
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Consolidated production of coniferol and other high-value aromatic alcohols directly from lignocellulosic biomass†
Robson
Tramontina
ab,
James L.
Galman
 a,
Fabio
Parmeggiani
a,
Sasha R.
Derrington
a,
Timothy D. H.
Bugg
c,
Nicholas J.
Turner
a,
Fabio M.
Squina
d and
Neil
Dixon
*a
a,
Fabio
Parmeggiani
a,
Sasha R.
Derrington
a,
Timothy D. H.
Bugg
c,
Nicholas J.
Turner
a,
Fabio M.
Squina
d and
Neil
Dixon
*a
aManchester Institute of Biotechnology (MIB), School of Chemistry, The University of Manchester, 131 Princess Street, M1 7DN Manchester, UK. E-mail: neil.dixon@manchester.ac.uk
bPostgraduate program in Biosciences and Technology of Bioactive Products (BTPB). Institute of Biology, State University of Campinas – UNICAMP, 500 Albert Einstein Av, Zip Code 13083-852, Campinas, SP, Brazil
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
dProgram of Technological and Environmental Processes, Universidade de Sorocaba, Raposo Tavares Road, km 92.5 – Vila Artura, Zip code 18023-000, Sorocaba, SP, Brazil
First published on 25th November 2019
Abstract
Sustainable production of fine chemicals and biofuels from renewable biomass offers a potential alternative to the continued use of finite geological oil reserves. However, in order to compete with current petrochemical refinery processes, alternative biorefinery processes must overcome significant costs and productivity barriers. Herein, we demonstrate the biocatalytic production of the versatile chemical building block, coniferol, for the first time, directly from lignocellulosic biomass. Following the biocatalytic treatment of lignocellulose to release and convert ferulic acid with feruloyl esterase (XynZ), carboxylic acid reductase (CAR) and aldo-keto reductase (AKR), this whole cell catalytic cascade not only achieved equivalent release of ferulic acid from lignocellulose compared to alkaline hydrolysis, but also displayed efficient conversion of ferulic acid to coniferol. This system represents a consolidated biodegradation–biotransformation strategy for the production of high value fine chemicals from waste plant biomass, offering the potential to minimize environmental waste and add value to agro-industrial residues.
Introduction
Alternative chemical production processes are of increasing global importance, driven by the need to balance sustainable and efficient resource utilization. Several strategies are being explored to reduce the long-term environmental impacts often incurred in the production of materials, additives and pharmaceuticals.1 Among them, the use of lignocellulose from sugarcane bagasse and wheat straw (WS) as a starting material is a field of intense study, since plant biomass is a renewable source of polysaccharides and phenolic compounds that can generate a wide range of molecules with industrial interest.2,3Lignocellulosic biomass is a suitable source of aromatic compounds comprising a range 10–30% of its content,4 and one of the most abundant aromatic units of lignocellulose is ferulic acid, composing more than 2% yield (wt/wt) of some plant biomass.5–8 Ferulic acid is an important structural component of the plant cell wall, responsible for cross-linking between hemicellulose and lignin,6 providing physical and mechanical protection to the cell wall. Ferulic acid, along with other aromatic carboxylic acids, their reduced products and derivatives were identified as “Top Value-Added Chemicals from Biomass”. For instance, aromatic aldehydes and alcohols (monolignols), such as cinnamaldehyde, caffeyl alcohol and coniferol, are essential building blocks for high value-chemicals, including pharmaceutical compounds.9–12 However, these high value monolignols derivatives are generally found in low concentrations in plants, and their total chemical synthesis involves complex or non-ecofriendly routes.11,13
Coniferyl alcohol (coniferol), a compound with high market value, is synthesized via the phenylpropanoid biochemical pathway in plants. Coniferol is widely used as a metabolic intermediate to elucidate chemical structures of and bio-synthetic pathways, including compounds derived from lignins and lignans.14,15 In addition to direct applications, coniferol is also used to synthesize various valuable chemicals, such as pinoresinol (hypoglycemic agent), sesamin (several bioactive properties such as anticholesterolemic and antihypertensive properties16) and silibinin (used as a hepatoprotective and recently shown to have therapeutic effect for both cancer and arthritis treatment17,18) (Fig. 1). Moreover, coniferol has also been reported as a precursor of the floral scents, dihydroconiferyl alcohol, coniferyl acetate and iso-eugenol from Petunia axillaris,19 and therefore is a potential starting material for the cosmetic fragrance industry.
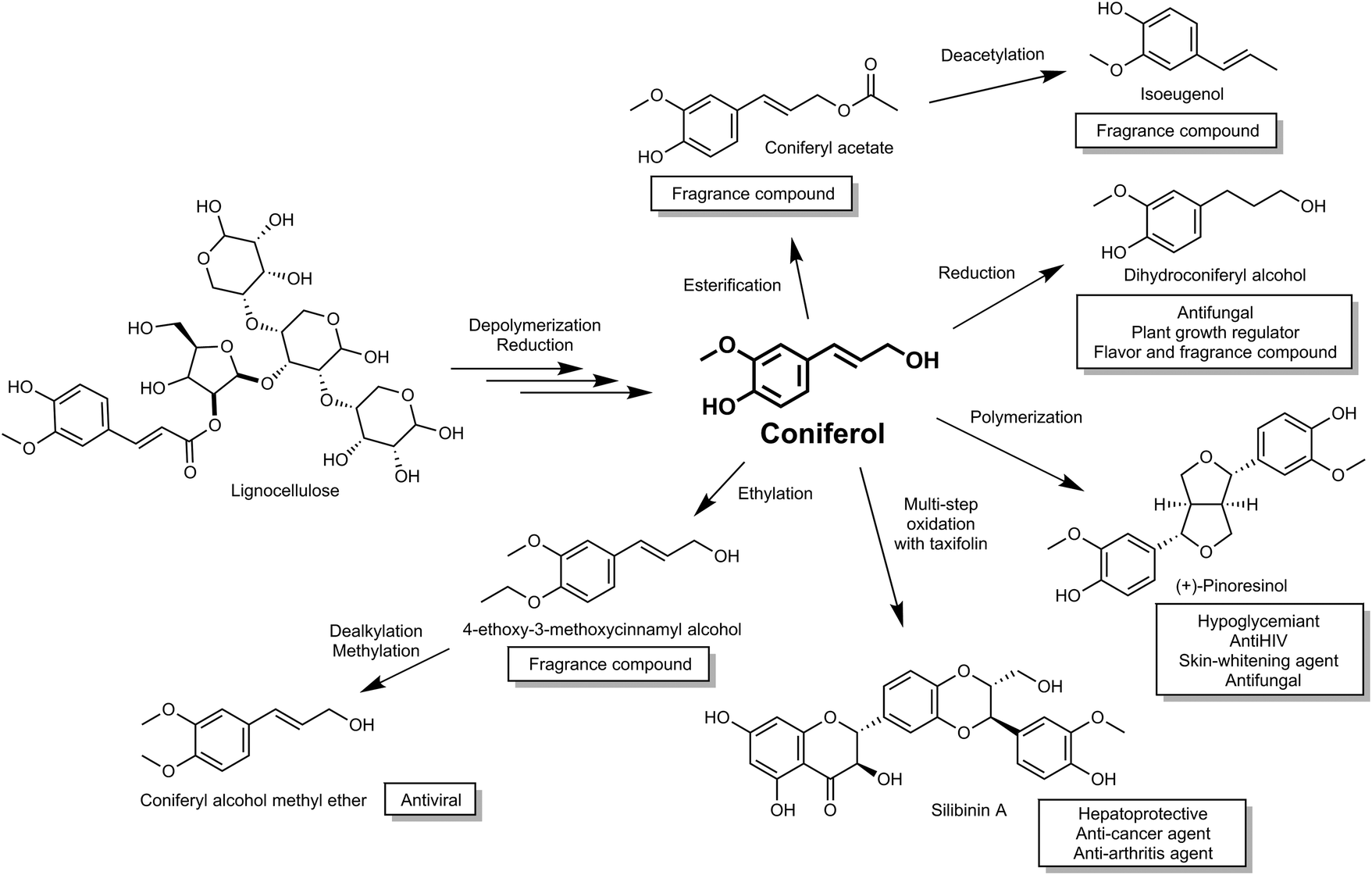 | ||
| Fig. 1 Activity and use of compounds derived from plant biomass via the building block intermediate coniferol. | ||
The biotechnological production of coniferol, from inexpensive precursors such as agro-industrial residues presents a promising alternative to supply this compound. The phenylpropanoid-dependent synthesis pathway of coniferol involves eight enzymes, including two cytochrome P450 enzymes, which are difficult to actively express in prokaryotic microorganisms.20 According to previous studies, 124.9 mg L−1 coniferol can be obtained by reconstructing the de novo pathway from tyrosine in microorganisms using rich growth media.10
