPR in the temperature range of 150 to 250 °C (Fig. 4). These are corresponded to reduction of CuO to metallic copper.43,44 The low temperature peak is attributed to dispersed CuO particles strongly interacting with the support and the high temperature reduction peak is attributed to bulk CuO particles weakly interacting with the support.44 Both these reduction peaks shifted to lower temperature when WOx was present in the catalyst composition (10CuO–4WOx/Al2O3). The low temperature peak shifted from 187 to 178 °C and the high temperature peak shifted from 230 to 221 °C (Fig. 4). Thus, WOx facilitated the reduction of CuO. The strong interaction between CuO and WOx particles supported on Al2O3 is the reason for the appearance of H2-TPR peaks at lower temperatures.
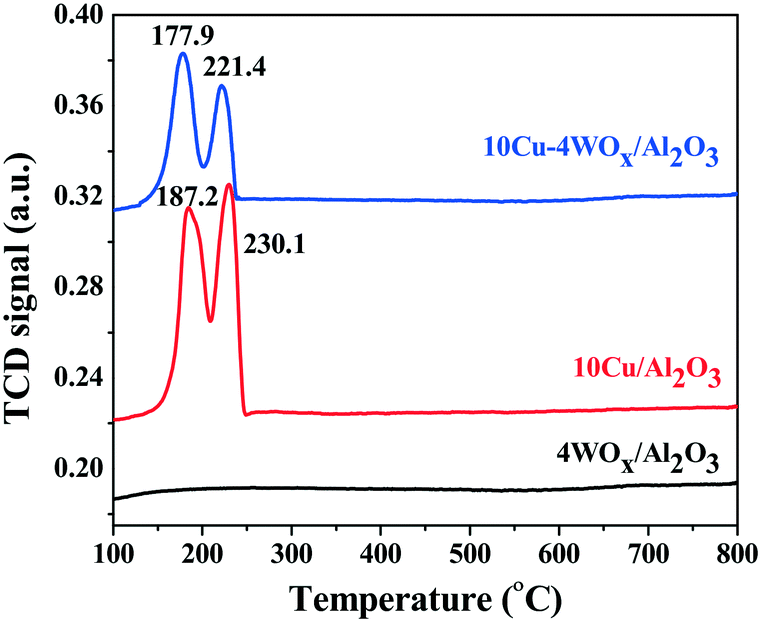 | ||
| Fig. 4 H2-TPR profiles of as-synthesized 4WOx/Al2O3, 10Cu/Al2O3 and 10Cu–4WOx/Al2O3. | ||
No prominent reduction peak in H2-TPR was observed for 4WOx/Al2O3 in the temperature range of 100 to 800 °C. Thus, WOx on Al2O3 did not reduce in hydrogen consumption below 800 °C. The hydrogen consumption by the catalysts in this temperature range is therefore attributed solely to CuO. The overall hydrogen consumption for CuO reduction was lower for 10CuO–10WOx/Al2O3 (0.273 mmol per g catalyst) than for 10CuO/Al2O3 (0.596 mmol per g catalyst). Since the amount of Cu is the same in both the catalysts and all Cu should be reduced below 800 °C, we expect the same hydrogen consumption value for 10CuO/Al2O3 and 10CuO–10WOx/Al2O3 (considering that tungsten is not reduced in this temperature range). However, the lower value observed for the latter catalyst tempts us to postulate that a some Cu is perhaps covered with WOx particles or Al2O3 as in the case of Pt/WOx/ZrO2 (ref. 39) and hence, not reduced. The active copper content participating in the reactions was lower in the former than in the latter. Based on hydrogen consumption we estimated that only 30 mol% of total Cu in the catalyst was reduced to metallic Cu in 10CuO/Al2O3, while it was only 17 mol% in 10CuO–4WOx/Al2O3. The TPR profile was deconvoluted and the low and high temperature peaks were in the proportion of 7% and 93%, respectively, in 10CuO/Al2O3 and 62.7% and 37.3%, respectively, in 10CuO–4WOx/Al2O3. Thus, upon modification with WOx, a greater amount of strongly interacting CuO species had formed in 10CuO–4WOx/Al2O3, while the proportion of weakly interacting CuO particles had decreased. In a related Pt system (4Pt–8WOx/Al2O3),23 we found earlier that with WOx addition, the reduction peak of PtO shifts to lower temperatures due to the increased crystallite size of the platinum. WOx did not show any reduction peak below 800 °C. The present results are in line with these observations. Yang et al.45 found that when the tungsten loading was higher (10 wt%, for example, as in the case of xNi–10W/Al-MCM-41), reduction of both NiO (368–483 °C) and WOx (500–735 °C) was observed. Unlike that observed for our system, the reduction peaks of NiO shifted to a higher temperature, revealing the formation of weakly interacting nickel species with the support. Perhaps, the difference in W content is the reason for the different reduction behaviors and metal–support interactions. A lesser amount of W leads to dispersed WOx species which require a higher temperature (>800 °C) as seen in the present study. Further, Yang et al.45 reported formation of a very active Ni17W3 species in their catalyst composition. However, similar such Cu–W and Cu–Al phases were not detected by XRD for our catalyst compositions (Fig. 2).
