
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fucose-modified thermoresponsive poly(2-hydroxypropyl methacrylate) nanoparticles for controlled doxorubicin release from an injectable depot†
Huayang
Yu
 ,
Jason V.
Rowley
,
David C.
Green
and
Paul D.
Thornton
*
,
Jason V.
Rowley
,
David C.
Green
and
Paul D.
Thornton
*
School of Chemistry, University of Leeds, Leeds, LS2 9JT, UK. E-mail: p.d.thornton@leeds.ac.uk
First published on 14th July 2020
Abstract
The modification of poly(2-hydroxypropyl methacrylate) with a single fucose group per polymer chain enabled macromolecular self-assembly, and the formation of thermoresponsive nanoparticles. In addition, poly(2-hydroxypropyl methacrylate)200 formed a self-healing material that may act as an injectable vehicle and depot for poly(2-hydroxypropyl methacrylate) nanoparticle delivery and localisation at a particular site. In combination, the injectable depot permits the controlled release of doxorubicin from the fucose presenting nanoparticles that it contains following injection. Such thermoresponsive materials are highly promising candidates for the treatment of diseases that may be remedied by exploiting targeted fucose-cell binding, such as pancreatic cancer.
Introduction
Effective cancer treatment in the absence of extremely intrusive side-effects remains a key challenge in contemporary medicine.1 Providing a remedy that destroys cancer cells, whilst leaving healthy cells relatively unaffected must be realised for this challenge to be overcome.2 Encapsulating a toxic anti-cancer drug within a shielding polymeric carrier protects the drug from interacting with cells, thus preventing the damage of non-cancerous cells, and the aggressive side-effects that are associated with this occurring during chemotherapy.3–5 Extensive drug loading into polymeric nanoparticles, and a mechanism for the programmed release of the drug from the nanoparticles, are essential features of effective treatments.6–10 Additionally, next-generation drug delivery vehicles must present cell-binding groups that enable extensive therapeutic interaction with target cells only.11–13Fucose plays a key role in mammalian development, immunity and cancer metastasis.14 Increased expression of fucosyltransferases is associated with pancreatic cancer, whereby enzymes accelerate malignant transformation through the fucosylation of sialylated precursors, and pancreatic cancer cells display enhanced fucose uptake. Pancreatic cancer is an extremely aggressive cancer with a dismal survival rate; it is estimated that the 5-year survival rate is 5% owing to its aggressiveness, and a lack of effective therapies.15 Currently, patients that undergo treatment with the chemotherapy drug Gemcitabine (Gemzar®) have a median survival time reported to be only 5.7 months.16 A plausible explanation for this abysmal inefficiency is the extremely ineffective delivery of anticancer drugs to the tumour site. There is enormous demand for the creation of fucose-presenting nanoparticles to be deployed as increasingly advanced drug delivery systems for the treatment of pancreatic cancer.
Poly(2-hydroxypropyl methacrylate) (PHPMA) is non-toxic and biocompatible.17,18 PHPMA-containing block copolymers can be readily formed by reversible addition–fragmentation chain-transfer (RAFT) aqueous dispersion polymerisation.19 Such polymers have been reported to self-assemble into a plethora of arrangements dependent on their macromolecular configuration, and the environment in which they are maintained. For instance, poly(glycidyl methacrylate)-b-PHPMA may be manipulated to form spheres, worms or vesicles dependent on the composition of the block copolymer, and the temperature of the aqueous solution in which the polymer is dispersed.20 PHPMA, and PHPMA-based materials have been applied to the thermally-triggered release of drug molecules,21 and controlled release applications.22
Implantable polymer-based materials for drug and cell delivery have attracted significant recent attention.23–27 Such materials may facilitate the immobilisation of drug-loaded nanoparticles at the target site, ensuring extensive drug release at diseased areas, and minimal drug release to healthy tissue. This enhances the efficacy of the treatment, reducing the side-affects that are caused by drug interaction with non-target, healthy cells. An implantable depot that enables the controlled and prolonged release of fucose-presenting nanoparticles for long-term pancreatic cancer treatment is a noteworthy long-term goal.
We report that the simple modification of PHPMA polymer chains with a single fucose unit enables the polymer to self-assemble, forming nanoparticles in aqueous solution. Such nanoparticles are stable at room temperature but undergo a morphological change upon heating to physiological temperature, which may be exploited to actuate therapeutic payload release. Unmodified PHPMA can form an injectable depot that undergoes a sol–gel transition at a temperature slightly above physiological temperature. Consequently, the depot, which is self-healing and can withhold guest molecules itself, may act as an injectable host for the controlled release of doxorubicin (Dox) from fucose-capped PHPMA nanoparticles at a localised site.
