
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-rechargeable zinc-ion batteries†
Buddha Deka
Boruah
 *a,
Angus
Mathieson
ab,
Bo
Wen
ab,
Sascha
Feldmann
c,
Wesley M.
Dose
ad and
Michael
De Volder
*a
*a,
Angus
Mathieson
ab,
Bo
Wen
ab,
Sascha
Feldmann
c,
Wesley M.
Dose
ad and
Michael
De Volder
*a
aInstitute for Manufacturing, Department of Engineering, University of Cambridge, Cambridge CB3 0FS, UK. E-mail: bd411@cam.ac.uk; mfld2@cam.ac.uk
bCambridge Graphene Centre, University of Cambridge, Cambridge CB3 0FA, UK
cCavendish Laboratory, University of Cambridge, JJ Thomson Ave, Cambridge CB3 0HE, UK
dDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 23rd June 2020
Abstract
Batteries that can be directly recharged by light would offer a new approach to balancing the unpredictable energy surpluses and deficits associated with solar energy. Here, we present a new aqueous zinc-ion battery (photo-ZIB) that can directly harvest sunlight to recharge without the need for external solar cells. The light charging process is driven by photo-active cathodes consisting of a mixture of vanadium oxide (V2O5) nanofibers, poly(3-hexylthiophene-2,5-diyl) and reduced graphene oxide, which provide the desired charge separation and storage mechanism. This process is studied using photodetectors, transient absorption spectroscopy and electrochemical analysis in dark and light conditions. The V2O5 cathodes have gravimetric capacities of ∼190 mA h g−1 and ∼370 mA h g−1 in dark and illuminated conditions respectively and photo-conversion efficiencies of ∼1.2%. Finally, we demonstrate a fully functional photo-ZIB with a ∼64 cm2 optical window in pouch cell format.
Broader contextThe development of reliable off-grid power supplies is essential to fight energy poverty in developing rural communities. Solar systems have been identified by the World Bank as a key technology for universal electricity access, yet solar energy is intermittent, and therefore solar cells usually have to be connected to rechargeable batteries or electrochemical capacitors. This increases the cost and complexity of these solutions. To address this issue, we propose a new cathode material for zinc-ion battery that can be recharged directly by light without the need for external solar cells. Instead, the light charging process in these batteries relies on a novel cathode formulation consisting of a mixture of vanadium pentoxide nanofibers mixed with poly(3-hexylthiophene-2,5-diyl) and reduced graphene oxide. These materials offer a good separation of charges as well as ion storage, which allows for direct light charging of the battery. In addition, the proposed zinc-ion batteries are using an aqueous electrolyte resulting in a cost effective and safe solution. This makes these systems suited for the intended deployment in rural communities. Finally, the fabrication of these electrodes is relatively straight forward, which we have demonstrated by fabricating a 100 cm2 pouch cell. |
Introduction
Development of reliable off-grid power supplies are essential to fight energy poverty in developing rural communities, as well as to power autonomous integrated devices (e.g. internet of things and smart city devices).1–5 Off-grid solar systems have been identified by the World Bank6 as a key technology for universal electricity access, yet solar energy is intermittent, and therefore solar cells are typically connected to rechargeable batteries or capacitors.7–11 This approach often requires additional electronics to match the voltage output of the energy harvester to the input voltage requirements of the energy storage units.7,12,13 This adds to the cost and energy losses in the system.14 Therefore, a material that is capable of harvesting and storing energy simultaneously could provide significant cost reduction while reducing the unpredictability of energy availability associated with solar technologies. It is therefore not surprising that in the past few years, a number of publications have started exploring photo-active battery electrodes that can be recharged directly by light. A Li-ion battery (LIB) electrode consisting of a physical mixture of photovoltaic and battery materials for harvesting solar energy and simultaneous energy storage has been reported,15 but such photo-rechargeable devices suffer from inefficient photo-induced charge carrier separation due to energy level mismatch between energy harvester and storage components, and blocking of light by the energy storage material. Alternatively, we proposed a LIB with a 2D perovskite chemistry that can simultaneously store energy and drive solar recharging, but unfortunately it offers only limited lifetime and low efficiency.14In this paper, we present the first photo-rechargeable zinc-ion battery (photo-ZIB) with a much improved efficiency relative to previously reported systems (∼1.2% in this work compared to 0.06% for LiFePO4–Ru dye LIB photo-cathode system,15 and 0.034% for 2D perovskite LIB system14). This system is using an aqueous electrolyte and Zn metal electrodes, which are cost effective as compared to other battery technologies.16–18 In addition, Zn metal has a high theoretical capacity (∼820 mA h g−1 or ∼5855 mA h cm−3)19,20 and has a redox potential making it a suitable anode for aqueous electrolyte (Zn2+/Zn −0.76 V vs. standard hydrogen electrode).21,22 The cathode is the photoactive part of our design, which consists of vanadium pentoxide (V2O5) nanofibers mixed with poly(3-hexylthiophene-2,5-diyl) (P3HT) and reduced graphene oxide (rGO) which allows for both solar energy harvesting and charge storage in the same electrode as depicted in the energy band diagram of Fig. 1a (the photo-charging mechanism is discussed further on). V2O5 is selected here because of its high reversible capacity (∼375 mA h g−1)23 and suitable bandgap energy for light harvesting in the visible light spectrum (∼2.2 eV).24 Further, V2O5 nanofibers allow charge conduction along the nanofiber length,25 resulting in lower recombination probabilities of photo-excited carriers before extraction.
