
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of a hydrogen peroxide-selective fluorogenic probe and its application in Parkinson's disease in vitro and vivo models†
Yao
Lu‡
a,
Bo
Peng‡
b,
Xinghan
Qiu‡
a,
Xin
Li
b,
Zheng
Li
a,
Duoteng
Zhang
a,
Wenhui
Ji
a,
Bin
Fang
b,
Qiong
Wu
a,
Chengwu
Zhang
*a,
Lin
Li
 *a and
Wei
Huang
ab
*a and
Wei
Huang
ab
aKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Nanjing Tech University (Nan-jingTech), 30 South Puzhu Road, Nanjing, 211816, P. R. China. E-mail: iamcwzhang@njtech.edu.cn; iamlli@njtech.edu.cn
bFrontiers Science Center for Flexible Electronics, Xi’an Institute of Flexible Electronics (IFE) and Xi’an Institute of Biomedical Materials & Engineering, Northwestern Polytechnical University, 127 West Youyi Road, Xi’an 710072, China
First published on 15th July 2020
Abstract
Parkinson's disease (PD) is the second most common neurodegenerative disease worldwide and the abnormal level of hydrogen peroxide (H2O2) in the dopaminergic neurons is strongly implicated in the progression of PD. Turn-on fluorogenic probes have been used to detect various biomarkers such as H2O2 due to their high sensitivity and low background fluorescence. However, the complicated design and synthesis of fluorogenic probes limit the development of such excellent biochemical tools. Herein, we proposed a multicomponent one-pot reaction system using POCl3 as the core and by changing specific receptor groups, different phosphate ester-based fluorogenic probes could be assembled to detect various target analytes. Based on this platform, we synthesized a hydrogen peroxide (H2O2) probe UFPS-1 that is able to selectively and sensitively detect H2O2 with near-red emission and large Stokes shift. More importantly, UFPS-1 monitored H2O2 in a quantitative manner in both in vitro and in vivo PD models, suggesting that UFPS-1 could potentially be utilized for the diagnosis of PD.
1. Introduction
With the continuous development of life sciences and medicine, the exploration of various life metabolism activities has become more and more in-depth.1 In recent years, the intersection of synthetic chemistry and biological imaging has continuously promoted the collaborative development of high-quality optical instruments and fluorogenic probes, thus providing a powerful chemical detection tool for the study of basic aspects of cell physiology.2 Fluorescence probes exhibit several advantageous properties, such as robust response, high sensitivity, real-time and intuitive, visible detection,3 and are powerful tools for studying biochemical processes and diagnosing disease biomarkers.4A turn-on fluorogenic probe generally contains a receptor group and a quenched fluorescent group. The probe itself emits no or weak fluorescence and exhibits fluorescence enhancement only when the recognition group interacts with the specific analyte.5 Small-molecule fluorogenic probes have shown to be selective and sensitive to specific intracellular analytes and can be used to detect physiological and pathological processes in cells with high fidelity in space and time.6 In recent years, Xiong et al. designed an acetate-based NIR fluorogenic probe for the in situ detection of hydrogen peroxide in tumors.7 Huang et al. modified the NIR hemicyanine framework with a benzil moiety and designed a two-photon fluorogenic probe that can monitor hydrogen peroxide in an organism in real time.8 Fluorogenic probe imaging technology has developed into one of the most effective methods for real-time detection of life activities.
However, different types of probes for various target analytes have been designed and proposed in recent decades but in the face of a complex system of disease biomarkers, the design and synthesis of fluorogenic probes in a convenient, simple, and universal way has always been missing.
One-pot multicomponent reaction has been widely used in compound library constructions. For instance, phosphorus oxychloride (POCl3) as the core is able to produce a structurally diverse phosphate ester-based small molecule drug library.9 Compounds with nucleophilic groups (e.g., hydroxyl or amine groups) are able to rapidly replace the chloride group and form a tri-ester phosphate.10 Therefore, POCl3-based one-pot multicomponent reaction is versatile to various functional groups and can facilitate the generation of a structurally-diverse phosphate ester library. Notably, phosphate esters are the key components of important molecules, such as DNA, RNA, and ATP. In addition, upon activation, phosphate esters undergo hydrolysis rapidly. Therefore, phosphate esters are commonly used as the core in various types of fluorogenic probes, all of which possess high bioavailability, water solubility, and excellent reaction kinetics.11 Although one-pot multicomponent reactions are extremely useful in drug discovery,12 they have been rarely used in the synthesis of fluorogenic probe library.
Herein, using phosphate esterification reaction of POCl3 with a fluorophore and different receptor groups, we were able to utilize a one-step multicomponent reaction to efficiently provide the fluorogenic probes (Scheme 1) and we term this novel platform as a universal fluorogenic probe system (UFPS). As a proof-of-concept, we conjugated the borate ester group as the receptor group and pyrimidine as the fluorophore, and by using POCl3 one-pot reaction, we prepared our first “prototype” UFPS-1 for H2O2 detection (Scheme 1 and Fig. S1, ESI†). The UFPS-1 probe exhibits remarkable sensitivity and selectivity towards H2O2. We observed that H2O2, as a kind of reactive oxygen species (ROS), is closely related to many diseases such as Parkinson's disease (PD) in the process of oxidative stress.13 Therefore, we tried to apply UFPS-1 to PD animal models and successfully detected endogenous H2O2 in the in vitro and in vivo PD models. Moreover, the fluorescence dye of UFPS-1 shows a large Stokes shift at near-red emission and excellent turn-on fluorescent property. Therefore, we speculate that by changing the specific receptor groups, this UFPS platform will be able to robustly generate a phosphate ester-based fluorogenic probe library for detecting various biochemical processes and analytes.
