
Iridium complex-linked porous organic polymers for recyclable, broad-scope photocatalysis of organic transformations†
Zi-Yue
Xu
,
Yi
Luo
,
Dan-Wei
Zhang
 *,
Hui
Wang
,
Xing-Wen
Sun
* and
Zhan-Ting
Li
*
*,
Hui
Wang
,
Xing-Wen
Sun
* and
Zhan-Ting
Li
*
Department of Chemistry, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Fudan University, 2205 Songhu Lu, Shanghai 200438, China. E-mail: zhangdw@fudan.edu.cn; sunxingwen@fudan.edu.cn; ztli@fudan.edu.cn
First published on 22nd November 2019
Abstract
Two rigid porous organic polymers (Ir-POP-1 and Ir-POP-2) were prepared from the coupling reactions of tetraphenylmethane tetraborate and two [Ir(ppy)2(dtbbpy)]+-based bitopic linkers and applied as heterogeneous visible-light photocatalysts for organic transformations. Ir-POP-2 was found to exhibit high catalytic activity for a wide range of organic reactions, which include Smiles–Truce rearrangement of alkyliodides, desulfurative conjugate addition to Michael acceptors, and aerobic oxidations of sulfides and arylboronic acids. For all the transformations, Ir-POP-2 could achieve heterogeneous photocatalytic efficiency that rivals that of the homogeneous prototype iridium complexes. This remarkably high photocatalytic performance has been attributed to the large pore size of the conjugated backbone. The new heterogeneous photocatalyst was also highly stable to achieve good recyclability for all the studied reactions and could be reused eight to nineteen times.
Introduction
The past decade has witnessed the rapid development of visible light photoredox catalysis of organic reactions due to its mild reaction conditions and the potential of harvesting solar energy for chemical transformations.1 In a general working model, this strategy utilizes metal complexes and organic dyes to engage in single-electron transfer processes with organic substrates upon photoexcitation with visible light. The most widely used metal complex photocatalysts are polypyridyl complexes of ruthenium and iridium.2 In comparison with ruthenium complexes, a library of ligands with discrete electronic properties has been developed for iridium,3 which allows for the design of tunable iridium-based photoredox catalysts. Most of the reported studies have focused on their applications in homogeneous organic transformations. In contrast, examples of heterogeneous reactions catalyzed by iridium complexes immobilized to rigid porous polymers or frameworks are very rare.4 In 2011, Lin and co-workers reported the first example which involved the utilization of the Sonogashira cross-coupling reaction to introduce Ir(ppy2)(bpy)+ complexes into a cross-linked polymer for heterogeneous photocatalysis of the aza-Henry reaction with four cycles of the catalyst.4a Recently, Han and co-workers demonstrated that [Ir(ppy)2(bpy)]+-cross-linked polycarbazole networks could work as heterogeneous photocatalysts for the oxidation of sulfides, hydroxylation of arylboronic acids, and cross-dehydrogenative coupling reactions.4b For sulfide oxidation, the catalyst realizes five cycles of high conversion. Given that loading precious-metal catalysts to solid supports represents one of the most efficient strategies for their green and cost-lowering applications,5 it is highly valuable to develop new iridium complex-incorporated recyclable photocatalysts to expand their catalysis for important organic transformations.In the past decade, conjugated porous polymers and related structures have emerged as promising platforms for the construction of recyclable heterogeneous catalysts.6 Tetrakisphenylmethane and its derivatives are synthetically accessible and easily allow cross-coupling polymerization and exhibit permanent backbone porosity. Incorporation of multipyridyl-iridium complexes into tetrakisphenyl-methane-based polymers through conjugated cross-linkers would lead to highly stable heterogeneous photocatalysts that maintain the porosity of the backbones and enable the dispersion of the catalytic sites.7 Herein we present the synthesis of two highly stable [Ir(ppy)2(dtbbpy)]+ (ppy: phenylpyridine, dtbbpy: 4,4′-di(tert-butyl)-2,2′-bipyridine)-linked porous organic polymers, Ir-POP-1 and Ir-POP-2, from Pd-catalyzed coupling reactions of a tetrakisphenyl-methane borate and two [Ir(ppy)2(dtbbpy)]+-derived bromides. We demonstrate that the iridium complexes in the new porous coordination polymers can be applied as heterogeneous photoredox catalysts for four discrete organic transformations with previously unattainable recyclability.