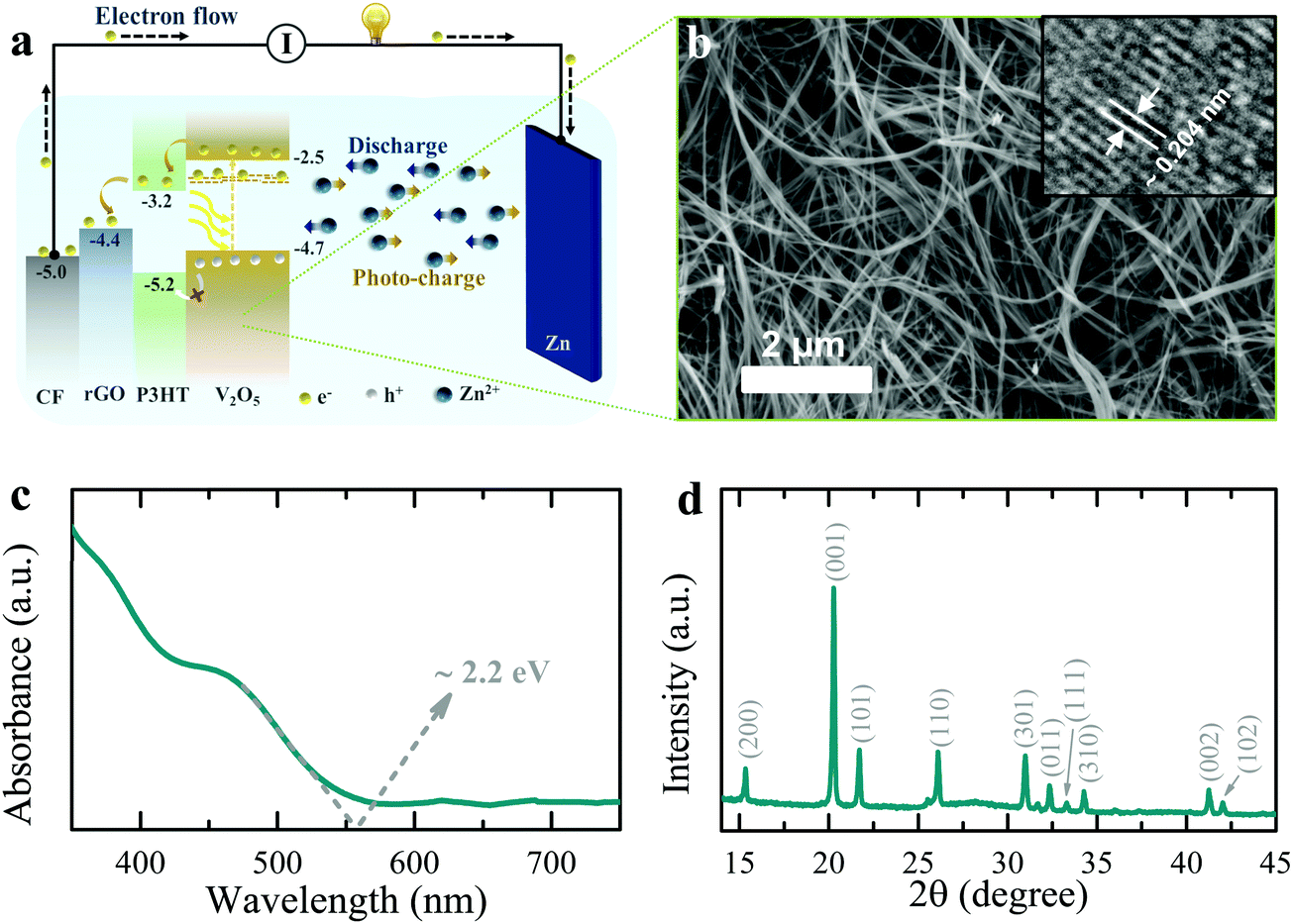 | ||
| Fig. 1 (a) Schematic illustration of the photo-charging mechanism of photo-ZIBs. (b) SEM image of V2O5 nanofibers (scale bar ∼2 μm) and inset showing a high-resolution TEM image. (c) UV-Vis spectrum of V2O5 nanofibers with optical energy band edge of ∼2.2 eV. (d) XRD pattern showing the V2O5 nanofibers have an orthorhombic crystal structure (space group: Pmmn (59); JCPDS card no: 03-065-0131). | ||
Fig. 1b shows an SEM image of the V2O5 nanofibers used in this work, which have a typical diameters of 50 to 100 nm (see synthesis process in methods section) and the TEM inset shows an interplanar spacing of ∼0.204 nm, which corresponds to (202) planes. The absorption spectrum of the V2O5 nanofibers in Fig. 1c shows an optical energy band edge of ∼2.2 eV and XRD data in Fig. 1d confirms the expected orthorhombic crystal structure (space group: Pmmn (59); JCPDS card no: 03-065-0131). Finally, the specific surface area of the V2O5 nanofibers is ∼44.8 m2 g−1 with the BET N2 adsorption/desorption isotherms shown in Fig. S1 (ESI†).
As depicted in Fig. 1a, the energy levels of P3HT and rGO can support the transport of photo-excited electrons from V2O5 nanofibers to the current collector. On the other hand, unpaired photo-induced holes are blocked by P3HT and accumulate in the photo-cathode. To confirm this photo-charge generation and separation process, we measure the photo-current of a photodetector with a layer-by-layer fluorine doped tin oxide coated glass substrate (FTO)/rGO/P3HT/V2O5/Ag structure. An increase in the response current of the photodetector under illumination (λ ∼ 455 nm, intensity ∼2 mW cm−2) in absence of an external bias voltage is observed (see Fig. 2a, ΔI = Ilight − Idark; where Idark and Ilight represent currents in dark and illuminated conditions). This confirms that processes internally drive the separation of photo-excited electrons from V2O5 into FTO through P3HT and rGO. At the same time, P3HT blocks the photo-excited holes to prevent recombination. We verified this by photo-current measurement of a layer-by-layer FTO/P3HT/V2O5/rGO/Ag photodetector (Fig. 2b), where no distinct increase in response current is detected under illumination of light (λ ∼ 455 nm, intensity ∼2 mW cm−2) at 0 V bias voltage. This indicates that permutation of the order of the layers, such that the energies do not align as shown in Fig. 1a, results in no photo-current. Blocking photo-excited holes is important for photo-rechargeable ZIBs as we anticipate they help drive the de-intercalation of Zn2+ ions from the cathode (see further).