The oxidation states of copper and tungsten in reduced 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 catalysts were determined using XPS technique. The spectra of Cu in the 2p core level region and of W in the 4f core level region are depicted in Fig. 5. Spectral fitting of Cu 2p peaks revealed the presence of copper in +2, +1 and zero oxidation states. The shake-up satellite peak centered at around 944 eV confirmed the presence of Cu2+ species.46,47 Thus, not all copper was present in the metallic state in the measured catalysts. The intensity of the shake-up satellite was low for 10Cu–4WOx/Al2O3. The relative percentages of surface Cu species were calculated from the ratio of the areas of corresponding characteristic peaks and listed along with the binding energy values in Table 2. For 10Cu/Al2O3, the 2p3/2 and 2p1/2 lines of Cu2+ species occurred at 936.4 and 956.2 eV. Those of the Cu1+ species appeared at 934.8 and 954.6 eV and those of the Cu0 species appeared at 932.7 and 952.8 eV.48 These XP lines of Cu (2+, 1+ and 0) for 10Cu–4WOx/Al2O3 appeared at lower binding energy values (by 0.2–0.6 eV, Table 2) indicating that copper in the bimetallic catalyst is electron rich and hence more metallic in nature than that in the monometallic catalyst. The relative concentration ratio of 0, +1 and +2 Cu species is 33:37:30 (for 10Cu/Al2O3) and 56:26:18 (for 10Cu–4WOx/Al2O3). Thus, the WOx-modified catalyst contained a higher proportion of reduced copper than the unmodified catalyst. The reduction curves in H2-TPR (appearing at lower temperature) have also indicated that copper on WOx-promoted Al2O3 reduces more easily than on pure Al2O3.
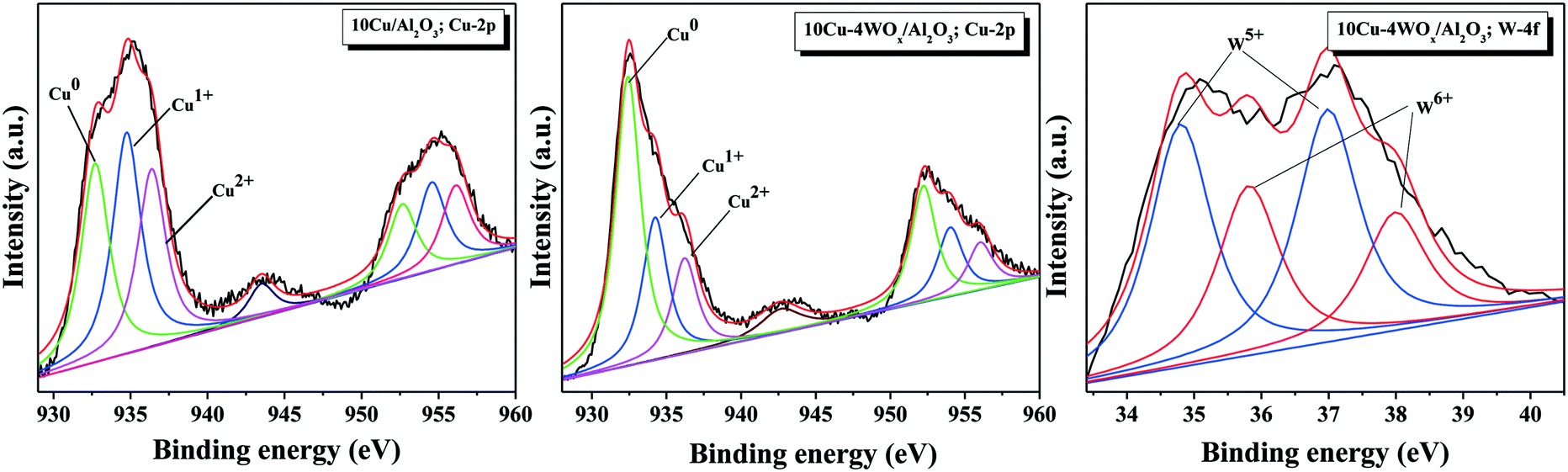 | ||
| Fig. 5 XPS profiles of reduced 10Cu/Al2O3 in the Cu 2p core level region and 10Cu–4WOx/Al2O3 in the Cu 2p and W 4f core level regions. The deconvoluted peaks of Cu0 (green), Cu1+ (blue) and Cu2+ (pink) as well as of W5+ (blue) and W6+ (red) are indexed. | ||
| Catalyst | Cu 2p peak positions (eV) | Relative intensity (%) | W 4f peak positions (eV) | Relative intensity (%) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cu0 | Cu1+ | Cu2+ | Cu0 | Cu1+ | Cu2+ | W5+ | W6+ | W5+ | W6+ | ||||||
| 2p3/2 | 2p1/2 | 2p3/2 | 2p1/2 | 2p3/2 | 2p1/2 | 4f7/2 | 4f5/2 | 4f7/2 | 4f5/2 | ||||||
| 10Cu/Al2O3 | 932.7 | 952.8 | 934.8 | 954.6 | 936.4 | 956.2 | 33 | 37 | 30 | — | — | — | — | — | — |
| 10Cu–4WOx/Al2O3 | 932.2 | 952.2 | 934.2 | 954.0 | 936.2 | 956.0 | 56 | 26 | 18 | 34.8 | 37.0 | 35.9 | 38.0 | 60 | 40 |
This difference in the concentration and electronic structure of Cu speciation in supported Cu catalysts can bring about variation in catalytic activity.22–24 The H2-TPR curves of 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 (Fig. 4) showed complete reduction of copper at 230.1 and 221.4 °C, respectively. However, detection of Cu1+ and Cu2+ species in XPS for the samples reduced at 350 °C for 2.5 h under a hydrogen flow of 20 ml min−1 reveals that the catalyst samples were exposed to air during handling, allowing some of the copper to oxidize to +1 and +2 states during sample preparation for the XPS measurement. Alternatively, copper covered with WOx and Al2O3 particles is perhaps not fully reduced and showed peaks corresponding to +1 and +2 species. Further, Cu–WOx and Cu–Al2O3 phases in the catalysts can explain the presence of Cu1+ and Cu2+ species in the reduced catalysts. Such phases were, however, not detected in the XRD (Fig. 2). Thus, even if they were there, they might be present as amorphous phases not showing XRD peaks.
