DOI:
10.1039/D0MA00382D
(Paper)
Mater. Adv., 2020,
1, 1858-1865
Asymmetric-donor (D2D2′)–acceptor (A) conjugates for simultaneously accessing intrinsic blue-RTP and blue-TADF†
Received
5th June 2020
, Accepted 16th July 2020
First published on 21st July 2020
Abstract
The development of new photoluminescent (PL) materials with simultaneous room-temperature phosphorescence (RTP) and thermally activated delayed fluorescence (TADF) features is highly desirable for bio-imaging, security applications and sensors due to the involvement of both singlet and longer-lived triplet states. Here, we report two carbazolyl-phenoxy-phthalonitrile conjugates (CPPN, CPPNF). Spectroscopic studies combining two daughter compounds (PPN, PPNF) in polar and nonpolar hosts confirmed efficient blue-RTP from the higher-energy triplet state (TPPN) due to the phenoxy-phthalonitrile (PPN) part, and blue-TADF via reverse intersystem crossing from the low-lying triplet state (TCzPN) of the carbazolyl-phthalonitrile (CzPN) part to the singlet (S1) state of the same CzPN part, utilizing the TPPN state that acts as an intermediate for spin–vibronic coupling. Such PL characteristics are observed due to the energetic proximity of 3LEPPN, 1CTCzPN and 3CTCzPN. In the hydrogen-bonded matrix and crystals, we found faint persistent green-RTP characteristics of the PPNF due to supramolecular interactions and aggregation of the molecule. This study could pave the way to understanding the involvement of different excited states associated with TADF and RTP processes of asymmetric-donor–acceptor systems.
1. Introduction
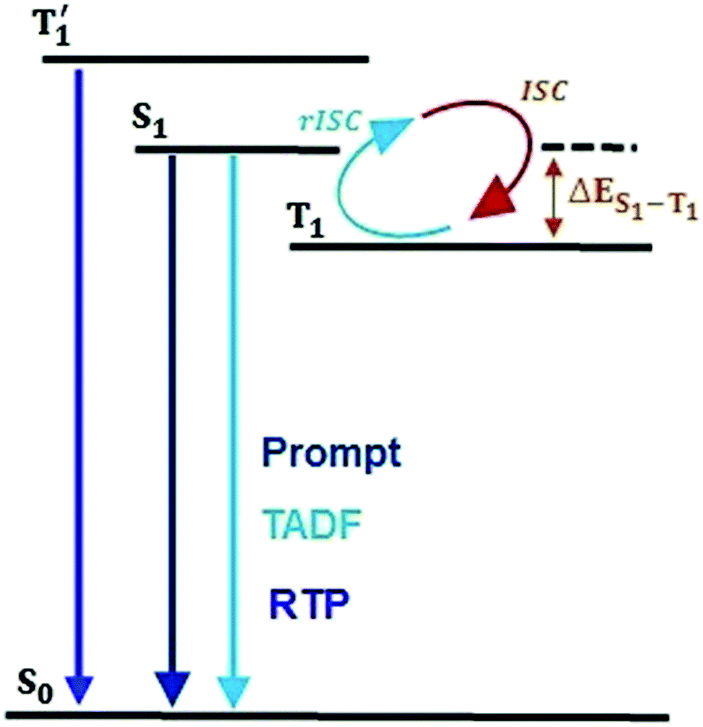
Single-component organic luminescent materials (SCOLMs) that show emission features with high photoluminescence quantum yield (PLQY) utilizing both singlet and triplet excited states have attracted increasing attention for their potential use in organic chemical sensing,1,2 and bio-imaging3 (Fig. 1). Efficient emission via fluorescence4 can occur from the singlet excited states where the relaxation conserves spin. On the other hand, phosphorescence from the lowest triplet (T1) state is typically very weak because it involves a spin flip, which is quantum mechanically forbidden.5 Therefore, a search for emitters that show simultaneous radiative decay of both singlet and triplet states and high PLQY are of particular interest.
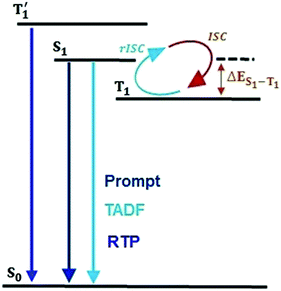 |
| | Fig. 1 Jablonski diagram for emission via RTP and TADF in the proposed molecular design. | |
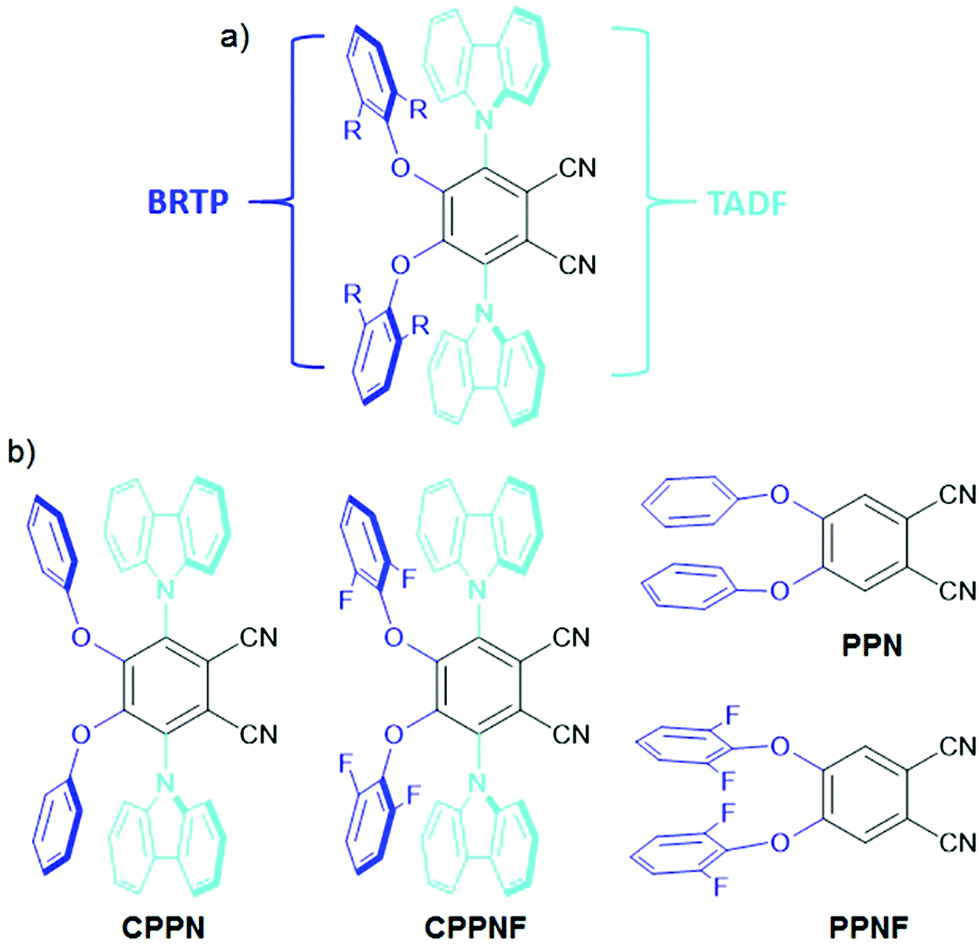
Recently, organic donor–acceptor (D–A) based emitters with a thermally activated delayed fluorescence (TADF)6–11 feature that occurs via reverse intersystem crossing (rISC) from either the higher lying triplet state (Tn, n > 1)6–8 or the lowest triplet (T1) state to the lowest singlet (S1) state,9–11 have showed exciting capabilities for energy-efficient light emission by harvesting of both singlet and triplet states. To harness the triplet exciton via TADF, numerous research studies have been undertaken, which have established that apart from the low S1–T1 gap between the singlet and triplet states, the nonadiabatic effect between the low-lying excited states along with mixing of the intermediate excited states plays a crucial role in the process of ISC and rISC.12–17 On the other hand, the efficient room-temperature phosphorescence (RTP) from the triplet state of heavy-metal based systems has been widely investigated.18–22 These systems show up to 100% internal electroluminescence efficiency due to the use of noble metals allowing strong spin–orbit coupling (SOC).18 Despite their high efficiency electroluminescence, these systems are both expensive and toxic. An alternative approach to substitute such systems with cheaper organic materials which can be easily processed and functionalized using a wide variety of substituents, is therefore desirable. Recently, purely organic RTP materials23–29 have boosted tremendous interest in data security protection30 and invisible ink,31 and photodynamic therapy.29 However, these systems show weak RTP due to inefficient ISC between the S1 and T1 states caused by the low SOC of the lighter elements present in the dye.32–35 In order to enhance the ISC