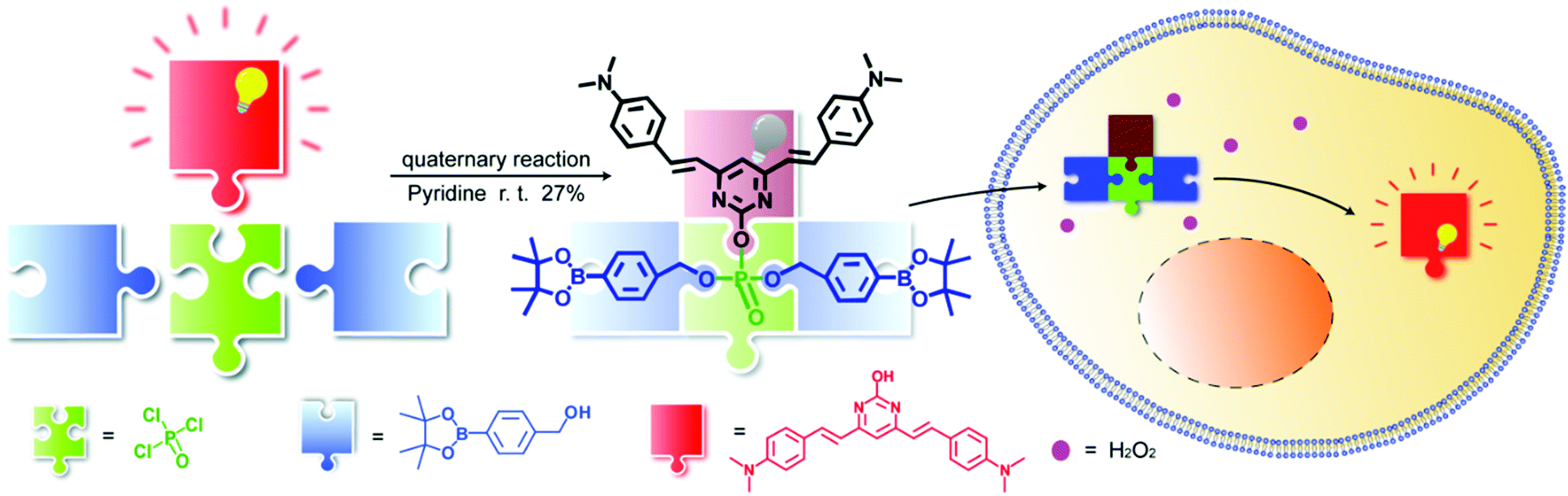 | ||
| Scheme 1 A multicomponent one-step reaction system with POCl3 as the core to synthesize UFPS-1. | ||
2. Experimental section
2.1. Materials and methods
All the chemicals were purchased from Aldrich or TCI based in China and used without further purification, unless otherwise stated. All the non-aqueous reactions were carried out at nitrogen atmosphere in oven-dried glassware. The reaction progress was monitored by thin layer chromatography (TLC) on pre-coated silica plates (250 μmol L−1 thickness, purchased from Qingdao Haiyang Chemical Co.) and the spots were visualized by UV light or iodine. Flash column chromatography (200–300 mesh) was carried out using silica gel bought from Qingdao Haiyang Chemical Co. The 1H NMR and 13C NMR spectra were acquired over a Bruker AV-500 spectrometer using CDCl3, CD3OD, CD3CN, or (CD3)2SO as the solvent. Chemical shifts were reported in parts per million referenced with respect to the residual solvent (CDCl3 = 7.26 ppm, CD3OD = 3.31 ppm, CD3CN = 1.94 ppm, and (CD3)2SO = 2.50 ppm for 1H NMR; CDCl3 = 77.16 ppm, CD3OD = 49.00 ppm, CD3CN = 1.32 ppm and (CD3)2SO = 39.52 ppm for 13C NMR). 1H NMR coupling constants (J) were reported in Hertz (Hz) and the multiplicity is indicated as follows: s (singlet), d (doublet), t (triplet), m (multiplet), and dd (doublet of doublets). High resolution mass spectra (HRMS) were obtained using an LTQ Orbitrap XL hybrid FTMS (Fourier Transform Mass Spectrometer) by Thermo Fisher Scientific. Fluorescence spectra were recorded using a HITACHI F4600 fluorescence spectrophotometer with excitation slit widths of 5 nm and emission slit widths of 10 nm. Analytical HPLC (Shimadzu LC-20AD) and mass spectra were recorded on a Shimadzu LC-ESI system equipped with a prominence RF-20A fluorescence detector, an SPD-M20A UV-Vis detector, and an autosampler, using reverse-phase Hypersil Gold 5 μm C18 100 Å 250 × 4.6 mm columns. 0.1% FA/H2O and 0.1% FA/acetonitrile were used as the eluents (flow rate was 1 mL min−1). All measurements were performed at room temperature. All the images were acquired on a Zeiss LSM880 NLO (2 + 1 with BIG) confocal microscope system equipped with objective LD C-Apochromat 63×/1.15 W Corr M27, cell incubator with temperature control resolution ±0.1 °C, 405 nm diode laser, argon ion laser (458, 488, and 514 nm), HeNe laser (543 and 594 nm), Rack LSM 880 including a 633 nm laser, and a spectra physics femtosecond Ti:sapphire laser (∼4 W at 800 nm), which corresponded to approximately 1% (∼40 mW at 800 nm, the output laser pulses have a tunable center wavelength in the range from 690 nm to 1040 nm with a pulse duration of 150 < fs and a repetition rate of 80 MHz) average power in the focal plane as the excitation source, with the main beam splitter wheel VIS equipped for ROGB lasers/Axio imager beam coupling optics for NLO and 405 nm laser and 8 channels AOTF for simultaneous control of 8 laser lines. Band-pass filters were used in one-photon imaging. Internal photomultiplier tubes were used to collect the signals in 8-bit unsigned 1024 × 1024 pixels at a scan speed of 200 Hz. The images were processed with a Zeiss User PC Advanced for the LSM system (BLUE).As for the cell imaging, the 543 nm laser was used with a 1AU pinhole to get higher resolution. The voltage was 18% and a band-pass filter for the 543 nm laser was used. As for Drosophila brain imaging, 543 nm wavelength was used, the voltage was 10%, and a band-pass filter for the 543 nm wavelength was used. Z-Stack model was used and the frame time was 0.63 s for one photo. All the photos have two channels, the fluorescence field and the bright field. We used the fluorescence channel for analysis.
