
Podoviridae bacteriophage for the biocontrol of Pseudomonas aeruginosa in rainwater†
Brandon
Reyneke
a,
Sehaam
Khan
b,
Pilar
Fernández-Ibáñez
c and
Wesaal
Khan
 *a
*a
aDepartment of Microbiology, Faculty of Science, Stellenbosch University, Private Bag X1, Stellenbosch, 7602, South Africa. E-mail: wesaal@sun.ac.za; Fax: +27218085846; Tel: +27218085804
bFaculty of Health Sciences, University of Johannesburg, PO Box 17011, Doornfontein, 2028, South Africa
cNanotechnology and Integrated BioEngineering Centre, School of Engineering, University of Ulster, Newtownabbey, Northern Ireland, UK
First published on 13th November 2019
Abstract
Bacteriophages targeting Pseudomonas spp. were isolated and characterised for the biocontrol pre-treatment of rainwater. Bacteriophages PAW33 and PFW25 were characterised as members of the Podoviridae and Myoviridae families, respectively. Bacteriophage PAW33 displayed specific activity against Pseudomonas aeruginosa (P. aeruginosa) strains and was applied in small-scale bacteriophage pre-treatment trials (8 h and 24 h) to restrict the proliferation of an environmental P. aeruginosa S1 68 strain. Hereafter, the respective samples (bacteriophage pre-treated and non-pre-treated samples) were subjected to treatment in small-scale SODIS compound parabolic collector (SODIS-CPC) systems for 4 h under natural sunlight. For the 8 h pre-treatment and SODIS-CPC trials, similar total log reductions in colony forming units (CFU) per mL and gene copies (GC) per mL were obtained for the bacteriophage pre-treated [3.68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log (CFU) and 2.34log (GC)] and non-pre-treated [3.74log (CFU) and 2.33log (GC)] samples. In contrast, for the 24 h trial (followed by SODIS) a higher CFU per mL log reduction was recorded for the pre-treated sample (4.61log) in comparison to the non-pre-treated sample (3.91log), with comparable results obtained using EMA-qPCR. Gene expression analysis indicated that PAW33 pre-treatment for 24 h influenced the ability of P. aeruginosa S1 68 to initiate stress response mechanisms (decreased expression of the recA and lexA genes) during the SODIS-CPC treatment and resulted in the decreased expression of the phzM gene (virulence factor responsible for pyocyanin production). Bacteriophage PAW33 thus displays promise as a pre-treatment strategy of rainwater as it restricts the proliferation of P. aeruginosa and may increase the efficiency of primary disinfection methods.
log (CFU) and 2.34log (GC)] and non-pre-treated [3.74log (CFU) and 2.33log (GC)] samples. In contrast, for the 24 h trial (followed by SODIS) a higher CFU per mL log reduction was recorded for the pre-treated sample (4.61log) in comparison to the non-pre-treated sample (3.91log), with comparable results obtained using EMA-qPCR. Gene expression analysis indicated that PAW33 pre-treatment for 24 h influenced the ability of P. aeruginosa S1 68 to initiate stress response mechanisms (decreased expression of the recA and lexA genes) during the SODIS-CPC treatment and resulted in the decreased expression of the phzM gene (virulence factor responsible for pyocyanin production). Bacteriophage PAW33 thus displays promise as a pre-treatment strategy of rainwater as it restricts the proliferation of P. aeruginosa and may increase the efficiency of primary disinfection methods.
Water impactVarious opportunistic pathogens have been shown to survive conventional rainwater treatment strategies, including solar pasteurization and solar disinfection. Bacteriophage biocontrol displays promise to be used as a rainwater pre-treatment strategy to increase the removal of opportunistic pathogens and thereby reduce the health risk associated with the use of rainwater. |
1. Introduction
Pseudomonas aeruginosa (P. aeruginosa) is an opportunistic pathogen that is primarily associated with nosocomial infections where it may cause pneumonia, urinary tract and skin infections.1 However, the ubiquitous distribution of Pseudomonas spp. in the environment significantly increases the health risk associated with this organism and John et al. recently reported on an outbreak of community-acquired P. aeruginosa pneumonia in Cape Town, South Africa.2 The authors noted that while no cases of community-acquired P. aeruginosa were reported at a local government hospital over a 10 year period (2007 to 2016); over a period of three months (March to May 2017), 9 cases were reported. This outbreak coincided with a severe drought in Cape Town (2017 to 2018), where stringent water restrictions were implemented and residents were increasingly using alternative water sources (e.g. rainwater and grey water). It was hypothesised that the use of these alternative water sources may have led to the exposure of the community members to P. aeruginosa.Although various treatment methods have been implemented to reduce the levels or remove pathogens and opportunistic pathogens from environmental water reservoirs,3 many microorganisms employ survival strategies and are capable of persisting. Certain strains of P. aeruginosa have been shown to survive conventional disinfection strategies including sub-optimal chlorination, ultra-violet (UV) radiation and heat treatment.4–7 Using EMA-qPCR, Strauss et al. reported on the detection of Pseudomonas spp. in rainwater samples treated at solar pasteurization temperatures >90 °C and in water samples treated by solar disinfection (SODIS; UV radiation and solar mild-heat) for 8 h.5 Similarly, using culture-based methods, Clements et al. isolated Pseudomonas spp. from rainwater samples pasteurized at temperatures >70 °C, with certain isolates identified as P. aeruginosa using species-specific primers.7Pseudomonas aeruginosa has also readily been detected in grey water,8,9 with Gross et al. and Gilboa and Friedler reporting on the detection of viable P. aeruginosa in grey water samples treated in a vertical flow bioreactor and a rotating biological contractor followed by UV disinfection, respectively.10,11 The survival of Pseudomonas spp. during water treatment has subsequently been attributed to the initiation of stress response mechanisms, including heat-shock proteins and deoxyribonucleic acid (DNA) repair mechanisms, the ability of the organisms to form biofilms and survive intracellularly within protozoa.5,7 As bacteria are able to undergo an adaptive response and build-up resistance to conventional disinfection treatments,12 alternative treatment strategies targeting pathogenic species directly are required.
Bacteriophage therapy or bacteriophage biocontrol has gained increased interest in recent years, due to the specificity with which pathogens may be targeted.13–15 Bacteriophages are bacterial viruses, which are ubiquitously distributed in the environment and may interact with bacteria by either causing lysis of the host cell (lytic phages) or integrating their phage genome into the host cell (temperate phages).16 Numerous studies have subsequently reported on the potential of bacteriophage biocontrol to target food-borne pathogens (food safety);17 biofilm formation on medical devices or treat infectious diseases (human and veterinary medicine);18 to reduce economic losses in agriculture (targeting plant pathogens) and aquaculture (targeting fish pathogens);19,20 or in bioremediation strategies for the selective removal of bacteria from water.21 While investigating the use of bacteriophages for the biocontrol of Salmonella spp. in wastewater, Turki et al. showed that the isolated bacteriophages were able to reduce the proliferation of the target pathogen (reduction in sample optical density) over time in co-culture experiments.22 Additionally, using DNA fingerprinting analysis [PCR based amplification of enterobacterial repetitive intergenic consensus (ERIC) sequences], the authors reported on the decreased detection of the enterobacterial community and Salmonella spp. in the treated wastewater over time (0 h to 8 h). Similarly, Goldman et al. reported that bacteriophage treatment could reduce membrane fouling caused by the opportunistic pathogens Acinetobacter johnsonii, Bacillus subtilis and P. aeruginosa, by 40 to 60%.23 An added benefit of using bacteriophages to target persisting pathogens is the relative ease with which this treatment can be combined with existing disinfection methods. For example, Zhang et al. applied a mixture of P. aeruginosa bacteriophages to selectively remove this organism from water passing through granular activated carbon and anthracite biofilters.24 Results indicated that the bacteriophage treatment was able to reduce P. aeruginosa concentration by 55 to 75% in the two biofilter systems, with minimal impact on the beneficial microorganisms and thereby contribute to an improvement in effluent water quality.
The primary aim of the current study was thus to isolate and characterise bacteriophages targeting Pseudomonas spp. and apply the best performing bacteriophage as a biocontrol pre-treatment (8 h and 24 h) of roof-harvested rainwater. Following the completion of the bacteriophage pre-treatment trial, the respective samples (bacteriophage pre-treated samples and non-pre-treated control samples) were subjected to treatment in small-scale SODIS-CPC systems. Culture- and molecular-based (EMA-qPCR) analysis were used to quantify P. aeruginosa S1 68 and bacteriophage PAW33 during the pre-treatment and SODIS trials, while gene expression assays were used to monitor the expression of P. aeruginosa S1 68 stress response and virulence genes.
2. Materials and methods
2.1 Bacterial strains and growth conditions
Pseudomonas aeruginosa (P. aeruginosa) ATCC 27853, P. fluorescens ATCC 13525 and P. protegens ATCC 17386 reference strains were obtained from Microbiologics® (St Cloud, Minnesota, USA) and were used for the isolation, propagation and characterisation of the bacteriophages. The bacterial strains (target and non-target bacterial species) utilised for the host range determination of the isolated bacteriophages are indicated in Table S1 (ESI†). All strains were cultured at 30 °C in tryptic soy broth (TSB; Biolab, Merck, Wadeville, South Africa) or on tryptic soy agar (TSA; Biolab, Merck), with the exception of Legionella spp. (see ESI† for Legionella spp. growth conditions). For the double-layer plaque assays (double-layer overlays), the TSA medium contained 1.2% (w/v) Agar Bacteriological (Biolab, Merck) in the bottom layer and 0.6% agar (w/v) in the soft top layer.252.2 Isolation, purification and propagation of bacteriophages
Bacteriophages were isolated by screening various environmental water sources including influent wastewater collected from the Stellenbosch Wastewater Treatment Plant (GPS co-ordinates: −33.943505, 18.824584), river water from the Plankenburg River (GPS co-ordinates: −33.927761, 18.850544) and roof-harvested rainwater from a rainwater harvesting tank connected to the JC Smuts building at Stellenbosch University (GPS co-ordinates: −33.930858, 18.865611). Selection for Pseudomonas spp. bacteriophages was performed as previously described by Sillankorva et al.,25 with minor modifications. Briefly, following the centrifugation step (10000 × g; 10 min; 4 °C), the supernatant from each sample was filtered through a sterile GN-6 Metricel® S-Pack Membrane Disc Filter (Pall Life Sciences, Michigan, USA) with a pore size of 0.45 μm, to remove residual host bacteria from the sample.19 The filtered supernatant was tested for the presence of bacteriophages,25 whereafter five repeated rounds of plaque purification and re-infection were performed.26
The bacteriophages were selected for further studies based on their initial lysis profiles during bacteriophage purification (number and consistency of plaque formation, plaque clarity and plaque size).25 Code identifiers were assigned to the isolated bacteriophages based on the bacteria from which they were isolated (i.e. PA – P. aeruginosa; PF – P. fluorescens and PP – P. protegens), the source of the bacteriophage (e.g. W – wastewater; R – river water) and the plaque number. For example, PAW1 indicates that the bacteriophage was isolated using P. aeruginosa (PA), from wastewater (W) and was the first plaque isolated (1).
The purified bacteriophages were used to prepare concentrated bacteriophage solutions for use in subsequent experiments using the small-scale liquid culture method as described by Sambrook and Russell,27 with minor modifications. Briefly, following the onset of bacterial cell lysis, the samples were centrifuged (10000 × g; 10 min; 4 °C) and filtered through a 0.2 μm Acrodisc® PF syringe filter (Pall Life Sciences). The filtered supernatants were centrifuged at 25000 × g for 60 min using an Avanti J-E Centrifuge with a JA 20 fixed angle rotor (Beckman Coulter, California, USA). Following centrifugation, the supernatant was removed and the obtained pellet was re-suspended in 1 mL SM-buffer [5.8 g L−1 sodium chloride (NaCl; Saarchem, Durban, South Africa), 2 g L−1 magnesium sulphate heptahydrate (MgSO4·7H2O; Saarchem), 50 mL 1 M Tris, pH 7.5]. The plaque forming units (PFU) per mL concentrated sample were determined by serial dilution (10−1 to 10−5) in SM-buffer and double-layer plaque assays. The concentrated bacteriophage samples in SM-buffer were stored at 4 °C until further use.