rate, heteroatoms (N, O, S) and halogens are introduced as they can provide radiationless ISC transition between the orbitals of different symmetries i.e.1(π–π*) to 3(n–π*) and vice versa (El-Syed rule).34 However, the RTP efficiency of such molecular systems is very low due to the slow rate of ISC that causes a high lifetime of the triplet states. In addition, the molecular vibrations that readily increase the non-radiative pathways of the localized triplet excitons become another major bottleneck to harvest efficient phosphorescence yield.36 To circumvent the above issues, crystal formation,37,38 aggregation,39,40 host–guest interactions41 and deuteration42 have been studied to reduce the nonradiative pathways under ambient conditions. To observe both blue-TADF (BTADF) and blue-RTP (BRTP) simultaneously with high PLQY from the SCOLMs under ambient conditions becomes a difficult task in the field of chemical physics. To harvest triplet excitons simultaneously from a single molecule, fine-tuning of the singlet–triplet energy gap is required for both TADF and RTP, while intra- and/or intermolecular interactions (lp⋯π, π⋯π, hydrogen bonding)27,30 and heavy elements are the prerequisites for RTP.19 Recently, several design principles have been used to observe both TADF and RTP. Huang et al. synthesized angular and linear isomers, and tuned the energy gaps using the host materials to harvest triplet excitons through TADF and RTP.43 Ward et al. showed TADF and RTP separately through chemical modification.44 Additionally, mechanical force,45,46 structural modification,47 conformational switching48 and change in host matrices49 have successfully been used to demonstrate both TADF and RTP. Despite these achievements, the major issue is the lack of a proper structural design and understanding of the origin of simultaneous BRTP and BTADF in a single molecule under ambient conditions. In addition, to observe such emission features with persistent-RTP (PRTP), the materials need to show longer-lived triplet emissions under ambient conditions in host matrices. Due to these bottlenecks, the mechanism of simultaneously observing BRTP and BTADF along with PRTP in the SCOLMs under ambient conditions remains murky. Hence, the development of such SCOLMs is of prime importance. Taking a cue from our previous reports of D4–A molecular systems30,31 where the phenoxy or substituted phenoxy donors are covalently attached to either the phthalonitrile (PN) or the terephthalonitrile ring, here we attached both phenoxy or 2,5-difluorophenoxy and carbazole donors at the 4,5 and 3,6-positions of the PN core respectively, to get mixed donor–acceptor (D2D2′–A) geometries (CPPN, CPPNF) (Fig. 2). We hypothesized that incorporation of asymmetric donors in the PN core would play a crucial role in observing both BRTP and BTADF due to the involvement of the TPPN state caused by the phenoxy-phthalonitrile (PPN) part and upconversion of the triplet state (TCzPN) to the singlet state (SCzPN) caused by the traditional carbazolyl-phthalonitrile (CzPN) part. The possible advantage of such a system with asymmetric donors motivated us to access the role of the intermediate state in the RTP and TADF processes. Furthermore, 2,5-difluorophenoxy donors are selected because they may favor both SOC via lp(O/F)⋯π interactions due to the presence of multiple O and F atoms in the molecular backbone and hydrogen-bonding (H-B) interactions with the host to facilitate faint persistent-RTP. Our studies revealed that both compounds (0.1% poly(methyl methacrylate), PMMA) under ambient conditions show both-BRTP (CPPN: phosphorescence quantum yield, ΦP = 3%; CPPNF: ΦP = 3.5%) and BTADF (CPPN: fluorescence quantum yield, ΦF = 43%; CPPNF: ΦF = 44%) due to the radiative decay of the upper triplet (TPPN) state (higher-energy) and rISC from the lower-lying triplet (TCzPN) state to the singlet (SCzPN) state. In contrast to PMMA, bis[2-(di-(phenyl)phosphino)-phenyl]ether oxide (DPEPO) films (0.1%) of both compounds showed an increase in ΦP (∼10%) while a decrease in ΦF (∼10%) was observed. Moreover, photoluminescence (PL) studies of CPPNF in both polyvinyl alcohol (PVA) matrix and crystals revealed faint-green-persistent RTP characteristics under ambient conditions.