2.2. Synthesis of probe UFPS-1
Compound PB-OH was synthesized according to the reported literature.14PB-OH (116 mg, 0.3 mmol) was dissolved in 10 mL pyridine and phosphorus oxychloride (85 μL, 0.9 mmol) was added dropwise, and the mixture was stirred for 2 h until the solution was clarified. The flask was then placed in a cold trap at −40 °C, and 4-(hydroxymethyl)phenylboronic acid pinacol ester (0.69 g, 3 mmol) was dissolved in pyridine with slow dropwise addition to the reaction system, and the mixture was stirred at room temperature for 3 h. The reaction system was placed at −20 °C, quenched by adding 2 mL of water, extracted with ethyl acetate, and the organic phase was evaporated. The residue was purified with silica gel column chromatography to afford UFPS-1 as a pale orange-yellow solid (72.7 mg, yield 27%), as shown in Fig. S1, ESI.†
1H NMR (500 MHz, CDCl3, ppm) δ = 7.82 (m, 6H), 7.45 (d, J = 8.75 Hz, 4H), 7.39 (d, J = 7.85 Hz, 4H), 6.98 (s, 1H), 6.80 (d, J = 15.8 Hz, 2H), 6.69 (d, J = 8.75 Hz, 4H), 5.39 (d, J = 7.25 Hz, 4H), 3.01 (s, 12H), 1.33 (s, 24H). 13C NMR (125 MHz, CDCl3, ppm) δ = 166.29, 151.25, 138.95, 138.88, 138.40, 134.98, 129.36, 126.80, 123.58, 120.14, 112.23, 112.02, 83.80, 70.02, 40.21, 24.87.
2.3. General procedure for fluorescence measurement
The stock solution of UFPS-1 (10 mM) was prepared in DMSO. The 2, 10, and 20 μM solutions of UFPS-1 were prepared in phosphate buffered saline (PBS) buffer (10 mM, pH = 7.4, 0.02% Triton X-100). Deionized water was used throughout the experiment. Various ROS and RNS species were prepared according to the reported literature.15 The solutions of anions and amino acid were prepared from the corresponding salts and the concentration was varied in the range from 10 to 100 μM.2.4. Quantum yield measurement
Quantum yield was determined using rhodamine B as a standard. Fluorescence quantum yield was determined by the following equation:wherein, Φ is the quantum yield, A is the absorbance at the excitation wavelength (A was kept at ≤0.05 during the fluorescence measurements to avoid self-quenching), F is the fluorescence intensity at the excitation wavelength, and η is the refractive index of the solvent. The subscripts ST and X refer to the standard and unknown, respectively. The Φ of rhodamine B is 0.65 in ethanol.16Φ of UFPS-1 was 0.007 and Φ of UFPS-1 with H2O2 was 0.037 (λex = 540 nm).
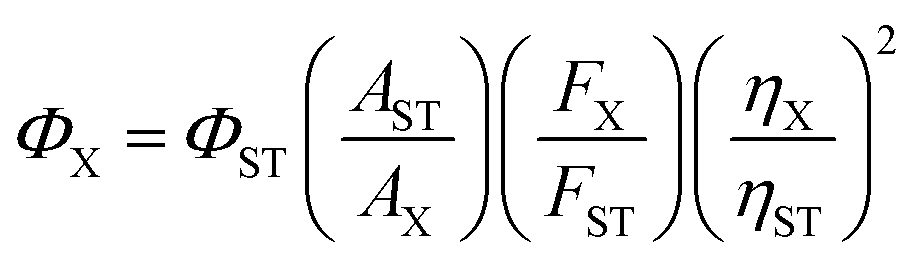
2.5. Absorption and fluorescence spectrum studies of UFPS-1
The absorption and emission spectra of UFPS-1 in PBS buffer solution (pH = 7.4, 10 mM, 0.2% v/v DMSO, and 0.02% v/v Triton X-100) were measured in the presence of H2O2. UFPS-1 (2 μM) was placed in the PBS buffer solution mixture and shaken on a shaker at 37 °C for 2 h, and then the absorption and emission spectra of UFPS-1 reacting with different concentrations of H2O2 were measured.2.6. Selectivity and ion interference
To measure the selectivity of UFPS-1 in the presence of common ions and amino acids in PBS buffer solution (pH = 7.4, 10 mM, 0.2% v/v DMSO, and 0.02% v/v Triton X-100) in the presence of H2O2, UFPS-1 (2 μM) was put into the PBS buffer solution mixture, then 43 analytes (200 μM) were added and shaken on a 37 °C shaker for 2 h, followed by measurement of the emission value of the reaction solution at 640 nm. λex = 540 nm.In order to measure whether common ions and amino acids interfere with the detection of H2O2 by UFPS-1, UFPS-1 (2 μM) was put in the PBS buffer solution mixture, 100 equiv. of H2O2 was added, 43 analytes (200 μM) were added and shaken at 37 °C for 2 h, followed by measurement of the emission value of the reaction solution at 640 nm. λex = 540 nm.
2.7. Cell culture and MTT assay
HepG2 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 100.0 mg mL−1 streptomycin, and 100 IU mL−1 penicillin. The cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C.The cytotoxicity of UFPS-1 was determined by using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) colorimetric cell proliferation kit (Roche) by following the manufacturer's guidelines. Briefly, HepG2 cells were grown to 70–80% confluence in 96-well plates. The medium was aspirated and then replaced with 100 μL of the medium containing different concentrations of UFPS-1 (0.1, 0.5, 1, 5, 10, 20, 30, 40, and 50 μM). UFPS-1 was dissolved in DMSO and the final concentration in DMSO was lowered. The same volume of DMSO was used as a negative control.