Experimental
Materials and methods
L(−)-Fucose 97% was purchased from ACROS Organics. Chloroform (HPLC grade), dichloromethane (DCM) (HPLC grade) and acetone were obtained from VWR Chemicals. HPMA, Phosphate buffered saline tablet, N,N-dimethylformamide, 2,2′-azobis(2-methyl-propionamidine) dihydrochloride (AAPH) granular 97%, 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid N-succinimidyl ester (SCPDB), dialysis tubing benzoylated 2000 Da, deuterium oxide 99.9 atom D% and acetic acid 99–100% were purchased from Sigma-Aldrich. Triethylamine and tetrahydrofuran (THF, analytical grade) were obtained from Fisher Chemical. Sodium cyanoborohydride 95%, and ethylenediamine 99% were purchased from Alfa Aesar. Sodium meta-periodate was obtained from Thermo Scientific. 4-Cyano-4-(phenylcarbonothioyl)thiopentanoic acid, dimethyl sulfoxide and doxorubicin hydrochloride were purchased from Fluorochem. Dimethyl sulfoxide (99.80% D) was purchased from EURISO-TOP.Chemical structures and functional groups were identified by Nuclear Magnetic Resonance spectroscopy (NMR, Bruker AVANCE III HD-400) and Attenuated Total Reflection (ATR-PLATINUM) Fourier Transform Infrared Spectroscopy (FTIR, BRUKER ALPHA). Scanning Electron Microscopy (SEM, FEI NanoSEM 450, elemental composition by energy-dispersive X-ray analysis) was employed to analyse size and topography of particle and particle surface. UV-vis spectroscopy (VARIAN 50 Probe UV-visible Spectrometer) was used to measure drug release from doxorubicin loaded polymer nanoparticles. Advanced Polymer Chromatography (APC) was employed to measure molecular weight of the synthesised polymers. APC was conducted on a Waters Acquity APC system using an Acquity column (Acquity APC TM 200 2.5 μm, 4.5 × 150 mm) calibrated against poly(methyl methacrylate) standards in THF. Dynamic Light Scattering (DLS) analysis was carried out in triplicate using a Malvern Zetasizer Nano ZSP series instrument with a 4 mW He–Ne laser at a wavelength at 633 nm. An avalanche photodiode (APD) detector and DTS software were used. The light was scatted at 173° and collected by a back-scatter-optic arrangement. Samples were equilibrated for 2 minutes prior to analysis at room temperature and at 37 °C in disposable polystyrene cuvettes (1.5 mL to 3.0 mL). Glass transition temperatures were measured by Differential Scanning Calorimetry (DSC), using a TA Instruments DSC Q20 instrument with a heating rate of 5 °C min−1 between 0–100 °C and an inert gas flow of N2 at a rate of 50 mL min−1. Rheology experiments were conducted using a rheometer (Anton Paar, MCR 302) with a 25 mm parallel plate. Frequency sweeps were recorded at 100–0.1 rad s−1 with a constant amplitude (1%) at room temperature, using a 1.2 g PHPMA depot and a 1.9 mm gap.
Fucose conjugation of ethylenediamine
0.100 g of L(−)-fucose was dissolved in 10.0 mL of deionised water. Then 0.391 g of sodium meta-periodate (mole ratio of fucose![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sodium meta periodate = 1:3) was added to the fucose solution (colourless) and stirred overnight in the dark to activate the aldehyde group. 41.0 μL of ethylenediamine was added into the mixture which was then stirred overnight (mole ratio of fucose:ethylenediamine = 1:1). 0.038 g of sodium cyanoborohydride was then added to the mixture which was stirred for 3 hours followed by freeze-drying for 48 hours (mole ratio of fucose:sodium cyanoborohydride = 1:1).
:sodium meta periodate = 1:3) was added to the fucose solution (colourless) and stirred overnight in the dark to activate the aldehyde group. 41.0 μL of ethylenediamine was added into the mixture which was then stirred overnight (mole ratio of fucose:ethylenediamine = 1:1). 0.038 g of sodium cyanoborohydride was then added to the mixture which was stirred for 3 hours followed by freeze-drying for 48 hours (mole ratio of fucose:sodium cyanoborohydride = 1:1).
SCPDB conjugation to amine-bearing fucose
0.118 g of fucose-amine and 0.213 g of SCPDB RAFT agent were dissolved in 5.0 mL of deionised water and 20.0 mL of acetone, respectively. The SCPDB RAFT agent solution was added dropwise into the fucose-amine solution with stirring followed by the addition of 3.0 mL of acetic acid. The mixture was stirred at 60 °C overnight. Following this, the solution underwent rotary evaporation at 60 °C to remove acetone and freeze-dried for 48 hours. The fucose-RAFT agent was washed multiple times with DCM, being recovered by centrifugation for 30 minutes at 4500 rpm, before finally being dried in a vacuum oven at room temperature overnight.HPMA Polymerisation from the fucose-RAFT agent
0.052 g of fucose-RAFT agent was dissolved in 3.0 mL and 2.0 mL of acetone and deionised water. Then, 0.091 g of AAPH was dissolved in the mixture (mole ratio of fucose-RAFT agent:AAPH = 1:3). 1.608 g of HPMA was added dropwise into the reaction which was stirred at 55 °C overnight (mole ratio of fucose-RAFT agent:HPMA = 1:100). The product underwent rotary evaporation at 50 °C for 20 minutes and the recovered product was freeze-dried overnight. Finally, fucose-PHPMA was washed multiple times with deionised water, being recovered by centrifugation for 30 minutes at 4500 rpm, before finally being freeze-dried for 48 hours.