Curve fitting of the W 4f XP spectrum confirmed the presence of tungsten in both +5 and +6 oxidation states. The 4f7/2 and 4f5/2 lines of W5+ species appeared at 34.8 and 37.0 eV and those of W6+ species appeared at 35.9 and 38.0 eV.49–51 Neat WOx/Al2O3 contained W in the +6 oxidation state only. However, in the presence of Cu and in a hydrogen environment, some of the tungsten oxide reduced to acidic W5+OxH species. These reduced tungsten oxide species are known to activate fatty acids (OA, for example) and alter the deoxygenation mechanism toward HDO.21–24 The relative surface concentration of W5+:W6+ was in the ratio of 60:40. The reduction of tungsten from the +6 to +5 oxidation state was found higher in Pt (ref. 23) than in Cu–WOx/Al2O3 catalyst. Higher temperature (>800 °C) is needed to reduce “neat” WO3. However, tungsten oxide in the presence of a metal and in a hydrogen environment can reduce at lower temperatures. XPS confirmed such a reduction of W6+ to W5+ species in 10Cu–4WOx/Al2O3 treated with hydrogen at 350 °C for 2.5 h. However, H2-TPR did not show the corresponding tungsten reduction peak (Fig. 4) perhaps due to the low sensitivity of the thermal conductivity detector used or the baseline of the curve covered the broad and tiny reduction peaks of tungsten.
Catalytic activity
The catalytic activity data of Al2O3-supported xCu and 10Cu–yWOx catalysts for deoxygenation of oleic acid (OA) are presented in Table 3. Deoxygenation of OA yielded n-octadecane (C18) formed by the HDO path and n-heptadecane (C17) formed by the DCO path. Octadecanal and octadecanol were the intermediate (incomplete deoxygenation) products (referred to as others). C10–16 HCs were the cracking products. A control experiment at 300 °C and 20 bar H2 pressure after 5 h of reaction revealed that very little deoxygenation occurred (OA conversion = 4 mol%) in the absence of a catalyst (Table 3, run no. 1). Over 8WOx/Al2O3, the conversion of OA at these conditions was only 8 mol% (run no. 2). However, with the xCu/Al2O3 catalyst, OA conversion increased to 38–97 mol% depending on the copper content (x) (run no. 3–6). 10Cu/Al2O3 enabled the highest OA conversion of 97 mol% after 5 h of reaction (run no. 4). Others (octadecanal and octadecanol) were the major products over this catalyst. It gave a higher amount of C18 than the other Cu compositions. The turnover frequency (TOF = moles of C18 formed per mole of surface Cu atoms (estimated from metal dispersion obtained from CO-chemisorption) per hour) was the highest (19 h−1) for the 10Cu/Al2O3 catalyst.| Run no. | Catalyst | Reaction temp. (°C) | H2 pressure (bar) | Reaction time (h) | OA conv. (mol%) | TOFb (h−1) | Product selectivity (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| C18 | C17 | C10–16 | Others | |||||||