Results and discussion
The synthetic routes for Ir-POP-1 and Ir-POP-2 are shown in Scheme 1. The dichloro-bridged iridium(III) dimers 2a and 2b were first prepared from the reaction of 1a or 1b with iridium chloride in methoxyethanol and water. The two dimers were then treated with 3 in dichloromethane and methanol to afford [Ir(ppy)2(dtbbpy)]+ complexes 4a and 4b. Finally, the two iridium complexes were coupled with tetraborate 5 in dioxane and water to produce Ir-POP-1 and Ir-POP-2 after being treated with hot water and organic solvents to remove soluble species. Starting from 6a and 6b, iridium complexes Ir-Ph-1 and Ir-Ph-2 were also prepared as controls. Single crystals suitable for X-ray diffraction analysis were obtained for complexes 4a, 4b and Ir-Ph-2 by slow evaporation of their solution in a suitable solvent. All their crystal structures showed that the two 2-phenyl-substituted pyridine ligands were oriented in the opposite direction of the octahedral complex core (Fig. 1). Thus, complexes 4a and 4b resembled rod-like 4,4′-dibromobiphenyl or longer analogues in their coupling reactions with 5 to form rigid porous polymers Ir-POP-1 and Ir-POP-2. Both polymers were insoluble in common solvents including water, DMF, DMSO, acetonitrile and acetone, whereas the controls Ir-Ph-1 and Ir-Ph-2 were soluble in the above organic solvents. Thermogravimetric analyses revealed that the two polymers were stable up to 350 °C (10% weight loss) (Fig. S1, ESI†). | ||
| Scheme 1 The synthesis of polymers Ir-POP-1 and Ir-POP-2 and complexes Ir-Ph-1 and Ir-Ph-2. | ||
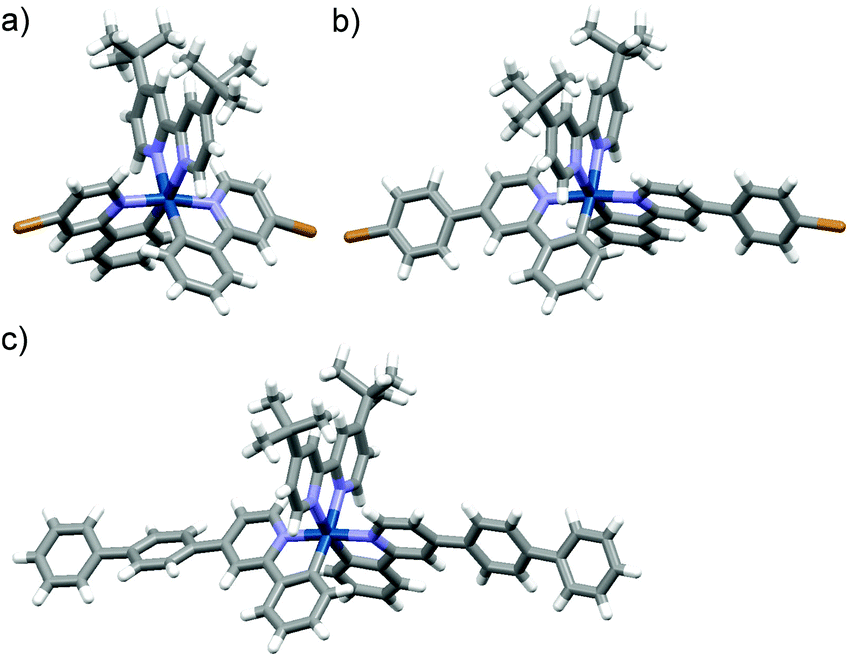 | ||
| Fig. 1 The crystal structure of complexes (a) 4a, (b) 4b, and (c) Ir-Ph-2 (CCDC deposit no. 1961159–1961161†). | ||
Fourier transform infrared spectroscopy of polymers Ir-POP-1 and Ir-POP-2 showed the absence of the C–B (around 1020 cm−1) or B–O (around 1320 cm−1) vibration absorption of the borate group8 and the C–Br (around 1068 cm−1) vibration absorption of 4a or 4b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8b (Fig. S2 and S3, ESI†), indicating that the precursors were consumed completely in the coupling reactions. Scanning electron microscopy (SEM) images and powder X-ray diffraction (PXRD) patterns indicated that Ir-POP-1 existed as amorphous solids (Fig. S4 and S5, ESI†), whereas Ir-POP-2 featured spherical aggregates with sizes around 1 μm (Fig. S4 and S5, ESI†). Energy dispersive X-ray spectroscopy (EDS) confirmed the existence of the C, N, F, P and Ir elements (Fig. S6 and S7, ESI†). X-ray photoelectron spectroscopy (XPS) further confirmed the composition of the above elements (Fig. 2a). The oxygen peak may be attributed to the partial oxidation of iridium during oxidative polymerization and the coordinated solvent molecules.9 The local spectrum of the Ir 4f peak showed a division of two emission peaks (Ir 4f5/2: 64.8 eV and Ir 4f7/2: 62.0 eV), which indicated the +3 oxidation state of iridium atoms (Fig. 2b).4b,10