After 24 h incubation, 10 μL MTT solution (5 mg mL−1) was added to the cells. After further incubation at 37 °C for 4 h in a dark environment, the colored MTT-formazan crystals were produced. The solution was replaced by 100 μL DMSO to dissolve the crystals. After 10 min, the absorbance was measured at 560 nm by using a microplate reader.
2.8. Living cell imaging
HepG2 cells were seeded at a density of 5 × 105 cells per dish in a 35 mm confocal laser imaging dish and grown for 24 h. The cell cultures were incubated with the UFPS-1 probe at the concentration of 5 μM and maintained at 37 °C in an atmosphere of 5% CO2 for 3 h, followed by washing of the cells with PBS (3 × 2 mL per dish). For exogenous control experiments, the cells were incubated with 20 μM H2O2 for 3 h at 37 °C and then washed 3 times with PBS. The cells were further incubated with the 5 μM UFPS-1 probe for 3 h. For endogenous H2O2 experiments, the cells were incubated with 10 μM rotenone (ROS inducer17) in the culture medium for 3 h at 37 °C, and after washing 3 times with PBS solution, the cells were further incubated with the 5 μM UFPS-1 probe for 3 h. To verify that the fluorescence enhancement was attributed to intracellular H2O2, we applied N-acetylcysteine (NAC), a H2O2 scavenger, as the control.18All the cells were washed 3 times with PBS before imaging. Finally, the cells were imaged using a confocal laser scanning microscope (ZEISS LSM 880) and a single photon laser (63× water immersion objective) with 543 nm excitation and recorded emission at 570–700 nm.
2.9. Fluorescent imaging of H2O2 in the Drosophila brain
Parkin null and wild-type Drosophila were obtained from Neurodegeneration Research Laboratory (NDRL) of National Neuroscience Institute (NNI) of Singapore. All the Drosophila were grown on corn flour-molasses medium and maintained at 25 °C. We selected parkin null and wild-type Drosophila and divided them into the experimental group and the control group. In addition, we chose glutathione (GSH) as an inhibitor. Unlike the antioxidant mechanism of NAC, GSH is directly involved in the detoxification of H2O2 in the brain.19 After dissection, the experimental group of Drosophila brains were placed in UFPS-1 (25 μM) in PBS (pH 7.4), the control Drosophila brains were immersed in PBS buffer, and the inhibition group was placed in UFPS-1 (25 μM) and GSH (250 μM) in PBS buffer, and incubated for 5 h at 37 °C. Subsequently, each group of brain tissues was washed three times with PBS. The confocal laser scanning fluorescence images of the Drosophila brain were obtained under the excitation laser with 543 nm wavelength and recorded at 570–700 nm.3. Results and discussion
3.1. UFPS-1 design and synthesis
Pyrimidine derivatives are widely used as photoluminescence materials due to their excellent biocompatibility, photostability, and deep-red emission.20 Moreover, the fluorescence of pyrimidine-based dyes can be easily quenched by the intramolecular charge transfer (ICT) effect.14 Aryl borates are considered to be species with complementary amphiphilic reactivity on H2O2, which have been widely used in the detection of H2O2.21 We believe that an H2O2 probe with excellent performance can be obtained by assembling the two substances with POCl3. UFPS-1 was synthesized under multicomponent one-pot reaction, conjugating one molecule of pyrimidine and two molecules of borate ester. After the successful synthesis of UFPS-1, the structure of the product was characterized and confirmed by 1H NMR, 13C NMR, and HRMS (Fig. S11–S13, ESI†). When the borate ester group reacts with H2O2, the phosphate ester structure would be hydrolyzed to restore the fluorescence (Fig. S4 and S5, ESI†).22 The mechanism of UFPS-1 in H2O2 sensing was further confirmed by high performance liquid chromatography (HPLC). As shown in Fig. S6 (ESI†), at time 0, a single peak with a retention time of 23 min in the HPLC chromatogram was assigned to UFPS-1. Upon H2O2 treatment, the peak area of UFPS-1 decreased and a new peak for PB-OH with a retention time of about 3 min emerged. The HPLC results indicated that PB-OH was indeed the final product of the reaction between UFPS-1 and H2O2, which proved that our design (H2O2 can lead to the hydrolysis of phosphate group, and release PB-OH) is successful.3.2. Optical characterizations of probe UFPS-1