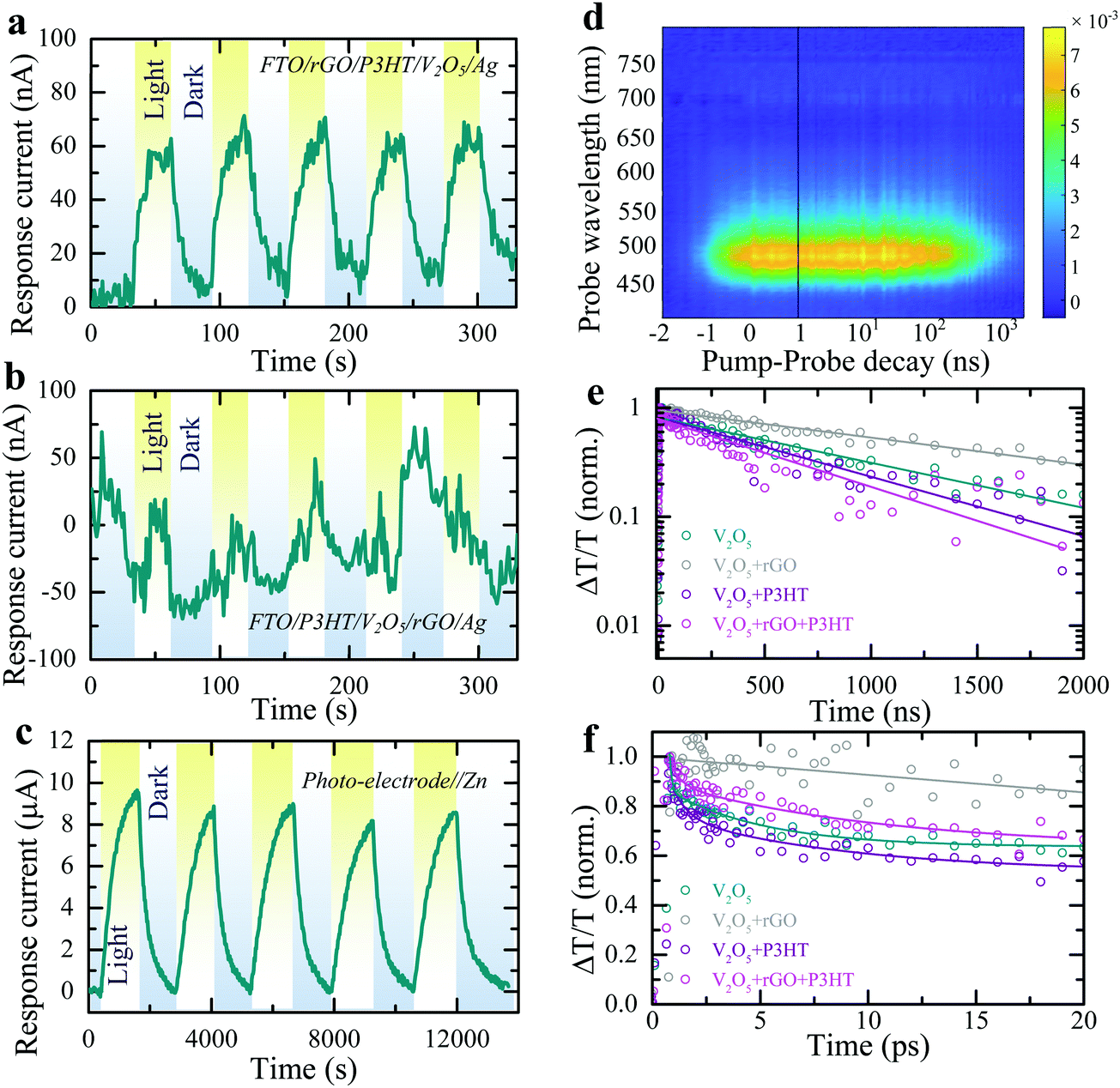 | ||
Fig. 2 Cyclic response current plots of (a) FTO/rGO/P3HT/V2O5/Ag and (b) FTO/P3HT/V2O5/rGO/Ag (layer-by-layer structure) photodetectors at 0 V bias voltage under periodic illumination (λ ∼ 455 nm, intensity ∼2 mW cm−2). (c) Absolute response current plot of the photo-ZIB (photo-electrode//Zn) under dark and light illuminated (λ ∼ 455 nm, intensity ∼12 mW cm−2) conditions at 0 V applied voltage. (d) TA map of a pristine V2O5 film, excited with a 400 nm pump pulse. (e) Normalised TA kinetics of the main ground state bleach region compared to different combinations of pristine V2O5, V2O5 + rGO (V2O5 and rGO in a 98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio), V2O5 + P3HT (V2O5 and P3HT in a 98:2 ratio) and V2O5 + rGO + P3HT (V2O5, rGO and P3HT in a 98:1:1 ratio), indicating an enhanced charge carrier lifetime in the photo-electrode composition containing rGO. (f) Ultrafast normalised kinetics integrated spectrally over the same region, taken at the same fluence as in Fig. 2d and Fig S4b, c (ESI†). :2 ratio), V2O5 + P3HT (V2O5 and P3HT in a 98:2 ratio) and V2O5 + rGO + P3HT (V2O5, rGO and P3HT in a 98:1:1 ratio), indicating an enhanced charge carrier lifetime in the photo-electrode composition containing rGO. (f) Ultrafast normalised kinetics integrated spectrally over the same region, taken at the same fluence as in Fig. 2d and Fig S4b, c (ESI†). | ||
Practical photo-battery electrodes are prepared by mixing V2O5, P3HT, rGO and polyvinylidene fluoride (PVDF) binder in a 93:1:1:5 ratio, followed by drop-casting on a carbon felt (CF) current collector (Fig. S2 shows SEM image, ESI†) rather than using the above layer-by-layer process (see methods). The form factor of photo-batteries inherently needs to be different from thick classic battery electrodes and instead require large surface area to collect light, akin to solar cells, and therefore have a low areal loading, which is 0.8–1.2 mg cm−2 in this work. The device configuration is depicted in Fig. S3 (ESI†). As shown in Fig. 2c, chronoamperometry measurements of these mixed electrodes against a Zn metal anode in dark and light conditions show the response current increases consistently from 0 μA to ∼9 μA when illuminated (λ ∼ 455 nm). This demonstrates that the desired charge transport properties are maintained in the mixed electrodes. Note that the higher response current in the electrochemical cell test (Fig. 2c) as compared to that of electrical measurement test (Fig. 2a and b) is due to a difference in illuminated light intensity (2 mW cm−2vs. 12 mW cm−2) and the difference in conductivity of the current collector (CF vs. FTO).