Chemical methods for the reduction of carboxylic acids require hazardous reagents such as sodium borohydride, expensive metals or complex catalyst formulations.21 Accordingly a green alternative for the reduction of aromatic acids is the use of enzymes. In particular a class of enzymes called carboxylic acid reductases (CARs) are noted as important catalysts in the toolbox for sustainable chemistry for the selective reduction of acids to aldehydes.22 The substrate scope of CARs is exceptionally broad, which offers potential for their application in diverse synthetic processes, i.e. for the preparation of aldehydes as end products for the flavor and fragrance sector and the integration of CARs in cascade reactions.23 CARs have a clear advantage over other enzymes that are able to perform carboxylic acid reduction, since this reaction is thermodynamically favoured because it is coupled to ATP hydrolysis.21 Further chemical reduction of aromatic aldehydes to alcohols often requires fine control of reaction conditions, especially when the compound also contains alkene functional groups, such as ferulic acid, due to the possible non-selective reduction of both the carbonyl and alkenyl functionalities. Thus, enzymatic reduction with alcohol dehydrogenases (ADH) and aldo-keto reductases (AKR) are preferable since they act on aldehydes in more mild conditions and with greater chemoselectivity.24–26 For example, the recently reported termite Coptotermes gestroi aldo-keto reductase (CgAKR-1), was shown to be active on several fermentation inhibitor aldehydes commonly found in lignocellulose and therefore presents a great potential for the synthesis of monolignols and related alcohols.24 Previously an enzymatic cascade has been reported for the de novo production of cinnamyl alcohol from phenylalanine via cinnamic acid using a CAR from Mycobacterium marinum (mCAR) and an ADH from Saccharomyces cerevisiae (ScADH).25 However, the reduction of ferulic acid to coniferol has not been explored using this proposed catalytic pathway, and indeed to our knowledge there are no reports of such a cascade using heterologous AKR enzymes.23,27–30
The generation of these chemicals from abundant biomass sources has many advantages including use of mild reaction conditions, compatibility with aqueous media, sourcing of catalysts from renewable feedstocks and chemo-, regio- and enantioselectivity. In this study we explored the use of both an in vitro enzymatic and whole-cell biocatalytic route to release ferulic acid from biomass sources and convert it directly into coniferol (Fig. 2).
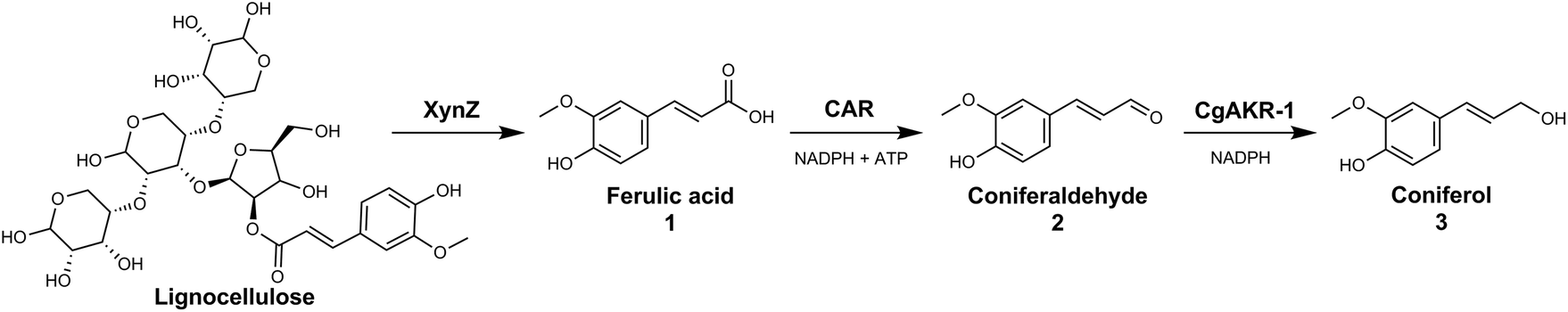 | ||
| Fig. 2 Proposed biocatalytic route from lignocellulosic biomass to coniferol (3) via ferulic acid (1) and coniferyl aldehyde (2). Biocatalytic steps performed with (i) chimeric xylanase (XynZ), that contains a glycoside-hydrolase domain (GH10) and a feruloyl esterase domain (CE1), (ii) carboxylic acid reductase (CAR), and (iii) aldo-keto reductase (CgAKR-1). Both reductions require NADPH, and CAR requires ATP for enzyme activity. | ||
Results and discussion
Enzymatic reduction of biomass derived aromatic acids
In order to test the feasibility of the enzyme cascade proposed in Fig. 2, the enzymes were recombinantly produced in E. coli, purified, and tested for activity in vitro (Methods). Initially, the carboxylic acid reductase (CAR) step was assessed by monitoring reduction of ferulic acid 1 to coniferyl aldehyde 2 using five different CARs, monitored by LC-MS. Although these CARs had their activities evaluated against diverse acids previously, none of these studies provided a detailed comparison of diverse CARs using ferulic acid as a substrate.23,27–29 The relative activity of the enzyme candidates for the single reduction of 1 to 2 were, Nocardia Iowensis (NiCAR),27Segniliparus rugosus (SrCAR),25,28Segniliparus rotuduns (SroCAR),25Mycobacterium marinum (mCAR)25 and Tsukamurella paurometabola (TpCAR)21 with 100%, 100%, 91%, 80% and 42% respectively (Table 1). Both, NiCAR and SroCAR have been previously demonstrated to reduce 1, and NiCAR was reported to have greater activity against this substrate.25,28 Previously SrCAR, mCAR activity against cinnamic acid and benzoic acid respectively has been reported, however neither enzyme has previously been assessed against ferulic acid.22,25|
|
|||
|---|---|---|---|
| Enzyme | CAR | CAR + CgAKR-1 | |
| 2 (%) | 2 (%) | 3 (%) | |
| MCAR | 80 | 13 | 87 |
| TpCAR | 42 | 3 | 75 |
| NiCAR | 100 | 0 | 100 |
| SroCAR | 91 | 7 | 85 |
| SrCAR | 100 | 6 | 94 |
Next, the substrate specificity of CgAKR-1 was assessed by monitoring NADPH co-factor depletion, revealing CgAKR-1 to be active against a wide-range of aromatic aldehyde substrates including 2 (Fig. S1†). Next, we developed a two-step enzyme cascade using different CARs and CgAKR-1 (Table 1). From this, the most efficient double reduction was observed with NiCAR-CgAKR-1 and SrCAR-CgAKR-1, producing coniferol 3 from 1 with excellent conversions of 100% and 94% respectively. A detailed time course experiment was performed to examine the relative activities of NiCAR and SrCAR in the absence and presence of CgAKR-1 for the generation of 2 and 3 (Fig. 3). NiCAR presented faster conversion of 1 to 2 with 88% conversion in 5 h, whereas with SrCAR only 20% conversion was observed after 5 hours. In comparison, the NiCAR-CgAKR-1 cascade pushed forward the initial reduction reaction relative to NiCAR alone with >62% depletion (vs. 7% for NiCAR-alone) of 1 within 30 min. Enhanced depletion of 1 was also observed for the SrCAR-CgAKR-1 cascade relative to SrCAR alone with 50% depletion of 1 within 5 h. Both cascades produced 3 in excellent yields 98 and 81% with greater productivity observed for the NiCAR-CgAKR-1 cascade. Minimal intermediate aldehyde 2 was observed for the SrCAR-CgAKR-1 cascade (<10%), compared to the NiCAR + CgAKR-1 cascade (up 40%), this relative productivity was consistent with relative production levels of 2 for the CAR-alone single reduction results.