2.3 Characterisation of the isolated bacteriophages
In order to increase bacteriophage retention on the electron microscopy grids and thereby increase bacteriophage visualisation during electron microscopy analysis, the hydrophilicity and “stickiness” of the 200 mesh carbon-coated Formvar grids (Agar Scientific, Essex, United Kingdom) were increased by using an alcian blue (Electron Microscopy Sciences, Pennsylvania, USA) pre-treatment (1% alcian blue in 1% acetic acid in water) as described by Laue and Bannert.28 Concentrated bacteriophage samples with a titre of >109 PFU per mL were then mixed with glutaraldehyde (2.5% v/v; Agar Scientific) for 5 min and 25 μL of the glutaraldehyde treated concentrated bacteriophage sample was loaded onto the alcian blue pre-treated grids and were allowed to settle for 10 min. Hereafter, the sample was stained with 1% uranyl acetate for 2 min. Excess stain was removed using filter paper and the grids were allowed to air dry. The grids were visualised with a Zeiss MERLIN field emission scanning electron microscope (FE-SEM; Zeiss, Germany) at the Electron Microbeam Unit of the Central Analytical Facility (CAF) at Stellenbosch University. A Zeiss five-diode scanning transmission electron detector (Zeiss aSTEMA Detector) and Zeiss Smart SEM software was used to generate STEM images. Beam conditions during analysis on the Zeiss MERLIN FE-SEM were 20 kV accelerating voltage, 150 pA probe current, with a working distance of approximately 3.9 to 4.0 mm. Images were acquired in bright fields mode with the S1 diode activated.2.4 Analysis of bacteriophage nucleic acids – restriction enzyme digestion and molecular analysis
To ensure that no potential residual host bacterial DNA was analysed in the subsequent bacteriophage nucleic acid determination, 500 μL of the concentrated bacteriophage samples was treated with DNaseI (Thermo Scientific, Lithuania) as outlined in Reyneke et al.29 Following the DNase treatment, bacteriophage nucleic acid was extracted from the concentrated samples using the NucleoSpin® Tissue kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions.The type of nucleic acid was confirmed by treatment with DNaseI (dsDNA; Thermo Scientific), S1 nuclease (ssDNA; Thermo Scientific) and RNase [ribonucleic acid (RNA); Fermentas, Thermo Scientific],19 while the purified bacteriophage nucleic acids were digested with either EcoRI (Roche Diagnostics, Risch-Rotkreuz, Switzerland) or ClaI (Fermentas).26 All nuclease and restriction enzyme digestions were performed according to the manufacturer's instructions.
In order to confirm the preliminary classification of the isolated bacteriophages, primers targeting families within the Caudovirales order, namely Podoviridae and Myoviridae, were designed. The PhiSiGns online bacteriophage genes and primers tool as described by Dwivedi et al. was used to identify signature genes within the respective bacteriophage families.30 The identified gene sequences were retrieved from the Genbank sequence database of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/genbank/) and were aligned using ClustalX version 2.0.10 and visualised using GeneDoc version 2.7.00.31,32 Primers targeting the specific gene sequences were designed based on the sequence alignments and are presented in Table 1 along with the respective PCR cycling parameters. Each PCR assay was performed in a final volume of 25 μL and consisted of 1× Green GoTaq® Flexi buffer (Promega, Madison, WI, USA), 2.0 mM MgCl2 (Promega), 0.1 mM dNTP mix (Thermo Scientific Fisher, Finland), 0.1 μM of the respective forward and reverse PCR primers (Table 1), 1.5 U GoTaq® Flexi DNA polymerase (Promega) and 5 μL template DNA. Sterile distilled H2O was used as a negative control.
| Target gene | Primers | Primer sequence (5′-3′) | Cycling parameters | Ref. [NCBI accession no.] |
|---|---|---|---|---|
| a A final elongation step of 5 min at 72 °C was included for each PCR assay. b Combined annealing and elongation step; NCBI – National Center for Biotechnology Information. | ||||
| Hypothetical protein (225 bp) (Podoviridae) | Podo-Hypo-F | CGACTTCCTCATCGACAACG | 95 °C for 3 min, 30 cycles of 94 °C for 30 s, 48 °C for 30 s, 72 °C for 45sa | Designed using vB_PaeP_MAG4 [KR052142.1] LP14 [MH356729.1] and vB_PaeP_DEV [MF490238.1] genomes (NCBI) |
| Podo-Hypo-R | GCGAAGTTGTCAGGGTATTTTC | |||
| Hypothetical protein (254 bp) (Myoviridae) | Myo-Hypo-F | CGATGAGCTGCGAGACCTTA | 95 °C for 3 min, 30 cycles of 94 °C for 30 s, 50 °C for 30 s, 72 °C for 45sa | Designed using vB_Kpn_F48 [MG746602.1] genome (NCBI) |
| Myo-Hypo-R | GACCTTTGTGCTCGAACGGC | |||
| rpoS (gene expression reference gene) | rpoS-F | CTCCCCGGGCAACTCCAAAAG | 95 °C for 10 min; 50 cycles of 95 °C for 15 s, 60 °C for 10 s and 72 °C for 15 s | 33 |
| rpoS-R | CGATCATCCGCTTCCGACCAG | |||
| phzM (gene expression target gene) | phzM-F | AGACTTCTACAGCTACCTGAAGC | 95 °C for 10 min; 50 cycles of 95 °C for 15 s, 61 °C for 20 s and 72 °C for 30 s | 34 |
| phzM-R | GATGGCCTTGGTCAATTCGC | |||
| recA (gene expression target gene) | RECA-RT1 | CGCGGCCCTGGGACAGATCG | 95 °C for 10 min; 50 cycles of 95 °C for 15 s, 60 °C for 60 sb | 35 |
| RECA-RT2 | GATGCCGAGGGCGATGTCCAG | |||
| lexA (gene expression target gene) | LEXA-RT1 | CTCCTTCATCAAGCGCTGCCTGG | 95 °C for 10 min; 50 cycles of 95 °C for 15 s, 60 °C for 60 sb | 35 |
| LEXA-RT2 | GTTCGGCGACTTGAAGCCGAGTT | |||
All samples (digested nucleic acids and PCR products) were analysed by agarose gel electrophoresis in 0.8% agarose (SeaKem® LE Agarose; Lonza, Rockland, ME, USA) containing 0.5 μg mL−1 ethidium bromide, at 50 volts for 180 min (digested nucleic acids) or 80 volts for 80 min (PCR products) with the use of 1× tris/acetic acid/ethylenediaminetetraacetic acid (EDTA) (TAE) buffer followed by visualisation on a Vilber Lourmat gel documentation system (Vilber Lourmat, Collegien, France). The digested samples were compared to undigested bacteriophage nucleic acid and the GeneRuler 1 kb Plus DNA ladder (Thermo Scientific). The genome size of the isolated bacteriophages was estimated by compiling the DNA fragment sizes using the standard ladder.36 The obtained PCR products were cleaned and concentrated using the Wizard® SV Gel and PCR Clean-up System (Promega) and were sent for DNA sequencing to the CAF at Stellenbosch University. Sequences were examined using FinchTV version 1.4.0 software and identification completed using the NCBI Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
2.5 Host range determination
The host range of the isolated bacteriophages was determined by spotting 10 μL of the concentrated bacteriophage stock solutions (106 to 107 PFU per mL) on TSA (with the exception of Legionella spp.; ESI†) with 5 mL freshly prepared soft top-agar, which had been inoculated with 50 to 100 μL of the strain to be tested and incubating the plates at 30 °C for 18 h.25,26 The host range for each bacteriophage was determined in triplicate for each bacteriophage–host combination and consisted of screening 20 Pseudomonas spp. (target bacterial strains; Table S1, ESI†) and 57 non-target bacterial strains representative of 14 genera (Table S1, ESI†). Reference, environmental and clinical isolates of both the target and non-target bacterial strains were included in the host range determination analysis.2.6 One-step growth curve and bacteriophage sensitivity to physical parameters
Based on the results obtained during the host range determination, K. pneumoniae ATCC 10031 was used as the host for PFW25 during subsequent experimentation, while P. aeruginosa ATCC 27853 was used as the host for PAW33. One-step growth curves were subsequently performed in order to determine the latency period and burst size of the isolated bacteriophages, as previously described by Sillankorva et al.,25 with minor modifications. Briefly, a multiplicity of infection (MOI) of 0.0003 was used and samples were collected every 10 min over a period of 2 h. Following overnight incubation at 30 °C, the PFU for each time point was recorded and results were reported as the average number of bacteriophages released per infected host cell. The bacteriophage burst size (number of bacteriophages released per infected host cell) was computed as the ratio between the final bacteriophage count and the initial bacteriophage count recorded during the latency period.37The influence of pH on the isolated bacteriophages' stability was evaluated by suspending bacteriophages at ∼106 PFU per mL in 1 mL SM-buffer aliquots, with a pH range of 4.0 to 10.0 (intervals of 1 unit).37,38 The bacteriophage solutions were then incubated at room temperature for 1 h, whereafter the bacteriophage titre (PFU per mL) in the solutions were determined using double-layer plaque assays. The effect of temperature on bacteriophage stability was assessed by incubating 2 mL bacteriophage suspensions (∼105 PFU per mL in SM-buffer) at 30 °C (control), 40 °C, 50 °C, 60 °C and 70 °C for 2 h. Samples were collected for bacteriophage titre (PFU per mL) determination at each temperature, using double-layer plaque assays, at 0, 10, 30, 60 and 120 min.