8b (Fig. S2 and S3, ESI†), indicating that the precursors were consumed completely in the coupling reactions. Scanning electron microscopy (SEM) images and powder X-ray diffraction (PXRD) patterns indicated that Ir-POP-1 existed as amorphous solids (Fig. S4 and S5, ESI†), whereas Ir-POP-2 featured spherical aggregates with sizes around 1 μm (Fig. S4 and S5, ESI†). Energy dispersive X-ray spectroscopy (EDS) confirmed the existence of the C, N, F, P and Ir elements (Fig. S6 and S7, ESI†). X-ray photoelectron spectroscopy (XPS) further confirmed the composition of the above elements (Fig. 2a). The oxygen peak may be attributed to the partial oxidation of iridium during oxidative polymerization and the coordinated solvent molecules.9 The local spectrum of the Ir 4f peak showed a division of two emission peaks (Ir 4f5/2: 64.8 eV and Ir 4f7/2: 62.0 eV), which indicated the +3 oxidation state of iridium atoms (Fig. 2b).4b,10
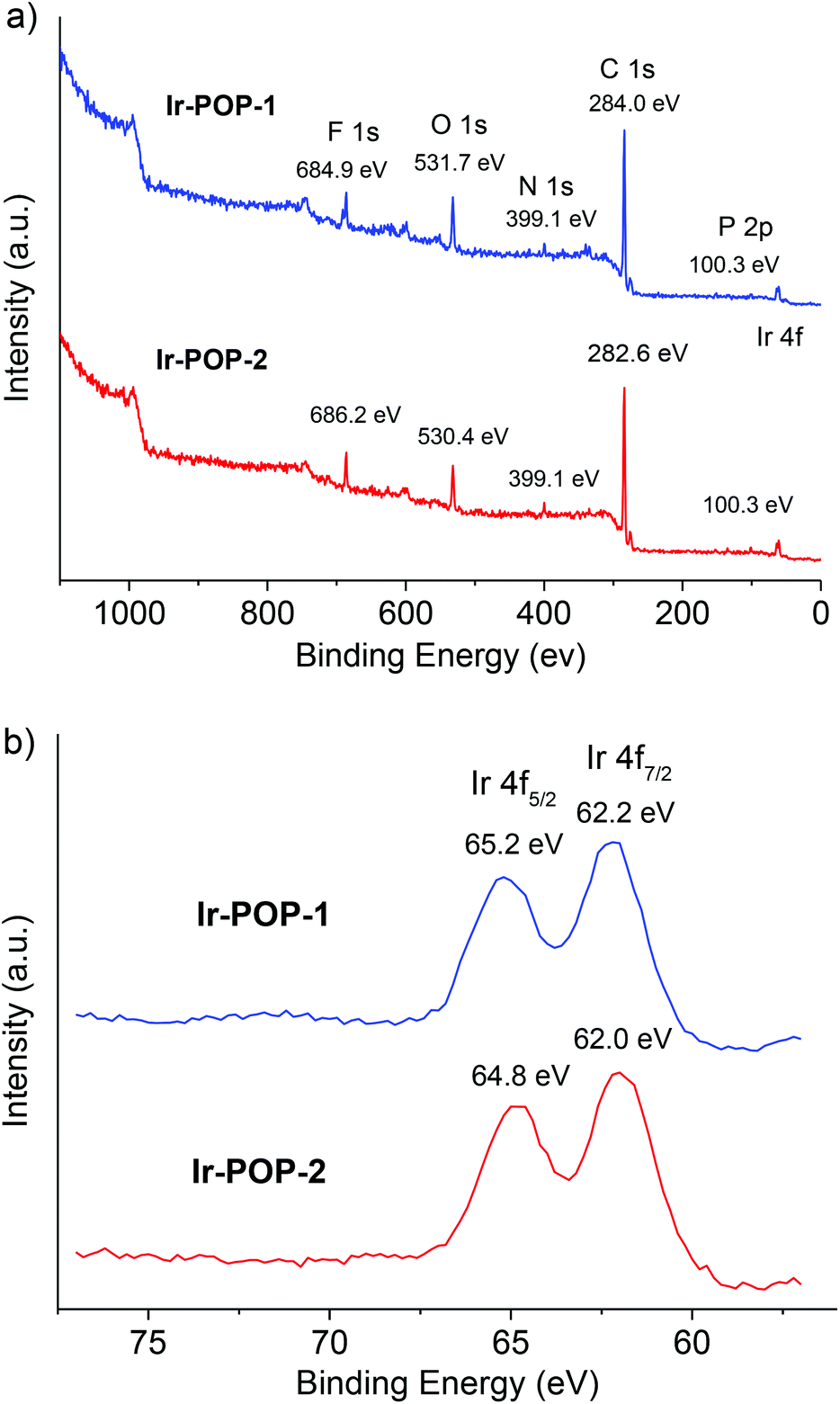 | ||
| Fig. 2 (a) The XPS survey spectra and (b) the Ir 4f spectra of polymers Ir-POP-1 and Ir-POP-2. | ||
The permanent porosities of Ir-POP-1 and Ir-POP-2 were investigated by nitrogen sorption measurement at 77 K. Their Brunauer–Emmett–Teller (BET) surface areas were determined to be 29 and 124 m2 g−1, respectively (Fig. 3). The value of Ir-POP-2 was significantly larger than that of Ir-POP-1, which was consistent with the elongated linkers of its backbone and suggested that Ir-POP-2 might exhibit higher photocatalytic activity.
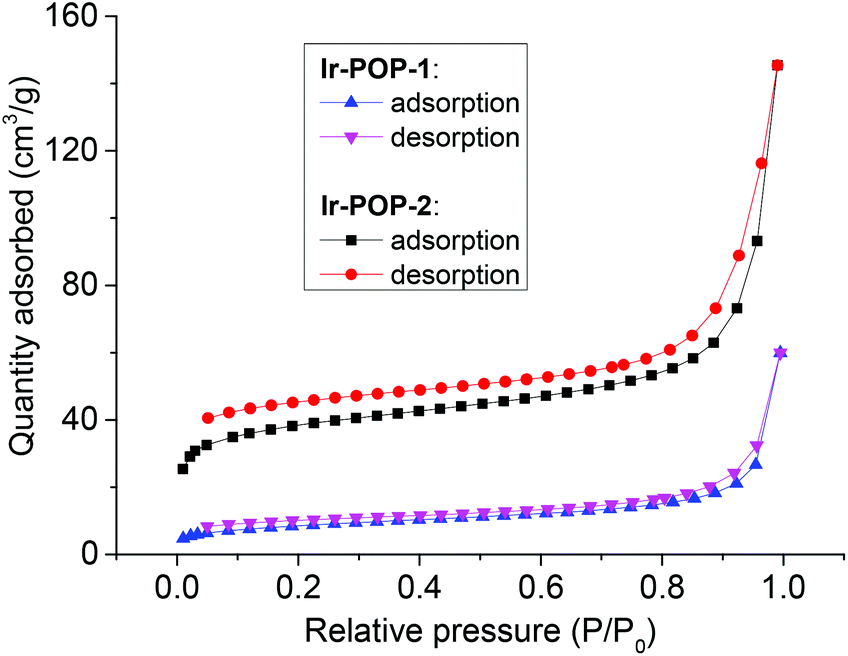 | ||
| Fig. 3 N2 adsorption and desorption isotherms of Ir-POP-1 and Ir-POP-2 at 77 K. | ||
With the two highly stable porous Ir-complex-incorporated polymers in hand, we investigated their potential for heterogeneous visible light photocatalysis of organic transformations. The intramolecular Smiles–Truce rearrangement reaction of aryl amines was first studied (Table 1). Stephenson et al. recently reported that this reaction can be realized homogeneously in acetonitrile through visible light mediated photocatalysis with the prototype iridium complex [Ir(ppy)2(dtbbpy)]PF6.11 The product is a useful substrate for the synthesis of tetrahydrothienoazepinone, which has received significant attention in drug development.11a We first studied the rearrangement reaction of model molecule 8a to produce 9a, using the reported reaction conditions with Ir-POP-1, Ir-POP-2, Ir-Ph-1 or Ir-Ph-2 as the photocatalyst. Under standard conditions, both control complexes Ir-Ph-1 (E1/2(M/M−) = −1.50 V vs. SCE) and Ir-Ph-2 (E1/2(M/M−) = −1.53 V vs. SCE) (Fig. S8, ESI†) could realize quantitative transformation through homogeneous photocatalysis (entries 1 and 2, Table 1) in 1.5 hours. This reaction time was substantially shorter than that (23 hours) required by the prototype complex [Ir(ppy)2(dtbbpy)]PF6 for quantitative conversion of 8a into 9a, suggesting that the two aryl units of Ir-Ph-1 and Ir-Ph-2 increased their catalytic activity. Insoluble IR-POP-2, which contained an identical molar amount of the Ir complex, catalyzed the reaction quantitatively in a heterogeneous manner in 24 hours (entry 4, Table 1), which was very close to that required by the prototype complex [Ir(ppy)2(dtbbpy)]PF6, indicating that the pores of Ir-POP-2 are large enough to avoid significant obstruction by the backbone for the catalytic