PHPMA synthesis: HPMA polymerisation from 4-cyano-4-(phenylcarbonothioyl)thiopentanoic acid
0.020 g of 4-cyano-4-(phenylcarbonothioyl)thiopentanoic acid RAFT agent was dissolved in 3.0 mL and 2.0 mL of acetone and deionised water, respectively. Then, 0.059 g of AAPH was dissolved into the mixture (mole ratio of RAFT agent:AAPH = 1:3). 1.025 g of HPMA was added dropwise into the reaction and stirred at 55 °C overnight (mole ratio of fucose-RAFT agent:HPMA = 1:100). The product was rotary evaporated at 50 °C for 20 minutes and freeze-dried for 48 hours. Finally, PHPMA was washed with deionised water, centrifugation for 30 minutes at 4500 rpm and freeze-dried for 48 hours.
PHPMA gel synthesis: HPMA polymerisation from 4-cyano-4-(phenylcarbonothioyl)thiopentanoic acid
As a representative procedure to produce PHPMA200:0.020 g of 4-cyano-4-(phenylcarbonothioyl)thiopentanoic acid RAFT agent was dissolved in 3.0 mL and 2.0 mL of acetone and deionised water, respectively. Then, 0.059 g of AAPH was dissolved into the mixture (mole ratio of RAFT agent:AAPH = 1:3). 2.050 g of HPMA was added dropwise into the reaction, which was stirred at 55 °C overnight (mole ratio of fucose-RAFT agent:HPMA = 1:200).
Results and discussion
The overall route to fucose-capped PHPMA nanoparticles that are able to withhold, and gradually release Dox is presented in Fig. 1, and detailed reaction schemes provided in Schemes S1 and S2 (ESI†). Fucose was successfully oxidised by sodium meta-periodate, as confirmed by the proton corresponding to the aldehyde group being present in the 1H NMR spectrum (Fig. S1, ESI†). Fucose-amine was produced by sequential reaction with ethylene diamine and sodium cyanoborohydride, and the structure confirmed by 13C NMR (Fig. S3, ESI†). The peaks present in the region 40–55 ppm are assigned to the conjugated diamine. RAFT agent conjugation to fucose-amine, to enable RAFT polymerisation to occur from the fucose-amine initiator, was confirmed by the peaks in the aromatic region of Fig. S5 (ESI†), which remained after thorough washing with DCM. Different chain lengths (20, 40, 60, 80 and 100 repeat units) of fucose-PHPMA were synthesised to ascertain the optimal polymer for stable nanoparticle formation. The average molecular weight values of fucose-PHPMA with 20, 40, 60, 80 and 100 repeat units were assessed by 1H NMR spectroscopy using the proton environments of the RAFT agent aromatic group and peaks f, g, h, e, d and i to confirm that the desired polymers were produced (Fig. S6–S10, ESI†). APC was used to further evaluate polymer molecular weight (Table S1, ESI†).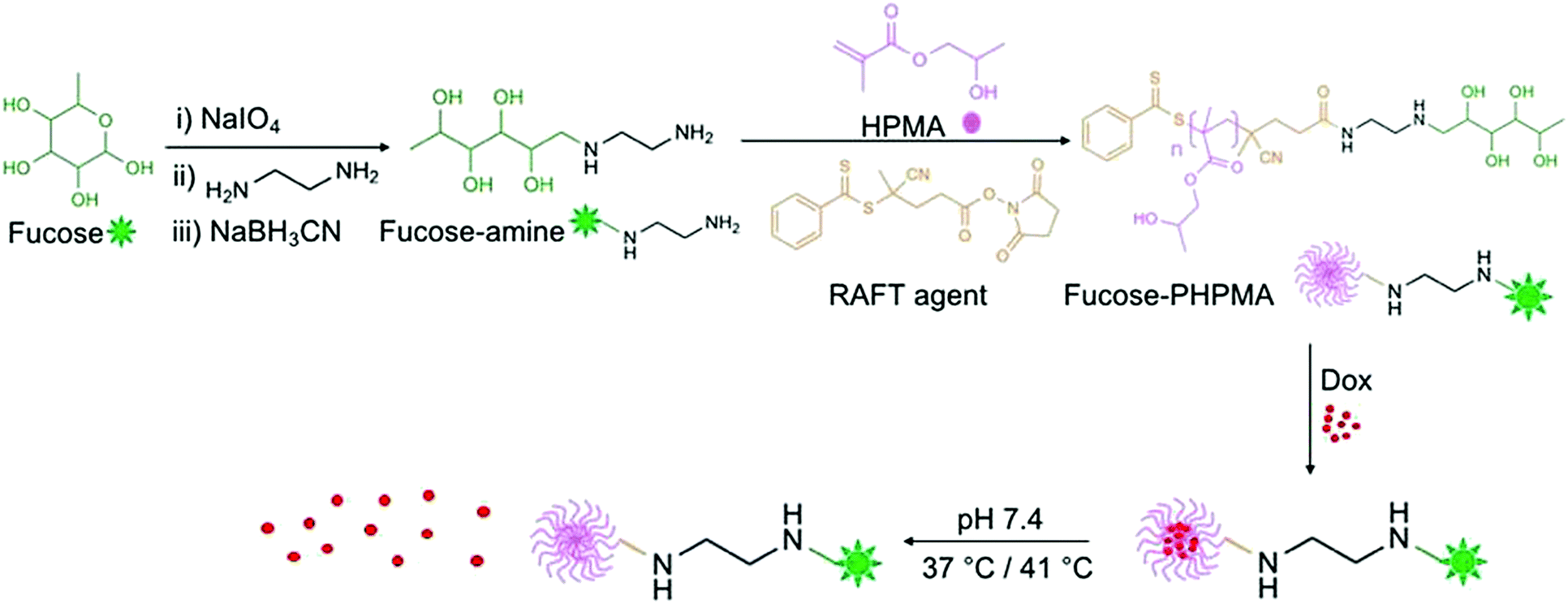 | ||