Photoluminesence (PL) emissions and transient absorption (TA) spectroscopy measurements of the photo-cathodes are used to understand photo-charge carrier dynamics. The steady state PL emission spectra of pure V2O5 as well as different mixtures with P3HT and rGO are shown in Fig. S4a (ESI†). The spectra show emission at ∼720 nm from the oxygen deficiency state, which may originate from charge channeling, i.e. photo-excited charges generated across the bandgap of V2O5 at ∼520 nm relaxing to lower energy state at ∼720 nm where they recombine radiatively resulting in a PL enhancement at that energy.26 A small enhancement of the lower energy emission is observed upon the addition of P3HT – this is due to P3HT's inherent PL at this wavelength bolstering the aggregate signal. TA spectroscopy was used to probe the charge carrier dynamics of the various electrode compositions to investigate the transport properties between the various components. Fig. 2d shows the TA map of a pristine V2O5 film, with the prominent ground-state bleach around 500 nm present. The absorption spectra are shown in more detail at various pump–probe time delays in Fig. S4b (ESI†) which also displays a small bleach feature at ∼720 nm corresponding to oxygen deficiency states.26 The extraction of the relative charge carrier dynamics and absorption strength is demonstrated for the two states in Fig. S4c (ESI†). Similar data sets are obtained for thin films comprising the V2O5 with rGO and P3HT both individually and combined. The corresponding charge carrier dynamics are shown in Fig. 2e for the various compositions over micro-second timescales. Fig. 2f shows the same TA at ultrafast timescales over the first 20 ps after the pump pulse (in all cases 400 nm). On both the ultrafast and micro-second timescales the addition of rGO results in an increase in the observed charge carrier lifetime due to efficient photo-excited charge carrier transport from the V2O5 to the rGO, as depicted in Fig. 1a. This confirms the effectiveness of rGO as a conductive additive in the final electrode composition in transporting photo-excited electrons throughout the electrode and to the current collector. In contrast, the addition of P3HT alone has the opposite effect. The slight reduction of the TA lifetime in the V2O5 with P3HT sample indicates poor charge transfer between the V2O5 and the P3HT and helps to confirm its role as an effective hole-blocking layer between the V2O5 and the current collector. The full electrode composition (photo-cathode), comprising V2O5, rGO and P3HT exhibits similar TA lifetimes to the V2O5 with P3HT sample, implying that the P3HT effectively coats the V2O5 and/or rGO – preventing effective hole transfer between the two materials. Similar results are demonstrated at ultrafast timescales, shown in Fig. 2f, except that the TA lifetime for the photo-cathode is observed to be slightly longer than the pure V2O5 or V2O5 with P3HT samples at these timescales – indicating that the electron transfer from the V2O5 to the rGO is present at these timescales but is obscured by other processes at longer timescales. Note that in addition to an effective hole-blocking layer, P3HT it is also capable of contributing to additional photo-excited charges. Photons of insufficient energy to excite charges in the V2O5 may still be absorbed by the P3HT (bandgap of ∼2.0 eV) and transfer to the rGO (electrons) and V2O5 (holes), increasing the overall device performance.
Next, the electrochemical responses of the photo-ZIBs are analyzed using cyclic voltammetry (CV) and galvanostatic charge discharge (GCD) techniques in dark and illuminated conditions. As shown in Fig. 3a, the CV curves at different scan rates (0.1 mV s−1 to 1.0 mV s−1) over the potential window of 0.2–1.6 V show two pairs of reduction/oxidation peaks at 0.52/0.72 V (weak peaks) and 0.85/1.1 V (strong peaks), which are caused by the Zn2+ intercalation/de-intercalation reactions.23,27,28 Fig. S5a (ESI†) shows CV curves of the initial five cycles at scan of 0.5 mV s−1 in dark. CV tests of the photo-ZIB in dark and light show an increase in current in illuminated conditions as well as a slight reduction in over potentials (Fig. S5b, ESI,† shows CVs at 0.5 mV s−1 in dark and illuminated). The CV profiles (Fig. 3b) at scan rate of 1.0 mV s−1 show an increase of ∼54% in the swept CV area when