2.7 Bacterial challenge tests
To determine the activity of the isolated bacteriophages against the respective host bacterial species (PAW33 – P. aeruginosa ATCC 2785; PFW25 – K. pneumoniae ATCC 10031), bacterial challenge tests were performed as described by Turki et al. with minor modifications.22 Briefly, 50 mL TSB was inoculated with an overnight culture of the respective host bacterial species and corresponding bacteriophage to achieve MOI values of 1, 0.1 and 0.01. A non-infected culture of the respective host bacterial species was included as a negative control. All challenge tests were performed in triplicate. The flasks were incubated at 30 °C on a rotary shaker (New Brunswick Scientific, NY, USA) at 120 rpm for 24 h. Samples were collected every 2 h for the first 8 h and every 4 h thereafter to monitor host bacterial growth within the samples by measuring the optical density at 650 nm with a T60 visible spectrophotometer (PG-Instruments Limited, Leicester, UK).To determine whether bacteriophage resistant mutants had emerged during the bacterial challenge tests, 2 mL aliquots were collected after the 24 h incubation and were centrifuged at 10000 × g for 5 min (Spectrafuge™ 24D Digital Microcentrifuge, Labnet International, Edison, USA). The bacterial pellet was re-suspended and was used to inoculate 5 mL freshly prepared soft top-agar, which was poured onto TSA plates. Ten microliters of concentrated bacteriophage stock solutions (106 to 107 PFU per mL) were spotted onto the surface of the plate and the plates were incubated at 30 °C for 18 h. Following incubation, the plates were examined for plaque formation to determine whether the respective host bacterial species were still susceptible to the isolated bacteriophages. The concentration of the respective host bacterial species within the samples, after the 24 h bacterial challenge tests, was determined by preparing serial dilutions (10−1 to 10−4) and spread-plating 100 μL of the samples onto TSA, incubating at 30 °C for 18 h and then enumerating the colony forming units (CFU) per mL. Additionally, based on the results obtained for the bacterial challenge test performed on the P. aeruginosa ATCC 27853 strain, agglutination tests were performed to screen for lipopolysaccharide (LPS) defective mutants.39
2.8 Small-scale bacteriophage pre-treatment of spiked rainwater and SODIS-CPC trials
Two small-scale bacteriophage pre-treatment (8 h or 24 h) and SODIS-CPC trials were performed using bacteriophage PAW33 and the environmental isolate P. aeruginosa S1 68. Roof-harvested rainwater was collected from a rainwater tank located on a local farm (GPS coordinates: 33°56′38.5′′S 18°46′26.3′′E), whereafter four 1 L rainwater aliquots were autoclaved three times. An overnight culture of the environmental P. aeruginosa S1 68 strain was spiked into each of the four 1 L rainwater aliquots to achieve a final concentration of 1.0 × 107 CFU per mL. Two of the 1 L rainwater aliquots were simultaneously spiked with 500 μL of a concentrated bacteriophage solution to achieve an MOI of 0.01, with one of the samples incubated for 8 h and the other aliquot incubated for 24 h at 30 °C at 120 rpm on a rotary shaker (New Brunswick Scientific). The remaining two 1 L rainwater aliquots served as the no bacteriophage treatment controls and were spiked with 500 μL sterile SM-buffer and were also incubated for 8 and 24 h at 30 °C at 120 rpm on a rotary shaker. Ten millilitre aliquots were collected at 0 and 8 h time intervals from the 8 h pre-treatment and corresponding no pre-treatment control samples and at 0, 1, 2, 4, 8, 16 and 24 h time intervals from the 24 h pre-treatment and corresponding no pre-treatment control samples.Four SODIS-CPC reactors (described in Waso et al.40), with a treatment capacity of 500 mL, were filled with the 8 h or 24 h pre-treated samples or the 8 h or 24 h no pre-treatment control samples, respectively, and were exposed to natural sunlight for 4 h. The remaining rainwater from each sample (±400 mL) was incubated at room temperature in the dark over the same time period to serve as the “dark controls” for the SODIS-CPC trial. Samples (10 mL) were collected from each SODIS-CPC system at 0, 1, 2, 3 and 4 h. The temperature of the collected samples was monitored with a hand-held mercury thermometer (ALLA France®, Chemillé, France).
However, as Pseudomonas spp. may enter a viable but non-culturable state during unfavourable conditions (such as those experienced during disinfection treatments), EMA-qPCR assays were included to monitor the gene copy numbers of P. aeruginosa S1 68. Briefly, 2 mL of a collected sample was centrifuged at 10000 × g for 10 min. The obtained pellet was re-suspended in 1 mL saline (0.85% NaCl), whereafter the sample was treated with 6 μM ethidium monoazide bromide (EMA; Biotium, Hayward, CA, USA) as outlined in Reyneke et al.29 Deoxyribonucleic acid extraction was completed using the Quick-DNA™ Fecal/Soil Microbe Miniprep Kit (Zymo Research, Inqaba Biotech, South Africa) according to the manufacturers' instructions.
Similarly, in order to ensure that only DNA from intact (infective) PAW33 virions were quantified during qPCR analysis, 2 mL of a collected sample was treated with 5 U per mL DNase (Thermo Scientific) as outlined in Reyneke et al.,29 whereafter the sample was centrifuged at 25000 × g for 60 min. The obtained pellet was re-suspended in the pre-lyse mixture (Buffer T1, B3 and Proteinase K) of the NucleoSpin® Tissue kit (Macherey-Nagel) and the DNA extraction was completed as outlined by the manufacturer.
For the relative quantification of the rpoS, phzM, recA and lexA genes of P. aeruginosa S1 68 the primer pairs and cycling parameters as outlined in Table 1 were utilised. The cycle quantification values (Cq) of the reference gene (rpoS) were utilised to normalise the calculated Cq values of the target genes (phzM, recA and lexA) amplified from the corresponding samples (ΔCq), and the fold change (2−ΔΔCq) was compared to the baseline sample (0 h samples).43recA and lexA were selected as target genes as they are involved in the global SOS response initiated by bacteria upon exposure to adverse environmental conditions, particularly those associated with DNA damage (inactivation mechanism of SODIS),44 while phzM is a virulence-associated gene involved in the production of pyocyanin [secondary metabolite (blue-green pigment) produced by P. aeruginosa] which has also been hypothesised to protect P. aeruginosa from oxidative stress (inactivation mechanism of SODIS).34 High-resolution melt curve analysis was included for each qPCR assay in order to verify the specificity of the primer set by ramping the temperature form 65 to 97 °C at a rate of 0.2 °C s−1 with continuous fluorescent signal acquisition at 15 readings per °C. Standard curves for the quantification of P. aeruginosa S1 68 and PAW33 were generated using the methodology outlined in Reyneke et al.29
3. Results
3.1 Bacteriophage isolation and characterisation
Bacteriophages were selected for further analysis based on the number and consistency of plaque formation, plaque clarity (clear plaques selected over turbid plaques) and plaque size. Consequently, bacteriophages PAW33 (isolated using P. aeruginosa ATCC 27853) and PFW25 (isolated using P. fluorescens ATCC 13525) were selected for further analysis.Based on the STEM micrographs (Fig. 1), both bacteriophages were classified as members of the order Caudovirales (tailed bacteriophages). The morphological features of PAW33 (Fig. 1a) indicated that this bacteriophage belongs to the Podoviridae family. Bacteriophages belonging to this family are characterised as having short, “stubby” non-contractile tails.48 It was observed that the capsid of PAW33 had a hexagonal outline indicating an icosahedral nature, with the capsid diameter recorded as ∼63 nm and the tail measuring <20 nm in length (tapering) (Fig. 1). PFW25 was identified as a Myoviridae bacteriophage based on its morphological features (Fig. 1b). Bacteriophages belonging to this family are characterised as having contractile tails.48 As was observed for PAW33, the capsid of PFW25 had a hexagonal outline indicating an icosahedral nature; however, the capsid was slightly elongated. The capsid was ∼72 nm wide and ∼88 nm long, with the contractile tail measuring ∼125 nm (Fig. 1).
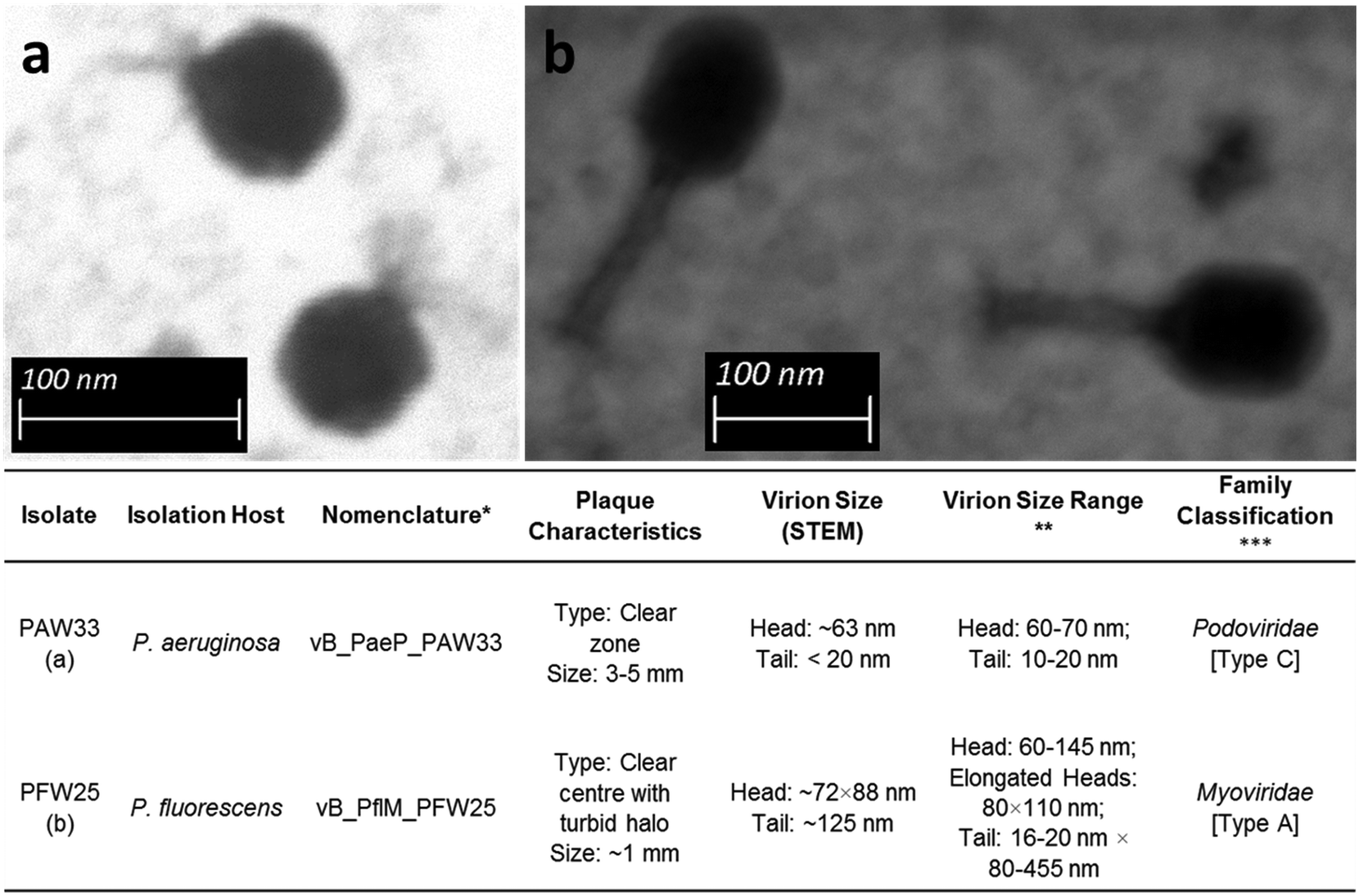 | ||
| Fig. 1 Scanning transmission electron micrographs and general characteristics of (a) PAW33 at a magnification of ×159000 and (b) PFW25 at a magnification of ×100000. *Bacteriophage nomenclature as proposed by Kropinski et al.;45 **9th Report of the International Committee on Taxonomy of Viruses;46 ***classification as described by Bradley47 indicated in brackets. | ||
3.2 Nucleic acid analysis and PCR-based identification of PAW33 and PFW25
Results obtained following restriction endonuclease digestion with DNaseI (dsDNA), S1 nuclease (ssDNA) and RNase (RNA) confirmed that the bacteriophages were dsDNA viruses, which corresponds to their classification in the Caudovirales order. The DNA fragments obtained after digestion with EcoRI or ClaI indicated that PAW33 had an estimated molecular weight of 73 kb, while the molecular weight of PFW25 could not be estimated following digestion; however, this may potentially be explained by resistance mechanisms employed by PFW25 to protect its genome during host cell infection (outlined in ESI†).Following the design and optimisation of numerous primer sets, the Podo-Hypo-F/R (targeting Podoviridae) and Myo-Hypo-F/R primer sets (targeting Myoviridae) (Table 1), were selected to confirm the preliminary classification of the isolated bacteriophages and enable the quantification of the bacteriophages during the pre-treatment and SODIS-CPC trials. Conventional PCR analysis of DNA obtained from PAW33 using the Podo-Hypo-F/R primer set resulted in the amplification of a 225 bp product. Sequencing analysis of the amplicon indicated that PAW33 shared sequence similarity with Pseudomonas bacteriophages LP14 (GenBank accession no: MH356729.1), YH30 (GenBank accession no: KP994390.1), phi176 (GenBank accession no: KM411960.1) and Pa2 (GenBank accession no: NC_027345.1), respectively (Table S2, ESI†). These bacteriophages are listed as belonging to the Podoviridae family, which further corroborates the preliminary classification of PAW33. The Podo-Hypo-F/R primer set did not amplify DNA from PFW25.