| Fig. 1 Reaction outline for the creation of fucose-PHPMA100 nanoparticles that are capable of encapsulating, and releasing Dox at physiological temperature. | ||
The products from each step of the overall reaction (Scheme S1, ESI†) were analysed by FTIR spectroscopy. The aldehyde group of opened fucose (Fig. S17 black, ESI†) confirms fucose oxidation by sodium meta-periodate. Peaks corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S, C–S and aromatic groups can be found in the FTIR spectra of the fucose-RAFT agent, and fucose-PHPMA, confirming successful synthesis of activated fucose and the desired final product, respectively (Fig. S17 and S18, ESI†). Additionally, a strong and broad peak corresponding to the alcohol group is present in the fucose-PHPMA FTIR spectrum further confirming HPMA polymerisation from the fucose-RAFT agent (Fig. S17, ESI†). This route to product, in which the RAFT agent is covalently conjugated to fucose, ensures that each PHPMA chain possesses a desired terminal fucose unit.
S, C–S and aromatic groups can be found in the FTIR spectra of the fucose-RAFT agent, and fucose-PHPMA, confirming successful synthesis of activated fucose and the desired final product, respectively (Fig. S17 and S18, ESI†). Additionally, a strong and broad peak corresponding to the alcohol group is present in the fucose-PHPMA FTIR spectrum further confirming HPMA polymerisation from the fucose-RAFT agent (Fig. S17, ESI†). This route to product, in which the RAFT agent is covalently conjugated to fucose, ensures that each PHPMA chain possesses a desired terminal fucose unit.
Nanoparticles were produced from the various fucose-PHPMA batches by coacervation. Nanoparticle size increased with polymer chain length when Dox was absent from the nanoparticles, and also when Dox was loaded into the nanoparticles (Tables 1 and 2). At room temperature, the particle sizes of blank and Dox-loaded nanoparticles were comparable both in terms of dimensions and PDI values. In all instances, the PDI values were less than 0.3, confirming a stable dispersion formed. Conversely, PHPMA of the same number of repeat units, but lacking fucose, did not form stable nanoparticles (Table S2, ESI†), emphasising the necessity for fucose conjugation to PHMPA for stable nanoparticle assembly. Fucose-PHPMA100 with and without Dox-loading were shown as spherical nanoparticles by SEM analysis (Fig. 2(a) and (b), respectively).