illuminating the cell (λ ∼ 455 nm, intensity ∼12 mW cm−2). From the current peak position at different scan rates, we calculate that light increases the diffusion constant of this system by ∼43% and ∼32% for intercalation and de-intercalation respectively (see calculations in ESI†).29 This increase in the diffusion constant under illumination is in agreement with rate enhancements observed in LIBs under illumination.30 CV curves taken at different light intensities (λ ∼ 455 nm, 5 mW cm−2 and 12 mW cm−2) show that current increases with light intensity (Fig. S5c, ESI†) because of the increasing numbers of photo-charge charge carriers available to contribute to the photo-charging mechanism and CV curves as well as electrical photo-responses measured at different wavelengths (420 nm, 455 nm, 470 nm, 528 nm and white light) confirm the photo-response over a large wavelength range (Fig. S5d and S6, ESI†). Finally, CV responses of V2O5–rGO (V2O5, rGO and PVDF in a 93:2:5 ratio) photo-cathodes without P3HT in dark and illuminated conditions show a lesser increase in CV swept area of ∼12% (Fig. S7, ESI†) and hence, the low photo-charge response also confirm the benefits of P3HT as a hole blocking layer in the proposed photo-charging mechanism.
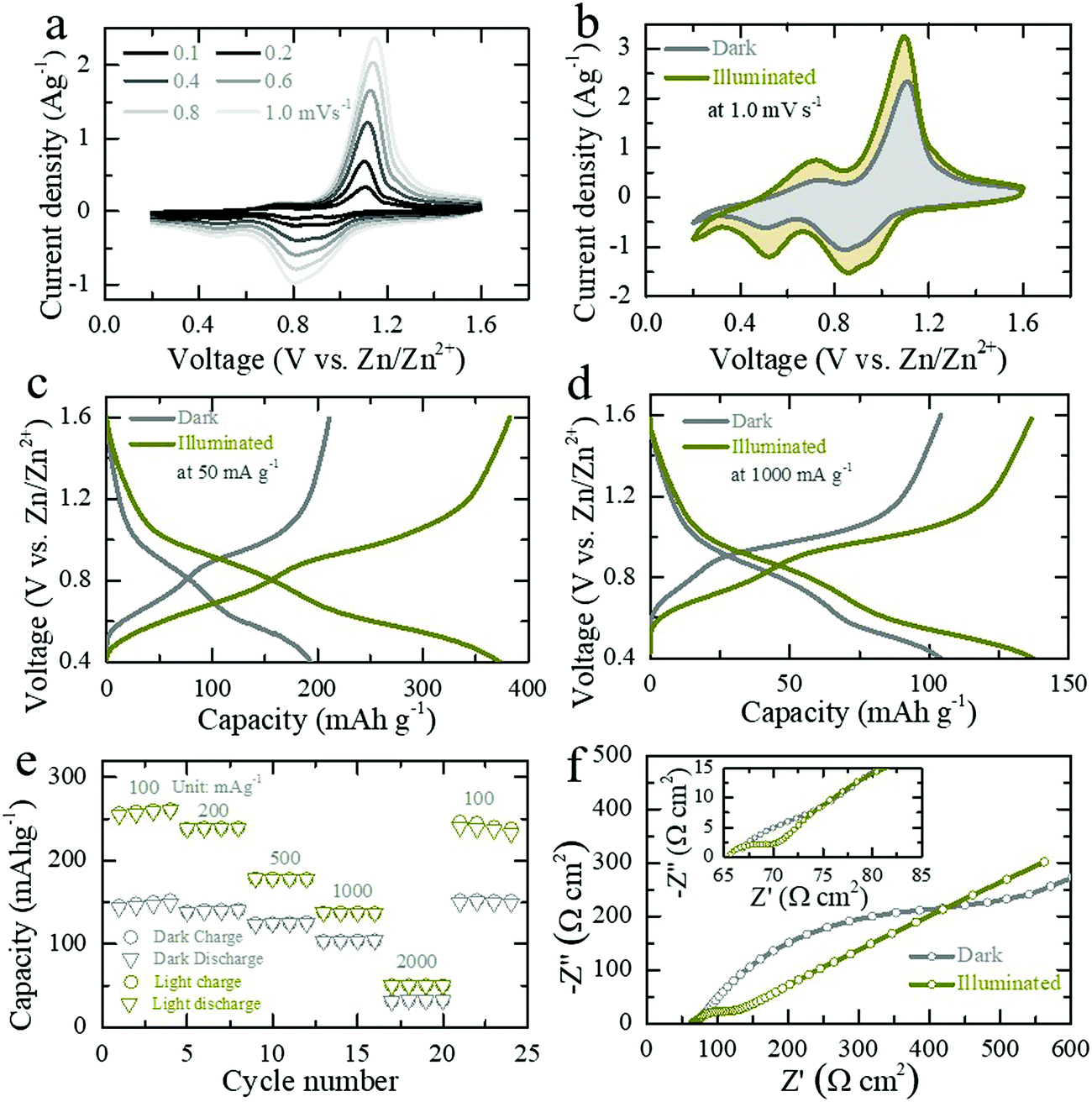 | ||
| Fig. 3 (a) CV curves at scan rates of 0.1 to 1.0 mV s−1 over a working voltage range of 0.2–1.6 V. (b) CV curves at 1.0 mV s−1 in dark and illuminated (λ ∼ 455 nm, intensity ∼12 mW cm−2) conditions. (c and d) GCD profiles at low (50 mA g−1, 5th GCD cycles) and high (1000 mA g−1, 19th GCD cycles) specific currents in dark and illuminated conditions (λ ∼ 455 nm, intensity ∼12 mW cm−2). (e) Rate capacity tests of the photo-ZIB in dark and illuminated conditions. (f) AC impedance spectra of the photo-ZIB in dark and illuminated (λ ∼ 455 nm, intensity ∼12 mW cm−2) conditions. | ||
The GCD curves Fig. 3c and d show that the capacity of photo-ZIBs almost doubles from ∼190 mA h g−1 to ∼370 mA h g−1 when exposed to light (λ ∼ 455 nm, intensity ∼12 mW cm−2) at a discharge specific current of 50 mA g−1 and increases from ∼103 mA h g−1 to ∼137 mA h g−1 at faster discharge rates of 1000 mA h g−1, which leave less time for photo harvesting. For completeness, Fig. S8a–c (ESI†) shows the GCDs at 100 mA g−1, 200 mA g−1 and 500 mA g−1 in dark and light. Fig. 3e shows the rate capacities of the photo-ZIB in dark and light, demonstrating that even at very high specific currents of 2000 mA g−1, illumination still results in a ∼60% of capacity increase. Electrochemical impedance spectroscopy (EIS 10 mHz to 100 kHz at voltage amplitude of 10 mV) was carried out after the second galvanostatic discharge cycle to 1.0 V with 1 h rest, and in dark and light (λ ∼ 455 nm, intensity ∼12 mW cm−2). As shown in Fig. 3f the charge transfer resistance (Rct) decreases from ∼446 Ω cm2 to ∼123 Ω cm2 when illuminated, whereas the high frequency series resistance decreases only from 67 Ω cm2 to ∼65.5 Ω cm2. Further, ex situ XRD (Fig. S9b, ESI†) and Raman (Fig. S9c, ESI†) at different states of discharge and charge confirm the charge storage reversibility of the photo-electrode during cycling. Moreover, Fig. 4a shows the long-term cycling measurement at a specific current of 500 mA g−1. The increase in the capacity at initial few cycles could be due to the activation of photo-cathode material, where similar characteristics are also observed in previous reports of V2O5 based ZIBs.27,31 Moreover, the subsequent capacity fading after 30 cycles could be due to direct drop casting of the photo-cathode material on the CF current collector without used of standard battery electrode conductive additive (e.g. SuperP). Moreover, the lower coulombic efficiencies at initial few cycles (∼98% in the first cycle) could be attributed from severe dendrite growth and self-corrosion of the Zn anode in aqueous electrolyte.32,33
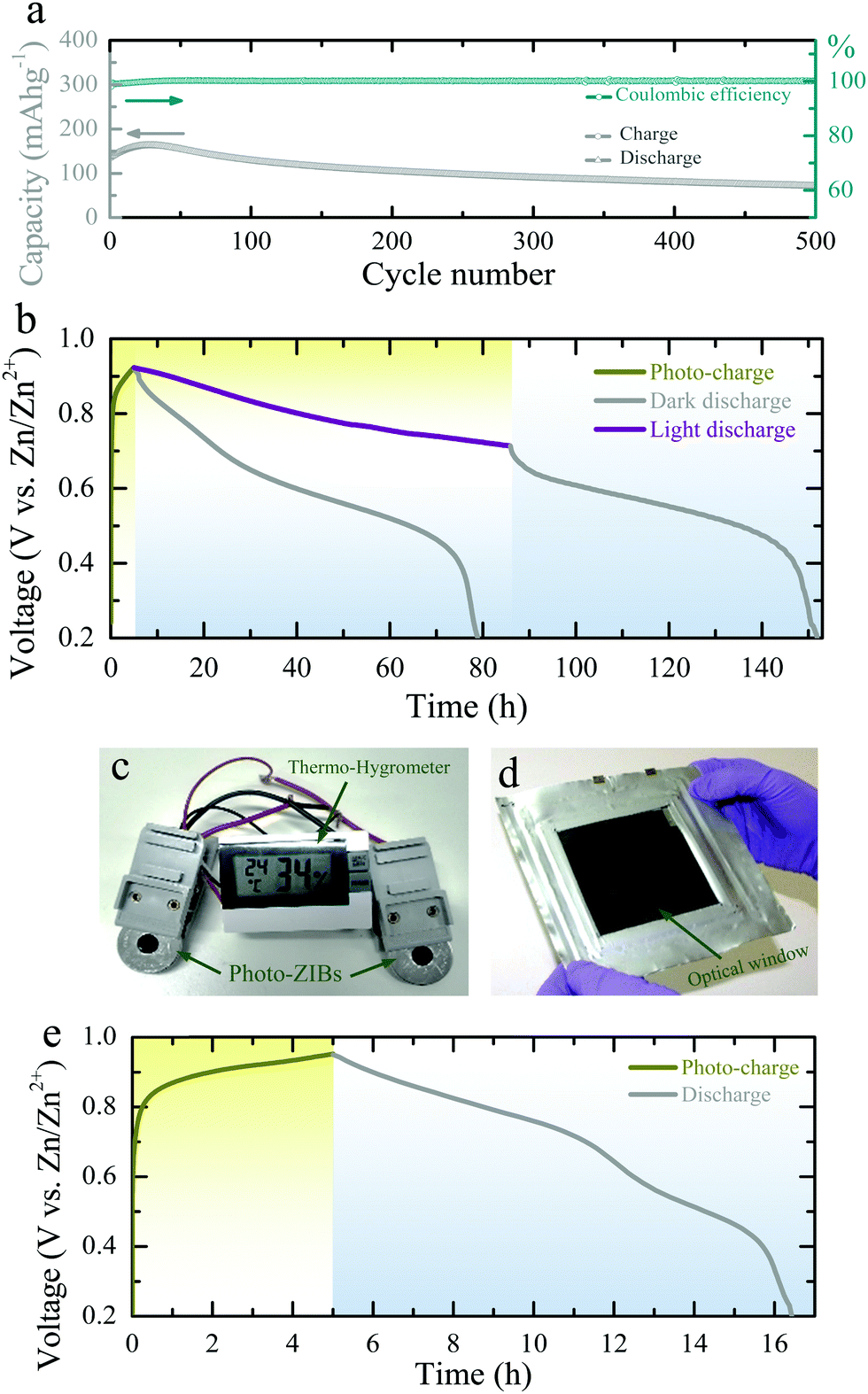 | ||
| Fig. 4 (a) Long-term photo-ZIB cycling in the dark at a specific current of 500 mA g−1. The GCDs are measured between 0.4–1.6 V in a coin cell. (b) Photo-charge (λ ∼ 455 nm, intensity ∼12 mW cm−2) and galvanostatic discharge of the photo-ZIB (specific current of 100 mA m−2) in dark and illuminated conditions. (c) Photograph showing a 1.5 V Thermo-hygrometer powered by two photo-ZIBs charged by light. (d and e) Photograph of a photo-ZIB pouch cell (∼100 cm−2) with a ∼64 cm−2 optical window and a photo-charge (λ ∼ 455 nm, intensity ∼12 mW cm−2) and discharge of the pouch cell. | ||