After getting UFPS-1, we performed the optical response test of H2O2. As shown in the Fig. 1a and Fig. S2 (ESI†), the main absorption peak of UFPS-1 appeared at 455 nm in PBS, and the absorption peak gradually shifted to 490 nm with the addition of H2O2. Subsequently, we studied the fluorescence response of UFPS-1 to H2O2. UFPS-1 itself only showed weak deep-red fluorescence at 640 nm when excited by light at 540 nm (Fig. 1b and Fig. S3, ESI†). Upon the addition of different concentrations of H2O2 (0 to 200 μM), the fluorescence at 640 nm reached the maximum intensity, and about 4-fold of fluorescence increment was observed within 2 h (Fig. 1b). The detection limit of the probe was calculated to be 92 nM (Fig. S7 and Table S1, ESI†), and the cellular concentration of H2O2 was reported as 10−8–10−4 M23 which is much higher than the detection limit of UFPS-1. It indicated that our probe should be able to detect cellular H2O2. Additionally, we also checked the reaction kinetics of UFPS-1 to H2O2 (Fig. 1c). The fluorescence intensity increased rapidly after the addition of H2O2 (0 to 200 equiv.) and saturated after 75 min (200 equiv.), which suggested a rapid response of UFPS-1 to H2O2.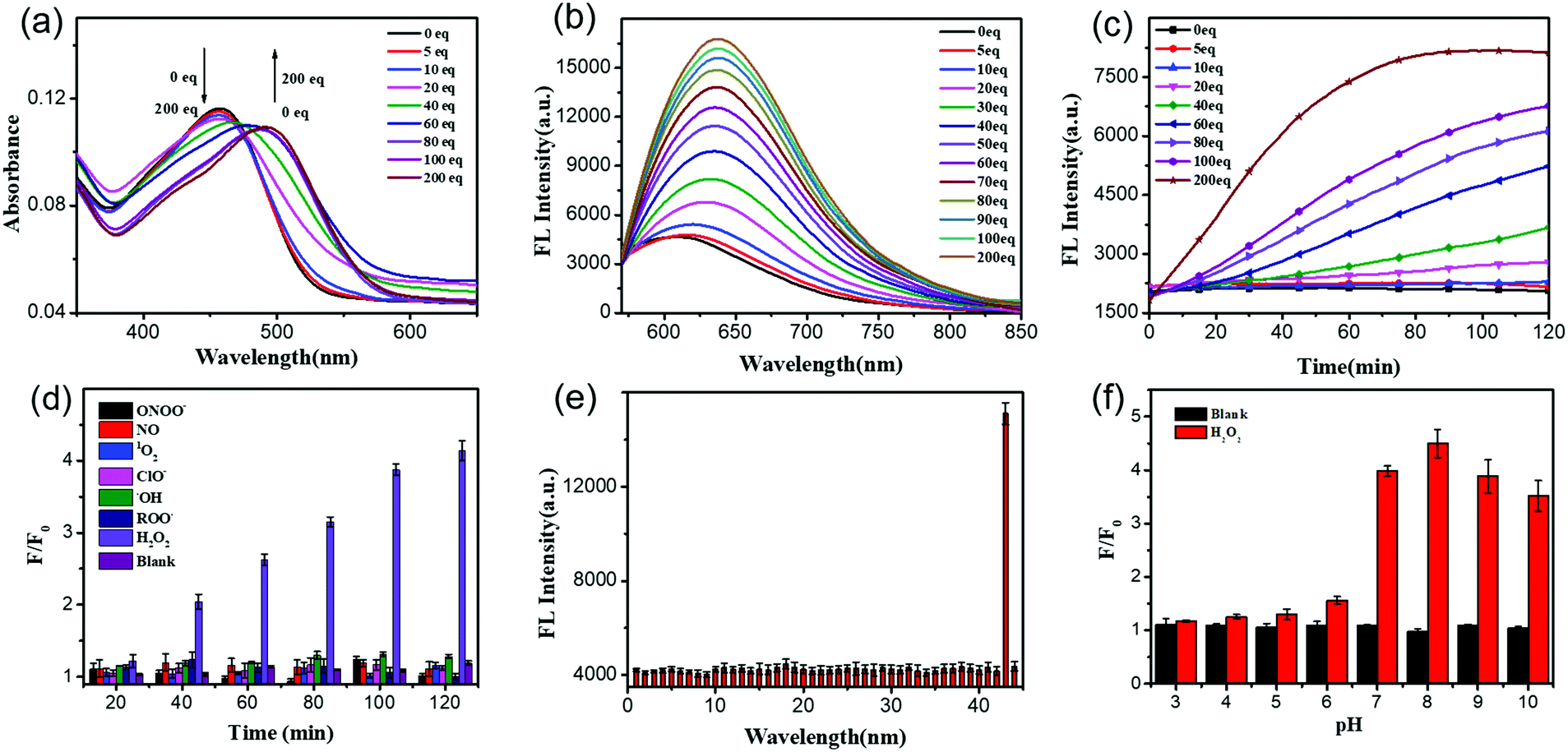 | ||
Fig. 1 (a) Absorbance and (b) emission spectra of UFPS-1 (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) μM) in PBS (0.2% v/v DMSO, 0.02% v/v Triton-X100) in the presence of H2O2 with different concentrations (0 to 200 equiv.) incubated for 2h; (c) fluorescence response of UFPS-1 (2 μM) to various H2O2 (0 to 200 equiv.) at different time points (0 to 120 min); (d) fluorescence response of UFPS-1 (2 μM) to various ROS and RNS species at the single concentration of 200 μM in 120 min; (e) fluorescence response of UFPS-1 (2 μM) to 43 analytes at the single concentration of 200 μM; (f) fluorescence response of UFPS-1 (2 μM) at the single concentration of H2O2 (200 μM) to various pH in 2 h. λex = 540 nm, λem = 640 nm. F/F0: the real time fluorescence intensity compared to the initial fluorescence intensity. μM) in PBS (0.2% v/v DMSO, 0.02% v/v Triton-X100) in the presence of H2O2 with different concentrations (0 to 200 equiv.) incubated for 2h; (c) fluorescence response of UFPS-1 (2 μM) to various H2O2 (0 to 200 equiv.) at different time points (0 to 120 min); (d) fluorescence response of UFPS-1 (2 μM) to various ROS and RNS species at the single concentration of 200 μM in 120 min; (e) fluorescence response of UFPS-1 (2 μM) to 43 analytes at the single concentration of 200 μM; (f) fluorescence response of UFPS-1 (2 μM) at the single concentration of H2O2 (200 μM) to various pH in 2 h. λex = 540 nm, λem = 640 nm. F/F0: the real time fluorescence intensity compared to the initial fluorescence intensity. | ||
An ideal fluorogenic probe should be able to selectively respond to the target analyte without interference by other substances. H2O2 is a product of cellular oxidative stress and belongs to ROS. Since some of the substances in the cells such as ROO˙ and ONOO− contain an O–O structure similar to H2O2, it is possible to interfere with the detection. In order to verify the selectivity of UFPS-1 toward H2O2, we selected six reactive oxygen/nitrogen species (ROS/RNS) substances (i.e., 1O2, ClO−, ˙OH, ROO˙, NO, and ONOO−) that were common in cellular metabolism. Impressively, these 6 most prominent potential interferents, at various concentrations, scarcely induced any fluorescence increment in UFPS-1. As shown in Fig. 1d, at the concentration of 200 μM for each analyte, only H2O2 exhibited significant fluorescence increment. In addition, 42 different ions and amino acids that are commonly found in physiological environments were tested as well. Similarly, no obvious fluorescence change had been observed upon the addition of these ions and amino acids to UFPS-1 (Fig. 1e and Fig. S8, ESI†). The above results demonstrated that UFPS-1 possessed excellent selectivity towards H2O2 among the potential interferents, ions, and amino acids. It could be applied for the specific detection of H2O2 even in complex biological environments.