| a Reaction conditions: OA = 2 g, n-heptane (solvent) = 30 g, catalyst = 0.2 g. b Turnover frequency (TOF) = moles of C18 formed per mole of surface Cu atoms (estimated from CO-chemisorption) per hour. | ||||||||||
| 1 | Nil | 300 | 20 | 5 | 4.0 | — | 5.0 | 8.0 | 0.4 | 86.6 |
| 2 | 8WOx/Al2O3 | 300 | 20 | 5 | 9.0 | — | 9.0 | 43.2 | 0.8 | 47.0 |
| 3 | 5Cu/Al2O3 | 300 | 20 | 5 | 38.0 | 2 | 13.3 | 12.7 | 0.5 | 73.5 |
| 4 | 10Cu/Al2O3 | 300 | 20 | 5 | 97.0 | 19 | 48.4 | 12.5 | 0.1 | 39.0 |
| 5 | 15Cu/Al2O3 | 300 | 20 | 5 | 87.0 | 14 | 32.5 | 3.6 | 0.1 | 63.8 |
| 6 | 20Cu/Al2O3 | 300 | 20 | 5 | 67.0 | 10 | 20.0 | 4.4 | 0.1 | 75.5 |
| 7 | 10Cu–2WOx/Al2O3 | 300 | 20 | 1 | 35.0 | 7 | 10.4 | 8.9 | 0.2 | 80.5 |
| 8 | 10Cu–4WOx/Al2O3 | 240 | 20 | 1 | 10.0 | 1 | 2.9 | 1.4 | 0.2 | 95.5 |
| 9 | 10Cu–4WOx/Al2O3 | 260 | 20 | 1 | 22.0 | 4 | 8.0 | 3.7 | 0.3 | 88.0 |
| 10 | 10Cu–4WOx/Al2O3 | 280 | 20 | 1 | 72.0 | 114 | 77.6 | 3.6 | 0.7 | 18.1 |
| 11 | 10Cu–4WOx/Al2O3 | 300 | 5 | 1 | 39.0 | 10 | 13.7 | 7.6 | 0.4 | 78.3 |
| 12 | 10Cu–4WOx/Al2O3 | 300 | 10 | 1 | 51.0 | 27 | 25.6 | 8.9 | 0.5 | 65.0 |
| 13 | 10Cu–4WOx/Al2O3 | 300 | 20 | 1 | 97.0 | 182 | 91.9 | 3.4 | 0.6 | 4.0 |
| 14 | 10Cu–4WOx/Al2O3 | 300 | 30 | 1 | 100 | 190 | 93.0 | 4.5 | 0.4 | 2.1 |
| 15 | 10Cu–8WOx/Al2O3 | 300 | 20 | 1 | 91.0 | 191 | 93.2 | 4.0 | 0.3 | 2.5 |
| 16 | 10Cu–8WOx/Al2O3 | 300 | 20 | 5 | 100.0 | 40 | 89.4 | 2.0 | 0.5 | 8.1 |
| 17 | 10Cu–12WOx/Al2O3 | 300 | 20 | 1 | 56.0 | 132 | 84.0 | 5.4 | 0.6 | 10.0 |
| 18 | 10Cu–16WOx/Al2O3 | 300 | 20 | 1 | 53.0 | 126 | 85.0 | 4.5 | 0.5 | 10.0 |
To monitor the effect of WOx on the catalytic activity of Cu, bimetallic catalysts (with 10 wt% Cu and varying amounts (y) of W) were evaluated for 1 h at 300 °C and 20 bar H2 pressure (run no. 7, 13, 15, 17 and 18). OA conversion increased with increasing W content up to 4 wt% and above that the OA conversion decreased. 10Cu–4WOx/Al2O3 exhibited OA conversion of 97% at the end of 1 h, which the monometallic catalyst (10Cu/Al2O3) yielded only after 5 h (compare run no. 13 with 4). At similar conversions (ca. ∼97%), C18 is the major product over the bimetallic catalyst (run no. 13), while significant amounts of others (39.0%) and C17 (12.5%) formed on the monometallic Cu catalyst (Fig. 6). Thus, WOx enhanced the catalytic HDO activity and selectivity of Cu/Al2O3.
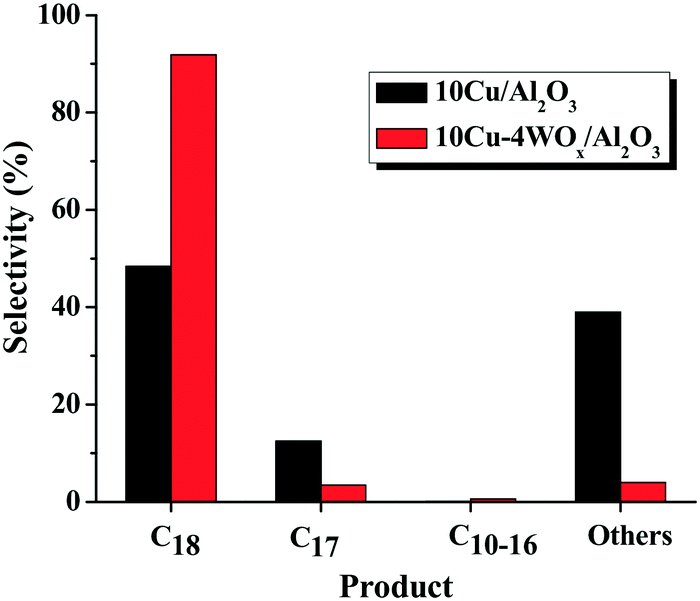 | ||
| Fig. 6 Plot showing the influence of WOx on product selectivity at similar conversions of OA (97.0%) over 10Cu/Al2O3 and 10Cu–4WOx/Al2O3 catalysts. Reaction conditions: see Table 3, run no. 4 and 13. | ||
Huang et al.52 employed a coprecipitation approach for preparing bifunctional Ru–W/SiAl catalysts for the hydrogenolysis of phenols and phenyl ethers to arenes. Coprecipitation is a simpler method for catalyst preparation. However, we followed sequential wet impregnation, as this method lends most of the Cu to locate on WOx-modified Al2O3 while coprecipitation can lead to Cu with little contact with WOx. Note that the catalytic activity tests (Table 3) revealed that Cu located on WOx-modified Al2O3 shows higher HDO selectivity (C18) while that on unmodified Al2O3 is active for hydrogenation and DCO (C17) reactions.