process. Under the same conditions, the reaction catalyzed by Ir-POP-1 afforded 9a in 52% yield (entry 3, Table 1), which may be attributed to its smaller porosity that did not allow free access of the substrate molecules to the Ir complexes in the polymer. The Ir catalyst was indispensable for the reaction, and decreasing the loading of the Ir complex (0.5 mol%) also caused significant reduction of the reaction yield (entries 5 and 6, Table 1). With the use of a smaller amount of DIPEA or a light source of lower power, the yield decreased significantly (entries 7–10, Table 1). The reaction could occur in other organic solvents (entries 11–15, Table 1), including N,N-dimethylacetamide (DMF), N,N-dimethylacetamide (DMAC), dimethyl sulfoxide (DMSO), acetone or N,N′-dimethylpropyleneurea (DMPU). However, the yield was generally notably lower than that in MeCN. With NEt3, tetramethylethylenediamine (TMEDA), Hantzsch ester (5.0) or 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU) as a base, the reaction also afforded a lower yield of 9a or even did not occur (entries 16–19, Table 1).
a
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst [mol%] | Base [equiv.] | Solvent | Yieldb [%] |
| a Standard conditions: 8a (48 mg, 0.1 mmol, 1.0 equiv.), Ir photocatalyst (1 μmol, 1.0 mol%), base (0.5 mmol, 5.0 equiv.), two 34 W blue LED lamps, 25 °C. b Yield determined by GC using dodecane as the internal standard. c Reaction time: 1.5 h. d Using one 23 W compact fluorescent lamp (CFL) as the light source. e No light source. f 2,2,6,6-Tetramethylpiperidinyl-1-oxide (TEMPO, 1.0 equiv.) was added as a free radical scavenger. | ||||
| 1c | Ir-Ph-1 (1.0) | DIPEA (5.0) | MeCN | >99 |
| 2c | Ir-Ph-2 (1.0) | DIPEA (5.0) | MeCN | >99 |
| 3 | Ir-POP-1 (1.0) | DIPEA (5.0) | MeCN | 52 |
| 4 | Ir-POP-2 (1.0) | DIPEA (5.0) | MeCN | >99 |
| 5 | Ir-POP-2 (0.5) | DIPEA (5.0) | MeCN | 57 |
| 6 | — | DIPEA (5.0) | MeCN | n.d. |
| 7 | Ir-POP-2 (1.0) | DIPEA (3.0) | MeCN | 11 |
| 8 | Ir-POP-2 (1.0) | — | MeCN | n.d. |
| 9d | Ir-POP-2 (1.0) | DIPEA (5.0) | MeCN | 9 |
| 10e | Ir-POP-2 (1.0) | DIPEA (5.0) | MeCN | n.d. |
| 11 | Ir-POP-2 (1.0) | DIPEA (5.0) | DMF | 65 |
| 12 | Ir-POP-2 (1.0) | DIPEA (5.0) | DMAC | 59 |
| 13 | Ir-POP-2 (1.0) | DIPEA (5.0) | DMSO | 80 |
| 14 | Ir-POP-2 (1.0) | DIPEA (5.0) | Acetone | 22 |
| 15 | Ir-POP-2 (1.0) | DIPEA (5.0) | DMPU | 20 |
| 16 | Ir-POP-2 (1.0) | NEt3 (5.0) | MeCN | 68 |
| 17 | Ir-POP-2 (1.0) | TMEDA (5.0) | MeCN | 26 |
| 18 | Ir-POP-2 (1.0) | Hantzsch ester (5.0) | MeCN | 40 |
| 19 | Ir-POP-2 (1.0) | DBU (5.0) | MeCN | n.d. |
| 20f | Ir-POP-2 (1.0) | DIPEA (5.0) | MeCN | n.d. |

After 6 hours and with Ir-POP-2 (1.0 mol%) as the catalyst, the reaction of 8a afforded 9a in 56% yield. After seven cycles, 9a was still formed in 50% yield (Fig. S9, ESI†). Inductively coupled plasma-optical emission spectroscopy (ICP-OES) experiment revealed that Ir-POP-2 had a loading of 73.9 wt% for [Ir(ppy)2(dtbbpy)]+ catalytic center, which was very close to the calculated value of 75.0 (wt%). After recycling, the Ir complex only had ≤0.4% leaching. These results clearly supported the high stability of the new heterogeneous catalyst.