For in vivo applications, excellent spectral response and stability over a specific pH range is the fundamental property of H2O2 probes. We examined the changes in the fluorescence intensities of UFPS-1 at 640 nm with or without the addition of H2O2 (200 μM) in the pH range of 3–10 (Fig. 1f). Without H2O2, UFPS-1 remained non-fluorescent at all the tested pH values. With the analyte, in the range of pH 7–10, the fluorescence of UFPS-1 was independent of the change in the pH, while it did not emit fluorescence in the pH range of 3–7. Despite the relatively poor activity in acidic conditions, the fluorescent intensity of UFPS-1 in the presence of H2O2 reached the maximum at pH 7–9, which was favorable for H2O2 detection in physiological environments.
3.3. Fluorescence imaging of UFPS-1 in living cells
Inspired by the excellent spectral properties of UFPS-1, especially the high selectivity and sensitivity towards H2O2, the potential application of UFPS-1 in the detection of cellular H2O2 was examined in the living cells. Firstly, the cytotoxicity of UFPS-1 in HepG2 cells was evaluated using the MTT assay. 94.1% of HepG2 cells remained viable after incubation with 50 μM UFPS-1 for 24 h (Fig. S9, ESI†), which indicated the relatively low cytotoxicity of UFPS-1 at the working concentrations. To confirm that UFPS-1 is also safe for normal somatic cells, we also performed the same test in LO2 cells. After incubating with 50 μM UFPS-1 for 24 h, more than 90% of the LO2 cells remained viable (Fig. S10, ESI†), which further proves that our probes are safe for living organisms.For fluorescent imaging, we first tested the turn-on ability of UFPS-1 using HepG2 cells, as shown in Fig. 2a(2); UFPS-1 successfully detected intracellular H2O2 under the normal conditions, which demonstrated the excellent cell permeability and outstanding sensitivity of UFPS-1 towards relatively low cellular concentrations of H2O2. We next evaluated the ability of UFPS-1 in detecting the change in the exogenous and endogenous level of H2O2 under different conditions. As expected, the addition of exogenous H2O2 (20 μM) significantly enhanced the fluorescent signal of UFPS-1 (Fig. 2a(3)). In order to detect the variation in the endogenous H2O2 concentrations, rotenone (Rot, 10 μM), an inhibitor of the oxidative phosphorylation chains in mitochondrial respiration, was administered to the HepG2 cells to stimulate the production of endogenous H2O2. With the stimulation of Rot, the fluorescence of UFPS-1 was enhanced as well (Fig. 2a(4)). It is noteworthy that the fluorescence of UFPS-1 was attenuated when NAC, the antioxidant, was applied (Fig. 2a(5)). Based on the above experimental results, we have demonstrated that UFPS-1 was able to detect exogenous and endogenous H2O2 in living cells.
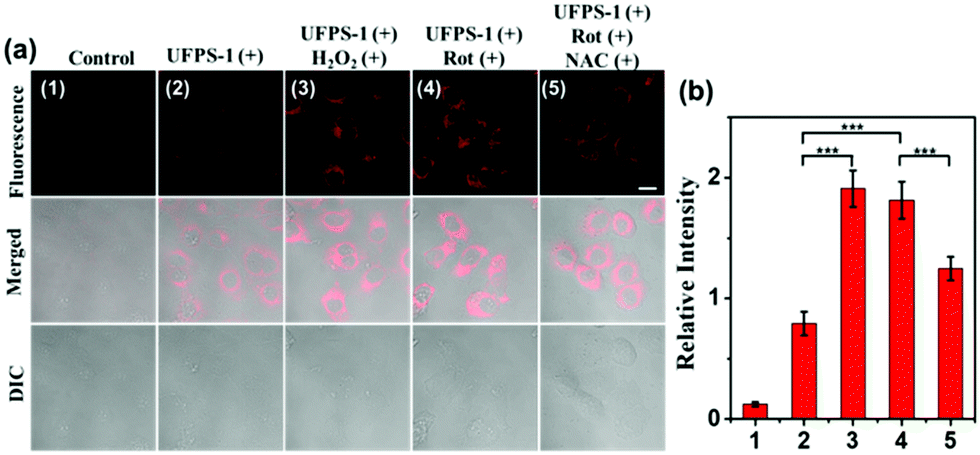 | ||
| Fig. 2 (a) Fluorescence imaging of the exogenous/endogenous H2O2 level in living HepG2 cells with UFPS-1. The first column: control. Second column: cells incubated with UFPS-1 (5μM) only for 3h. Third column: cells incubated with UFPS-1 (5μM) for 3h and then treated with H2O2 (20μM) for 3h. Fourth column was incubated with UFPS-1 (5μM) for 3h and then treated with Rot (10μM) for 3h. Fifth column: cells incubated with UFPS-1 (5μM) for 3 h and then treated with Rot (10μM) and NAC (10μM) for 3h. λex = 543 nm, λem = 570–700 nm. Scale bar = 10μm; (b) relative fluorescence intensities of (1–5). Statistical analyses were performed with the Student's t-test. ***P < 0.001. | ||
3.4. The application of UFPS-1 in PD Drosophila model