Conventional PCR analysis of DNA obtained from PFW25 using the Myo-Hypo-F/R primer set resulted in the amplification of a 254 bp product. Sequencing analysis of the amplicon indicated that PFW25 shared sequence similarity with Klebsiella phage vB_Kpn_F48 (GenBank accession no: MG746602.1) (Table S2, ESI†). Although the sequence similarity corresponded to a Klebsiella bacteriophage, vB_Kpn_F48 was classified as a Myoviridae bacteriophage by Ciacci et al.37 This result thus corroborates the preliminary classification of PFW25 as a Myoviridae bacteriophage. Additionally, the Myo-Hypo-F/R primer set did not amplify DNA from PAW33.
3.3 Host range determination for PAW33 and PFW25
The host range of the isolated bacteriophages was assessed against various target (Pseudomonas spp.) and non-target bacterial species (Table S1, ESI†), with activity recorded as the presence of clear zones or plaques (++), turbid zones or plaques (+) or no growth inhibition (−). While no activity was observed for PAW33 against the 57 non-target bacterial species analysed, lytic activity was observed against 92% (n = 11) of the P. aeruginosa strains and 25% (n = 2) of the other Pseudomonas spp. tested (Fig. 2). In contrast, PFW25 displayed activity against the non-target bacterial species, K. pneumoniae ATCC 10031 and ATCC 333305 (results not shown), but none of the other non-target bacterial species analysed. PFW25 also displayed lytic activity against 75% (n = 3) of the P. fluorescens strains analysed and against 25% (n = 3) of the other Pseudomonas spp. tested (Fig. 2).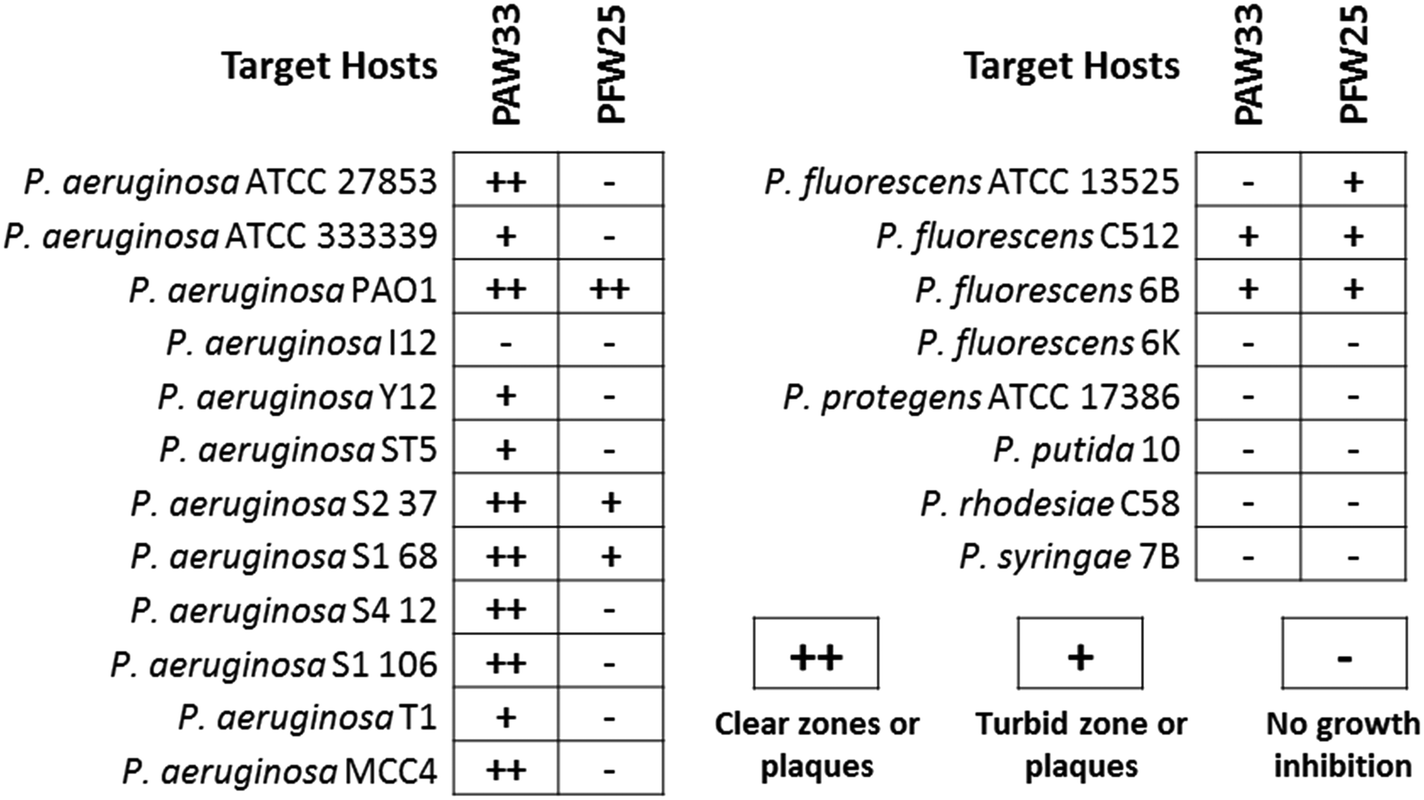 | ||
| Fig. 2 Host range of PAW33 and PFW25 against the target Pseudomonas species. | ||
3.4 Bacteriophage growth characteristics and sensitivity to physical parameters
Under the conditions studied (ambient temperature of 20 to 22 °C and aerobic conditions), PAW33 displayed a latency period of ∼80 min, a rise period of ∼50 min and a burst size of ∼136 PFU per infected cell, when co-cultured with P. aeruginosa ATCC 27853 (results not shown). As indicated, K. pneumoniae ATCC 10031 was used to elucidate the life cycle of bacteriophage PFW25 during the one-step growth experiments and subsequent experiments. Under the conditions studied, PFW25 displayed a latency and rise period of ∼30 min each, while the burst size of PFW25 was ∼47 PFU per infected cell (results not shown).Results from the temperature stability tests indicated that the infectivity of PAW33 remained stable between 30 °C and 50 °C; however, a significant decrease (∼5log) in infectivity was observed after 10 min at 70 °C and 60 min at 60 °C (Fig. S1a, ESI†). A similar temperature sensitivity profile was observed for PFW25 as its infectivity remained stable between 30 °C and 50 °C. PFW25 infectivity gradually decreased by ∼1log after 60 min at 60 °C and then remained relatively constant for the remaining 60 min. In comparison, a significant decrease (∼ 4log) in PFW25 infectivity was recorded after 30 min at 70 °C (Fig. S1b, ESI†). Results for the pH stability tests indicated that PAW33 retained infectivity after incubation at pH values ranging from 6.0 to 9.0, while a 0.19log, 0.41log and 0.51log decrease in infectivity was observed following incubation at pH 4, 5 and 10 (as compared to the mean PFU recorded for pH 6.0 to 9.0) (Fig. S1c, ESI†). In comparison, the infectivity of PFW25 remained relatively constant after incubation at pH values ranging from 5.0 to 8.0; however, at pH 4.0, 9.0 and 10.0, a 0.43log, 0.25log and 0.55log decrease, in PFW25 infectivity was observed (as compared to the mean PFU recorded for pH 5.0 to 8.0), respectively (Fig. S1d, ESI†).
3.5 Efficiency of PAW33 and PFW25 to control target host growth
Results for the bacterial challenge tests indicated that the untreated P. aeruginosa control increased significantly (p = 0.00004) during hour 2 to 6 as an increase in sample turbidity was observed, whereafter bacterial growth started to plateau, remaining relatively constant over the next 18 h (Fig. S2a, ESI†). In comparison, at all three MOI's analysed, PAW33 was effectively able to inhibit the proliferation of P. aeruginosa ATCC 27853 during the first 12 h of co-culture, whereafter steady increases in P. aeruginosa growth was observed (Fig. S2a, ESI†). Although, PAW33 was not able to completely eliminate the P. aeruginosa population, culture-based analysis following the 24 h co-culture indicated that the P. aeruginosa CFU were 1.30log (p = 0.0038), 1.08log (p = 0.0048) and 1.06log (p = 0.0046) lower in the samples treated at an MOI of 1, 0.1 and 0.01, respectively, in comparison to the untreated bacterial control (results not shown). In order to determine whether the increase in P. aeruginosa growth in the PAW33 treated samples was due to the emergence of resistance to the bacteriophage, bacterial cells were harvested and susceptibility to PAW33 was assessed using the spot-test method. Results indicated that the P. aeruginosa population were still susceptible to PAW33; however, bacteriophage resistant mutants had emerged. These colonies were characterised by the production of a red pigment, which resulted in a red mutant phenotype observed on the TSA plates (results not shown). Visualisation of these colonies using microscopy and comparison to the untreated control samples (not treated with PAW33 during co-culture) revealed that these bacteriophage resistant mutants clumped together following the agglutination test, indicating that their bacterial cell surface was LPS defective.
Results for the bacterial challenge tests indicated that limited growth was observed in the untreated K. pneumoniae ATCC 10031 control during the first 4 h, whereafter bacterial growth increased significantly (p = 0.00003) during the next 6 h and then started to plateau, remaining constant over the next 12 h (Fig. S2b, ESI†). In comparison, PFW25 was effectively able to inhibit the proliferation of K. pneumoniae during the first 16 h of co-culture for all three MOI ratio's tested; however significant increases in K. pneumoniae growth was observed between 16 and 24 h (Fig. S2b, ESI†). Culture-based analysis following the 24 h co-culture indicated that the K. pneumoniae CFU were 0.94log (p = 0.0122), 1.05log (p = 0.0129) and 0.85log (p = 0.0187) lower in the samples treated at an MOI of 1, 0.1 and 0.01, respectively, as compared to the untreated bacterial control (results not shown). Spot test analysis of the culture following completion of the co-culture experiments indicated that the K. pneumoniae population was still susceptible to PFW25; however, bacteriophage resistant mutants had also emerged as turbid plaques (in comparison to clear plaques observed when the untreated K. pneumoniae controls were subjected to PFW25 during the spot test analysis) were visible.