Having found that 10Cu–4WOx/Al2O3 is the best HDO catalyst, it was evaluated at different temperatures and pressures. OA conversion and C18 selectivity increased with increasing reaction temperature from 240 to 300 °C (run no. 8, 9, 10 and 13). Also, with increasing H2 pressure, OA conversion and HDO selectivity increased (run no. 11–14). At 300 °C and 30 bar H2, the bimetallic catalyst (10Cu–4WOx/Al2O3) showed complete conversion of OA in 1 h with 93% selectivity for the C18 product (run no. 14). TOF was 190 h−1. The HDO/DCO product ratio over this catalyst was 93.0/4.5. To the best of our knowledge, this is the highest HDO selectivity reported to date in deoxygenation of vegetable oils. Non-noble metal NiMo and NiW catalysts are known to yield an HDO:DCO product ratio in the range of 69:21 to 20:80.14,15 Pt–WOx/Al2O3 (ref. 23) and Pt–MoOx/ZrO2 (ref. 24) gave an HDO:DCO product ratio of 88:9 and 91:7.3, respectively. The highest activity and HDO selectivity over the non-sulfided, non-noble metal catalyst of the present study is therefore expected to lead to an eco-friendly, economically beneficial fatty acid deoxygenation process producing renewable HCs (green diesel). The optimal reaction conditions for the HDO reaction were 300 °C and 30 bar H2 (Table 3), but when comparing the catalytic performances of the catalysts with varying W loadings (entries 7, 13, 15, 17 and 18) we employed 20 bar H2 instead of 30 bar because better differentiation can be obtained when the catalysts are compared at conditions below the optimum level.
Irrespective of reaction conditions and composition, a correlation existed between OA conversion and C18 yield over 10Cu–yWOx/Al2O3 catalysts (Fig. 7). The higher the OA conversion, the higher was the selectivity/yield of C18 product. Any deviation from the correlation is perhaps due to changes in the crystallite size and acidity of the catalysts. In fact, we found a correlation (Fig. 8) between the TOF data (Table 3) and the average crystallite size of Cu as well as the acidity of the catalysts (Table 1). There seems to be an optimum value for the crystallite size of Cu (10–10.5 nm) and acidity (1.56 mmol g−1) to get the highest TOF value. The TOF of the catalyst was lower above and below that critical value. The catalyst with 10 wt% Cu and 4–8 wt% W had the optimum composition. Too low or high content of W leads to catalysts with inferior catalytic performance.