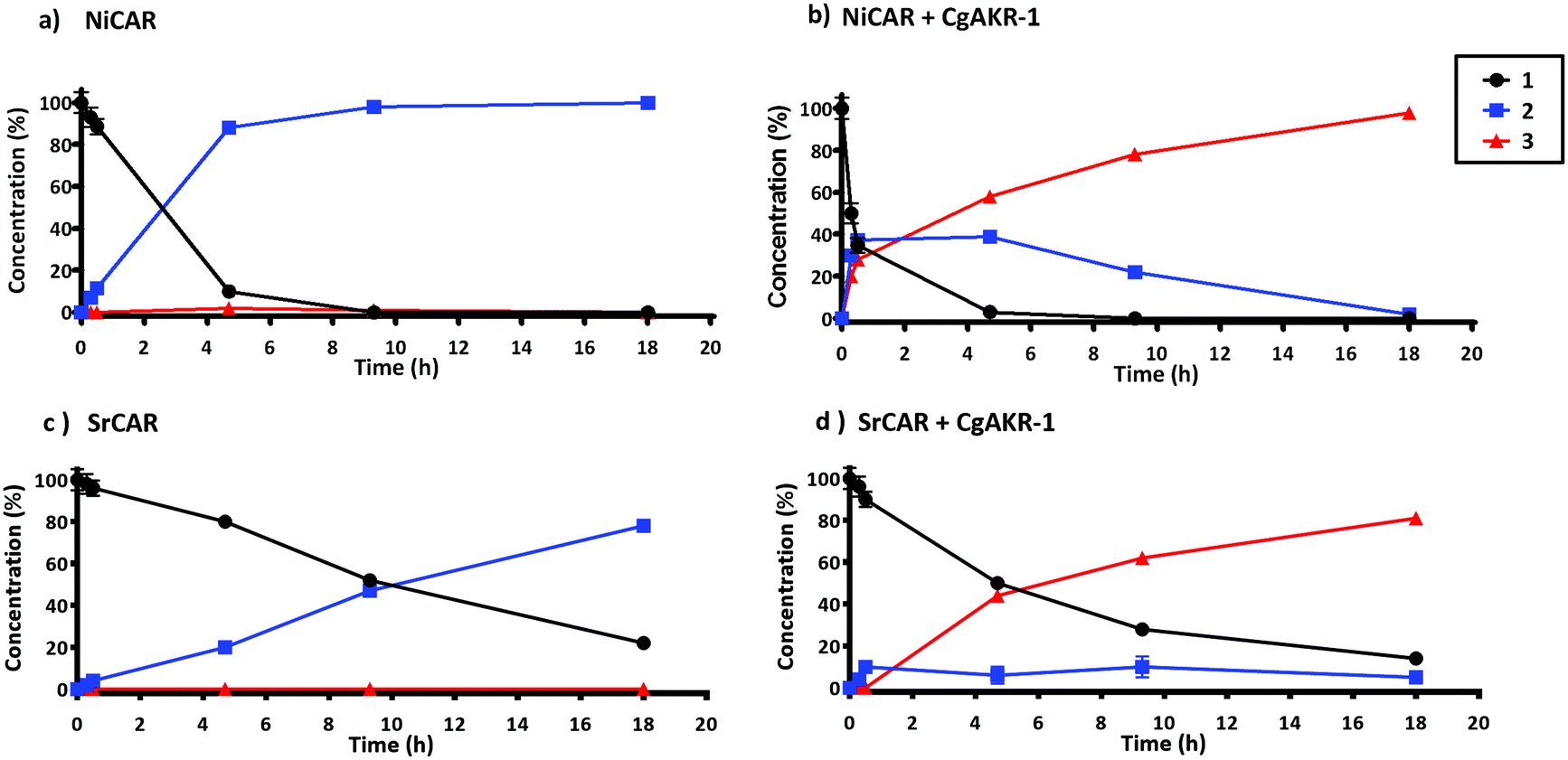 | ||
| Fig. 3 Time course of CAR and CAR-CgAKR-1 production of 2 and 3 from substrate 1. Reaction performed in 100 mM potassium phosphate buffer (pH 7.5), 1 mM 1, 10 mM MgCl2, 250 μg NiCAR (a and b) or SrCAR (c and d), CgAKR-1 100 μg (b–d), 10 mM ATP, 6 mM NADPH, final volume 0.5 ml at 30 °C, 250 rpm. Error bars represent standard deviation (SD) of two biological replicates. For raw values see Table S6.† | ||
Whole cell biotransformation of biomass derived aromatics
The application of whole cell biocatalysis for reductive biotransformations can be advantageous over in vitro multi-enzyme cascades because the addition of the expensive cofactors ATP and NADPH are unnecessary, rendering the whole cell biocatalytic process more practical at large scales, as well as, reducing costs and negative environmental implications associated with enzyme isolation.23,31 Thus, in order to convert 1 to 3 the genes encoding the appropriate biocatalysts were transferred to a whole cell system. E. coli strains were transformed with DNA vectors containing the recombinant genes encoding the enzymes required to convert 1 into 3 (Methods). Following induction of gene expression and substrate incubation, quantification of the resulting compounds from the culture medium was performed using LC-MS. To evaluate the role of recombinant and endogenous aldehyde reductase enzymes present in the E. coli host, NiCAR and SrCAR alone or in combination with CgAKR-1 were evaluated using E. coli strains BL21 and RARE, the latter is a strain that lacks the endogenous aldehyde reductase enzymes.32 In BL21 cells harboring CAR-only 67% and 62% of 1 was reduced by SrCAR and NiCAR respectively (equivalent to 582 and 605 mg L−1), affording predominately 3 (47 and 45%), with small amounts of 2 (19 and 5% respectively) (Fig. 4). The observed yields demonstrate enhanced productivity in comparison to a previous study that reported just 11% conversion of 1 to 3 using a whole cell SrCAR-dependent biotransformation.28 In the RARE E. coli strain, 1 was reduced to 2 using either NiCAR or SrCAR and gave 68% and 89% product conversion respectively after 24 hours. As such, if the desired product is an aldehyde compound, SrCAR in the RARE host should be used, as this combination minimizes the double reduction to the alcohol 3. Unexpectedly NiCAR in the RARE strain also led to a small production of 3 (12%).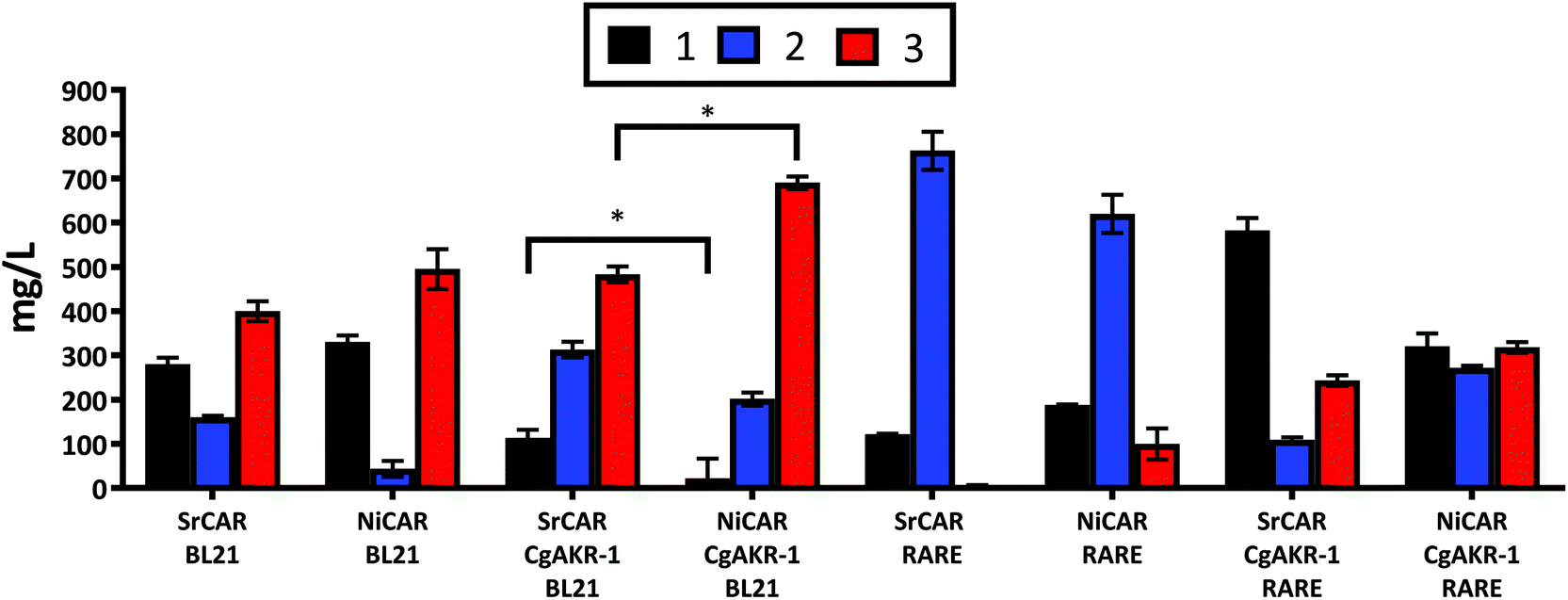 | ||
| Fig. 4 Quantification of whole cell biotransformation producing 2 and 3 from 1 using CAR and CAR-CgAKR-1, enzymes expressed in BL21 or RARE (non-reducing) E. coli strains. The results were given in mg L−1, medium supplemented with 5 mM (970 mg L−1) 1, and quantified after 24 h. Error bars represent standard deviation (SD) of two biological replicates. *The p-value was <0.05 between SRCAR-CgAKR-1 BL21 and NiCAR-CgAKR-1 BL21 according to the student test. For raw values see Table S6.† | ||
Next we evaluated the whole cell biocatalytic reduction of 2 to 3 using the CgAKR-1 in comparison with the alcohol dehydrogenase (KRED1) from Paraburkholderia phytofirmans, previously reported to act on cinnamyl aldehyde in a whole cell biocatalytic reaction (Fig. S2†).25 CgAKR-1 and KRED1 were assessed in RARE strains, and both presented efficient reduction of 2. Slightly enhanced activity was observed for CgAKR-1, and so this enzyme was selected for subsequent whole cell cascade reactions with CARs (Fig. S2†).