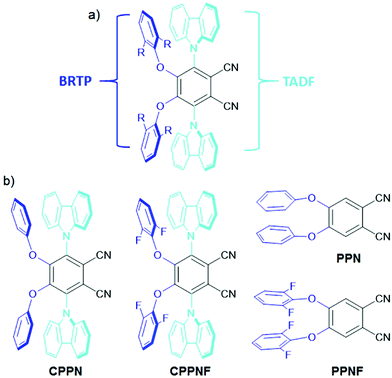 |
| | Fig. 2 (a) Design strategy for creating materials with simultaneous blue-RTP and blue-TADF. (b) Structure of the molecules (CPPN and CPPNF) including the two control compounds (PPN and PPNF) synthesized in this study. | |
Considering two control compounds we speculate that the BRTP originates due to the PPN part of the molecule while BTADF occurs due to the vibronic coupling between the lowest S1(CT) (CzPN) and T1(CT) (CzPN) states via the upper TPPN (3LE) state. The photoluminescence studies of CPPNF in the hydrogen-bond (H-B) matrix further show that it exhibits PRTP with lifetimes of 39.51 ms and 269.21 ms.
2. Experimental
2.1. Synthesis and characterization
The compounds were synthesized via the traditional nucleophilic aromatic substitution reaction (SNR) between the phenol derivatives and 3,4,5,6-tetrafluorophthalonitrile.30,31 The isolated intermediates were reacted separately with the sodium salt of carbazole to obtain the final compounds (CPPN, CPPNF) (Fig. 2). Control compounds (PPN, PPNF) were synthesized following a similar method. All the compounds were characterized using NMR spectroscopy, HRMS, and X-ray analysis (see the ESI†).
2.2. Photophysical measurements
Absorption measurements were performed using a Cary 8454 UV-vis instrument from Agilent Technologies. Steady-state emission, phosphorescence, and time resolved analysis of the samples were recorded on Horiba Fluorolog. All the phosphorescence spectra were recorded by giving the detector a delay of 0.05 ms. The measurement details can be found in the reported literature.8,32,33 Temperature-dependent PL measurements were performed using a liquid N2 Dewar assembly. All the energy levels have been calculated from the onset of the emission spectra.
3. Results and discussion
3.1. X-ray crystallography
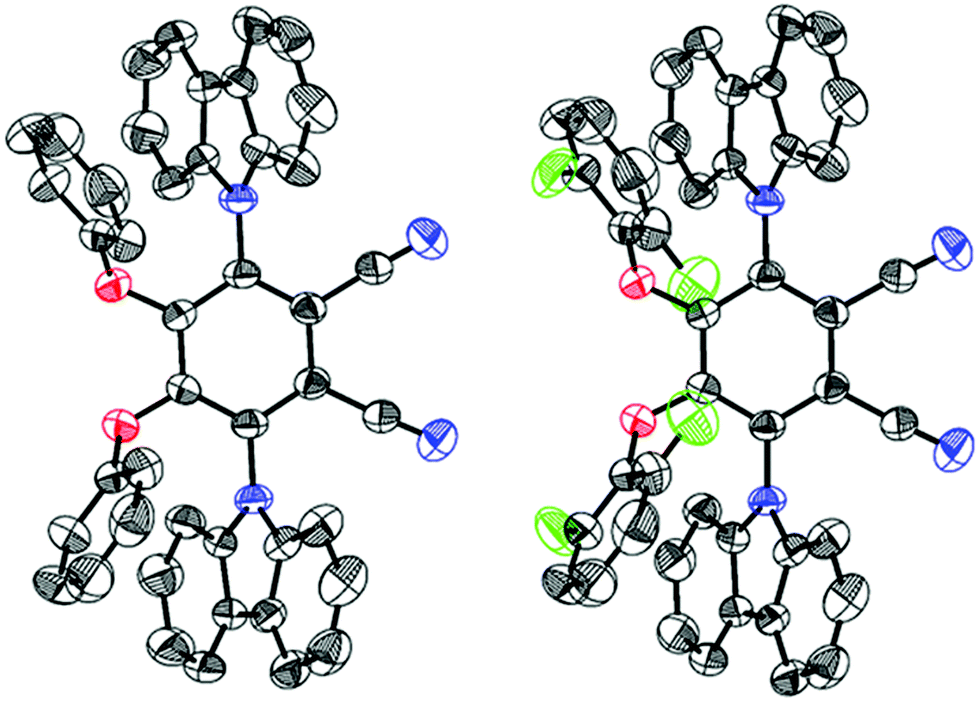
X-ray analysis (Fig. 3) of both compounds revealed that two adjacent phenoxy donors are covalently attached to the 4,5-positions of the PN core with torsions of −54.51° (CPPN) and −50.39° (CPPNF) when viewed along the C(2)–C(3)–O(1)–C(17) atoms while torsions of 120.83° (CPPN) and 120.48° (CPPNF) viewed along the atoms C(1)–C(2)–N(2)–C(5) are measured when the 3,6-positions of the PN are substituted with the carbazolyl rings. Interestingly, the two phenyl rings in both CPPN and CPPNF are in face-to-face orientation with the carbazolyl donors (centroid-to-centroid; CPPN, 3.608 Å; CPPNF, 3.600 Å). Furthermore, strong intramolecular lp(F)⋯π (centroid) interactions (3.270 Å) are also observed. Thus, X-ray analysis unambiguously confirms our structural model.