The above optimized conditions were then applied for a variety of structurally related starting materials (Scheme 2). It was found that tosyl (Ts), trifluorotosyl (TsF3, for 9b and 9g) and tert-butyloxycarbonyl (Boc, for 9c) all could be used as a protecting group (PG) for this transformation, but tosyl was the most suitable. Both (CH2)3 and (CH2)4 linkers could achieve good to excellent reaction yields, and the substrates bearing a –(CH2)4– group at the 4- and 5-positions of the thiophene ring also gave high yields of the corresponding products (9d and 9f). When the positions of the two substituents were exchanged, the corresponding starting materials still underwent the rearrangement reaction to afford the corresponding products (9h and 9i) in high yields. The anisole derivative also underwent a similar rearrangement. However, the yield of the reaction was considerably lowered (9j). In contrast, the reaction of the cyclopentene and cyclohexene-derived substrates afforded the corresponding products (9k–m) in good to excellent yields. The polymer Ir-POP-1 could catalyze all the above reactions. However, the yields were considerably lower (Fig. S10, ESI†). The recyclability of Ir-POP-2 for the reaction of 8a and 8d was further investigated and 90% conversion was achieved after eight or five cycles, respectively (Fig. 4a and b). Reducing the amount of Ir-POP-2 to 0.5 mol% (entry 5, Table 1) led to lowered yields, but the catalyst could exhibit good recyclability (five times, in 57%, 56%, 57%, 54%, and 53% yields), reflecting the high stability of the polymeric catalyst.
 | ||
| Scheme 2 Substrate scope for the Ir-POP-2-catalyzed rearrangement of alkyliodides 8a–m to form the related alkylamines 9a–m. The yield represents the isolated one, and the value in the bracket represents the GC yield. Standard conditions: 8 (0.1 mmol, 1.0 equiv.), Ir-POP-2 (1 μmol, 1.0 mol%), DIPEA (85 μL, 0.5 mmol, 5.0 equiv.), 25 °C. | ||
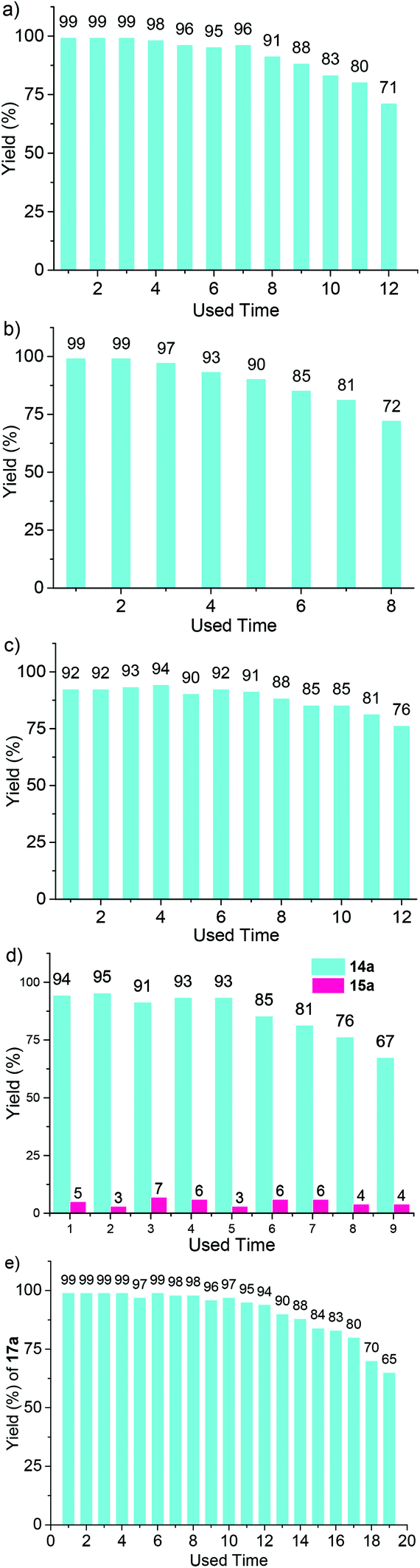 | ||
| Fig. 4 Recyclability of Ir-POP-2 in the reactions for the generation of (a) 9a, (b) 9d, (c) 12a, (d) 14a and 15a, and (e) 17a. The catalysts were recycled from the reaction mixture by centrifugation, washed with acetonitrile and dichloromethane and dried, and reused in a fresh reaction solution. | ||
With the addition of 1.0 equiv. of TEMPO as a radical trapping agent,12 the reaction was inhibited completely (entry 20, Table 1). Stern–Volmer quenching experiments indicated that DIPEA rather than 8a was the appropriate quencher for the photosensitizer (Fig. S11, ESI†).13 Cyclic voltammetry (CV) experiments revealed that the redox potential of the Ir-complex species (E1/2(IrIII/IrII) = −1.53 V vs. SCE for Ir-Ph-2 and E1/2(IrIII/IrII) = −1.50 V vs. SCE for Ir-Ph-1) matched well with that of 8a (E1/2 = −1.26 V vs. SCE) (Fig. S8, ESI†). Because Ir-POP-1 and Ir-POP-2 contained the Ir-Ph-1 and Ir-Ph-2 units, respectively, all the above results supported that the reaction mediated by Ir-POP-1 and Ir-POP-2 proceeded also through a radical mechanism as reported previously for the homogeneous photocatalytic reaction.11a On–off experiments confirmed that a continuous light source was indispensable for the reaction (Fig. S12a, ESI†), while a leaching experiment supported the heterogeneity of the procedure (Fig. S12b and S13, ESI†).14
Catalytic activities of polymers Ir-POP-1 and Ir-POP-2 were also investigated for intermolecular reactions. Qin et al. recently reported the visible-light photoredox catalysis for the conjugate addition reaction between N-benzoyl alkylsulfinamides and Michael acceptors in the presence of [Ir(ppy)2(dtbbpy)]PF6,15 which provides a new synthetic utility of N-acyl alkylsulfinamides as alkyl radical precursors. We first conducted the reaction of 10a and 11a in the presence of Ir-Ph-1, Ir-Ph-2, Ir-POP-1 and Ir-POP-2. The heterogeneous photocatalytic activity of Ir-POP-2, with an identical molar amount of the iridium complex, was comparable to that of homogeneous Ir-Ph-1 or Ir-Ph-2, whereas the photocatalytic activity of Ir-POP-1 was considerably lower (Table S1, ESI†). Reaction conditions were then screened with Ir-POP-2 as the heterogeneous photocatalyst (Tables S2 and S3, ESI†), and the optimized reaction conditions were applied for alkyl sulfinamides 10a and 10b and a variety of Michael acceptors 11a–f to form the corresponding products 12a–l (Scheme 3). The reaction tolerated different kinds of Michael acceptors including 2-methylenemalonate diesters, acrylic acids, acrylates and α- and β-methylene butyrolactones. In most cases, radical addition products were obtained in good to excellent yields. The recyclability of Ir-POP-2 for the photocatalytic reaction of 10a and 11a to produce 12a was further investigated. After seven cycles, 12a still could be obtained in 91% yield (Fig. 4c). Stern–Volmer quenching experiments indicated that the deprotonated sulfonamide 10a could be oxidized by polymers Ir-POP-1 and Ir-POP-2 to generate tert-butyl radicals which subsequently coupled with 11a to afford 12a (Fig. S14, ESI†), as reported for the homogeneous reactions.15 An on–off experiment confirmed the indispensability of continuous irradiation (Fig. S15a, ESI†), whereas a leaching experiment supported that the reaction was catalyzed heterogeneously by the insoluble polymer catalyst, rather than the possibly available trace amount of iridium species dissolved in the solvent (Fig. S15b and S16, ESI†).