In the presence of the CAR-CgAKR-1 cascades in BL21 cells, the double reduction was enhanced, relative to the respective CAR-alone, to afford 3 with conversion yields of 75% and 53% using NiCAR-CgAKR-1 and SrCAR-CgAKR-1 respectively (equivalent to 690 and 483 mg L−1) (Fig. 4). Collectively, NiCAR-CgAKR-1 exhibited higher activities for the production of 3 compared to SrCAR-CgAKR-1 for both in vitro (Table 1) and the whole cell biocatalytic cascades (Fig. 4) 5.1-fold (p = 6.8 × 10−6) greater depletion of 1 and 1.4-fold (p = 2.0 × 10–2) for the production of 3.
Comparison of the heterologous recombinant enzymes and endogenous aldehyde reductase activity indicates that as expected, little or no aldehyde reduction occurs in the E. coli RARE strain with CAR (Fig. 4). In the RARE E. coli strain, with CAR-CgAKR-1 (i.e. heterologous aldehyde reductase only) production of 3 between 26–35% was observed, and in BL21 strain with CARs present (i.e. endogenous aldehyde reductases only) gave 47–57% of 3, indicating that endogenous E. coli aldehyde reductase activity is better than CgAKR-1 or that RARE cells have less NADPH/ATP available. However, NiCAR-CgAKR-1 in BL21 cells (i.e. with both heterologous and endogenous aldehyde reductases) greater formation of the alcohol product 3 (75%) was observed. Indicating that both endogenous and heterologous recombinant aldehyde reducing enzymes acted in cooperation improving the conversions of 3 from 1.
Whole cell biotransformation process optimization
The use of whole cell biocatalysts can often require optimization of reaction conditions to increase product yield, minimize side reactions and improve enantioselectivity.31 Accordingly, we tested the impact of different media composition on the production of 3 using the whole cell NiCAR-CgAKR-1 biocatalytic route (Fig. 5A). TB medium was shown to be the best media, leading to 94% reduction of 1 with a 76% conversion rate into 3 (645 ± 19 mg L−1) in comparison with LB medium that converted 65% of 1 to 3. TB medium is a complex media rich in available carbon and nitrogen sources,33 these properties can lead to better protein expression and increased cell density per liter of medium, which may explain the enhanced production of 3. The production of 3 in minimal (M9) media was similar with either a glucose or glycerol carbon source (564 ± 67 and 512 ± 35 mg L−1). In addition to the reduced cost, one other major advantage of using minimal media is the ease of product identification and isolation from the less complex media composition.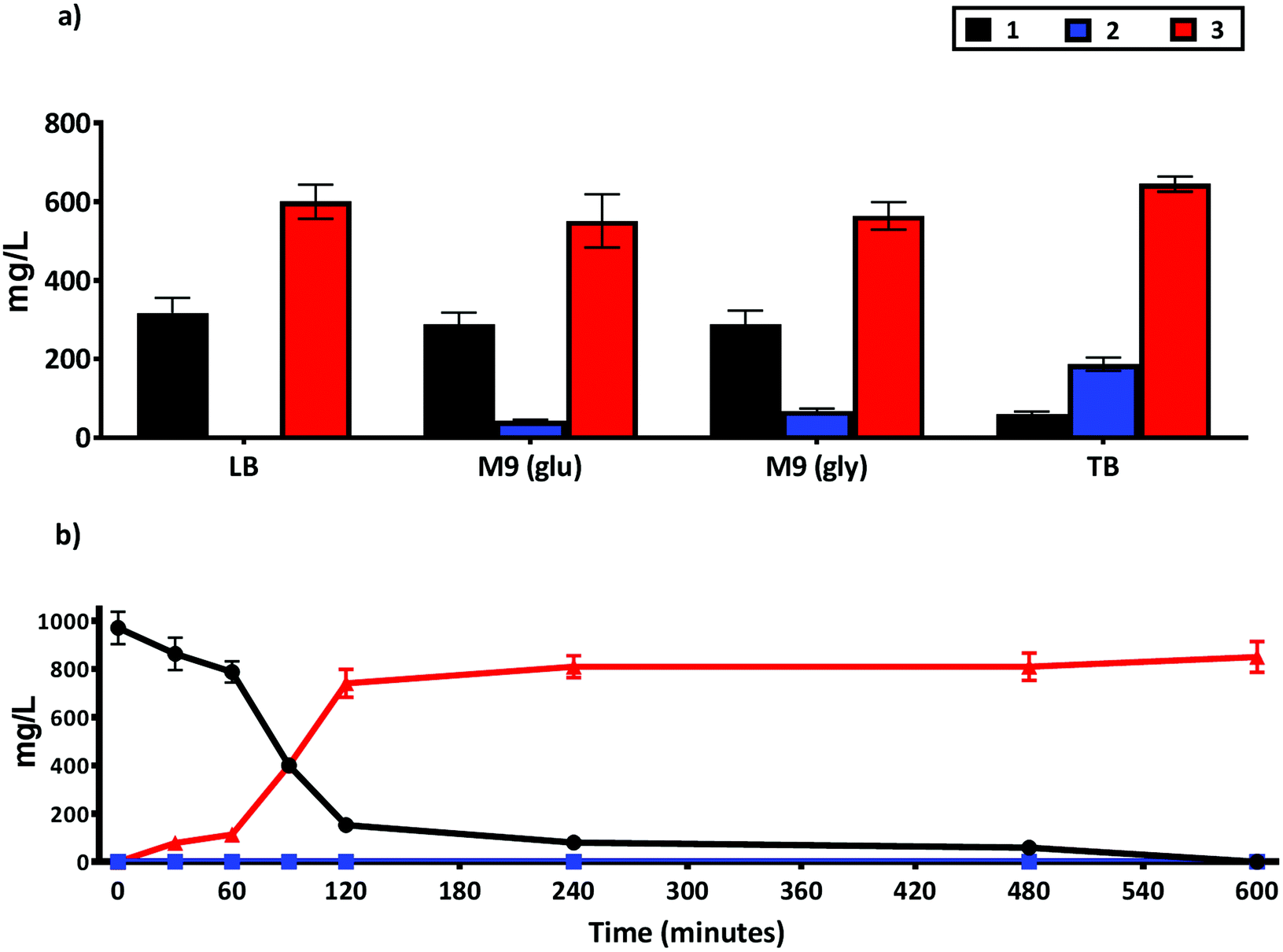 | ||
| Fig. 5 Production of 2 and 3 from 1 using a whole cell biotransformation using an E. coli strain BL21 expressing the NiCAR-CgAKR-1 enzyme cascade. (a) Activity compared in different growth media and carbon source TB, LB, M9-glucose and M9 glycerol. (b) Time course quantification of whole cell biotransformation of 1 into 2 and 3 in TB media. Medium supplemented with 5 mM 1 (970 mg L−1), samples were taken at 16 h in (a) and at times indicated in (b). The results were given in mg L−1. All the assays were performed in duplicate. For raw values see Table S6.† | ||
As efficient whole-cell biocatalytic processes involve both a cell growth phase and a substrate conversion phase, two major issues affecting biotransformation efficiency can be considered as the total amount of cellular biomass and the concentration of recombinant enzyme per cell.31 Thus, these factors were evaluated by setting three variables in the system; the incubation time pre-induction (2–4 hours); inducer concentrations (arabinose and rhammanose from 5 to 10 mM); and induction time before the addition of substrate 1 (2–4 hours) on the formation of 3 following incubation/biotransformation