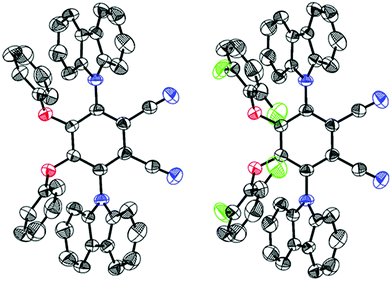 |
| | Fig. 3 Oak Ridge thermal ellipsoid plots (50% probability ellipsoids) of CPPN (left) and CPPNF (right). The protons are removed for the sake of clarity. | |
3.2. Absorption and emission characteristics in solutions
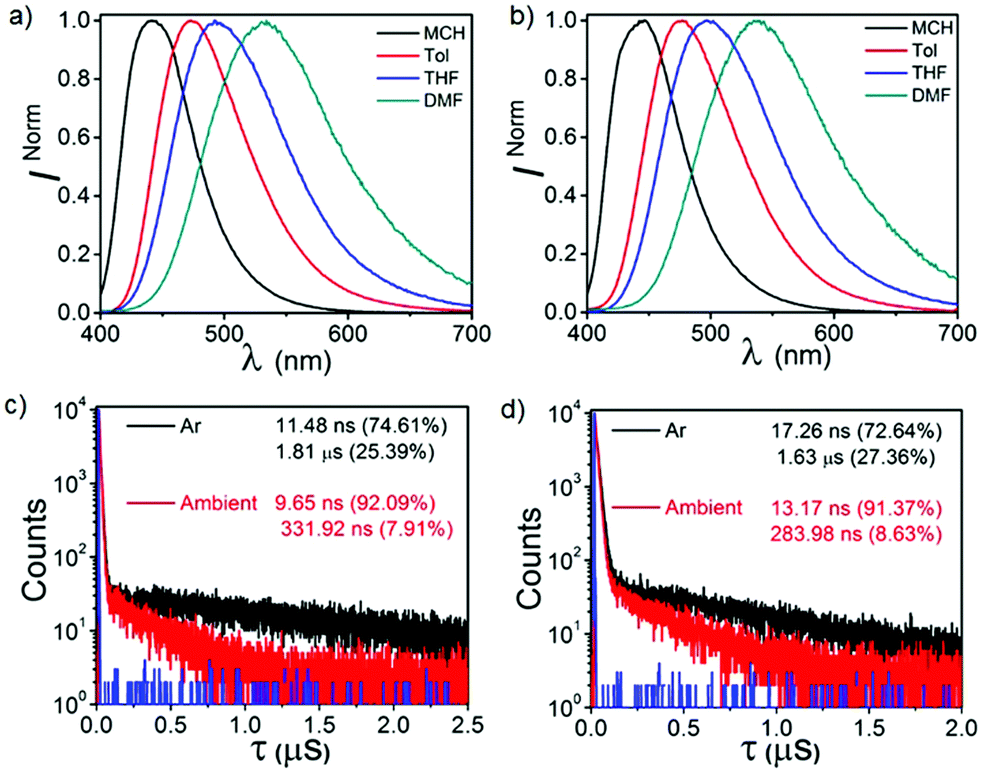
Fig. S5 (ESI†) shows the normalized ultraviolet-visible spectra of both CPPN and CPPNF in the solvents of disparate polarity (10 μM). Both compounds exhibit higher energy absorption bands at 285 nm, 315 nm and 325 nm which can be attributed to the π–π* transition. In addition, a broad tail (340–425 nm), which can be assigned to charge transfer (CT), is accompanied by the former bands. Likewise, steady-state emissions of both compounds were recorded in the solvents of disparate polarity (λex = 350 nm, 10 μM) (Fig. 4 and Fig. S6, ESI†). Emission spectra of both compounds show a gradual batho-chromic shift of the emission maxima along with reduced intensity of the broad emission band with increasing solvent polarity. Such emission features confirm that the excited state is CT in nature. Furthermore, substantial enhancement of emission intensity as well as lifetimes recorded in deoxygenated toluene solutions suggest an involvement of the triplet state that relaxes radiatively from the same singlet excited state (Fig. 4 and Fig. S7, ESI†). Moreover, the biexponential decay of both compounds (CPPN: λem = 473 nm, τPF = 9.65 ns, τDF = 331.92 ns; CPPNF: λem = 477 nm, τPF = 17.26 ns, τDF = 1.63 μs) under ambient conditions unambiguously confirms the TADF feature.
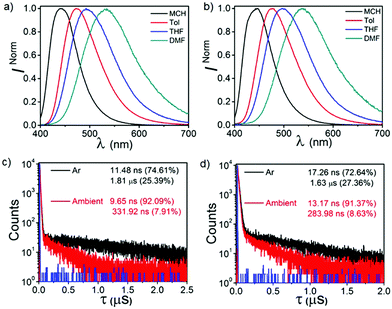 |
| | Fig. 4 Solvatochromic emissions of (a) CPPN and (b) CPPNF under ambient conditions. Lifetime analysis of (c) CPPN and (d) CPPNF in toluene under ambient and deoxygenated conditions. | |
3.3. Emission characteristics in PMMA films
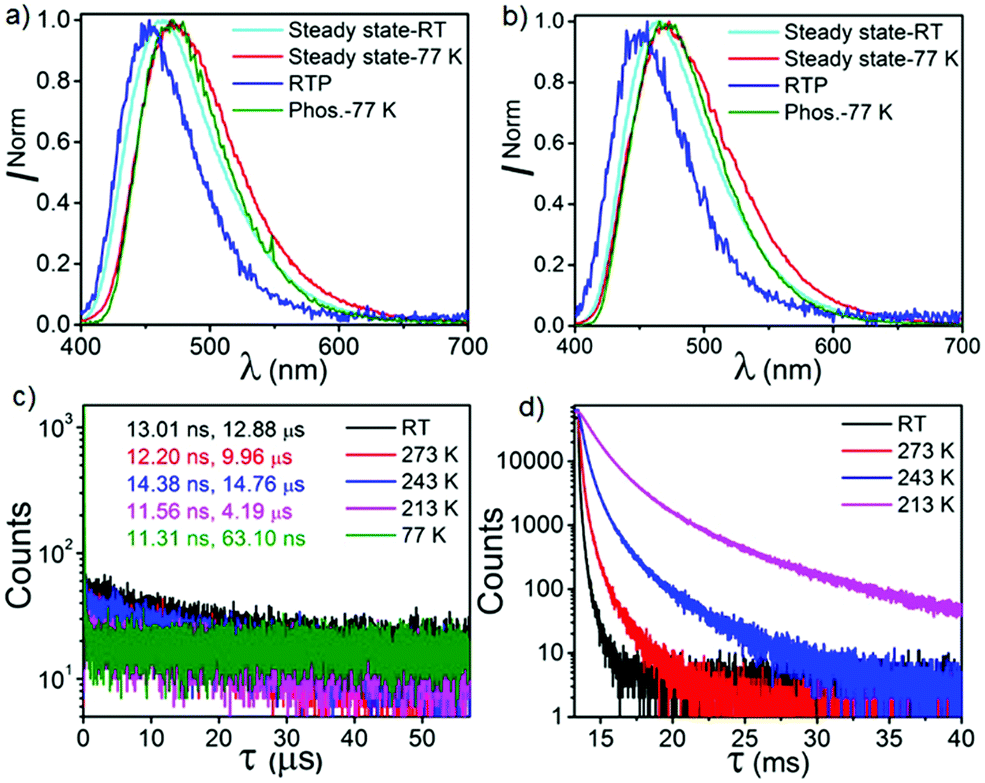
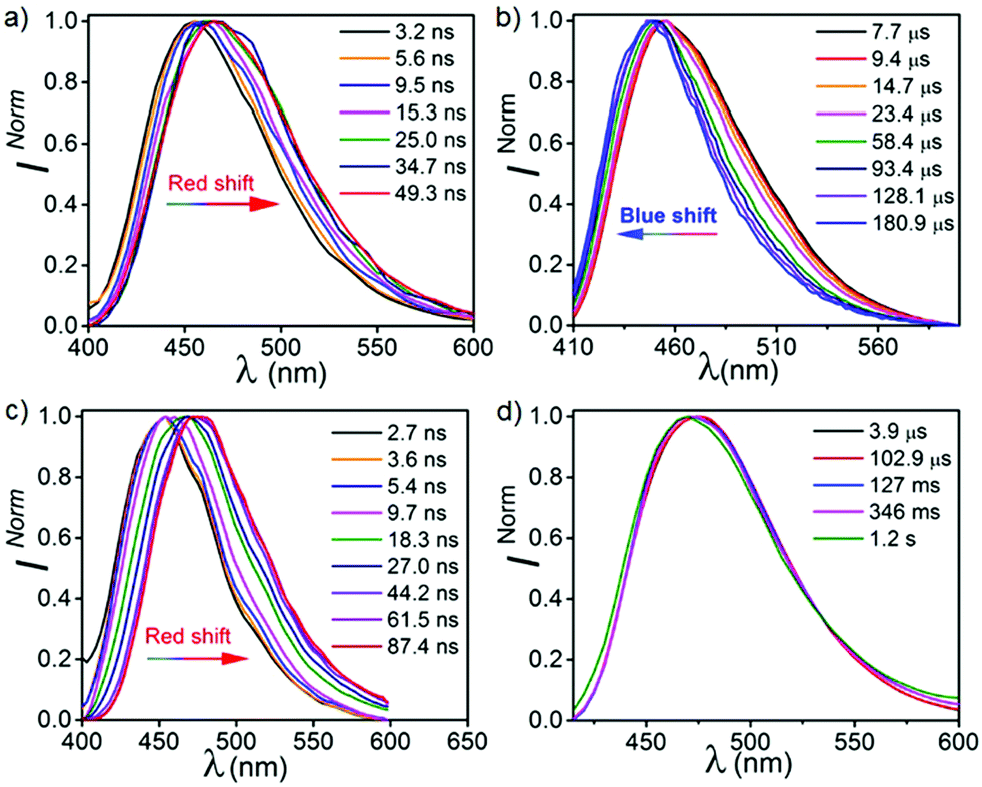
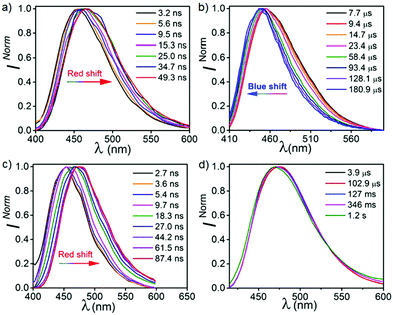