3.6 Small-scale bacteriophage pre-treatment of spiked rainwater prior to SODIS-CPC
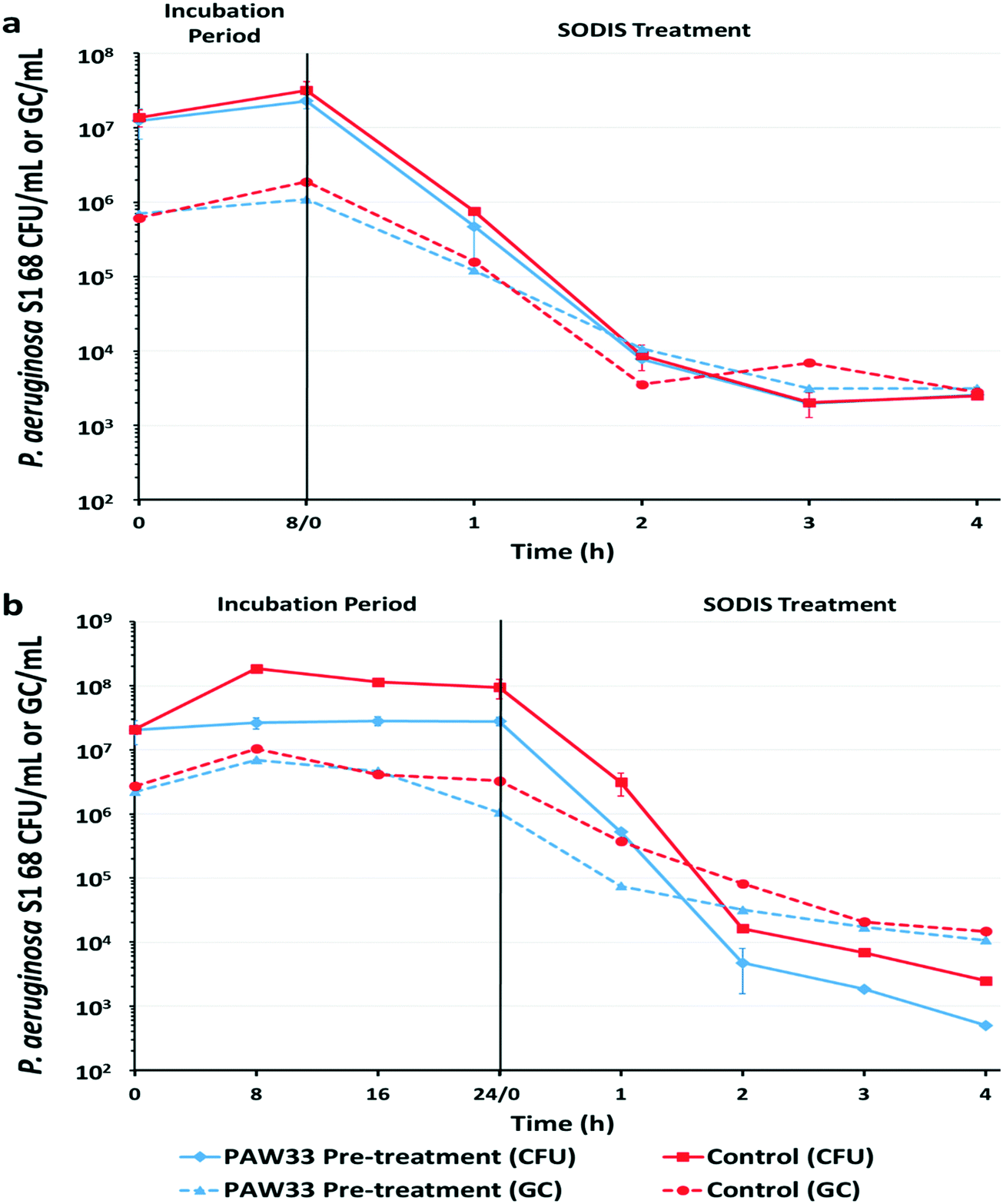 | ||
| Fig. 3 Pseudomonas aeruginosa concentration (CFU per mL and GC per mL) in sterile rainwater during an (a) 8 h bacteriophage pre-treatment and (b) 24 h bacteriophage pre-treatment, followed by a 4 h SODIS treatment. Red lines (■, ●) indicate the sample that was not pre-treated with PAW33 (control; target host only), while the blue lines (♦, ▲) indicate the sample that was treated with PAW33 at an MOI of 0.01. The CFU per mL (culture-based analysis) are indicated with a solid line, while the GC per mL (molecular-based analysis) are indicated by the dashed line. The data points represent the means of duplicate samples, the error bars indicate standard deviation (SD). | ||
For the 8 h trial (Fig. 3a), culture-based analysis of the non-pre-treated control sample indicated that the P. aeruginosa S1 68 CFU counts increased by 0.36log, from 1.38 × 107 CFU per mL to 3.19 × 107 CFU per mL, over the 8 h incubation period. Subsequent exposure of the non-pre-treated sample to a 4 h SODIS-CPC treatment resulted in a total log reduction of 3.74 (p = 0.0109) in P. aeruginosa S1 68 CFU counts (2.50 × 103 CFU per mL recorded after SODIS-CPC), from the initial concentration of 1.38 × 107 CFU per mL (Table S4, ESI†). Correspondingly, EMA-qPCR analysis indicated that the P. aeruginosa S1 68 GC increased by 0.49log over the 8 h incubation period, from 6.13 × 105 GC per mL to 1.88 × 106 GC per mL, with a reduction to 2.80 × 103 GC per mL recorded following SODIS-CPC treatment [2.33 total log reduction (p = 0.0087)] (Fig. 3a; Table S4, ESI†). Culture-based and EMA-qPCR analysis of the corresponding dark control sample (collected after the 4 h SODIS-CPC treatment), indicated that the concentration of P. aeruginosa S1 68 remained relatively constant with 2.13 × 107 CFU per mL and 1.81 × 106 GC per mL recorded, respectively (results not shown).
Culture-based analysis of the corresponding 8 h bacteriophage pre-treated sample indicated that the P. aeruginosa S1 68 CFU per mL increased by 0.26log from 1.24 × 107 CFU per mL to 2.28 × 107 CFU per mL, whereafter the SODIS-CPC treatment reduced the cell counts to 2.58 × 103 CFU per mL [3.68 total log reduction (p = 0.0299) from the initial CFU of 1.24 × 107] (Fig. 3a; Table S4, ESI†). Similarly, EMA-qPCR analysis indicated that the P. aeruginosa S1 68 gene copies (GC) only increased by 0.19log during the 8 h pre-treatment, from 6.98 × 105 GC per mL to 1.09 × 106 GC per mL, whereafter the gene copies were reduced to 3.15 × 103 GC per mL during the SODIS-CPC treatment [2.34 total log reduction (p = 0.0033)] (Table S4, ESI†). Monitoring of PAW33 in the 8 h pre-treated sample indicated that the PFU per mL decreased by 0.28log from 6.00 × 104 PFU per mL to 3.16 × 104 PFU per mL, while 1.20 × 102 PFU per mL were detected following the SODIS-CPC treatment [2.70 total log reduction (p = 0.0023)] (Fig. S3a, ESI†). In contrast, the PAW33 GC per mL increased by 0.48log (1.80 × 104 GC per mL to 5.37 × 104 GC per mL) during the 8 h pre-treatment, whereafter the gene copies remained relatively constant, as 1.42 × 104 GC per mL were recorded following the SODIS-CPC treatment [0.12 total log reduction (p = 0.1909)] (Table S4 and Fig. S3a, ESI†). Culture-based and EMA-qPCR analysis of the corresponding dark control sample (collected after the 4 h SODIS-CPC treatment), indicated that the concentration of P. aeruginosa S1 68 remained relatively constant with 8.63 × 106 CFU per mL and 1.23 × 106 GC/mL recorded, respectively, while PAW33 also remained constant as 3.16 × 104 PFU per mL and 1.92 × 104 GC per mL were recorded (results not shown).
For the 24 h trial, culture-based analysis of the non-pre-treated control sample, indicated that P. aeruginosa S1 68 increased by 0.67log, from 2.08 × 107 CFU per mL to 9.42 × 107 CFU per mL over the 24 h incubation period (Fig. 3b). The P. aeruginosa S1 68 cell counts were subsequently reduced to 2.5 × 103 CFU per mL (from an initial CFU of 2.08 × 107) following the SODIS-CPC treatment [3.91 total log reduction (p = 0.0101)] (Table S4, ESI†). Similarly, EMA-qPCR analysis of the non-pre-treated control sample indicated that the P. aeruginosa S1 68 GC per mL marginally increased from 2.71 × 106 GC per mL to 3.28 × 106 GC per mL after 24 h (0.08log increase) (Fig. 3b). An overall total reduction of 2.26log (p = 0.0239) in GC was then observed following the SODIS-CPC treatment (GC reduced to 1.47 × 104 GC per mL) (Table S4, ESI†). Culture-based and EMA-qPCR analysis of the corresponding dark control sample (collected after the 4 h SODIS-CPC treatment), indicated that the concentration of P. aeruginosa S1 68 remained relatively constant with 9.63 × 107 CFU per mL and 7.98 × 106 PFU per mL recorded, respectively (results not shown).
Culture-based analysis of the corresponding bacteriophage pre-treated sample from the 24 h trial indicated that PAW33 was able to restrict the proliferation of P. aeruginosa S1 68 in the pre-treated sample, as the P. aeruginosa S1 68 CFU counts only increased by 0.14log, from 2.03 × 107 CFU per mL to 2.79 × 107 CFU per mL (Fig. 3b). Subsequent SODIS-CPC treatment of the pre-treated sample reduced the P. aeruginosa S1 68 CFU counts to 5.0 × 102 CFU per mL [4.61log reduction overall (p = 0.0079)], from the initial count of 2.03 × 107 (Table S4, ESI†). Similarly, EMA-qPCR analysis of the pre-treated sample indicated a 0.30log increase (5.31 × 105 GC per mL to 1.06 × 106 GC per mL) in P. aeruginosa S1 68 GC per mL during the 24 h pre-treatment, whereafter the GC were reduced to 1.07 × 104 GC per mL due to the SODIS-CPC treatment [2.32log reduction overall (p = 0.0128)] (Fig. 3b; Table S4, ESI†). Enumeration of the PAW33 plaque counts in the pre-treated sample indicated that the PFU per mL increased from 8.0 × 104 PFU per mL to 4.0 × 105 PFU per mL (0.70log increase) during the 24 h pre-treatment, whereafter a decrease to 1.3 × 102 PFU per mL was recorded following the SODIS-CPC treatment [2.79log reduction overall (p = 0.0115)] (Fig. S3b, ESI†). In comparison, the PAW33 GC per mL remained relatively constant in the pre-treated sample during the 24 h trial, as 2.50 × 104 GC per mL were detected at both 0 and 24 h, while 4.8 × 103 GC per mL were detected following SODIS-CPC treatment [0.72log reduction overall (p = 0.0270)] (Table S4, ESI†). Culture and EMA-qPCR analysis of the corresponding dark control sample (collected after the 4 h SODIS-CPC treatment), indicated that the concentration of P. aeruginosa S1 68 remained relatively constant with 4.62 × 107 CFU per mL and 6.45 × 106 GC per mL recorded, respectively, while PAW33 also remained relatively constant as 1.93 × 105 PFU per mL and 2.10 × 104 GC per mL were recorded (results not shown).