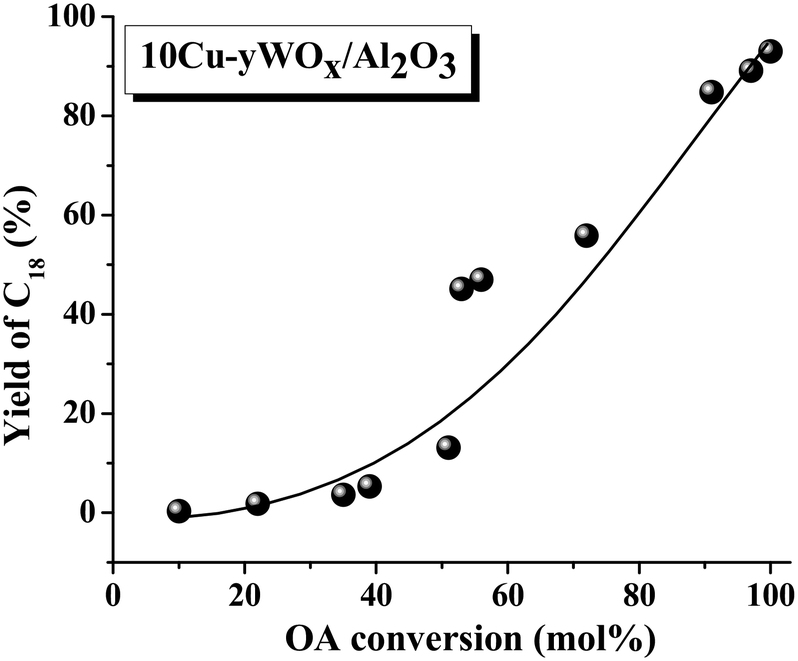 | ||
| Fig. 7 Correlation between OA conversion and yield of C18 over 10Cu–yWOx/Al2O3 catalysts. Data taken from Table 3 (run no. 7–15, 17 and 18). | ||
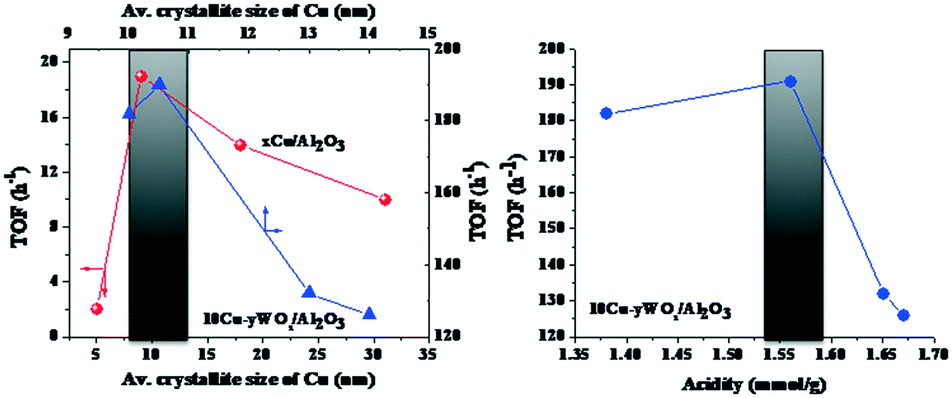 | ||
| Fig. 8 Dependence of TOF on the average crystallite size of Cu for xCu/Al2O3 and 10Cu–yWOx/Al2O3 catalysts (left) and on the acidity of the 10Cu–yWOx/Al2O3 catalysts (right). Data taken from Tables 1 and 3. | ||
10Cu–4WOx/Al2O3 was stable and reusable. After the catalytic run, it was separated from the liquid product by centrifugation/filtration. Then, it was washed with acetone, dried (110 °C), reduced in hydrogen (350 °C) and used in the recycle run (conducted at 300 °C and 30 bar H2 for 1 h). Quantitative conversion of OA with C18, C17, C10–16 and other products selectivity of 93.5%, 4.2%, 0.3% and 2.0%, respectively, was yielded. The spent catalyst showed similar structural characteristics (XRD and CO-chemisorption) to that of the fresh catalyst, confirming its stability in the reaction.
The XPS study pointed out that the proportion of metallic copper (Cu0) was higher in the WOx-promoted bimetallic Cu catalyst (56%) than in the unpromoted monometallic Cu catalyst (33%) (Table 2). In the presence of copper, a number of W species (60%) were reduced from the +6 to the +5 oxidation state, creating additional acidity (Table 1). For the related Pt catalysts,22–24 the reduced WOx species (instead of Al2O3) activate OA and direct the reaction path from DCO to HDO. If the role of Cu–WOx is the same as that of Pt–WOx in our earlier studies,22–24 the HDO/DCO selectivity would have remained unchanged. The higher HDO/DCO selectivity obtained over the Cu–WOx catalyst points out a mechanism other than that over the Pt–WOx catalyst (OA adsorption on reduced WOx, resulting in the formation of an ester between WOx–H and OA and following that the hydrogenation of the CC bond and successive deoxygenation forming octadecane with no other intermediates like aldehyde and alcohol). In the well-established reaction network (as on NiMo catalysts), the reduction of OA to aldehyde and its rapid equilibrium with the corresponding alcohol, the parallel decarboxylation of aldehyde to heptadecane and dehydration of alcohol to octadecane is the reaction pathway. Observation of a significant amount of others (aldehyde + alcohol) over the monometallic xCu/Al2O3 catalysts (Table 3, run no. 3–6), infers that the reaction on Cu–WOx may proceed the same as that on the NiMo catalysts. But then, the difference is that the dispersed, acidic (mainly moderate and strong acidity), reduced WOx–H facilitates OA adsorption (as in the case of Pt–WOx) and may increase the dehydration of the intermediate alcohol. The electronically richer Cu0 may accelerate the hydrogenation of the intermediate octadecene to octadecane. The dependence of TOF on the Cu crystallite size (Fig. 8) suggests that hydrogen adsorption and hydrogenation could be the rate-limiting step of the deoxygenation reaction. Higher acidity and reduced Cu0 species are, therefore, responsible for the higher HDO activity of the Cu–WOx/Al2O3 catalysts. Formation of considerable amounts of intermediate esters by reaction of the intermediate aldehyde and alcohol was reported for Ni catalysts and mainly for the deoxygenation under solvent-free conditions.15 The absence of these ester intermediate compounds in the present work is due to the different catalytic behavior of copper with respect to nickel or due to the presence of solvent. Detailed kinetic and reaction mechanism studies are needed for further confirmation, which will be the topic of our further study.