To elaborate our understanding of the emission characteristics in rigid medium, photophysical studies were performed in the poly(methyl methacrylate) (PMMA) matrix (0.1% (w/w)) (λex = 350 nm). It should be noted that the energy of all the emission peaks were calculated taking the on-set of the spectra. Both CPPN and CPPNF show broad emission bands at 463 nm in PMMA films with photoluminescence quantum yields (PLQY) of 47.5% and 45.3%, respectively (Fig. 5). We found that emission intensity of the λ463 significantly increases under argon environment at RT, indicating a triplet state involvement for the emission (Fig. S8, ESI†). The emission peak at 463 nm for both cases is assigned as typical TADF, which was confirmed by the temperature-dependent lifetime measurements (Fig. 5, Fig. S9 and Table S1, ESI†). Furthermore, time resolved emission spectra (TRES) of both compounds were collected from 3.2 ns to 49.3 ns at RT. The emission spectra continuously red shifts from 450 nm (onset, 3.10 eV) to 465 nm (onset, 3.03 eV) in both cases (Fig. 6) due to the relaxation of the excited states, while no further red shift was observed in 34 ns onward, which indicates that no other lower energy emissive states are present at RT (Fig. S10, ESI†). The phosphorescence measurements were performed with a high detector delay (0.05 ms) to remove the DF component. Surprisingly, phosphorescence measurements of both compounds under ambient conditions exhibit blue shifted narrow RTP bands at 455 nm (CPPN: 3.06 eV, τ = 58.49 μs, 175.06 μs, 619.3 μs, ΦP = 3.26%) and 452 nm (CPPNF: 3.05 eV, τ = 148.66 μs, 570.09 μs, 44.58 μs, ΦP = 2.24%) with a FWHM of ∼67 nm (Fig. 5 and Table 1) as compared to the FWHM of 80 nm (CPPN) and 78 nm (CPPNF) for delayed fluorescence, indicating different natures of excited states. Moreover, temperature-dependent lifetime measurements show a gradual enhancement of phosphorescence lifetimes50 (τp) with lowering of temperature, indicating BRTP characteristics of the molecules (Table S2 and Fig. S9, ESI†). Interestingly, at 77 K (λex = 350 nm), both steady state and phosphorescence emission bands of the compounds closely overlap (∼473 nm, red shifted), and they appear as a broadband with an on-set of 2.95 (CPPN) and 2.98 eV (CPPNF), indicating an involvement of the 3CT state (Fig. 5). The disappearance of the high-energy band (∼455 nm) at 77 K is discussed in a later section of the manuscript. Comparing emission and decay analysis recorded at both RT and 77 K, we confirm that two triplet states contribute to the emission under ambient conditions.