:H2O, 1:1 volume ratio), maintained at room temperature. The nanoparticles were measured both when vacant and loaded with Dox
| Polymer | Size (nm) | PDI |
|---|---|---|
| a Denotes Dox-loaded nanoparticles. | ||
| Fucose-PHPMA20 | 93 ± 5 | 0.170 |
| Fucose-PHPMA40 | 106 ± 6 | 0.205 |
| Fucose-PHPMA60 | 121 ± 1 | 0.169 |
| Fucose-PHPMA80 | 163 ± 6 | 0.165 |
| Fucose-PHPMA100 | 217 ± 3 | 0.151 |
| Fucose-PHPMA20 (Dox)a | 91 ± 2 | 0.209 |
| Fucose-PHPMA40 (Dox)a | 108 ± 4 | 0.218 |
| Fucose-PHPMA60 (Dox)a | 122 ± 1 | 0.186 |
| Fucose-PHPMA80 (Dox)a | 165 ± 2 | 0.217 |
| Fucose-PHPMA100 (Dox)a | 219 ± 2 | 0.181 |
| Polymer | Size (nm) | PDI |
|---|---|---|
| a Denotes Dox-loaded nanoparticles. | ||
| Fucose-PHPMA20 | 12 ± 0.2 | 0.203 |
| Fucose-PHPMA40 | 16 ± 2.5 | 0.215 |
| Fucose-PHPMA60 | 19 ± 1.2 | 0.217 |
| Fucose-PHPMA80 | 22 ± 1.3 | 0.218 |
| Fucose-PHPMA100 | 32 ± 3.3 | 0.232 |
| Fucose-PHPMA20 (Dox)a | 11 ± 1.6 | 0.238 |
| Fucose-PHPMA40 (Dox)a | 16 ± 0.8 | 0.251 |
| Fucose-PHPMA60 (Dox)a | 19 ± 1.3 | 0.256 |
| Fucose-PHPMA80 (Dox)a | 22 ± 1.5 | 0.233 |
| Fucose-PHPMA100 (Dox)a | 32 ± 1.0 | 0.201 |
 | ||
| Fig. 2 SEM images of fucose-PHPMA100 nanoparticles (a) and Dox-loaded fucose-PHPMA100 nanoparticles (b). | ||
To assess the thermoresponse of fucose-PHPMA nanoparticles, their dimensions were assessed by DLS at 37 °C. The particle size of all types of fucose-PHPMA nanoparticles decreased significantly at 37 °C, signifying a thermo-responsive behaviour (Table 2). PDI values of the particles at 37 °C were slightly higher than those at room temperature, however all samples had PDI values less than 0.3, confirming their stability against aggregation. Significantly smaller artefacts were present in the SEM images of Dox-loaded fucose-PHPMA100 nanoparticles at 37 °C that are assigned to released Dox (Fig. S20(d), ESI†).
0.033 mg (11%), 0.093 mg (31%), 0.158 mg (53%), 0.221 mg (74%) and 0.245 mg (82%) of Dox was then loaded into fucose-PHPMA20, fucose-PHPMA40, fucose-PHPMA60, fucose-PHPMA80 and fucose-PHPMA100 nanoparticles in PBS buffer solution, respectively. The extent of Dox release from each configuration of fucose-PHPMA nanoparticles was assessed at room temperature and at 37 °C (Fig. 3). Negligible release of Dox after 168 hours (≈1.6% for all nanoparticle samples) from the series of particles demonstrated that Dox can largely be held within all formed nanoparticles at room temperature (Fig. 3(a)). Particles created from fucose-PHPMA20 revealed the most rapid release of Dox at 37 °C, with complete release occurring after 29 hours (Fig. 3(b)). Restricted Dox release corresponded with greater HPMA content within the nanoparticles, enabling the release rate to be tuned by polymer composition. The extent of Dox release was determined to be 100%, 66%, 46% and 31% for fucose-PHPMA40, fucose-PHPMA60, fucose-PHPMA80 and fucose-PHPMA100 nanoparticles, respectively, after 168 hours (Fig. 3(b)).
 | ||
| Fig. 3 Dox release from fucose-PHPMA20, fucose-PHPMA40, fucose-PHPMA60, fucose-PHPMA80 and fucose-PHPMA100 nanoparticles (a) at room temperature PBS buffer solution and (b) at 37 °C in PBS buffer solution. | ||
Since fucose-PHPMA100 nanoparticles (with and without Dox)possessed the lowest PDI values, this material was taken forward for further analysis. The initial release studies revealed that 13% of Dox was released from fucose-PHPMA100 particles after 24 hours at 37 °C, the lowest release of all nanoparticle samples (Fig. 3(b)). Detailed release studies were then conducted in order to investigate the specific temperature that lead to complete Dox release, with studies conducted that measured Dox release at various temperatures. To begin with, the Dox release study ran over a temperature range from 22 °C to 50 °C and the solution temperature was increased by 1 °C every 15 minutes (Fig. 4(a)). Extensive Dox release occurred between 39 °C (19%) and 46 °C (47%) (Fig. 4(a)). The temperature range of greatest Dox release was then narrowed to between 35 °C and 46 °C, by increasing the solution temperature by 1 °C every 40 minutes. Extensive Dox release (19.2%, 0.041 mg) occurred between 41 °C and 42 °C (Fig. 4(b)), demonstrating the potential of the reported materials to release Dox more extensively under moderate heating. Further Dox release studies were carried out to determine the time required for complete Dox release to occur at 41 °C. 22% (0.048 mg) of Dox was released in the first hour (Fig. 4(c)), with continuous release culminating in 51% (0.126 mg), 74% (0.183 mg) and 90% (0.221 mg) Dox release after 24 hours, 48 hours and 72 hours, respectively (Fig. 4(d)). Dox release from the fucose-PHPMA100 nanoparticles reached 100% (0.245 mg) after 96 hours.