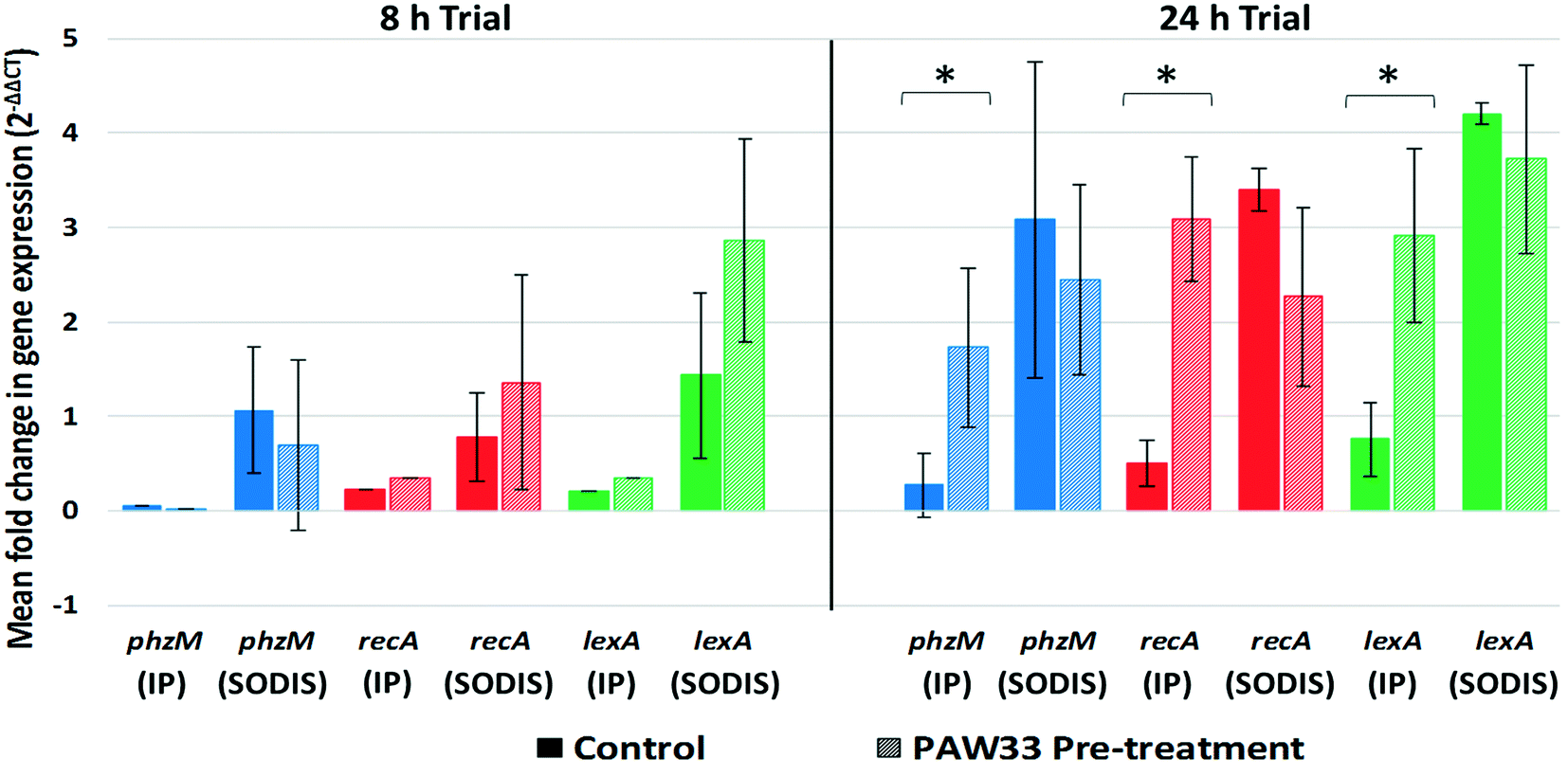 | ||
| Fig. 4 Mean fold change in phzM, recA and lexA gene expression by P. aeruginosa S1 68 after an 8 h or 24 h incubation period (IP) and after a 4 h SODIS-CPC treatment (SODIS). Solid columns indicate the sample that was not pre-treated with PAW33 (target host only), while the dashed columns indicate the sample that was treated with PAW33 at an MOI of 0.01. The data represent the means of multiple time points analysed for each phase (IP or SODIS) (duplicate samples analysed for each time point), the error bars indicate standard deviation (SD). Significant differences (p < 0.05) between samples are indicated by an asterisk. | ||
For the 24 h trial, phzM (0.27-fold), recA (0.50-fold) and lexA (0.76-fold) were up-regulated in the non-pre-treated control sample during the 24 h incubation period, whereafter increased expression of the phzM (3.08-fold), recA (3.41-fold) and lexA (4.20-fold) genes were observed following SODIS-CPC treatment (Fig. 4). Similarly, for the PAW33 pre-treated samples, phzM (1.73-fold), recA (3.08-fold) and lexA (2.92-fold) were up-regulated during the 24 h incubation trial. However, in comparison to the non-pre-treated control, while still up-regulated, decreased expression of phzM (2.44-fold), recA (2.27-fold) and lexA (3.72-fold) was observed during the SODIC-CPC treatment (Fig. 4). Overall, the mean expression level for phzM, recA and lexA in the pre-treated sample was thus lower as compared to the non-pre-treated control sample during the SODIC-CPC treatment (Fig. 4).
4. Discussion
Solar disinfection is widely investigated as a disinfection method that helps reduce the presence of pathogens in water and the incidence of diarrheal disease.3 One of the main disadvantages of SODIS however, is the low efficiency of the treatment of resistant pathogens.49 These bacteria are able to undergo an adaptive response and build-up resistance to stressful environments, such as those experienced during conventional water treatment methods. As bacteriophages may allow for the selective removal of problematic pathogens within water samples,22–24 bacteriophage biocontrol was investigated and combined with SODIS-CPC in order to reduce the concentration and limit the proliferation of P. aeruginosa in rainwater.Lytic bacteriophages displaying activity against Pseudomonas spp. were subsequently isolated from numerous environmental sources, with PAW33 (isolated using P. aeruginosa) and PFW25 (isolated using P. fluorescens) selected for further characterisation. Electron microscopy and nucleic acid analysis indicated that both PAW33 and PFW25 belong to the order Caudovirales and more specifically the Podoviridae and Myoviridae families, respectively. Subsequently, the pH and temperature sensitivity of PAW33 and PFW25 was assessed as various chemical and physical parameters (such as those associated with rainwater harvesting systems) may influence the viability/infectivity of bacteriophages by damaging their structural elements (e.g. head and tail structures).50 Results however, indicated that both bacteriophages were stable and retained their infectivity upon exposure to the physico-chemical parameters commonly associated with untreated harvested rainwater (pH 6.2 to 8.4; 19 to 26 °C) and temperatures experienced within large-scale SODIS systems (39 to 59 °C).6,51
Caudovirales bacteriophages are associated with more than 140 prokaryotic genera with varying degrees of host specificity reported.46,52 The host range determination then indicated that PAW33 was able to infect reference, environmental and clinical isolates of P. aeruginosa, with notable activity displayed against the multidrug-resistant P. aeruginosa T1 clinical isolate53 and numerous environmental strains previously isolated from a solar pasteurization system connected to a rainwater harvesting tank (Table S1, ESI†). Additionally, as PAW33 was able to infect two environmental P. fluorescens strains, it was classified as having a broad host range against P. aeruginosa strains, with limited activity against other Pseudomonas spp. and no activity against the non-target bacteria. In comparison, PFW25 was able to infect three P. fluorescens strains, two environmental and one clinical isolate of P. aeruginosa (Fig. 2) and two K. pneumoniae ATCC strains (results not shown). The activity displayed against K. pneumoniae and the efficiency of plating when PFW25 was cultured with K. pneumoniae ATCC 10031, coupled with the sequence similarity (hypothetical protein targeted by the Myo-Hypo-F/R primer set) displayed to bacteriophage vB_Kpn_F48, indicated that PFW25 may be better suited to target K. pneumoniae strains. Similarly, Wu et al. reported on the isolation of a Myoviridae bacteriophage (Kpp95) using K. pneumoniae, which was subsequently classified as having a broad host range, as the bacteriophage displayed lytic activity against K. pneumoniae, K. oxytoca, Enterobacter agglomerans and Serratia marcescens.54
For the bacterial challenge tests, while the bacteriophages PAW33 and PFW25 were able to inhibit the growth of their respective target hosts (P. aeruginosa ATCC 27853 and K. pneumoniae ATCC 10031), bacteriophage resistant P. aeruginosa and K. pneumoniae mutants had emerged. Specifically, for P. aeruginosa ATCC 27853, the bacteriophage resistant mutants were characterised by the production of a red pigment and were classified as being LPS defective (based on an agglutination test). A similar observation was made by Le et al.,39 where it was demonstrated that a chromosomal DNA deletion (gene fragment containing the hmgA and galU genes) conferred bacteriophage resistance to P. aeruginosa, with the deletion of hmgA resulting in the accumulation of a red compound (homogentisic acid) and the deletion of galU resulting in the loss of the O-antigen (which is required for bacteriophage adsorption). Moreover, as LPS is an important virulence factor within Gram-negative bacterial pathogens, the authors reported that, in a mouse infection model, the bacteriophage resistant P. aeruginosa were significantly attenuated.39 Thus, while the emergence of bacteriophage resistant bacteria is a major concern when employing bacteriophage biocontrol, these bacteria (such as the P. aeruginosa obtained in the current study following exposure to PAW33) may be less virulent39 and thereby pose a lower health risk to the end-user. Bacteriophages do however, have the ability to develop counter strategies to by-pass bacterial resistance mechanisms and thereby ensure the survival of the bacteriophage population and in so doing continue to restrict the proliferation of the target bacterial population.55 These strategies include, amongst others, the modification of the bacteriophage receptor binding proteins, which recognise new receptors/adsorption sites on bacteria, the production of enzymes to degrade bacterial capsules or exopolysaccharides, and the modification of the bacteriophage genome to circumvent restriction–modification systems (restriction enzyme digestion) in bacteria.55
The efficiency of bacteriophage biocontrol as a rainwater pre-treatment strategy was ultimately assessed using PAW33 as the biocontrol agent and P. aeruginosa S1 68 (environmental isolate obtained from rainwater pasteurized at 70 °C) as the target organism. Based on observations from the bacterial challenge tests (Fig. S2a, ESI†) and a supplementary bacterial challenge test conducted in sterile rainwater on P. aeruginosa ATCC 27853 and P. aeruginosa S1 68 (results not shown), two pre-treatment times, namely 8 h and 24 h, were assessed. It was hypothesised that the bacteriophage pre-treatment would firstly restrict the proliferation of the target host pathogen during the pre-treatment period and secondly sensitise the overall bacterial population to the primary treatment strategy (i.e. SODIS-CPC).
Culture-based and EMA-qPCR analysis indicated that PAW33 was able to restrict the proliferation of P. aeruginosa S1 68 in the rainwater during both the 8 h and 24 h pre-treatment trials. However, while similar total CFU and GC log reductions were obtained for the pre-treated [3.68log (CFU) and 2.34log (GC)] and non-pre-treated control samples [3.74log (CFU) and 2.33log (GC)] for the 8 h trial (followed by SODIS-CPC); culture-based analysis indicated that a higher overall log reduction was recorded for the 24 h bacteriophage pre-treated sample (followed by SODIS-CPC) (4.61log) in comparison to the non-pre-treated sample (3.91log). Additionally, culture-based analysis indicated that after the 24 h bacteriophage pre-treatment trial, faster inactivation of P. aeruginosa S1 68 occurred during the first hour (1.73log reduction) of the SODIS-CPC treatment. A similar observation was recently reported by Al-Jassim et al. where the ability of bacteriophages to sensitise a pathogenic New Delhi metallo β-lactamase-positive E. coli to SODIS was investigated.56 Results from the study indicated that exposure to bacteriophages increased the susceptibility of E. coli to SODIS, with faster inactivation of the E. coli observed (treatment time reduced from 4 h to 2 h). Additionally, using gene expression analysis, the authors reported that the exposure of E. coli to the bacteriophage resulted in a downregulation of cell wall functions, the ability to scavenge reactive oxygen species and DNA repair mechanisms, effectively rendering the E. coli more susceptible to SODIS treatment. It is however important to note that the Al-Jassim et al. study utilised a combination of bacteriophages at a high treatment concentration (MOI = 1), the bacteriophage and SODIS treatment occurred simultaneously and an artificial light source was used to simulate SODIS.56 In contrast, in the current study a lower treatment concentration (MOI = 0.01) of a single bacteriophage was used as a pre-treatment strategy to SODIS-CPC under natural sunlight. Thus, while the bacteriophage pre-treatment for 24 h, followed by SODIS-CPC, resulted in the highest total log reduction (4.61log) of P. aeruginosa S1 68 CFU per mL, the target host could not be completely eradicated using this combination treatment strategy, as 5.0 × 102 CFU per mL was still recorded following SODIS-CPC treatment. Additionally, while EMA-qPCR analysis indicated that comparable total log reductions [2.32log (pre-treated) and 2.26log (non-pre-treated)] in P. aeruginosa S1 68 concentrations were obtained for the 24 h trial samples, gene copies were still detected after SODIS-CPC, indicating that viable but non-culturable cells may be present within the samples. The survival of the P. aeruginosa S1 68 following the combination treatment is however, not surprising as Pseudomonas spp. may initiate a range of stress responses during both the planktonic or biofilm life cycles, including the production of heat shock proteins and the initiation of DNA repair mechanisms, amongst others, and thereby switch to a more tolerant phenotype to facilitate its survival under adverse conditions.57,58 However, as highlighted by Al-Jassim et al.,56 the ability of bacteria to initiate these stress response mechanisms may be severely impaired following/during exposure to bacteriophages.