using design of experiments approach (Table S1 and Fig. S3†). Considering maximal production of 3 the optimal conditions were: 4 hours pre-induction incubation, 10 mM of both inducers, and 2 hours of recombinant gene expression prior to substrate 1 addition (ESI Table 1†). These optimized cultivation parameters led to near complete depletion of 1, and 97% production of 3. A full time course experiment was performed with 1 at 5 mM concentration, 81% and 97% conversion (as assessed by LC-MS) to 3 was achieved after a 2 and 10 hours reaction respectively (Fig. 5B). A further scale-up reaction was performed in order to isolate and chemically characterize 3 (Fig. S4 and S5†), here a slightly reduced 74% conversion rate was reported.
Other routes to produce 3, and related phenylpropenoic acids, have been reported using different enzymatic routes to those reported here.10,25,30,34 For example, a de novo biosynthetic route to 3 from tyrosine was developed using a 4-step enzyme cascade, consisting of a tyrosine ammonia lyase, 4-coumaroyl-CoA ligase, cinnamoyl-CoA reductase, and a cinnamyl alcohol dehydrogenase (TAL-4CL-CCR-CAD) affording 125 mg L−1.10 Two further studies used a similar 3-step enzyme cascade to produce 3 from 1 using a 4-coumaroyl-CoA ligase, a cinnamoyl-CoA reductase, and a cinnamyl alcohol dehydrogenase (4CL-CCR-CAD) yielding 327 mg L−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30 and 108 mg L−134 of 3 from 1. Finally, a de novo biosynthetic route to cinnamyl alcohol from phenylalanine was developed using a 3-step enzyme cascade, consisting of a phenylalanine ammonia lyase, carboxylic acid reductase and alcohol dehydrogenase (PAL-CAR-ADH) affording 300 mg L−1 of cinnamyl alcohol.25 In order to determine the relative cost and efficiency for these different enzymatic routes, a value added calculation was performed taking into consideration the cost and volume of the substrate and media used (including carbon-source, buffers, inducers and antibiotics), and comparing this to the product titre and value (Table S4†). From this analysis the CAR-AKR route, developed here, displayed the greatest value added (47-fold), the 4CL-CCR-CAD routes displayed lower value added (33 and 23-fold), and the TAL-4CL-CCR-CAD route displayed the least added value (7-fold). This simple analysis indicates that the CAR-AKR route is a favorable route for the production of 3, however full process analysis would be required in the future to accurately determine and compare costs at scale.
30 and 108 mg L−134 of 3 from 1. Finally, a de novo biosynthetic route to cinnamyl alcohol from phenylalanine was developed using a 3-step enzyme cascade, consisting of a phenylalanine ammonia lyase, carboxylic acid reductase and alcohol dehydrogenase (PAL-CAR-ADH) affording 300 mg L−1 of cinnamyl alcohol.25 In order to determine the relative cost and efficiency for these different enzymatic routes, a value added calculation was performed taking into consideration the cost and volume of the substrate and media used (including carbon-source, buffers, inducers and antibiotics), and comparing this to the product titre and value (Table S4†). From this analysis the CAR-AKR route, developed here, displayed the greatest value added (47-fold), the 4CL-CCR-CAD routes displayed lower value added (33 and 23-fold), and the TAL-4CL-CCR-CAD route displayed the least added value (7-fold). This simple analysis indicates that the CAR-AKR route is a favorable route for the production of 3, however full process analysis would be required in the future to accurately determine and compare costs at scale.