 | ||
| Fig. 4 (a) Dox release from fucose-PHPMA100 nanoparticles into solution increasing in temperature from 22 °C to 50 °C at a rate of 1 °C every 15 minutes. (b) Dox release from fucose-PHPMA100 nanoparticles into solution increasing in temperature from 35 °C to 46 °C at a rate of 1 °C every 40 minutes (c) Dox release from fucose-PHPMA100 nanoparticles, monitored every five minutes at 41 °C. (d) Dox release from nanoparticles maintained at 41 °C over 7 days. | ||
Next, a thermoresponsive PHPMA depot was created that may act as an injectable delivery vehicle, and host, for Dox-loaded fucose-PHPMA100 nanoparticles that enables localised drug release (Fig. S23, ESI†). PHPMA80 and PHPMA200 depots were synthesised by RAFT polymerisation (Scheme S2, ESI†). PHPMA depot formation occurred upon polymer/DMSO solution being added to PBS buffer solution. The glass transition temperature of the depots was greater with increased polymer chain length (Fig. S24, ESI†), and the water content of the two depots were 87.1% (PHPMA80) and 82.5% (PHPMA200), by mass (Table S3, ESI†). The loss modulus of PHPMA200 depot formed in PBS buffer solution was constant across the 0.1–100 frequency range indicating that the material was stable and inelastic (Fig. S26, ESI†). PHPMA80 and PHPMA200 depots were then created in the presence of Dox fucose-PHPMA100 nanoparticles (Fig. S27, ESI†). Due to the enhanced thermal and mechanical stability of the PHPMA200 depot, Dox release from nanoparticles embedded within this material was studied at room temperature and at 37 °C (Fig. 5). Dox release increased up to 168 hours in solution maintained at 37 °C, before 43.9% (0.011 mg) Dox release was recorded after 288 hours. Only a negligible amount of Dox release (2.3%, 0.0006 mg) occurred at room temperature after 288 hours. The polymer nanoparticles are unable to penetrate the PHPMA depot, ensuring that they remain at the injection site following their administration.
 | ||
| Fig. 5 The percentage of Dox released from fucose-PHPMA100 nanoparticles loaded in PHPMA200 depot maintained in PBS buffer solution at room temperature, and at 37 °C. | ||
Further studies revealed that the PHPMA200 depot possesses self-healing properties. Two batches of the PHPMA200 material were produced; one containing aqueous rhodamine b solution incorporated and the other containing water only, for illustrative purposes. The PHPMA200 material can be stretched to 24.0 cm unevenly, which corresponds to 1200% elongation (Fig. 6a). Rhodamine b-containing material was placed in contact with blank PHPMA200 material, in the presence and absence of PBS buffer solution at 25 °C and at 37 °C (Fig. 6c, g, k and o). Over 1 hour, the two independent materials connected to reveal the self-healing properties of the PHPMA200 depot, indicating its potential suitability to be maintained for extended periods, and self-heal if damaged, in vivo (Fig. 6e, i, m and q).
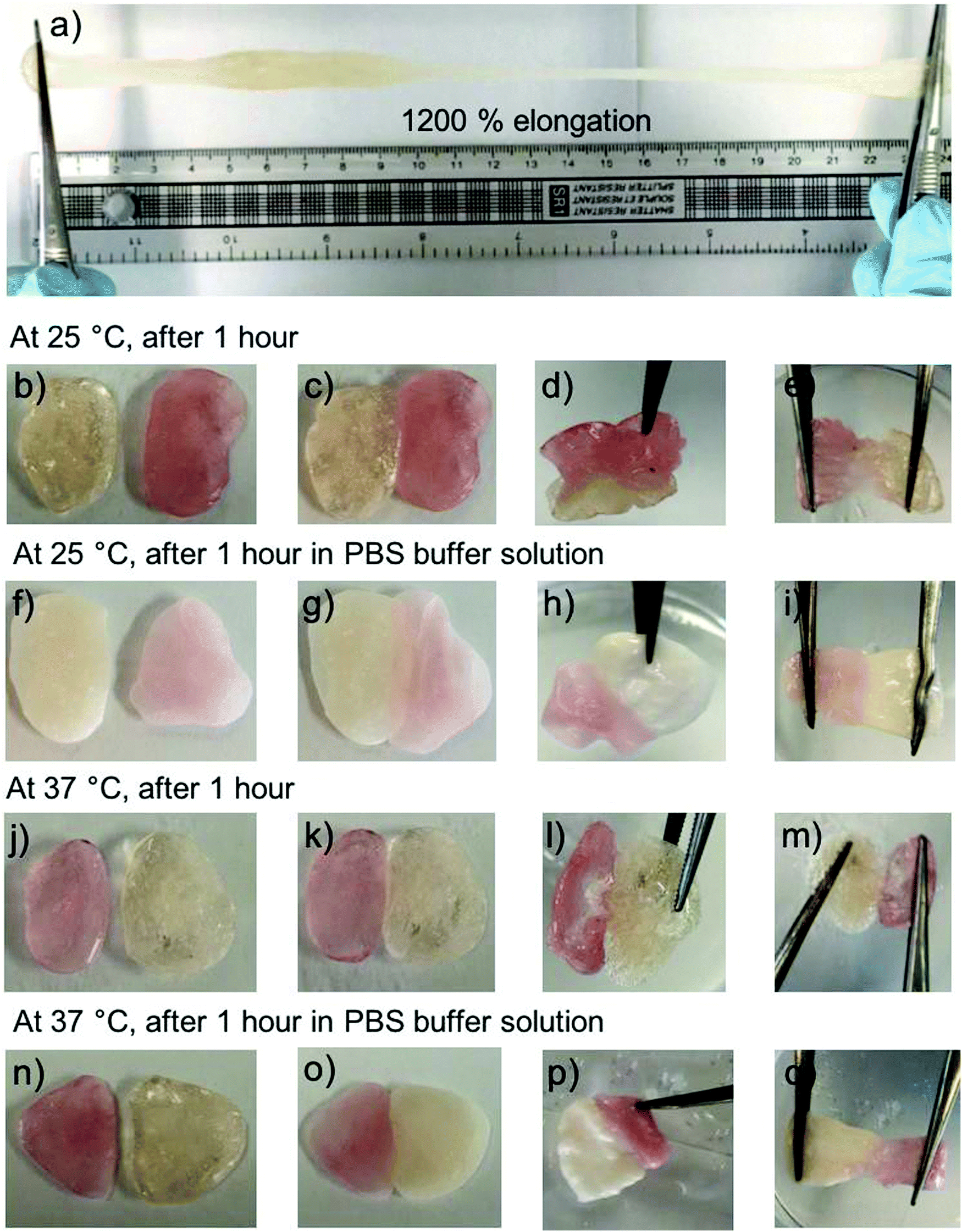 | ||