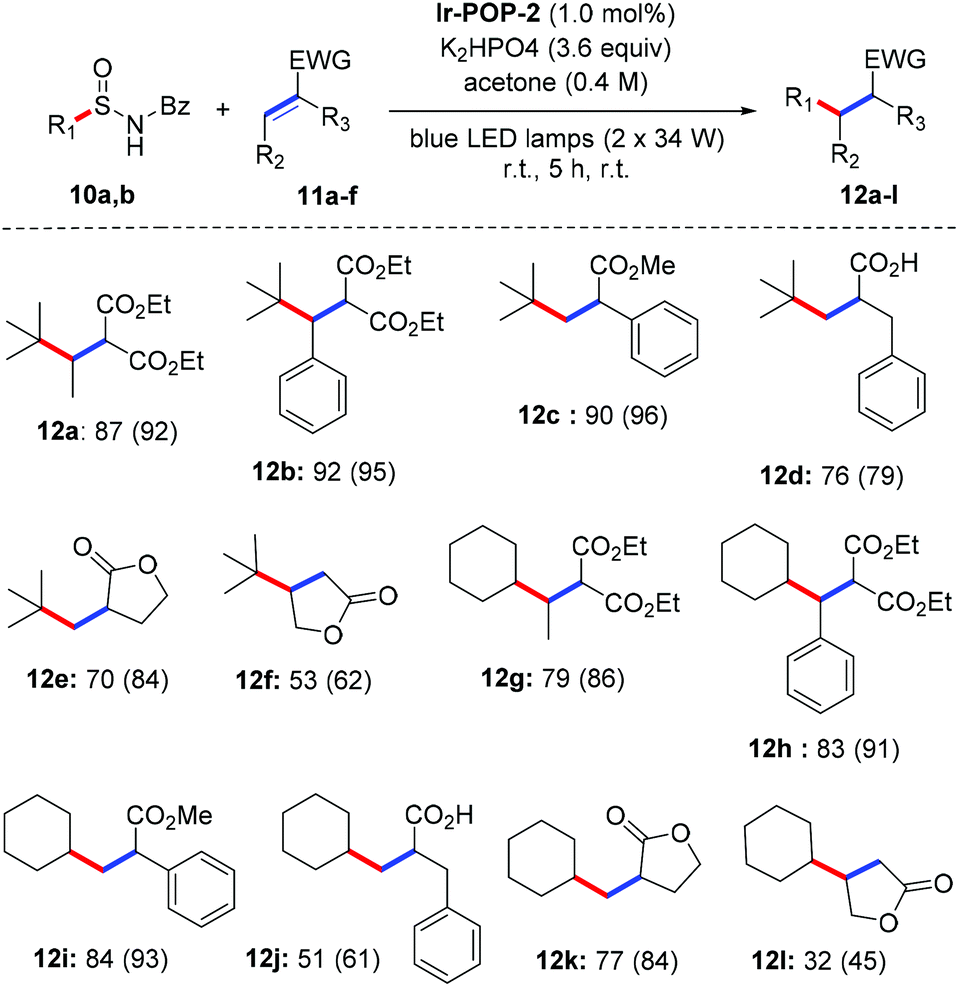 | ||
| Scheme 3 Substrate scope for the Ir-POP-2-catalyzed conjugate addition between N-benzoyl alkylsulfinamides 10a and b and Michael acceptors 11a–f to form 12a–l. The yield represents the isolated one and the value in the bracket represents the GC yield. EWG = electron-withdrawing group. Typical conditions: 10 (0.36 mmol, 1.8 equiv.), 11 (0.20 mmol, 1.0 equiv.), Ir-POP-2 (2 μmol, 1.0 mol%), K2HPO4 (125 mg, 0.72 mmol, 3.6 equiv.), 25 °C. | ||
Given the remarkably high activity of Ir-POP-2 as a heterogeneous photocatalyst, its further application for heterogeneous oxidation of sulfides was also performed (Table 2). All the studied substrates 13a–f, which bear an electron-donating group (EDG) such as the methoxy or methyl group or an EWG such as chlorine or bromine, were transformed into the corresponding sulfoxides 14a–f in excellent yields, with high selectivity over the corresponding sulfone products 15a–f. Again, light irradiation and the photocatalyst were indispensable for all the transformations (entries 7 and 8, Table 2). The recyclability of Ir-POP-2 was studied for the oxidation of 13a. After six cycles, 91% conversion of the substrate was still realized (Fig. 4d) and the catalyst could be recycled unprecedently 9 times.4b
a
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time [h] | Conversionb [%] | Selectivity [14:15]c |
| a Standard conditions: 13 (0.5 mmol, 1.0 equiv.), Ir-POP-2 (5 μmol, 1.0 mol%), air, two 34 W blue LED lamps, 25 °C. b Determined by 1H NMR. c Determined by 1H NMR. d No light source. e No photocatalyst. | |||||
| 1 | H | Me | 10 | >99 | 94:5 |
| 2 | OMe | Me | 20 | 94 | 92:2 |
| 3 | Me | Me | 18 | 95 | 90:5 |
| 4 | Cl | Me | 20 | 96 | 90:6 |
| 5 | Br | Me | 10 | 95 | 91:4 |
| 6 | H | Bn | 10 | 87 | 94:3 |
| 7d | H | Me | 25 | <1 | — |
| 8e | H | Me | 25 | <1 | — |

The aerobic oxidation of arylboronic acids represents a mild approach for the preparation of phenols.16 Recently, Han et al. reported that this reaction could be conducted heterogeneously with iridium complex-based conjugated polymers as photocatalysts.4b However, the recyclability of the polymeric catalysts was not demonstrated. We found that, in the presence of DIPEA as the sacrificial electron donor, Ir-POP-2 could efficiently catalyze this transformation in acetonitrile. Phenylboronic acid and the derivatives 16a–e that bear an EDG or EWG all underwent quantitative depletion to afford the phenols in excellent yields (Scheme 4), although a recent report showed that the introduction of an EDG group into the benzene ring would notably decrease the transformation.4b Moreover, the reaction of 2-naphthylboronic acid 16f also gave rise to naphthalene-2-ol 17f in a very high yield (96%). A study of the transformation of 16a revealed high recyclability of Ir-POP-2 (Fig. 4e), which could realize 90% conversion after thirteen cycles and retained considerable activity after nineteen cycles.