The developed whole cell biocatalytic system was also further evaluated for the reduction of other aromatic acids, and the reaction was performed using M9 media supplemented with glycerol in order to decrease GC-MS analysis complexity (Fig. S6†). Although different yields were observed for each tested substrate, the reactions were demonstrated to be effective across phenylpropenoic acid derivatives (Fig. 6). The chromatograms of the reactions showed conversion of vanillic 6 (64%–210 mg L−1), cinnamic 7 (76%–250 mg L−1), sinapinic 8 (18%–43 mg L−1), 3,4 dimethoxycinnamic 10 (47%–96 mg L−1) and coumaric acid 12 (49%–165 mg L−1), into their respective alcohols (Fig. 6). Whereas, the protocatechuic 4 (77%–275 mg L−1), syringic 5 (68%–185 mg L−1), caffeic 9 (9%–27 mg L−1), and 3,4,5-trimethoxycinnamic 11 (52%–116 mg L−1) acids were only converted to their respective aldehydes without any further reduction to alcohols observed (Fig. 6). In general, the previously identified substrate scope of CARs is exceptionally broad ranging from aliphatic compounds (from C2 to C18) to (hetero)aromatic compounds and a wide range of substituents is tolerated, where the major limitation is the presence of polar groups adjacent to the carboxylate moiety.36 Although 4, 5, 9 and 11 were not reduced to their respective alcohols it is difficult to assess any clear structure–activity relationship regarding CgAKR-1 substrate tolerance or indeed the endogenous aldehyde reductases. Overall, these results indicate that the whole cell biocatalyst reported here is useful for the production of biomass-derived aromatic alcohols and related molecules, and presents both more rapid and more complete conversion than other enzymatic strategies.23,25,30,34,35
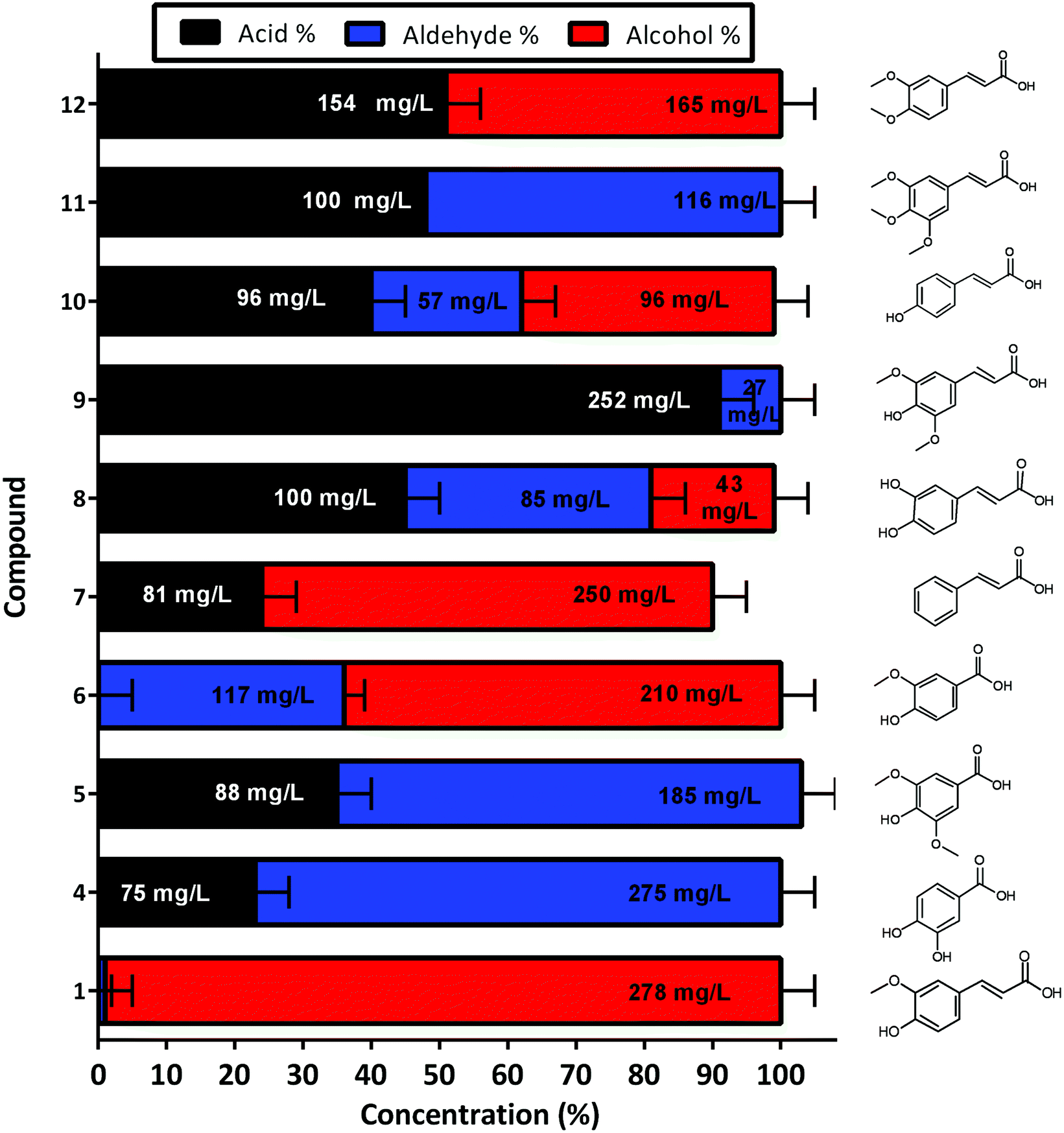 | ||
| Fig. 6 Production of other aromatic aldehydes and alcohols for the corresponding acid, via whole cell biotransformation using the BL21 expressing NiCAR-CgAKR-1 cascade. Activity was assessed 16 h after incubation/induction in M9 media supplemented with glycerol as a carbon source. Results reported in percentage of conversion of the 5 mM aromatic acid. All the assays were performed in duplicate. Ferulic acid 1, protocatechuic acid 4, syringic acid 5, vanillic acid 6, cinnamic acid 7, sinapic acid 8, caffeic acid 9, 3,4-dimethoxycinnamic acid 10, 3,4,5-trimethoxycinnamic acid 11, coumaric acid 12. For raw values see Table S6.† | ||
Consolidated biodegradation–biotransformation process
In plants, 1 is rarely found in free form and occurs widely linked to various carbohydrates as glycosidic conjugate ester.37 Thus we sought to develop a consolidated biodegradation–biotransformation process to first release ferulic acid 1 from insoluble lignocellulosic substrates, followed the conversion of 1 into 3 using the whole cell biocatalytic strains developed (Fig. 7). This was performed using wheat straw (WS) and wheat arabinoxylan (WAX). WAX is a partially purified polysaccharide and contains approximately 1.52 mg g−1 of 1 and similar amounts of coumaric acid (12), whereas WS contains less 1 and 12.7,38 WS or WAX biomass was incubated with E. coli BL21 expressing NiCAR-CgAKR-1 as described above in the presence of multi-domain β-xylanase (XynZ) from Clostridium thermocellum for 24 hours (Fig. 7). XynZ contains the feruloyl esterase domain (CE1), and has been reported to release phenolic compounds from biomass.38 Alkaline hydrolysis of 2% (w/v) WAX and WS was performed in order to compare the enzymatic activity of XynZ for the release of 1. The results demonstrate that XynZ efficiently releases both ferulic acid 1 and coumaric acid 12 from recalcitrant substrates after 24 hours incubation time (Fig. 7). Release of the aromatic acids from WAX was ∼3-fold higher than WS (WAX: 1 60 mg L−1 and 12 54 mg L−1 – WS: 1 26 mg L−1 and 12 17 mg L−1). Moreover, our results were consistent with previous findings that reported a feruloyl esterase from Aspergillus clavatus released phenolic compounds from WAX and sugarcane bagasse in similar yields.39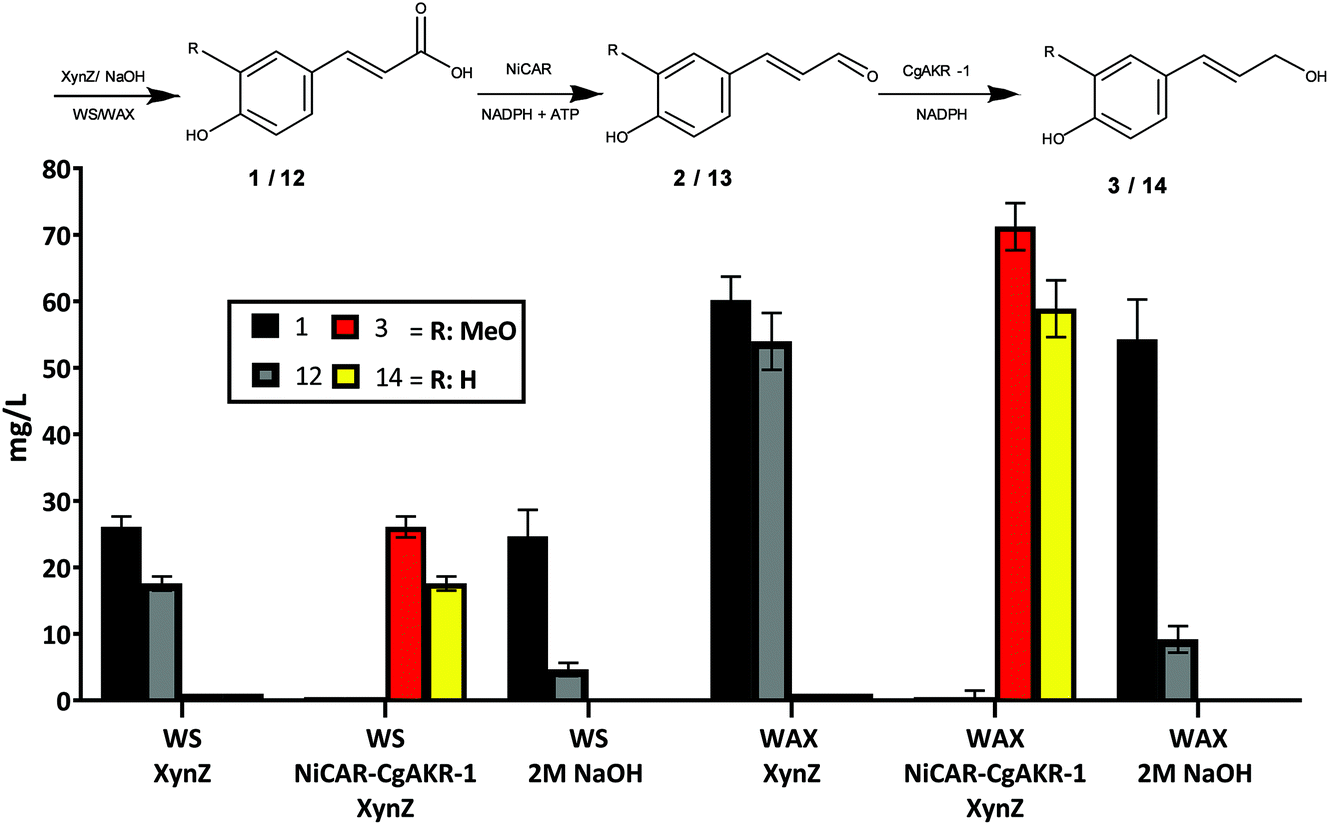 | ||
| Fig. 7 Consolidated biodegradation and biotransformation of wheat straw (WS) and wheat arabinoxylan (WAX) biomass to release 1 and 12 and produce coniferol 3 and coumaryl alcohol 14. The samples were taken 24 h after incubation with 2% (w/v) biomass and E. coli BL21 strain expressing NiCAR-CgAKR-1, and 1 mg g−1 XynZ/biomass (where indicated). The results were given in mg L−1. Neither aldehyde intermediate 2 nor 13 were detected. All the assays were performed in duplicate. For raw values see Table S6.† | ||