| Fig. 6 0.002 mg mL−1 of rhodamine b was used to partially colour PHPMA200. (a) Stretching PHPMA200; (b)–(e) demonstration of PHPMA200 self-healing at 25 °C for one hour; (f)–(i) demonstration of PHPMA200 self-healing at 25 °C for one hour in PBS buffer solution; (j)–(m) demonstration of PHPMA200 self-healing at 37 °C for one hour; (n)–(q) demonstration of PHPMA200 self-healing at 37 °C for one hour in PBS buffer solution. | ||
The PHPMA200 depot could also withhold rhodamine b in the absence of nanoparticles and be manipulated to form distinct and stable forms in PBS buffer solution. Rhodamine b-containing material could be formed upon injection onto a glass sheet as a viscous liquid (Fig. 7a). Upon the addition of PBS buffer solution, a depot formed that maintained both the loaded rhodamine b, and a defined structure. These materials could be rearranged and floated on the buffer solution, with the fusion of letters D and E occurring as further evidence of a self-healing material (Fig. 7c). The material dimensions and rhodamine b payload were both retained in PBS buffer solution, before the shapes were retrieved from solution and placed on a different glass sheet (Fig. 7d). The letters could be removed intact from solution and placed on a new glass sheet.
 | ||
| Fig. 7 (a) Writing “LEEDS” on a glass sheet covered with PBS solution using PHPMA200 DMSO solution containing 0.004 mg mL−1 rhodamine b; (b) writing complete; (c) upon the addition of further PBS buffer solution to the plate, the characters floated on the PBS buffer solution surface; (d) the letters could be removed and placed on another glass sheet. Recordings of the process are available online. | ||
Conclusions
Thermally-responsive fucose-modified PHPMA nanoparticles were synthesised by RAFT polymerisation. A single fucose group per polymer chain was sufficient to instigate polymer self-assembly to yield nanoparticles in aqueous solution. Spherical fucose-PHPMA nanoparticles that contained or lacked Dox were very stable at room temperature and at 37 °C, as determined by DLS and SEM analysis. Dox loading into the nanoparticles was achieved by coacervation; negligible Dox release from the nanoparticles occurred at room temperature, whilst extensive release occurred upon the solution temperature increasing to 41 °C. An injectable PHPMA200 depot was synthesised by RAFT polymerisation as a delivery vehicle for fucose-PHPMA100 nanoparticles. This material possessed self-healing properties in PBS buffer solution and offers great promise to enable nanoparticle localisation at a target site for the prolonged release of anticancer therapeutics.Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Jacob, J. T. Haponiuk, S. Thomas and S. Gopi, Mater. Today Chem., 2018, 9, 43–55 CrossRef CAS.
- X. R. Qiao, X. F. Wang, Y. Shang, Y. Li and S. Z. Chen,, Cancer Commun., 2018, 38, 13 CrossRef PubMed.
- F. Canfarotta, L. Lezina, A. Guerreiro, J. Czulak, A. Petukhov, A. Daks, K. Smolinska-Kempisty, A. Poma, S. Piletsky and N. A. Barlev, Nano Lett., 2018, 18, 4641–4646 CrossRef CAS PubMed.