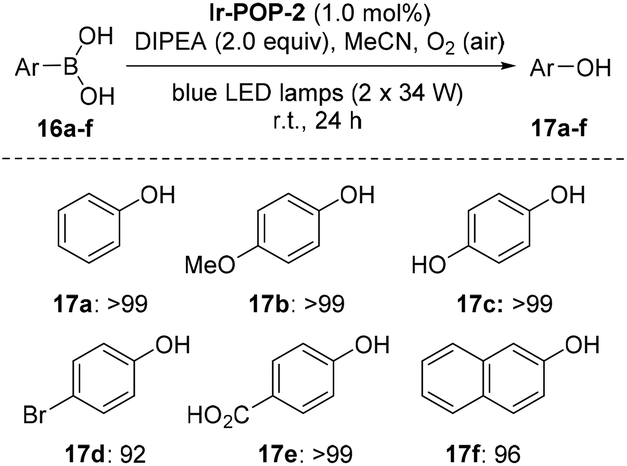 | ||
| Scheme 4 Photocatalytic hydroxylation of arylboronic acids catalyzed by Ir-POP-2. The yields were determined by 1H NMR. Standard conditions: 16 (0.1 mmol, 1.0 equiv.), Ir-POP-2 (1 μmol, 1.0 mol%), DIPEA (33 μL, 0.2 mmol, 2.0 equiv.), air, 25 °C. | ||
Conclusions
We have developed a convenient strategy for the preparation of iridium(III)-complex-connected porous organic polymers. One of the polymers, Ir-POP-2, exhibits highly efficient heterogeneous photocatalytic activity for broad-scope organic transformations including intramolecular Smiles–Truce rearrangement of alkyliodides, desulfurative conjugate addition for the C–C bond formation and aerobic oxidations of sulfides and arylboronic acids to form sulphones and phenols. For all the transformations, the new porous photocatalyst exhibits very high or previously unattainable recyclability. It is worth noting that, for all the organic transformations, Ir-POP-2 containing an identical amount of the iridium complex exhibits heterogeneous photocatalytic activities that are comparable with those of the homogeneous prototype complexes. Thus, we propose that the new catalytic polymer possesses pores that are large enough to allow electron transfer between the embedded, excited iridium complexes and discrete organic substrates. The work demonstrates that transition metal complex-embedded porous organic polymers can achieve very high stability of the backbones and high recyclability, a key feature for sustainable heterogeneous catalysis. Future studies will focus on the preparation of transition metal complex-embedded porous organic polymers that are expected to achieve large pores and the utility of highly modifiable conjugated linkers for loading chiral catalysts for important asymmetric catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21921003 and 21890732).Notes and references
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS; (b) Y. Xi, H. Yi and A. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 RSC; (c) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS; (d) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS; (e) J.-R. Chen, X.-Q. Hu, L.-Q. Lu and W.-J. Xiao, Acc. Chem. Res., 2016, 49, 1911–1923 CrossRef CAS; (f) B. König, Eur. J. Org. Chem., 2017, 1979–1981 CrossRef; (g) B. Chen, L.-Z. Wu and C.-H. Tung, Acc. Chem. Res., 2018, 51, 2512–2523 CrossRef CAS; (h) G. Zhang, Y. Liu, J. Zhao, Y. Li and Q. Zhang, Sci. China: Chem., 2019, 62, 1476–1491 CrossRef CAS; (i) X.-Q. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS; (j) Z. Jia, Y. Yuan, X. Zong, B. Wu and J. Ma, Chin. Chem. Lett., 2019, 30, 1488–1494 CrossRef CAS; (k) M.-J. Bu, C. Cai, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 1233–1237 RSC.