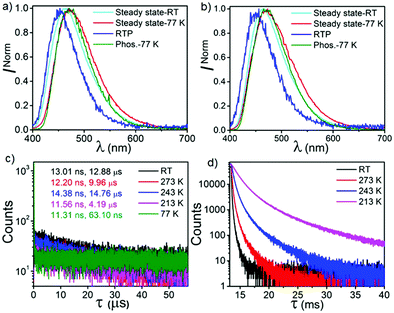 |
| | Fig. 5 Steady-state and phosphorescence spectra of (a) CPPN and (b) CPPNF at RT (ambient conditions) and 77 K in PMMA (0.1%). Temperature-dependent (c) fluorescence and (d) phosphorescence decay analysis of CPPN in 0.1% PMMA. | |
 |
| | Fig. 6 TRES of CPPN (a) without detector delay and (b) 6.8 μs detector delay under ambient conditions. TRES of CPPN (c) without detector delay and (b) varying detector delay from 0.034 to 110 ms at 77 K. | |
Table 1 Photophysical parameters of CPPN and CPPNF
|
|
PMMA
|
DPEPO
|
|
λ
F (nm) |
λ
P (nm) |
τ
F (ns, μs) |
τ
P (μs) |
Φ
F (%) |
Φ
P (%) |
λ
F (nm) |
λ
P (nm) |
τ
F (ns, μs) |
τ
P (ms) |
Φ
F (%) |
Φ
P (%) |
|
λ
ex = 350 nm (steady-state and phosphorescence spectra), 370 nm (lifetime).
Phosphorescence quantum yield under ambient conditions (λex = 350 nm).
Lifetimes (μs).
|
|
CPPN
|
463 |
455 |
13.01, 12.8c |
58.4, 175.0, 619.3 |
43.7 |
3.7 |
475 |
465 |
18.7, 5.9c |
0.49, 5.50, 39.30 |
10.9 |
10.1 |
|
CPPNF
|
463 |
452 |
15.5, 10.4c |
148.6, 570.0, 44.5 |
42.3 |
3.0 |
484 |
463 |
19.8, 4.9c |
40.4, 3.10 |
13.2 |
9.8 |
To understand the number of excited states involved in the process, TRES measurements were also performed at 77 K.
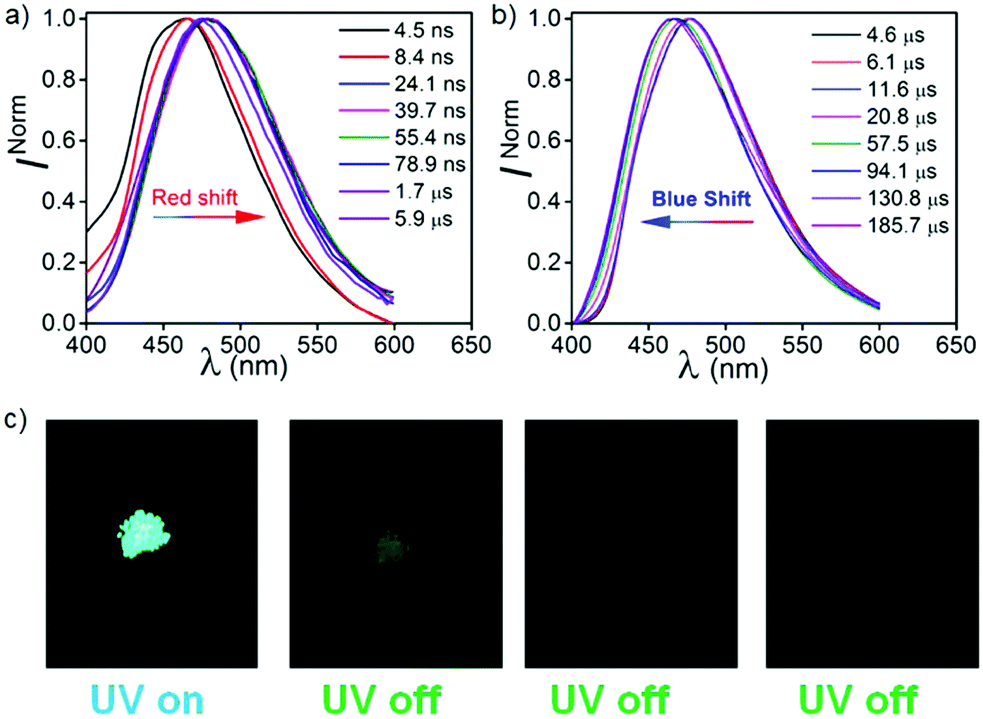
These studies show that the emission peaks of both compounds bathochromically shift from 455 to 475 nm (Fig. 6 and Fig. S11, ESI†). When we compare these results with the TRES data recorded at RT, we found that the spectrum obtained at ∼2 ns is closely overlapped by the unrelaxed TRES data at RT (3.2 ns) (Fig. S11, ESI†). Interestingly, on a later time scale (∼45 ns) at 77 K, a relatively larger bathochromic shift of the emission spectra was recorded (475 nm, 2.98 eV) as compared to the spectra recorded at RT (465 nm, 3.03 eV) at 49 ns. Such a red shifted emission band at 77 K can be explained by time resolved phosphorescence measurements, which is discussed in a later section of this paper. It is observed that the emission spectra recorded from 87 ns to 127 ms at 77 K is largely consistent with the previous spectra obtained at 45 ns, indicating that no new excited states are evolved.