- M. V. Novoselova, H. M. Loh, D. B. Trushina, A. Ketkar, T. O. Abakumova, T. S. Zatsepin, M. Kakran, A. M. Brzozowska, H. H. Lau, D. A. Gorin, M. N. Antipina and A. I. Brichkina, ACS Appl. Mater. Interfaces, 2020, 12, 5610–5623 CrossRef CAS PubMed.
- M. Miyazaki, E. Yuba, H. Hayashi, A. Harada and K. Kono, Bioconjugate Chem., 2018, 29, 44–55 CrossRef CAS PubMed.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- Y. Qu, B. Y. Chu, X. W. Wei, M. Y. Lei, D. R. Hu, R. Y. Zha, L. Zhong, M. Y. Wang, F. F. Wang and Z. Y. Qian, J. Controlled Release, 2019, 296, 93–106 CrossRef CAS PubMed.
- D. J. Price, M. Khuphe, R. P. W. Davies, J. R. McLaughlan, N. Ingram and P. D. Thornton, Chem. Commun., 2017, 53, 8687–8690 RSC.
- M. Khuphe, N. Ingram and P. D. Thornton, Nanoscale, 2018, 10, 14201–14206 RSC.
- B. S. McAvan, M. Khuphe and P. D. Thornton, Eur. Polym. J., 2017, 87, 468–477 CrossRef CAS.
- F. Araste, K. Abnous, M. Hashemi, S. M. Taghdisi, M. Ramezani and M. Alibolandi, J. Controlled Release, 2018, 292, 141–162 CrossRef CAS.
- W. Q. Cao, J. Zhou, A. Mann, Y. Wang and L. Zhu, Biomacromolecules, 2011, 12, 2697–2707 CrossRef CAS.
- M. Khuphe, C. S. Mahon and P. D. Thornton, Biomater. Sci., 2016, 4, 1792–1801 RSC.
- M. Schneider, E. Al-Shareffi and R. S. Haltiwanger, Glycobiology, 2017, 27, 601–618 CrossRef CAS PubMed.
- C. Lepage, R. Capacaccia, M. Hackl, V. Lemmens, E. Molina, D. Pierannunzio, M. Sant, A. Trama, J. Faivre and E. W. Grp, Eur. J. Cancer, 2015, 51, 2169–2178 CrossRef PubMed.
- S. Boeck, M. Haas, S. Kruger and V. Heinemann, In Vivo, 2014, 28, 1189–1192 Search PubMed.
- N. J. W. Penfold, J. R. Whatley and S. P. Armes, Macromolecules, 2019, 52, 1653–1662 CrossRef CAS.
- J. Madsen, S. P. Armes, K. Bertal, S. MacNeil and A. L. Lewis, Biomacromolecules, 2009, 10, 1875–1887 CrossRef CAS PubMed.
- S. Sugihara, S. P. Armes, A. Blanazs and A. L. Lewis, Soft Matter, 2011, 7, 10787–10793 RSC.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. D. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Macromolecules, 2018, 51, 8357–8371 CrossRef CAS PubMed.
- N. Orakdogen and B. Sanay, J. Appl. Polym. Sci., 2018, 135, 13 CrossRef.
- H. Y. Yu, N. Ingram, J. V. Rowley, S. Parkinson, D. C. Green, N. J. Warren and P. D. Thornton,, J. Mater. Chem. B, 2019, 7, 7795 RSC.
- W. D. Wang, L. Xiang, L. Gong, W. H. Hu, W. J. Huang, Y. J. Chen, A. B. Asha, S. Srinivas, L. Y. Chen, R. Narain and H. B. Zeng, Chem. Mater., 2019, 31, 2366–2376 CrossRef CAS.
- M. H. Chen, L. L. Wang, J. J. Chung, Y. H. Kim, P. Atluri and J. A. Burdick,, ACS Biomater. Sci. Eng., 2017, 3, 3146–3160 CrossRef CAS PubMed.
- A. R. Town, M. Giardiello, R. Gurjar, M. Siccardi, M. E. Briggs, R. Akhtar and T. O. McDonald, Nanoscale, 2017, 9, 6302–6314 RSC.
- B. Maity, S. Samanta, S. Sarkar, S. Alam and T. Govindaraju, ACS Appl. Bio Mater., 2020, 3, 3544–3552 CrossRef CAS.
- L. P. Datta, S. Manchineella and T. Govindaraju, Biomaterials, 2020, 230, 119633 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The synthesis steps to fucose-PHPMA and PHPMA, 1H NMR spectra, molecular weight of the synthesised polymers, FTIR spectra, DLS spectra, SEM images, percentage of water in PHPMA depot, procedures and calculations of Dox loading in the nanoparticles and depot DSC spectra, rheology spectra and calibration curve of Dox free base. See DOI: 10.1039/d0ma00280a |
| This journal is © The Royal Society of Chemistry 2020 |