- (a) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (b) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (c) S. Angerani and N. Winssinger, Chem. – Eur. J., 2019, 25, 6661–6672 CrossRef CAS; (d) L. Marzo, S. K. Pagire, O. Reiser and B. König, Angew. Chem., Int. Ed., 2018, 57, 10034–10072 CrossRef CAS PubMed; (e) Y.-P. Wu, M. Yan, Z.-Z. Gao, J.-L. Hou, H. Wang, D.-W. Zhang, J. Zhang and Z.-T. Li, Chin. Chem. Lett., 2019, 30, 1383–1386 CrossRef CAS; (f) R. Naumann and M. Goez, Green Chem., 2019, 21, 4470–4474 RSC.
- (a) T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC; (b) K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS; (c) M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC.
- (a) Z. Xie, C. Wang, K. E. de Krafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 2056–2059 CrossRef CAS; (b) H.-P. Liang, Q. Chen and B.-H. Han, ACS Catal., 2018, 8, 5313–5322 CrossRef CAS.
- (a) A. Savateev and M. Antonietti, ACS Catal., 2018, 8, 9790–9808 CrossRef CAS; (b) S. Yin, J. Li and H. Zhang, Green Chem., 2016, 18, 5900–5914 RSC.
- (a) N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC; (b) P. Kaur, J. T. Hupp and S. T. Nguyen, ACS Catal., 2011, 1, 819–835 CrossRef CAS; (c) J.-X. Jiang, C. Wang, A. Laybourn, T. Hasell, R. Clowes, Y. Z. Khimyak, J. Xiao, S. J. Higgins, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2011, 50, 1072–1075 CrossRef CAS; (d) Q. Sun, Z. Dai, X. Meng and F.-S. Xiao, Chem. Soc. Rev., 2015, 44, 6018–6034 RSC; (e) D. Becker, N. Konnertz, M. Boehning, J. Schmidt and A. Thomas, Chem. Mater., 2016, 28, 8523–8529 CrossRef CAS; (f) L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC; (g) N. B. McKeown, Sci. China: Chem., 2017, 60, 1023–1032 CrossRef CAS; (h) R.-R. Liang and X. Zhao, Org. Chem. Front., 2018, 5, 3341–3356 RSC; (i) F.-Y. Yuan, J. Tan and J. Guo, Sci. China: Chem., 2018, 61, 143–152 CrossRef CAS; (j) Q. Chen and B.-H. Han, Macromol. Rapid Commun., 2018, 39, 1800040 CrossRef; (k) C. Xie, J. Song, H. Wu, Y. Hu, H. Liu, Y. Yang, Z. Zhang, B. Chen and B. Han, Green Chem., 2018, 20, 4655–4661 RSC; (l) Y. Yuan and G. Zhu, ACS Cent. Sci., 2019, 5, 409–418 CrossRef CAS PubMed; (m) Z.-X. Low, P. M. Budd, N. B. McKeown and D. A. Patterson, Chem. Rev., 2018, 118, 5871–5911 CrossRef CAS PubMed.
- T. Muller and S. Bräse, RSC Adv., 2014, 4, 6886–6907 RSC.
- (a) S. Meng, X.-Q. Zou, C.-F. Liu, H.-P. Ma, N. Zhao, H. Ren, M.-J. Jia, J. Liu and G.-S. Zhu, ChemCatChem, 2016, 8, 2393–2400 CrossRef CAS; (b) J.-S. Sun, L.-P. Jing, Y.-Y. Tian, F.-X. Sun, P. Chen and G.-S. Zhu, Chem. Commun., 2018, 54, 1603–1606 RSC.
- R. Dorta, R. Goikhman and D. Milstein, Organometallics, 2003, 22, 2806–2809 CrossRef CAS.
- A. V. Bavykina, H.-H. Mautscke, M. Makkee, F. Kapteijn, J. Gascon and F. X. Llabrés i Xamena, CrystEngComm, 2017, 19, 4166–4170 RSC.
- (a) D. Alpers, K. P. Cole and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2018, 57, 12167–12170 CrossRef CAS; (b) J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J.-J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763–2767 CrossRef CAS.
- (a) H.-J. Chen, C. Liu, M. Wang, C.-F. Zhang, N.-C. Luo, Y.-H. Wang, H. Abroshan, G. Li and F. Wang, ACS Catal., 2017, 7, 3632–3638 CrossRef CAS; (b) Q. Chang, Z.-Y. Liu, P. Liu, L. Yu and P.-P. Sun, J. Org. Chem., 2017, 82, 5391–5397 CrossRef CAS; (c) T. Lei, W.-Q. Liu, J. Li, M.-Y. Huang, B. Yang, Q.-Y. Meng, B. Chen, C.-H. Tung and L.-Z. Wu, Org. Lett., 2016, 18, 2479–2482 CrossRef CAS.
- H.-W. Shih, M. N. V. Wal, R. L. Grange and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 13600–13603 CrossRef CAS.
- (a) Z. Guo, C. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. Li, D. Tesfagaber, A. Thiel and W. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS; (b) J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208–18216 CrossRef CAS PubMed.
- F. Xue, F.-L. Wang, J.-Z. Liu, J.-M. Di, Q. Liao, H.-F. Lu, M. Zhu, L.-P. He, H. He, D. Zhang, H. Song, X.-Y. Liu and Y. Qin, Angew. Chem., Int. Ed., 2018, 57, 6667–6671 CrossRef CAS PubMed.
- X. Zhang, K. P. Rakesh, L. Ravindar and H.-L. Qin, Green Chem., 2018, 20, 4790–4833 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization, TGA, FT-IR, SEM, PXRD, EDS, CV, fluorescence and recycling experiments (images, spectra and tables). CCDC 1961159–1961161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9gc03688a |
| This journal is © The Royal Society of Chemistry 2020 |