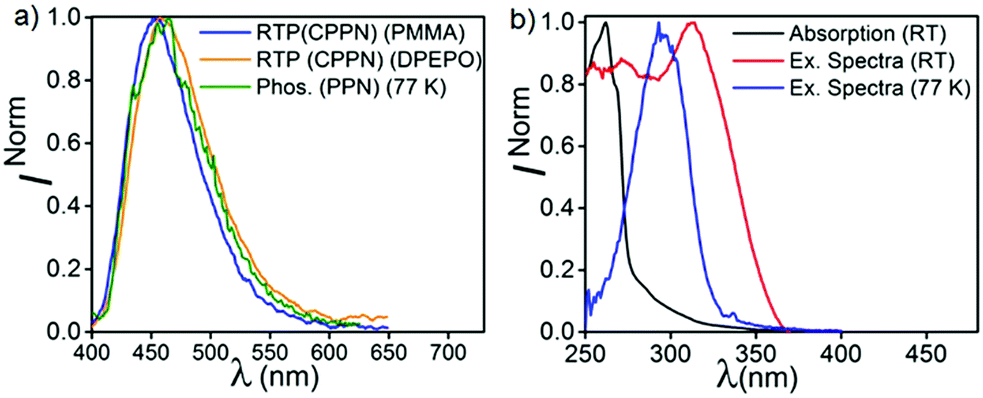
In order to understand the complex nature of both triplet emissions (BRTP/BTADF), we performed TRES at RT (λex = 370 nm) (Fig. 6). Both molecules (6.8 μs detector delay) show an emission band (FWHM = ∼72 nm) at ∼457 nm (3.0 eV) at an early decay time of 7.7 μs which matches well with the steady state emission observed at RT (Fig. S10, ESI†). On a later time scale (180 μs), the emission bands progressively blue shift to ∼3.06 eV and 3.03 eV with a reduction of FWHM (∼60 nm, 68 nm) in both cases (Fig. S12, ESI†). Moreover, the blue shifted emission spectra obtained on the later time scale resemble the BRTP peak obtained after 50 μs of the detector delay (Fig. S13, ESI†). This efficacy confirms that the excited species originated initially after 6.8 μs of detector delay is singlet in nature that contributes to the TADF, while the blue-shifted species observed on the later time scale originates from the higher-energy triplet state (BRTP). The time resolved area normalization spectra (TRANES) recorded in the time range of 7.7–180 μs also show an isoemissive point thus confirming the involvement of two emissive species due to DF and RTP (Fig. S14, ESI†). When we compare the TRES data at 7.7 μs and 49 ns (RT), we observed that both spectra are similar to each other, which indicates that the emission occurs from the same 1CT state that leads to delayed emission (Fig. S12, ESI†). At 77 K, the TRES measurements (3.4 μs) show no shift in the broad emission band (∼473 nm), while even after a further delay of 110 ms the phosphorescence emission band remained largely consistent with the emission recorded at a lower delay (3.4 μs), ensuring that only one 3CT state was involved in the emission (Fig. S15, ESI†). Based on the observations of narrow-RTP bands, delayed fluorescence (7.7 μs, RT) and phosphorescence recorded at 77 K, we confirmed that three radiative excited states (two triplets, one singlet) are involved in the emission. To understand the origin of the BRTP bands, we synthesized two control compounds (Fig. 2) in which only phenoxy (PPN) or 2,6-difluorophenoxy (PPNF) groups are covalently attached to the 4,5-positions of the PN core. 0.1% PMMA films of these compounds (λex = 330 nm, 50 μs delay) show an emission at ∼455 nm (τPPN = 56.20 μs and 343.47 μs and τPPNF = 197.77 μs and 1.58 ms) which closely overlaps with the BRTP peak of the parent compounds recorded at RT (Fig. 7 and Fig. S16, ESI†). At 77 K, in contrast to the parent compounds whose phosphorescence band was missing at ∼455 nm (λex = 350 nm), a clear vibrational feature (435, 455, 478 nm) was observed when they were excited at 290 nm, thus confirming the 3LE nature of the higher energy BRTP bands (PPN, 3.01 eV; PPNF, 3.03 eV) (Fig. S16, ESI†). When we recorded the absorption and excitation spectra of both PPN and PPNF, we found that (i) the excitation spectra (λem = 390–410 nm) recorded at RT do not overlap with the lower energy region of the absorption spectra, (ii) both absorption band (RT) and excitation spectra (77 K) do not overlap with each other, and (iii) the intensity of the excitation band at 350 nm is negligible (77 K) (Fig. 7 and Fig. S16, ESI†). We speculate that the excited states caused by PPN parts of the molecules are not populated when they are excited at 350 nm, while exciting at 290 nm a new weak phosphorescence emission band at 390–410 nm (77 K) was observed due to the spectral differences observed in the absorption and excitation spectra discussed above (Fig. S18, ESI†). Comparing the phosphorescence characteristics of both parent compounds measured at RT with the control compounds, we confirm that the higher energy BRTP peak originates from the PPN part (TPPN, ∼452–455 nm) of both parent molecular systems, while the CzPN part is responsible for the lower energy triplet state (TCzPN, ∼473 nm) that up-converts into the singlet state via the TADF process.15