Finally, in addition to the above CV and GCD measurements, we charge the photo-ZIBs by light only (i.e. without applied current) and discharge with fixed specific currents. Fig. 4b shows the photo-charging process (λ ∼455 nm, intensity ∼12 mW cm−2) as well as the discharge at a specific current of 100 mA m−2. Moreover, we can increase the capacity by illuminating the device with light while discharging (Fig. 4b). As shown in Fig. 4b, the voltage of the photo-charged photo-ZIB very slowly drops from 0.95 V to 0.715 V when discharged under light. This is due to the simultaneous actions of photo-charging and potentiostatic discharging. A nearly constant voltage response can be achieved when the photo-charge and discharge current rates are in equilibrium under illumination. Once the light is turned off, the voltage reduces to 0.2 V, following the discharge curve expected from the dark measurements. Fig. S10 (ESI†) shows the discharge curves at different specific currents in dark condition. The photo-conversion efficiency of the photo-ZIB is 1.2% using η = EAA1/PintA2 (where, EA is the areal energy density, Pin the light intensity, t is photo-charging time, A1 is the surface area of the photo-ZIB and A2 is the illuminated surface area).34 This value is higher than the previously reported efficiencies of 0.03–0.06% for photo-rechargeable LIBs.14,15 In addition, Fig. 4c shows that photo-ZIBs charged only by light can power a commercial sensor and its display (here a 1.5 V Digital Thermo-Hygrometer TFA, MPN: 30.5005). Finally, we demonstrate a larger scale implementation of photo-ZIBs in a ∼100 cm2 pouch cell with a ∼64 cm2 optical window (see Fig. 4d and photo-charging and discharge profiles in Fig. 4e).
This report demonstrates a high-performance photo-rechargeable aqueous photo-ZIB, which can be charged by light and whose capacity can almost be doubled under illumination. The photo-active cathodes of these photo-ZIBs consist of V2O5 nanofibers mixed with P3HT and rGO. These cathodes have a good absorption in the visible spectrum and allow for the separation and storage of charges needed for direct light charging without the use of solar cells. This mechanism is studied by testing photo-detectors, TA measurements, as well as GCD and CV measurements in dark and light conditions. The proposed photo-ZIBs achieve light conversion efficiencies of 1.2%, which is the highest reported for photo batteries to our knowledge, and we demonstrate that they can be implemented in both small coin cells and large pouch cells.
Experimental section
Material synthesis and characterization
For the synthesis of V2O5 nanofibers, we start from commercial V2O5 powder (Sigma-Aldrich). SEM images (Fig. S11a and b, ESI†) of the V2O5 precursor shows it consists of micrometer sized aggregates of nanoparticles (∼200–600 nm), EDS maps of V and O are shown in Fig. S11c–e (ESI†). Fig. S11f (ESI†) shows the XRD pattern of the V2O5 powder, which exhibits an orthorhombic structure. Further, the Raman spectrum of the V2O5 powder (Fig. S11g, ESI†) shows the characteristic peaks at 987 cm−1, 474 cm−1 and 398 cm−1 related to V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the vanadyl bond, V–O3–V symmetric stretching, and angle-bending of V–O3–V.25 The characteristic peaks belong to stretching of V–O–V bonds (∼694 cm−1), V3–Oc triply coordinated oxygen (∼521 cm−1), bending vibrational mode of V–Oc (∼299 cm−1), bending vibrations of Oc–V–Ob bonds (∼278 cm−1), and vibration mode of V–OV chains (∼142 cm−1), respectively of V2O5 powder are also observed.25 EDS mappings and Raman spectrum of the synthesized V2O5 nanofibers are shown in Fig. S12 and S13 (ESI†).
O stretching vibration of the vanadyl bond, V–O3–V symmetric stretching, and angle-bending of V–O3–V.25 The characteristic peaks belong to stretching of V–O–V bonds (∼694 cm−1), V3–Oc triply coordinated oxygen (∼521 cm−1), bending vibrational mode of V–Oc (∼299 cm−1), bending vibrations of Oc–V–Ob bonds (∼278 cm−1), and vibration mode of V–OV chains (∼142 cm−1), respectively of V2O5 powder are also observed.25 EDS mappings and Raman spectrum of the synthesized V2O5 nanofibers are shown in Fig. S12 and S13 (ESI†).