To prove that the deep red UFPS-1 probe holds the ability to detect the analyte in vivo, we selected the Drosophila PD model as an example. Drosophila shares 70% genome homology with that of humans. Parkin null Drosophila was developed by the gene deletion of endogenous parkin, which therefore displayed similar pathological features as that of the mammalian one, and were widely utilized as the PD model.24 In our previous work, we have confirmed the morphological changes in the mitochondria of Drosophila after gene knockout, which was well correlated to those of human PD patients.25 We applied UFPS-1 to the brain of parkin null Drosophila as well as the wide-type control. After incubation with UFPS-1, the brain of parkin null Drosophila showed enhanced fluorescence signal compared with that of wild-type control (Fig. 3a). To confirm whether the fluorescent signal was derived from H2O2, we applied GSH, which could directly neutralize H2O2 in the Drosophila brain. A significant decrease in the fluorescence intensity was observed in the GSH-treated PD group, indicating that the fluorescence signal indeed came from the turn-on reaction of UFPS-1 with H2O2 (Fig. 3a). Lastly, we quantified the fluorescence intensity derived from the Drosophila brain and the results showed that the fluorescence intensity of the PD Drosophila brain (39.76 (±0.77)) was more than twice of the intensity obtained for the wild-type Drosophila brain (18.26 (±0.21)) (Fig. 3b). The above data demonstrates the ability of UFPS-1 in the detection of H2O2in vivo.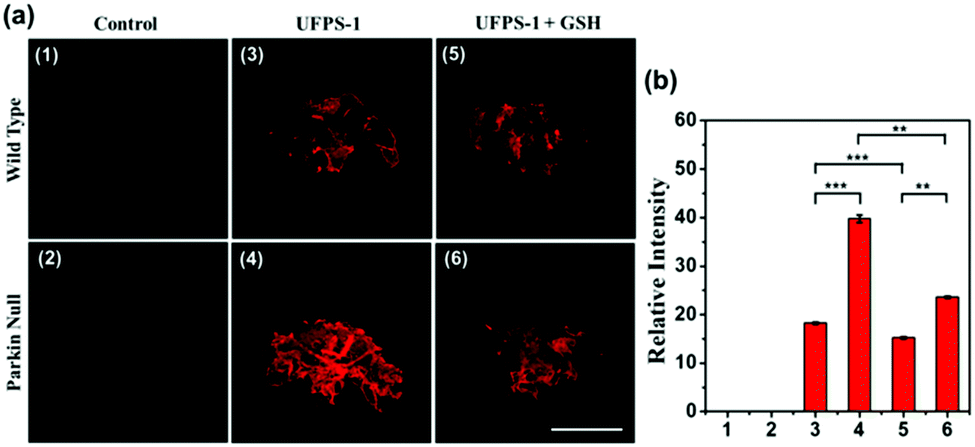 | ||
| Fig. 3 (a) Confocal fluorescence microscopy imaging of wild type (3) and parkin null (4) Drosophila brain with UFPS-1 (25 μM, 5 h) at room temperature. (5) Parkin null Drosophila brains incubated with UFPS-1 (25μM) and GSH (250 μM) for 5h. (1) and (2) are the corresponding control groups. λex = 543 nm, λem = 570–700 nm. Scale bar = 150 μm; (b) relative fluorescence intensities of (1–6). Statistical analyses were performed with the Student's t-test. ***P < 0.001, **P < 0.01, N = 5. | ||
4. Conclusion
In summary, we proposed a UFPS platform to synthesize a phosphate ester fluorogenic probe by multicomponent one-pot reaction with POCl3 as the core. Based on this, we designed and synthesized a fluorogenic turn-on probe UFPS-1, which displayed excellent selectivity to H2O2 among various potential interferents with near-red emission and a large Stokes shift. It was also able to quantitatively detect both endogenous and exogenous H2O2 in the in vitro and in vivo PD models. As a common and important ROS species, H2O2 is closely related to the pathogenesis of a series of diseases such as cancer, cardiovascular disease, and neurodegenerative diseases. Our work provides an effective strategy for the real-time monitoring of H2O2 in living organisms. It holds great potential in the early diagnosis and pathological mechanism study of the diseases mentioned above.More importantly, the excellent performance of UFPS-1 and the robust one-pot synthesis indicate the great potential of our UFPS platform. Utilizing this method to design a universal “toolbox” for fluorogenic probe is indeed intriguing and versatile. It is worthy to explore different receptor/recognition pairs that can be easily tuned into specific fluorogenic probes for the corresponding target biomolecules. Therefore, this platform holds great potential and will deliver a profound impact on chemical probe development and disease diagnosis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81672508), Jiangsu Provincial Foundation for Distinguished Young Scholars (BK20170041), Natural Science Basic Research Program of Shaanxi (Program No. 2019JM-016), Key Research and Development Program of Shaanxi (2020ZDLGY13-04), Open Research Fund of Anhui Key Laboratory of Tobacco Chemistry (20181140), China-Sweden Joint Mobility Project (51811530018), and Fundamental Research Funds for the Central Universities.Notes and references
- (a) K. D. Davis, N. Aghaeepour and A. H. Ahn, et al. , Nat. Rev. Neurol., 2020, 16, 381–400 CrossRef PubMed; (b) S. P. Haen, M. W. Löffler and H. Rammensee, et al. , Nat. Rev. Clin. Oncol., 2020 DOI:10.1038/s41571-020-0387-x.
- D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- D. Chen, W. J. Qin and H. X. Fang, et al. , Chin. Chem. Lett., 2019, 30, 1738–1744 CrossRef CAS.
- H. Cheng, Y. Li and B. Z. Tang, et al. , Chem. Soc. Rev., 2020, 49, 21–31 RSC.
- (a) M. E. Jun, B. Roy and K. H. Ahn, Chem. Commun., 2011, 47, 7583–7601 RSC; (b) X. Li, X. Gao and W. Shi, Chem. Rev., 2013, 114, 590–659 CrossRef PubMed.