Conclusions
Bimetallic WOx-promoted Cu/Al2O3 was found to be more active and HDO selective than the monometallic Cu/Al2O3 catalyst in converting OA into renewable fuel-grade HCs. A catalyst with 10 wt% Cu and 4 wt% W exhibited complete conversion of OA with 93% selectivity to n-octadecane (C18) at 300 °C, 30 bar H2, reaction time of 1 h and reactant oleic acid-to-catalyst weight ratio of 2 g/0.2 g. The HDO product yield over this catalyst is the highest known to date for a supported non-noble metal catalyst. The crystallite size of Cu, reducibility of metal and acidity of the catalyst influenced the catalytic HDO activity and selectivity. A bimetallic composition having copper with an optimum crystallite size of about 10 nm and acidity of 1.56 mmol g−1 was found to be the highest HDO product yielding catalyst.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SD thanks the Council of Scientific and Industrial Research (CSIR), New Delhi, for the CSIR-Bhatnagar Fellowship (SSB 000926; P90807). The authors thank Dr. Amitava Das and Dr. P. S. Subramanian (CSIR-Central Salt & Marine Chemicals Research Institute, Bhavnagar, India) for allowing us to use the nitrogen-physisorption facility.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS PubMed.
- G. W. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184 CrossRef CAS PubMed.
- T. V. Choudhary and C. B. Phillips, Appl. Catal., A, 2011, 397, 1 CrossRef CAS.
- C. Zhao, T. Bruck and J. A. Lercher, Green Chem., 2013, 15, 1720 RSC.
- Biomass to Biofuels: Strategies for Global Industries, ed. A. A. Vertés, N. Qureshi, H. P. Blaschek and H. Yukawa, John Wiley & Sons Ltd., 2010 Search PubMed.
- S. Lestari, P. Mäki-Arvela, H. Bernas, O. Simakova, R. Sjöholm, J. Beltramini, G. Max Lu, J. Myllyoja, I. Simakova and D. Y. Murzin, Energy Fuels, 2009, 23, 3842 CrossRef CAS.
- B. Donnis, R. G. Egeberg, P. Blom and K. G. Knudsen, Top. Catal., 2009, 52, 229 CrossRef CAS.
- http://www.nesteoil.com .
- J. A. Petri and T. L. Marker, US Pat., 7511181B2, 2009 – UOP LLC, US Search PubMed.
- T. L. Marker, P. Kokayeff, S. F. Abdo and T. N. Kalnes, US Pat., 8003836B2, 2011 – UOP LLC, US Search PubMed.
- http://www.uop.com/pr/release/PR.EniEcofiningFacility.pdf .
- http://www.tyson.com/RenewableEnergy/Iniotiative/Conoco/Default.aspx .
- D. Kubička and H. Horáček, Appl. Catal., A, 2011, 394, 9 CrossRef.
- R. W. Gosselink, S. A. W. Hollak, S.-W. Chang, J. van Haveren, K. P. de Jong, J. H. Bitter and D. S. van Es, ChemSusChem, 2013, 6, 1576 CrossRef CAS PubMed.
- C. Kordulis, K. Bourikas, M. Gousi, E. Kordouli and A. Lycourghiotis, Appl. Catal., B, 2016, 181, 156 CrossRef CAS.
- L. Simakova, O. Simakova, P. Mäki-Arvela, A. Simakova, M. Estrada and D. Y. Murzin, Appl. Catal., A, 2009, 355, 100 CrossRef.
- M. Amadi, A. Nambo, J. B. Jasinski, P. Ratnasamy and M. A. Carreon, Catal. Sci. Technol., 2015, 5, 380 RSC.
- M. V. Tsodikov, A. V. Chistyakov, M. A. Gubanov, P. A. Zharova, S. S. Shapovalov, A. A. Pasynskii, V. V. Kriventsov and I. I. Moiseev, Russ. Chem. Bull., 2015, 64, 2062 CrossRef CAS.
- L. Boda, G. Onyestyák, H. Solt, F. Lónyi, J. Valyon and A. Therrnesz, Appl. Catal., A, 2010, 374, 158 CrossRef CAS.
- S. Janampelli and S. Darbha, ChemistrySelect, 2017, 2, 1895 CrossRef CAS.
- S. Janampelli and S. Darbha, Catal. Today, 2018, 309, 219 CrossRef CAS.
- S. Janampelli and S. Darbha, Mol. Catal., 2018, 451, 125 CrossRef CAS.
- S. Janampelli and S. Darbha, Energy Fuels, 2018, 32, 12630 CrossRef CAS.
- S. Janampelli and S. Darbha, Catal. Commun., 2019, 125, 70 CrossRef CAS.
- M. Peroni, G. Maneino, E. Barath, O. Y. Gutierrez and J. A. Lercher, Appl. Catal., B, 2016, 180, 301 CrossRef CAS.