3 g of the V2O5 powder (Sigma-Aldrich) is mixed with a 2 M aqueous NaCl solution (100 ml) while stirring (300 rpm) at room temperature for 72 h. In this process, dissolution and recrystallization of V2O5 take place to form nanofibers.23 The resulting brownish suspension was washed with deionized water and ethanol followed by centrifugation and finally dried at ∼80 °C for 12 h in a vacuum oven.
The rGO was obtained by direct reduction of graphene oxide (Graphenea) at ∼350 °C (∼3 h) in hydrogen gas (∼100 sccm) and helium gas (∼100 sccm) environment using tubular atmospheric pressure CVD furnace.
Morphologies and crystal structures of the materials are characterized by SEM (FEI Magellan 400L with an acceleration voltage of 5 kV) and XRD (Bruker D8 Advance, Cu Kα radiation). Further, Raman spectroscopy and Brunauer–Emmett–Teller (BET) surface area characterizations are employed using Renishaw InVia and Micromeritics 3Flex (under nitrogen environment). The optical absorbance of the materials are studied using PerkinElmer UV/VIS/NIR Spectrometer (Lambda 750).
Preparation of electrodes
First, 1 mg rGO was dissolved into 2 ml N-methyl-2-pyrrolidone (NMP, Sigma-Aldrich) followed by ultra-sonication and mixing (VWR Analog Vortex Mister) processes. Then, 1 mg P3HT (Sigma-Aldrich) was added in the same solution and continued the dissolving processes. Thereafter, 93 mg V2O5 nanofibers was added followed by sonication and mixing for another 5 h. Finally, the photo-cathode solution was obtained by adding 5 mg PVDF (Solef 6020) binder followed by mixing for another 2 h. The photo-cathode is obtained by drop casting the electrode solution on CF current collector (Sigracet GDL 39 AA carbon graphite paper, SGL Carbon) followed by drying at ∼120 °C in a vacuum oven. For P3HT free photo-cathode, we added 93 mg V2O5 nanofibers to a 2 mg rGO solution (2 ml NMP) and then, 5 mg PVDF binder followed the same ultra-sonication and mixing processes. The active material mass (mass of V2O5) loading of the electrodes ranges from 0.8–1.2 mg cm−2.Designing of photo-batteries
First, we machined a hole on case of CR2450 coin cell with a ∼8 mm diameter followed by mounting optical glass using EPOXY (EVO-STIK) to allow for illumination. The photo-cathode is placed on the side of the coin cell having the optical window. The separator (Whatman glass microfiber filter paper) is placed on the top side of photo-cathode followed by adding ∼150 μl of 3 M Zn(CF3SO3)2 (Sigma-Aldrich) aqueous electrolyte. Then, the metallic Zn (Alfa Aesar, 0.25 mm thick) anode is placed on top of the separator. Finally, the optical coin cell is assembled by placing a stainless steel disk spacer with a spring on Zn anode side.Characterization of the photo-ZIBs
Electrochemical measurements of all the photo-ZIBs are measured by using a Biologic VMP-3 galvanostat. CV and GCDs are tested in dark and by illuminating light of wavelength of 455 nm (intensities ∼12 mW cm−2 and ∼5 mW cm−2) at equivalent scan rates and specific currents. Further, CV and GCD measurements are extended to dark and illuminated conditions with different light wavelengths of 420 nm, 470 nm, 528 nm, and white light. The AC impedance (EIS) measurement of the photo-ZIB is acquired in the frequency range 10 mHz to 100 kHz both in dark and illuminated conditions with voltage amplitude of 10 mV. The photo-charging performance of the photo-ZIBs are measured by recording open circuit voltage responses, and discharged by applying different currents.Ex situ XRD and Raman characterizations
To understand the charge storage reversibility of the photo-cathode, we assembled standard CR2450 coin cells (without optical windows) following the same procedures used for photo-ZIBs assembly. The photo-cathodes are cycled to different states of discharge and charge under constant current mode at 200 mA g−1. Then, the cells are disassembled and the photo-cathodes are washed with deionized water followed by drying at ∼120 °C for 12 h in a vacuum oven. Thereafter, Raman and XRD of these photo-electrodes are characterized by using Renishaw InVia and Bruker D8 Advance (Cu Kα radiation).Fabrication of photodetectors and electrical characterizations
The photodetectors with layer-by-layer structures are obtained by drop casting of pristine materials in sequence on FTO coated glass substrates (received from Sigma-Aldrich, surface resistivity ∼7 Ω sq−1) followed by drying at ∼120 °C in a vacuum oven. Silver conductive paste (Ag) was used as a top contact to measure the current–time responses of the photodetectors in absence of bias voltage (0 V) under periodic illumination (λ ∼ 455 nm). The electrical photo-responses are recorded using Agilent Sourcemeter integrated to Suss MicroTec Probe Station.The electrical photoresponses of V2O5 nanofibers are studied by drop casting the V2O5 nanofibers on Gold (Au)/Chromium (Cr) (40/10 nm) Inter Digitated Electrodes (IDEs) patterned on a Si3N4/Si wafer by standard UV lithographic technique. The current–voltage measurements are recorded in the voltage range −2 V to +2 V both in dark and illuminated conditions. Further, current–time photo-responses are measured by applying a 2 V bias voltage under periodic illumination of different light wavelengths of 455 nm, 528 nm, and white light.