The 3-step cascade (XynZ-CAR-AKR) for both biomass substrates showed complete conversion of the released acid intermediates 1 and 12 into their respective alcohols 3 and 14. Consistent with the content of the aromatic acids from the two different biomass substrates, the production of the alcohols was also 3-fold higher from WAX than WS at producing 3 or coumaryl alcohol (14) (WAX: 3 71 mg L−1 and 14 58 mg L−1 – WS: 3 26 mg L−1 and 14 17 mg L−1) (Fig. 7). No detection of 2 and coumaryl aldehyde (13) was reported, possibly because the lower amounts 1 available compared to a soluble feedstock, which permits more rapid and complete conversion to 3. Utilization of crude biomass by this method demonstrated high amounts of release of 1 and 12 and conversion to 3 and 14 achieving the maximum yield from the WS and WAX (Fig. 7). Beyond the applications of 3, the biomass derivate 14 also demonstrates potential as a building block of anti-oxidative and anti-inflammatory molecules such as p-coumaryl fatty acid esters and p-coumaryl alcohol γ-O-methyl ether.10 The results obtained with crude biomass are very promising in terms of value added, considering the relative cost of substrates 1 (€5.05 g−1) or wheat straw (€0.07 kg−1) and the product 3 (€275 g−1). As earlier the value added calculation was performed taking into consideration the substrate and media costs (in addition to enzymes production costs), and comparing this to the product titer and value (Table S4†). From this earlier analysis the best route to 3 from 1 was the CAR-AKR route, which displayed 47-fold value added, whereas here the XynZ-CAR-AKR route, using WS biomass directly, displayed 74-fold value added (Table S4†). Further scale up and techno-economic analysis would enable full evaluation of this process in a biorefinery context, and the viability of this route to out compete existing biotechnological production methods to monolignols.
Conclusions
We have demonstrated here that a whole cell biocatalytic cascade in E. coli can be applied to produce 3 directly from biomass via a consolidated biomass degradation–valorisation process. To the best of our knowledge, there are no reports in literature focusing on the production of 3 either from 1 using CARs or directly from biomass following biocatalytic degradation of lignocellulose. Therefore this novel method opens up a possibility to sustainably produce 3 and other compounds directly from biomass. The direct use of 1 and other aromatic acids with the whole cell biocatalytic cascade showed to be very efficient, simple, and ready to be explored at larger scales. Besides being present in untreated lignocellulosic biomass, 1 and related compounds are also a major component of lignin-hemicellulose fractions from pre-treated biomass. Process consolidation has the potential to increase production efficiency and also in this biocatalytic scenario reduces/removes the need for chemical pre-treatment of the biomass. The production of fine chemicals via this consolidated process approach has some potential major advantages, both in terms of sustainability but also the potential higher value of the products, due their naturally-derived sources and use of non-synthetic chemistry processes.40,41 Very few works in literature have demonstrated the direct release and biotransformation of aromatic acids from insoluble lignocellulose, previously only the production of vanillin from biomass has been reported.42In summary, the monolignol coniferol 3 possesses several applications, in lignin research, as a precursor to fragrance compounds, and as active pharmaceutical ingredients. Through the use of a three-enzyme cascade, we have demonstrated the simple production of this compound in high yield and purity with up to 97% conversion yield. The method presents several attractive features including use of plant biomass-derived feedstocks, use of biocatalytic route (XynZ, NiCAR, and CgAKR-1), ambient, low energy reaction conditions and significant production rates. Finally, this method opens up new routes for the conversion of biomass to useful products via synthetic biology approaches to create further designer organisms and biocatalytic pathways, and provides a toolbox platform for the generation of high-value aromatic molecules from renewable sources.
Author contributions
RT designed and carried out all the experiments. JG and FP supported the biochemical characterizations. SRD discovered and produced CAR enzymes. RT, FM and ND drafted the manuscript. TDH, NT, ND conceived the study and participated in data interpretation. All the authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. T. was supported a by grant from the São Paulo Research Foundation (FAPESP, proc number: 2017/23754-1). ND is supported by the BBSRC David Phillips Fellowship (BB/K014773/1) and BBSRC-FAPESP (FAPPA) grant (BB/L026244/1). ND, NT, JG, and TDHB are supported by BBSRC-FAPESP grant BB/P01738X/1. FMS is supported by the FAPESP grant (2015/50590-4).References
- Y.-H. P. Zhang, Biotechnol. Adv., 2015, 33, 1467–1483 CrossRef CAS.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS.