Gene expression analysis was subsequently included to monitor the SOS response-associated recA and lexA genes, while phzM (gene associated with pyocyanin production) was monitored as the bacterial challenge tests indicated that decreased pyocyanin was produced by bacteriophage resistant P. aeruginosa ATCC 27853. Results for the 8 h trial indicated that while phzM, recA and lexA expression levels were comparable in the PAW33 pre-treated and non-pre-treated control samples during the 8 h incubation period, the overall expression level for recA and lexA increased during the SODIS-CPC treatment, with higher overall expression levels for both genes observed in the PAW33 pre-treated sample. In comparison, although still up-regulated, phzM gene expression was lower in the PAW33 treated sample as compared to the non-pre-treated control sample during the SODIS-CPC treatment. Results for the 24 h trial then indicated that phzM, recA and lexA were up-regulated in the non-pre-treated control and PAW33 pre-treated samples during the 24 h incubation period, with continued upregulation observed following the SODIS-CPC treatment. However, decreased expression of phzM, recA and lexA were recorded in the PAW33 pre-treated sample as compared to the non-pre-treated control sample, during the SODIS-CPC treatment. The genes recA and lexA are known to be up-regulated in bacteria in response to adverse conditions as part of the SOS response mechanism and are primarily involved in DNA repair mechanisms.44 The decreased expression of recA and lexA in the PAW33 pre-treated P. aeruginosa S1 68 (in comparison to the non-pre-treated control sample), during the 4 h SODIS-CPC treatment, indicates that the bacteriophage pre-treatment for 24 h may have influenced the ability of the target host bacterium to initiate stress response mechanisms during the primary disinfection strategy (i.e. SODIS-CPC). Thus, based on the results obtained, a prolonged bacteriophage pre-treatment period may be required to sensitise the target host bacterium to the primary disinfection strategy (i.e. SODIS-CPC), by influencing the gene expression of recA and lexA during adverse conditions. Additionally, while assessing the influence of sub-lethal photodynamic inactivation [sPDI; photo-oxidative stress caused by the generation of reactive oxygen species (ROS) after a photosensitiser molecule was excited by visible light], Hendiani et al. reported that pyocyanin production (phzM expression) in P. aeruginosa ATCC 27853 as well as strains P2 and P3, increased during sPDI (±10-fold increase), with the authors hypothesising that the over-expression of pyocyanin played a possible protective role against sPDI-induced oxidative stress.34 As phzM gene expression was decreased in the 8 h and 24 h bacteriophage pre-treated samples (in comparison to the corresponding non-pre-treated samples) during the SODIS-CPC treatment, it is hypothesised that the decreased phzM expression may be due to the presence of bacteriophage resistant P. aeruginosa S1 68 cells within the sample (as was observed for the bacterial challenge tests). The bacteriophage pre-treatment may thus have influenced the ability of the bacteriophage resistant P. aeruginosa S1 68 cells to initiate pyocyanin production as a stress response mechanism, rendering the bacterial cells more susceptible to primary disinfection strategies (such as SODIS-CPC). Additionally, as pyocyanin is considered a virulence factor of P. aeruginosa,34 its decreased expression in the PAW33 pre-treated samples during SODIS-CPC treatment indicates that bacteriophage pre-treatment may decrease pathogen virulence. The overall results thus indicate that a longer bacteriophage pre-treatment may be required for the bacteriophages to adequately influence target host stress response mechanisms.
5. Conclusions
Results from the study indicate that PAW33 has the potential to be used in biocontrol strategies for the selective removal of P. aeruginosa from roof-harvested rainwater as this Podoviridae bacteriophage was able to effectively restrict the proliferation of P. aeruginosa S1 68 for up to 24 h. Additionally, an increase in the susceptibility of P. aeruginosa S1 68 to the SODIC-CPC disinfection treatment was observed after the 24 h bacteriophage pre-treatment trial, as a total log reduction of 4.61 was recorded. The implementation of the bacteriophage pre-treatment, as part of a large-scale point-of-use system, should however be investigated and optimised. Various strategies for implementation are recommended with the simplest strategy entailing the diversion of the water source (to be treated) into a secondary tank containing bacteriophages for pre-treatment, whereafter the treated water may be re-directed to the primary disinfection system (large-scale solar disinfection or solar pasteurization systems).Moreover, while gene copies and CFU were still detected after SODIS-CPC for both the 8 h and 24 h trials, it important to note that the efficiency of the bacteriophage pre-treatment may be improved by using a combination of bacteriophages,59 while the SODIS-CPC treatment efficiency may be further improved by increasing the SODIS treatment time (6 to 8 h SODIS exposures recommended in literature).5 Additionally, although the fold changes observed during gene expression analysis were not significant, results from the 24 h bacteriophage pre-treatment trial indicated that the P. aeruginosa S1 68 exhibited a reduced ability to initiate conventional stress response mechanisms (recA and lexA), while the expression of pyocyanin (phzM; virulence factor) was also decreased during the 8 and 24 h bacteriophage pre-treatment trial. The ability of bacteriophage biocontrol to influence pathogen stress response mechanisms and virulence during treatment should thus be further investigated. Moreover, as biofilm formation is a key survival strategy employed by P. aeruginosa, the biofilm disruption and anti-adhesive abilities of PAW33 should be investigated in future studies.
Author contributions
Conceived and designed the experiments: BR and WK. Performed the experiments: BR. Designed the primers: SK. Designed the small-scale SODIS-CPC systems: PFI. Analysed the data: BR and WK. Contributed reagents/materials/analysis tools: WK and SK. Compiled the manuscript: BR and WK. Edited the manuscript: SK and PFI.Funding
The financial assistance of the National Research Foundation (NRF) (Grant Number: 90320) of South Africa towards this research is hereby acknowledged. Opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to the NRF.Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors would like to thank Prof Lydia-Marie Joubert (Head of the Electron Microbeam Unit of the Central Analytical Facility at Stellenbosch University) for assistance with the uranyl acetate staining and STEM analysis.References
- J. A. Driscoll, S. L. Brody and M. H. Kollef, The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections, Drugs, 2007, 67(3), 351–368, DOI:10.2165/00003495-200767030-00003.
- T. J. John, U. Lalla, J. J. Taljaard, K. G. John, J. Slabbert and C. F. N. Koegelenberg, An outbreak of community-acquired Pseudomonas aeruginosa pneumonia in a setting of high water stress, QJM, 2017, 110(12), 855–856, DOI:10.1093/qjmed/hcx148.
- E. Ubomba-Jaswa, P. Fernández-Ibáñez, C. Navntoft, M. I. Polo-López and K. G. McGuigan, Investigating the microbial inactivation efficiency of a 25 L batch solar disinfection (SODIS) reactor enhanced with a compound parabolic collector (CPC) for household use, J. Chem. Technol. Biotechnol., 2010, 85(8), 1028–1037, DOI:10.1002/jctb.2398.
- R. Shrivastava, R. K. Upreti, S. R. Jain, K. N. Prasad, P. K. Seth and U. C. Chaturvedi, Suboptimal chlorine treatment of drinking water leads to selection of multidrug-resistant Pseudomonas aeruginosa, Ecotoxicol. Environ. Saf., 2004, 58(2), 277–283, DOI:10.1016/S0147-6513(03)00107-6.
- A. Strauss, P. H. Dobrowsky, T. Ndlovu, B. Reyneke and W. Khan, Comparative analysis of solar pasteurization versus solar disinfection for the treatment of harvested rainwater, BMC Microbiol., 2016, 16, 289, DOI:10.1186/s12866-016-0909-y.
- A. Strauss, B. Reyneke, M. Waso and W. Khan, Compound parabolic collector solar disinfection system for the treatment of harvested rainwater, Environ. Sci.: Water Res. Technol., 2018, 4, 976–991, 10.1039/c8ew00152a.
- T. L. Clements, B. Reyneke, A. Strauss and W. Khan, Persistence of viable bacteria in solar pasteurised harvested rainwater, Water, Air, Soil Pollut., 2019, 230, 130, DOI:10.1007/s11270-019-4184-z.
- G. P. Winward, L. M. Avery, R. Frazer-Williams, M. Pidou, P. Jeffrey, T. Stephenson and B. Jefferson, A study of the microbial quality of grey water and an evaluation of treatment technologies for reuse, Ecol. Eng., 2008, 32(2), 187–197, DOI:10.1016/j.ecoleng.2007.11.001.
- A. Maimon, E. Friedler and A. Gross, Parameters affecting greywater quality and its safety for reuse, Sci. Total Environ., 2014, 487, 20–25, DOI:10.1016/j.scitotenv.2014.03.133.
- A. Gross, D. Kaplan and K. Baker, Removal of chemical and microbiological contaminants from domestic greywater using a recycled vertical flow bioreactor (RVFB), Ecol. Eng., 2007, 31(2), 107–114 CrossRef.
- Y. Gilboa and E. Friedler, UV disinfection of RBC-treated light greywater effluent: kinetics, survival and regrowth of selected microorganisms, Water Res., 2008, 42(4–5), 1043–1050, DOI:10.1016/j.watres.2007.09.027.
- A. M. Wesche, J. B. Gurtler, B. P. Marks and E. T. Ryser, Stress, sub-lethal injury, resuscitation and virulence of bacterial foodborne pathogens, J. Food Prot., 2009, 72(5), 926–1138, DOI:10.4315/0362-028X-72.5.1121.
- M. R. J. Clokie, A. D. Millard, A. V. Letarov and S. Heaphy, Phages in nature, Bacteriophage, 2011, 1(1), 31–45, DOI:10.4161/bact.1.1.14942.
- N. K. Petty, T. J. Evans, P. C. Fineran and G. P. C. Salmond, Biotechnological exploitation of bacteriophage research, Trends Biotechnol., 2007, 25(1), 7–15, DOI:10.1016/j.tibtech.2006.11.003.
- A. Grant, F. Hashem and S. Parveen, Salmonella and Campylobacter: Antimicrobial resistance and bacteriophage control in poultry, Food Microbiol., 2016, 53, 104–109, DOI:10.1016/j.fm.2015.09.008.
- B. Wu, R. Wang and A. G. Fane, The roles of bacteriophages in membrane-based water and wastewater treatment processes: a review, Water Res., 2017, 110, 120–132, DOI:10.1016/j.watres.2016.12.004.
- G. G. Greer, Bacteriophage control of foodborne bacteria, J. Food Prot., 2005, 68(5), 1102–1111, DOI:10.4315/0362-028X-68.5.1102.
- J. R. Clark and J. B. March, Bacteriophages and biotechnology: vaccines, gene therapy and antibacterials, Trends Biotechnol., 2006, 24(5), 212–218, DOI:10.1016/j.tibtech.2006.03.003.
- M. G. Vinod, M. M. Shivu, K. R. Umesha, B. C. Rajeeva, G. Krohne, I. Karunasagar and I. Karunasagar, Isolation of Vibrio harveyi bacteriophage with a potential for biocontrol of luminous vibriosis in hatchery environments, Aquaculture, 2006, 255(1–4), 117–124, DOI:10.1016/j.aquaculture.2005.12.003.
- R. A. Frampton, A. R. Pitman and P. C. Fineran, Advances in bacteriophage-mediated control of plant pathogens, Int. J. Microbiol., 2012, 2012, 326452, DOI:10.1155/2012/326452.