- S. Foraita, Y. Liu, G. L. Haller, E. Baráth, C. Zhao and J. A. Lercher, ChemCatChem, 2017, 9, 195 CrossRef CAS.
- A. Kiméné, R. Wjcieszak, S. Paul and F. Dumeignil, J. Chem. Technol. Biotechnol., 2019, 94, 658 CrossRef.
- H. Imai, T. Kimura, K. Terasaka, X. Li, K. Sakashita, S. Asaska and S. S. Al-Khattaf, Catal. Today, 2018, 303, 185 CrossRef CAS.
- R. G. Kurushkkin, O. A. Bulavchenko, V. V. Kaichev and V. A. Yakovlev, Appl. Catal., B, 2015, 161, 531 CrossRef.
- E. Kordouli, L. Sygellou, C. Kordulis, K. Bourikas and A. Lycourghiotis, Appl. Catal., B, 2017, 209, 12 CrossRef CAS.
- E. Kordouli, b. Pawelec, K. Bourikkas, C. Kordulis, J. L. G. Fierro and A. Lycourghiotis, Appl. Catal., B, 2018, 229, 139 CrossRef CAS.
- J. Wu, J. Shi, J. Fu, J. A. Leidl, Z. Hou and X. Lu, Sci. Rep., 2016, 6, 27820 CrossRef CAS PubMed.
- A. S. Berenblyum, R. S. Shamsiev, T. A. Podoplelova and V. Y. Danyushevsky, Russ. J. Phys. Chem. A, 2012, 86, 1199 CrossRef CAS.
- C. Denk, S. Foraita, Y. Liu, K. Stoerzinger, Y. Liu, E. Baráth and J. A. Lercher, Catal. Sci. Technol., 2019, 9, 2620 RSC.
- https://nptel.ac.in/courses/103103026/14 .
- Gas sorption system operating manual, model 6, version 4.0, Quantachrome Instruments, Boynton Beach, FL, USA, 2015 Search PubMed.
- J. R. Regalbuto, T. H. Fleisch and E. E. Wolf, J. Catal., 1987, 107, 114 CrossRef CAS.
- W. Zhou, J. Luo, Y. Wang, J. Liu, Y. Zhao, S. Wang and X. Ma, Appl. Catal., B, 2019, 242, 410 CrossRef CAS.
- W. Zhou, Y. J. Zhao, S. P. Wang and X. B. Ma, Catal. Today, 2017, 298, 2 CrossRef CAS.
- D. G. Barton, S. L. Soled, G. D. Meitzner, G. A. Fuentes and E. Iglesia, J. Catal., 1999, 181, 57 CrossRef CAS.
- S. Garcia-Fernández, I. Gandarias, J. Requies, M. B. Güemez, S. Bennici, A. Auroux and P. L. Arias, J. Catal., 2015, 323, 65 CrossRef.
- Z. Si, D. Weng, X. Wu, J. Li and G. Li, J. Catal., 2010, 271, 43 CrossRef CAS.
- D. L. Hoang, T. T. H. Dang, J. Engeldinger, M. Schneider, J. Radnik, M. Richter and A. Martin, J. Solid State Chem., 2011, 184, 1915 CrossRef CAS.
- G. Vidya Sagar, P. V. Ramana Rao, C. S. Srikanth and K. V. R. Chary, J. Phys. Chem. B, 2006, 110, 13881 CrossRef PubMed.
- R. Yang, X. Du, X. Zhang, H. Xin, K. Zhou, D. Li and C. Hu, ACS Omega, 2019, 4, 10580 CrossRef CAS PubMed.
- T. Venkov, M. Dimitrov and K. Hadjiivonov, J. Mol. Catal. A: Chem., 2006, 243, 8 CrossRef CAS.
- J. Morales, A. Caballero, J. P. Holgado, J. P. Espinós and A. R. González-Elipe, J. Phys. Chem. B, 2002, 106, 10185 CrossRef CAS.
- D. Mukherjee, R. Singuru, P. Venkataswamy, D. Damma and B. M. Reddy, ACS Omega, 2019, 4, 4770 CrossRef CAS PubMed.
- T. Y. Kim, D. S. Park, Y. Choi, J. Baek, J. R. Park and J. Yi, J. Mater. Chem., 2012, 22, 10021 RSC.
- S. Zhu, X. Gao, Y. Zhu and Y. Li, J. Mol. Catal. A: Chem., 2015, 398, 391 CrossRef CAS.
- C. Liu, C. Zhang, S. Hao, S. Sun, K. Liu, J. Xu, Y. Zhu and Y. Li, Catal. Today, 2016, 261, 116 CrossRef CAS.
- Y.-B. Huang, L. Yan, M.-Y. Chen, Q.-X. Guo and Y. Fu, Green Chem., 2015, 17, 3010 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and table. See DOI: 10.1039/c9cy01939a |
| This journal is © The Royal Society of Chemistry 2020 |