Photoluminescence spectroscopy
PL spectra were recorded using a gated intensified CCD camera (Andor Star DH740 CCI-010) connected to a grating spectrometer (Andor SR303i). The pulsed output from a mode-locked Ti:Sapphire optical amplifier (Spectra-Physics Solstice, 1.55 eV photon energy, 80 fs pulse width) was frequency-doubled to 400 nm via second harmonic generation in a β-barium borate crystal and used as excitation source.Transient absorption spectroscopy
The third harmonic of a pulsed Nd:YVO laser (Picolo-AOT MoPa) was used as the pump beam (1 ns pulse width, 500 Hz repetition rate, 355 nm) for the ns regime measurements. The probe spectrum was generated using a white light quasi-continuum generated through pumping a CaF2 window with the 800 nm fundamental of a Ti:Sapphire amplifier (Spectra-Physics Solstice). A delay generator was used to electronically vary the pump–probe delay. For the short time fs regime, the pump was the 400 nm second harmonic generated by the 800 nm fundamental passing through a β-barium borate crystal. Transmitted probe and reference pulses were recorded with an NMOS linear image sensor (Hamamatsu S8381-1024Q) and processed by a customized PCI interface from Entwicklungsbüro Stresing.Designing of pouch cell-based photo-ZIB
An optical pouch cell is fitted with a glass window in order to illuminate a large area of the photo-cathode. The window was sealed to an opening ∼8 × 8 cm2 cut into the pouch foil (aluminium laminated film, MTI). The photo-cathode material was drop cast on a CF current collector (∼100 cm2) and placed directly against the window. Separator (Whatman glass microfiber filters paper) cut to size was soaked with 3 M Zn(CF3SO3)2 aqueous electrolyte. Zinc metal anode was placed against the photo-cathode. Nickel pouch cell tabs (MTI) were used to make direct electrical contact to the photo-cathode and zinc metal, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The B. D. B. acknowledges support from the Newton International Fellowship-Royal Society (UK) grant NIF\R1\181656. M. D. V acknowledges support from the ERC Consolidator grant MIGHTY – 866005. B. W. and A. M. acknowledge support from the EPSRC Graphene CDT EP/L016087/1.References
- K. Rose, S. Eldridge and L. Chapin, The internet of things: an overview, The Internet Society (ISOC), 2015, 80 Search PubMed
.
- X. Fan, X. Liu, W. Hu, C. Zhong and J. Lu, InfoMat, 2019, 1, 130–139 Search PubMed
- R. Fedele, M. Merenda and F. Giammaria, Energy harvesting for IoT road monitoring systems, Instrumentation, Mesure, Metrologie, 2018, 17, 605 CrossRef
- E. O’Dwyer, I. Pan, S. Acha and N. Shah, Appl. Energy, 2019, 237, 581–597 CrossRef
- M. Masera, E. F. Bompard, F. Profumo and N. Hadjsaid, Proc. IEEE, 2018, 106, 613–625 Search PubMed
- The World Bank, Report PAD2635, 2019.
- Q. Zeng, Y. Lai, L. Jiang, F. Liu, X. Hao, L. Wang and M. A. Green, Adv. Energy Mater., 2020, 10, 1903930 CrossRef CAS
- A. Gurung and Q. Qiao, Joule, 2018, 2, 1217–1230 CrossRef CAS
- Y. Sun and X. Yan, Sol. RRL, 2017, 1, 1700002 CrossRef
- H. Meng, S. Pang and G. Cui, ChemSusChem, 2019, 12, 3431–3447 CrossRef CAS PubMed
- R. Liu, Y. Liu, H. Zou, T. Song and B. Sun, Nano Res., 2017, 10, 1545–1559 CrossRef CAS
- H.-D. Um, K.-H. Choi, I. Hwang, S.-H. Kim, K. Seo and S.-Y. Lee, Energy Environ. Sci., 2017, 10, 931–940 RSC
- J. Xu, Y. Chen and L. Dai, Nat. Commun., 2015, 6, 1–7 Search PubMed
- S. Ahmad, C. George, D. J. Beesley, J. J. Baumberg and M. D. Volder, Nano Lett., 2018, 18, 1856–1862 CrossRef CAS PubMed
- A. Paolella, C. Faure, G. Bertoni, S. Marras, A. Guerfi, A. Darwiche and P. Hovington,
et al.
, Nat. Commun., 2017, 8, 14643 CrossRef CAS PubMed
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS
- Q. Pang, C. Sun, Y. Yu, K. Zhao, Z. Zhang, P. M. Voyles, G. Chen, Y. Wei and X. Wang, Adv. Energy Mater., 2018, 8, 1800144 CrossRef
- C. Liu, Z. Neale, J. Zheng, X. Jia, J. Huang, M. Yan, M. Tian, M. Wang, J. Yang and G. Cao, Energy Environ. Sci., 2019, 12, 2273–2285 RSC
- C. Xia, J. Guo, Y. Lei, H. Liang, C. Zhao and H. N. Alshareef, Adv. Mater., 2018, 30, 1705580 CrossRef PubMed
- G. Li, Z. Yang, Y. Jiang, C. Jin, W. Huang, X. Ding and Y. Huang, Nano Energy, 2016, 25, 211–217 CrossRef CAS
- Y. Li, Z. Huang, P. K. Kalambate, Y. Zhong, Z. Huang, M. Xie, Y. Shen and Y. Huang, Nano Energy, 2019, 60, 752–759 CrossRef CAS
- S. J. Lee, H. P. Kim, A. R. bin Mohd Yusoff and J. Jang, Sol. Energy Mater. Sol. Cells, 2014, 120, 238–243 CrossRef CAS
- T. Zhai, H. Liu, H. Li, X. Fang, M. Liao, L. Li, H. Zhou, Y. Koide, Y. Bando and D. Golberg, Adv. Mater., 2010, 22, 2547–2552 CrossRef CAS PubMed
- M. Kang, M. Chu, S. W. Kim and J.-W. Ryu, Thin Solid Films, 2013, 547, 198–201 CrossRef CAS
- P. Hu, T. Zhu, J. Ma, C. Cai, G. Hu, X. Wang, Z. Liu, L. Zhou and L. Mai, Chem. Commun., 2019, 55, 8486–8489 RSC
- J. Zhou, L. Shan, Z. Wu, X. Guo, G. Fang and S. Liang, Chem. Commun., 2018, 54, 4457–4460 RSC
- Y. W. Denis, C. Fietzek, W. Weydanz, K. Donoue, T. Inoue, H. Kurokawa and S. Fujitani, J. Electrochem. Soc., 2007, 154, A253–A257 CrossRef
- A. Lee, M. Vörös, W. M. Dose, J. Niklas, O. Poluektov, R. D. Schaller and H. Iddir,
et al.
, Nat. Commun., 2019, 10, 1–7 CrossRef PubMed
- M. Yan, P. He, Y. Chen, S. Wang, Q. Wei, K. Zhao, X. Xu, Q. An, Y. Shuang, Y. Shao and K. T. Mueller, Adv. Mater., 2018, 30, 1703725 CrossRef PubMed
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H.-Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 2–14 CrossRef
- H. Jia, Z. Wang, B. Tawiah, Y. Wang, C.-Y. Chan, B. Fei and F. Pan, Nano Energy, 2020, 70, 104523 CrossRef CAS
- R. Liu, J. Wang, T. Sun, M. Wang, C. Wu, H. Zou, T. Song, X. Zhang, S. T. Lee, Z. L. Wang and B. Sun, Nano Lett., 2017, 17, 4240–4247 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee01392g |
| This journal is © The Royal Society of Chemistry 2020 |