- S. Whitey, E. Cartmell, L. M. Avery and T. Stephenson, Bacteriophages - potential for application in wastewater treatment processes, Sci. Total Environ., 2005, 339(1–3), 1–18, DOI:10.1016/j.scitotenv.2004.09.021.
- Y. Turki, H. Ouzari, I. Mehri, A. B. Ammar and A. Hassen, Evaluation of a cocktail of three bacteriophages for the biocontrol of Salmonella of wastewater, Food Res. Int., 2012, 45(2), 1099–1105, DOI:10.1016/j.foodres.2011.05.041.
- G. Goldman, J. Starosvetsky and R. Armon, Inhibition of biofilm formation on UF membrane by use of specific bacteriophages, J. Membr. Sci., 2009, 342, 145–152, DOI:10.1016/j.memsci.2009.06.036.
- Y. Zhang, H. K. Hunt and Z. Hu, Application of bacteriophages to selectively remove Pseudomonas aeruginosa in water and wastewater filtration systems, Water Res., 2013, 47, 4507–4518, DOI:10.1016/j.watres.2013.05.014.
- S. Sillankorva, P. Neubauer and J. Azeredo, Isolation and characterization of a T7-like lytic phage for Pseudomonas fluorescens, BMC Biotechnol., 2008, 8, 80, DOI:10.1186/1472-6750/8/80.
- A. R. Stenholm, I. Dalsgaard and M. Middelboe, Isolation and characterization of bacteriophages infecting the fish pathogen Flavobacterium psychrophilum, Appl. Environ. Microbiol., 2008, 74(13), 4070–4078, DOI:10.1128/AEM.00428-08.
- J. Sambrook and D. W. Russell, Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, New York, USA, 3rd edn, 2001, vol. 1 Search PubMed.
- M. Laue and N. Bannert, Detection limit of negative staining electron microscopy for the diagnosis of bioterrorism-related micro-organisms, J. Appl. Microbiol., 2010, 109(4), 1159–1168, DOI:10.1111/j.1365-2672.2010.04737.x.
- B. Reyneke, T. Ndlovu, S. Khan and W. Khan, Comparison of EMA-, PMA- and DNase qPCR for the determination of microbial cell viability, Appl. Microbiol. Biotechnol., 2017, 101(19), 7371–7383, DOI:10.1007/s00253-017-8471-6.
- B. Dwivedi, R. Schmieder, D. B. Goldsmith, R. A. Edwards and M. Breitbart, PhiSiGns: an online tool to identify signature genes in phages and design PCR primers for examining phage diversity, J. Appl. Microbiol., 2012, 13(1), 37, DOI:10.1186/1471-2105-13-37.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Clustal W and Clustal X version 2.0, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS.
- K. B. Nicholas and H. B. Nicholas, GeneDoc: a tool for editing and annotating multiple sequence alignments, 1997, htttps://www.psc.edu/biomed/genedoc. Accessed 1 August 2018 Search PubMed.
- H. Savli, A. Karadenizli, F. Kolayli, S. Gundes, U. Ozbek and H. Vahaboglu, Expression stability of six housekeeping genes: a proposal for resistance gene quantification studies of Pseudomonas aeruginosa by real-time quantitative RT-PCR, J. Med. Microbiol., 2003, 52(5), 403–408, DOI:10.1099/jmm.0.05132-0.
- S. Hendiani, M. Pornour and N. Kashef, Quorum-sensing-regulated virulence factors in Pseudomonas aeruginosa are affected by sub-lethal photodynamic inactivation, Photodiagn. Photodyn. Ther., 2019, 26, 8–12, DOI:10.1016/j.pdpdt.2019.02.010.
- D. Hocquet, C. Llanes, M. Thouverez, H. D. Kulasekara, X. Bertrand, P. Plésiat, D. Mazel and S. I. Miller, Evidence for induction of integrin-based antibiotic resistance by the SOS response in a clinical setting, PLoS Pathog., 2012, 8(6), e1002778, DOI:10.1371/journal.ppat.1002778.
- Y. P. Yu, T. Gong, G. Jost, W. H. Liu, D. Z. Ye and Z. H. Luo, Isolation and characterisation of five lytic bacteriophages infecting a Vibrio strain closely related to Vibrio owensii, FEMS Microbiol. Lett., 2013, 348(2), 112–119, DOI:10.1111/1574-6968.12277.
- N. Ciacci, M. M. D'Andrea, P. Marmo, E. Demattè, F. Amisano, V. Di Pilato, M. Fraziano, P. Lupetti, G. M. Rossolini and M. C. Thaller, Characterisation of vB_Kpn_F48, a newly discovered lytic bacteriophage for Klebsiella pneumoniae of sequence type 101, Viruses, 2018, 10, 482, DOI:10.3390/v10090482.
- M. Jamal, T. Hussain, C. R. Das and S. Andleeb, Characterization of Siphoviridae phage Z and studying its efficacy against multidrug-resistant Klebsiella pneumoniae planktonic cells and biofilm, J. Med. Microbiol., 2015, 64(4), 454–462, DOI:10.1099/jmm.0.000040.
- S. Le, X. Yao, S. Lu, Y. Tan, X. Rao, M. Li, X. Jin, J. Wang, Y. Zhao, N. C. Wu, R. Lux, X. He, W. Shi and F. Hu, Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa, Sci. Rep., 2014, 4, 4738, DOI:10.1038/srep04738.
- M. Waso, S. Khan, A. Singh, S. McMichael, W. Ahmed, P. Fernández-Ibáñez, J. A. Byrne and W. Khan, Predatory bacteria in combination with solar disinfection and solar photocatalysis for the treatment of rainwater, Water Res., 2019 DOI:10.1016/j.watres.2019.115281 , In Press.
- C. Lambert, C.-Y. Chang, M. J. Capeness and R. E. Sockett, The first bite – profiling the predatosome in the bacterial pathogen Bdellovibrio, PLoS One, 2010, 5(1), e8599, DOI:10.1371/journal.pone.0008599.
- S. Roosa, C. V. Wauven, G. Billon, S. Matthijs, R. Wattiez and D. C. Gillan, The Pseudomonas community in metal contaminated sediments as revealed by quantitative PCR: a link with metal bioavailability, Res. Microbiol., 2014, 165, 647–656, DOI:10.1016/j.resmic.2014.07.011.
- P. H. Dobrowsky, S. Khan and W. Khan, Resistance of Legionella and Acanthamoeba mauritaniensis to heat treatment as determined by relative and quantitative polymerase chain reactions, Environ. Res., 2017, 158, 82–93, DOI:10.1016/j.envres.2017.06.003.
- J. E. Krebs, E. S. Goldstein and S. T. Kilpatrick, Lewin's Genes XII, Jones & Bartlett Learning, Burlington, MA, 12th edn, 2018, pp. 1363–1368 Search PubMed.
- A. M. Kropinski, D. Prangishvili and R. Lavigne, Position paper: the creation of a rational scheme for the nomenclature of viruses of bacteria and archaea, Environ. Microbiol., 2009, 11(11), 2775–2777, DOI:10.1111/j.1462-2920.2009.01970.x.
- Ninth Report of the International Committee on Taxonomy of Viruses, Virus Taxonomy – Classification and Nomenclature of Viruses. 2011, https://www.kau.edu.sa/Files/0011106/Subjects/Virus%20Taxonomy.pdf, Accessed 17 March 2019.
- D. E. Bradley, Ultrastructure of bacteriophages and bacteriocins, Bacteriol. Rev., 1967, 31(4), 230–314 CAS.
- O. Sepúlveda-Robles, L. Kameyama and G. Guarneros, High diversity and novel species of Pseudomonas aeruginosa bacteriophages, Appl. Environ. Microbiol., 2012, 78(12), 4510–4515, DOI:10.1128/AEM.00065-12.
- K. G. McGuigan, R. M. Conroy, H. Mosler, M. Du Preez, E. Ubomba-Jaswa and P. Fernandez-Ibañez, Solar water disinfection (SODIS): a review from bench-top to roof-top, J. Hazard. Mater., 2012, 235-236, 29–46, DOI:10.1016/j.jhazmat.2012.07.053.
- E. Jończyk, M. Klak, R. Międzybrodzki and A. Górski, The influence of external factors on bacteriophages – review, Folia Microbiol., 2011, 56(3), 191–200, DOI:10.1007/s12223-011-0039-8.
- B. Reyneke, T. E. Cloete, S. Khan and W. Khan, Rainwater harvesting solar pasteurization treatment systems for the provision of an alternative water source in peri-urban informal settlements, Environ. Sci.: Water Res. Technol., 2018, 4, 291–302, 10.1039/c7ew00392g.
- A. Kęsik-Szeloch, Z. Drulis-Kawa, B. Weber-Dąbrowska, J. Kassner, G. Majkowska-Skrobek, D. Augustniak, M. Łusiak-Szelachowska, M. Żaczek, A. Górski and A. M. Kropinski, Characterising the biology of novel lytic bacteriophages infecting multidrug resistant Klebsiella pneumoniae, Virol. J., 2013, 10(1), 100, DOI:10.1186/1743-422X-10-100.
- B. Havenga, T. Ndlovu, T. Clements, B. Reyneke, M. Waso and W. Khan, Susceptibility of the World Health Organisation critical priority list of antibiotic-resistant bacteria to surfactin, HonsBSc Thesis, Stellenbosch University, 2018 Search PubMed.
- L. T. Wu, S. Y. Chang, M. R. Yen, T. C. Yang and Y. H. Tseng, Characterization of extended-host-range pseudo-T-even bacteriophage KPP95 isolated on Klebsiella pneumoniae, Appl. Environ. Microbiol., 2007, 73(8), 2532–2540, DOI:10.1128/AEM.02113-06.
- J. E. Samson, A. H. Magadán, M. Sabri and S. Moineau, Revenge of the phages: defeating bacterial defences, Nat. Rev. Microbiol., 2013, 11, 675–687, DOI:10.1038/nrmicro3096.
- N. Al-Jassim, D. Mantilla-Calderon, G. Scarascia and P. Y. Hong, Bacteriophages to sensitize a pathogenic New Delhi metallo β-lactamase-positive Escherichia coli to solar disinfection, Environ. Sci. Technol., 2018, 2, 14331–14341, DOI:10.1021/acs.est.8b04501.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Survival strategies of infectious biofilms, Trends Microbiol., 2005, 13(1), 34–40, DOI:10.1016/j.tim.2004.11.010.
- E. B. M. Breidenstein, C. De La Fuente-Núñez and R. E. W. Hancock, Pseudomonas aeruginosa: all roads lead to resistance, Trends Microbiol., 2011, 19(8), 419–426, DOI:10.1016/j.tim.2011.04.005.
- J. Gu, X. Li, M. Yang, C. Du, Z. Cui, P. Gong, F. Xia, J. Song, L. Zhang, J. Li, C. Yu, C. Sun, X. Feng, L. Lei and W. Han, Therapeutic effect of Pseudomonas aeruginosa phage YH30 on mink hemorrhagic pneumonia, Vet. Microbiol., 2016, 190, 5–11, DOI:10.1016/j.vetmic.2016.03.016.
Footnote |
| † Electronic supplementary information (ESI) available: Legionella spp. growth conditions and host range determination. Nucleic acid analysis of PFW25. List of target and non-target bacterial species (host range determination). Sequencing results of the Podo-Hypo-F/R (Podoviridae) and Myo-Hypo-F/R (Myoviridae) primer sets. qPCR performance characteristics. Summary of cell counts gene copies and log reductions recorded for the pre-treatment/SODIS-CPC trials. Characterisation results for the isolated bacteriophages (i.e. temperature and pH sensitivity) and results obtained for the bacterial challenge tests. See DOI: 10.1039/c9ew00896a |
| This journal is © The Royal Society of Chemistry 2020 |