- A. Arevalo-Gallegos, Z. Ahmad, M. Asgher, R. Parra-Saldivar and H. M. N. Iqbal, Int. J. Biol. Macromol., 2017, 99, 308–318 CrossRef CAS.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3(11), 1227–1235 CrossRef CAS PubMed.
- J. P. N. Rosazza, Z. Huang, L. Dostal, T. Volm and B. Rousseau, J. Ind. Microbiol., 1995, 15, 457–471 CrossRef CAS PubMed.
- F. Xu, R.-C. Sun, J.-X. Sun, C.-F. Liu, B.-H. He and J.-S. Fan, Anal. Chim. Acta, 2005, 552, 207–217 CrossRef CAS.
- G. Niño-Medina, E. Carvajal-Millán, A. Rascon-Chu, J. A. Marquez-Escalante, V. Guerrero and E. Salas-Muñoz, Phytochem. Rev., 2010, 9, 111–120 CrossRef.
- P. J. Harris and J. A. K. Trethewey, Phytochem. Rev., 2010, 9, 19–33 CrossRef CAS.
- R. Gazak, D. Walterova and V. Kren, Curr. Med. Chem., 2007, 14, 315–338 CrossRef CAS.
- Z. Chen, X. Sun, Y. Li, Y. Yan and Q. Yuan, Metab. Eng., 2017, 39, 102–109 CrossRef CAS.
- S. Noda and A. Kondo, Trends Biotechnol., 2017, 35, 785–796 CrossRef CAS PubMed.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass - Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Richland, WA (United States), 2007 Search PubMed.
- S. Quideau and J. Ralph, J. Agric. Food Chem., 1992, 40, 1108–1110 CrossRef CAS.
- K. E. Achyuthan, P. D. Adams, S. Datta, B. A. Simmons and A. K. Singh, Molecules, 2010, 15, 1645–1667 CrossRef CAS PubMed.
- Y. Lv, X. Cheng, D. Wu, G. Du, J. Zhou and J. Chen, Bioresour. Technol., 2018, 267, 578–583 CrossRef CAS.
- L. Tarrago, C. Modolo, M. Yemloul, V. Robert, P. Rousselot-Pailley and T. Tron, New J. Chem., 2018, 42, 11770–11775 RSC.
- W. W. Tong, C. Zhang, T. Hong, D. H. Liu, C. Wang, J. Li, X. K. He and W. D. Xu, Sci. Rep., 2018, 8, 3241 CrossRef CAS.
- C. W. Y. Cheung, N. Gibbons, D. W. Johnson and D. L. Nicol, Anticancer Agents Med. Chem., 2010, 10(3), 186–195 CrossRef CAS.
- M. Kondo, N. OyamaA-Okubo, M. Sagae, T. Ando, E. Marchesi and M. Nakayama, Biosci. Biotechnol. Biochem., 2007, 71, 458–463 CrossRef CAS.
- Y. Wang, M. Chantreau, R. Sibout and S. Hawkins, Front. Plant Sci., 2013, 4, 220 Search PubMed.
- W. Finnigan, A. Thomas, H. Cromar, B. Gough, R. Snajdrova, J. P. Adams, J. A. Littlechild and N. J. Harmer, ChemCatChem, 2017, 9, 1005–1017 CrossRef CAS.
- M. K. Akhtar, N. J. Turner and P. R. Jones, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 87–92 CrossRef CAS.
- M. Winkler, Curr. Opin. Chem. Biol., 2018, 43, 23–29 CrossRef CAS.
- R. Tramontina, J. P. L. Franco Cairo, M. V. Liberato, F. Mandelli, A. Sousa, S. Santos, S. C. Rabelo, B. Campos, J. Ienczak, R. Ruller, A. R. L. Damásio and F. M. Squina, Biotechnol. Biofuels, 2017, 10, 4 CrossRef.
- E. Klumbys, Z. Zebec, N. J. Weise, N. J. Turner and N. S. Scrutton, Green Chem., 2018, 20, 658–663 RSC.
- P. Zucca, M. Literru, A. Rescigno and E. Sanjust, Biosci. Biotechnol. Biochem., 2009, 73, 1224–1226 CrossRef CAS.
- A. He, T. Li, L. Daniels, I. Fotheringham and J. P. N. Rosazza, Appl. Environ. Microbiol., 2004, 70, 1874–1881 CrossRef CAS PubMed.
- Y. Duan, P. Yao, X. Chen, X. Liu, R. Zhang, J. Feng, Q. Wu and D. Zhu, J. Mol. Catal. B: Enzym., 2015, 115, 1–7 CrossRef CAS.
- K. Napora-Wijata, G. A. Strohmeier and M. Winkler, Biotechnol. J., 2014, 9, 822–843 CrossRef CAS.
- J. Aschenbrenner, P. Marx, J. Pietruszka and J. Marienhagen, ChemBioChem, 2019, 20, 949–954 CrossRef CAS.
- B. Lin and Y. Tao, Microb. Cell Fact., 2017, 16, 106 CrossRef.
- A. M. Kunjapur, Y. Tarasova and K. L. J. J. Prather, J. Am. Chem. Soc., 2014, 136, 11644–11654 CrossRef CAS.
- K. E. Kram and S. E. Finkel, Appl. Environ. Microbiol., 2015, 81, 4442–4450 CrossRef CAS.
- S. Liu, J. Liu, J. Hou, N. Chao, Y. Gai and X. Jiang, Microb. Cell Fact., 2017, 16, 104 CrossRef.
- J. Aschenbrenner, P. Marx, J. Pietruszka and J. Marienhagen, ChemBioChem, 2019, 20, 949–954 CrossRef CAS.
- A. N. Khusnutdinova, R. Flick, A. Popovic, G. Brown, A. Tchigvintsev, B. Nocek, K. Correia, J. C. Joo, R. Mahadevan and A. F. Yakunin, Biotechnol. J., 2017, 12(11) DOI:10.1002/biot.201600751.
- D. M. de Oliveira, A. Finger-Teixeira, T. Rodrigues Mota, V. H. Salvador, F. C. Moreira-Vilar, H. B. Correa Molinari, R. A. Craig Mitchell, R. Marchiosi, O. Ferrarese-Filho and W. Dantas dos Santos, Plant Biotechnol. J., 2015, 13, 1224–1232 CrossRef CAS.
- F. Mandelli, L. B. Brenelli, R. F. Almeida, R. Goldbeck, L. D. Wolf, Z. B. Hoffmam, R. Ruller, G. J. M. Rocha, A. Z. Mercadante and F. M. Squina, Ind. Crops Prod., 2014, 52, 770–775 CrossRef CAS.
- A. R. L. Damásio, C. M. P. Braga, L. B. Brenelli, A. P. Citadini, F. Mandelli, J. Cota, R. F. de Almeida, V. H. Salvador, D. A. A. Paixao, F. Segato, A. Z. Mercadante, M. de Oliveira Neto, W. D. do Santos and F. M. Squina, Appl. Microbiol. Biotechnol., 2013, 97, 6759–6767 CrossRef.
- H. Yu, H. Tang and P. Xu, Sci. Rep., 2015, 4, 5397 CrossRef.
- S. Mishra, A. Sachan and S. G. Sachan, J. Ind. Microbiol. Biotechnol., 2013, 40, 545–550 CrossRef CAS.
- P. Chattopadhyay, G. Banerjee and S. K. Sen, J. Cleaner Prod., 2018, 182, 272–279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9gc02359c |
| This journal is © The Royal Society of Chemistry 2020 |