- (a) H. X. Fang, H. Zhang and L. Li, et al. , Angew. Chem., Int. Ed., 2020, 59, 7536–7541 CrossRef CAS PubMed; (b) Q. Sun, J. J. Xu and C. L. Ji, et al. , Anal. Chem., 2020, 92, 4038–4045 CrossRef CAS PubMed; (c) Z. J. Fang, Z. Su and W. J. Qin, et al. , Chin. Chem. Lett., 2020 DOI:10.1016/j.cclet.2020.03.063.
- J. Xiong, L. Xia and L. Li, et al. , Sens. Actuators, B, 2019, 288, 127–132 CrossRef CAS.
- X. Huang, Z. Li and Z. Liu, et al. , Dyes Pigm., 2019, 165, 518–523 CrossRef CAS.
- (a) U. Pradere, E. C. Garnier-Amblard and S. J. Coats, et al. , Chem. Rev., 2014, 114, 9154–9218 CrossRef CAS PubMed; (b) J. L. Wanga, Y. X. Wang and L. Xu, et al. , Carbohydr. Polym., 2018, 181, 19–26 CrossRef PubMed.
- B. G. Harvey, A. C. Chafin and M. D. Garrison, et al. , RSC Adv., 2015, 5, 74712 RSC.
- (a) P. Zhang, C. Fu and Q. Zhang, et al. , Anal. Chem., 2019, 91, 12377–12383 CrossRef CAS PubMed; (b) Y. Li, H. Wang and J. Li, et al. , Anal. Chem., 2011, 83, 1268–1274 CrossRef CAS PubMed.
- (a) R. O. Rocha, M. O. Rodrigues and B. A. Neto, ACS Omega, 2020, 5, 972–979 CrossRef CAS PubMed; (b) J. Y. Ge, X. M. Cheng and L. P. Tanb, et al. , Chem. Commun., 2012, 48, 4453–4455 RSC.
- (a) V. K. Lorraine and E. L. Anthony, Lancet, 2015, 386, 896–912 CrossRef; (b) T. Wu and M. Hallett, Brain, 2013, 136, 696–709 CrossRef PubMed; (c) S. J. Dixon and B. R. Stockwell, Nat. Chem. Biol., 2014, 10, 9–17 CrossRef CAS PubMed; (d) B. R. Stockwell, J. P. F. Angeli and H. Bayir, Cell, 2017, 171, 273–285 CrossRef CAS PubMed; (e) Y. Xie, W. Hou and X. Song, Cell Death Differ., 2016, 23, 369–376 CrossRef CAS PubMed.
- X. H. Qiu, C. Q. Xin and W. J. Qin, et al. , Talanta, 2019, 199, 628–633 CrossRef CAS PubMed.
- X. Li, R. R. Tao and L. J. Hong, J. Am. Chem. Soc., 2015, 137, 12296–12303 CrossRef CAS PubMed.
- R. F. Kubin and A. N. Fletcher, J. Lumin., 1982, 27, 455–462 CrossRef.
- (a) D. S. Higgins and J. T. Greenamyre, J. Neurosci., 1996, 16, 3807–3816 CrossRef CAS PubMed; (b) B. Chance, G. R. Williams and G. Hollunger, J. Biol. Chem., 1963, 238, 418–431 CAS; (c) T. B. Sherer, R. Betarbet and C. M. Testa, et al. , J. Neurosci., 2003, 23, 10756–10764 CrossRef CAS PubMed; (d) P. Caboni, T. B. Sherer and N. Zhang, Chem. Res. Toxicol., 2004, 17, 1540–1548 Search PubMed.
- A. Rahimmi, F. Khosrobakhsh and E. Izadpanah, Brain Res. Bull., 2015, 113, 34–40 CrossRef CAS PubMed.
- R. Dringen, Prog. Neurobiol., 2000, 62, 649–671 CrossRef CAS PubMed.
- Z. Liu, P. Shao and Z. Huang, et al. , Chem. Commun., 2008, 2260–2262 RSC.
- (a) A. R. Lippert, G. C. Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed; (b) R. Weinstain, E. N. Savariar and C. N. Felsen, J. Am. Chem. Soc., 2014, 136, 874–877 CrossRef CAS PubMed; (c) A. R. Lippert, K. R. Keshari and J. Kurhanewicz, J. Am. Chem. Soc., 2011, 133, 3776–3779 CrossRef CAS PubMed; (d) D. Srikun, A. E. Albers and C. I. Nam, J. Am. Chem. Soc., 2010, 132, 4455–4465 CrossRef CAS PubMed.
- (a) J. Kumamoto and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2515–2518 CrossRef CAS; (b) W. W. Butcher and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2420–2424 CrossRef CAS.
- M. Giorgio, M. Trinei and E. Migliaccio, Nat. Rev. Mol. Cell Biol., 2007, 8, 722–728 CrossRef CAS PubMed.
- (a) Y. Liu, L. Bai and Y. H. Li, et al. , Sens. Actuators, B, 2019, 279, 38–43 CrossRef CAS; (b) L. Li, C. W. Zhang and G. Y. Chen, et al. , Nat. Commun., 2014, 5, 3276 CrossRef PubMed; (c) H. Bolus, K. Crocker and G. Boekhoff-Falk, et al. , Int. J. Mol. Sci., 2020, 21, 3055 CrossRef PubMed; (d) E. Ziviani, R. N. Tao and A. J. Whitworth, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5018–5023 CrossRef CAS PubMed.
- H. Li, C. Q. Xin and G. B. Zhang, et al. , J. Mater. Chem. B, 2019, 7, 4227–4372 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The synthetic procedure and 1H NMR, 13C NMR, HRMS for UFPS-1, experimental details, Fig. S1–S13. See DOI: 10.1039/d0ma00318b |
| ‡ Authors are equally contributed. |
| This journal is © The Royal Society of Chemistry 2020 |