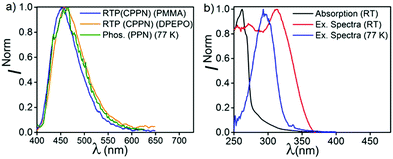 |
| | Fig. 7 (a) RTP of CPPN in PMMA and DPEPO under ambient conditions, and phosphorescence of PPN at 77 K. (b) Absorption and excitation spectra of PPN at RT, and excitation spectra of PPN at 77 K. | |
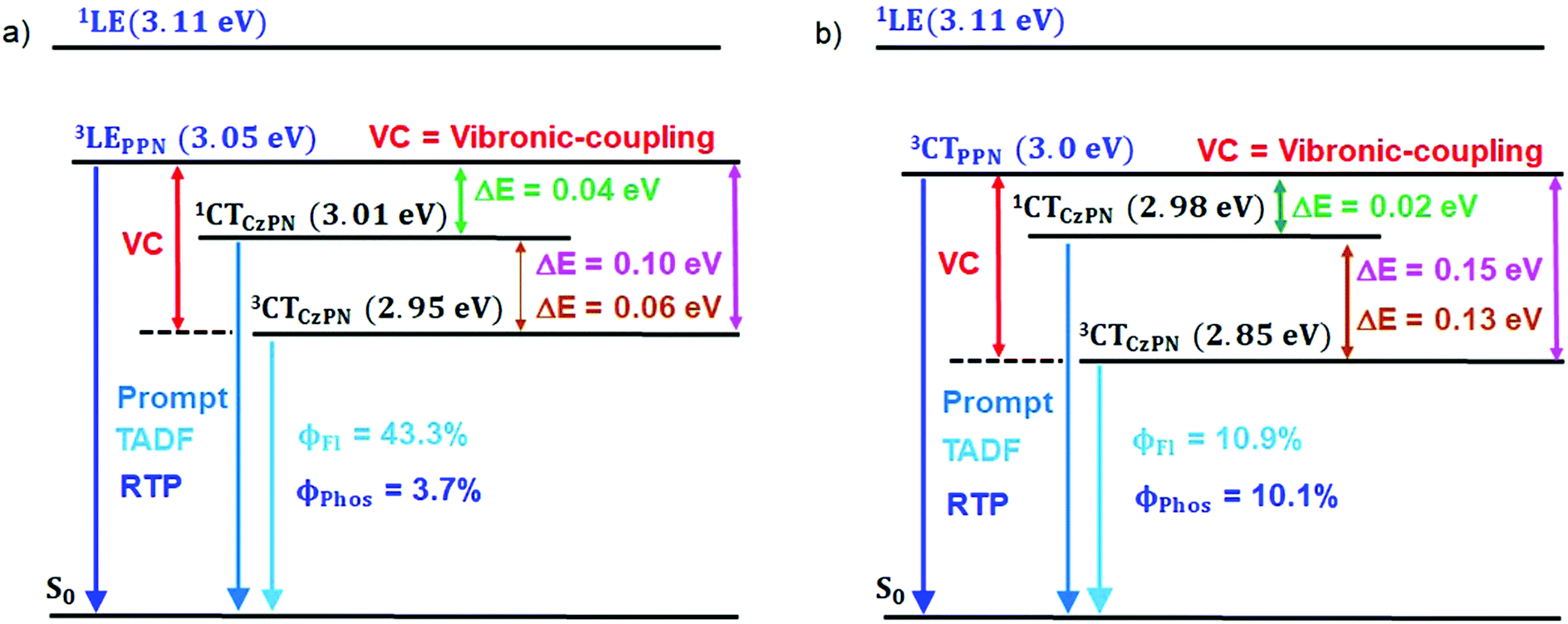
The PL measurements of the parent compounds (PMMA) are summarized in Table 1 and Table S3 (ESI†). The energy gaps (ΔES–T1: 1CTCzPN − 3LEPPN, ΔES–T2: 1CTCzPN − 3CTCzPN, ΔET–T: 3LEPPN − 3CTCzPN) between the singlet and triple states of both compounds were calculated to be 0.04, 0.06 and 0.10 eV (CPPN), and 0.05, 0.02 and 0.07 eV (CPPNF). We speculate that such close proximity of the energy levels (3LE, 1CT, 3CT) (Fig. 8) plays an important role to observe TADF via intermediate state-mixing caused by vibronic-coupling between the upper 3LE and lower-lying 3CT states.13,14
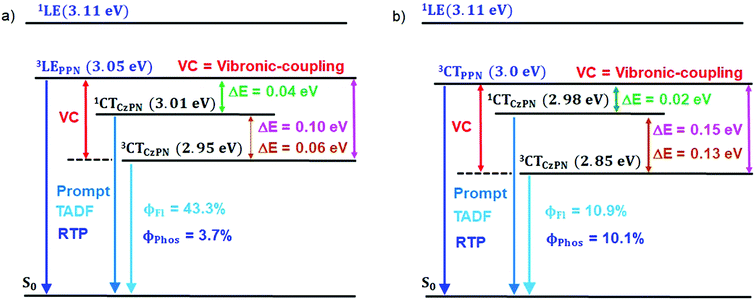 |
| | Fig. 8 Jablonski diagrams of CPPN in (a) PMMA (0.1%) and (b) DPEPO (0.1%) films. | |
3.4. Emission characteristics in DPEPO films
To gain more insight into the emission behavior, PL studies of bis[2-(di-(phenyl)phosphino)-phenyl]ether oxide (DPEPO) films (0.1%) were undertaken. The steady state PL characteristics of both compounds in DPEPO films under ambient conditions are very similar as compared to PMMA films. However, a bathochromic shift (CPPN, λssem = 475 nm, λPhosem = 465 nm; CPPNF, λssem = 484 nm, λPhosem = 463 nm) is observed as compared to PMMA due to the increased polarity of the host material (Fig. S19, ESI†). The PLQY values were found to be 21% (CPPN) and 23.2% (CPPNF) while the phosphorescence quantum yield (ΦP) was increased to 10.04% and 9.96%, respectively. Fluorescence lifetime analysis of the band (CPPN: λ475, τPF = 18.78 ns, τDF = 5.9 μs; CPPNF: λ475, τPF = 19.8 ns, τDF = 4.99 μs) under ambient conditions further confirms the TADF feature of the dyes (Fig. S20, ESI† and Table 1). The phosphorescence lifetimes of λ462 are found to be 59.61 ms and 5.16 ms (CPPN), and 68.17 ms and 5.4 ms (CPPNF) which are significantly long as compared to the values obtained in PMMA films under ambient conditions (Fig. S21, ESI†). Such long lifetimes of up to milliseconds further prove the origin of the RTP in these molecules.50 Interestingly, RTP lifetimes are substantially reduced with the increasing concentration of the dopants. Such increase in the ΦP values and decrease in the RTP lifetimes may originate due to the strong intermolecular interactions between the dopant and the host material which contribute to the reduction of the nonradiative decay through vibrations.41 Furthermore, the TRES measurements of 0.1% DPEPO films under ambient conditions are found to be similar to the results obtained in the PMMA films (Fig. S23–S25, ESI†). Interestingly, we observed that the emission band is red-shifted (460 nm) with a broadband signature (FWHM = 76–80 nm) as compared to the emission measured in PMMA (Fig. S19, ESI†), which suggests that the nature of the higher-lying triplet state (3LEPPN) in PMMA is changed to 3CTPPN (3.0 eV, both compounds) in DPEPO under ambient conditions. The phenoxy-phthalonitrile part of CPPN and CPPNF acts as the weak donor–acceptor part as compared to the carbazole–phthalonitrile part due to the presence of weak phenoxy donors. Therefore, a change in the environment from PMMA to DPEPO switches the nature of the triplet state from 3LE to 3CT. At 77 K, we observed a single red shifted phosphorescence band (50 μs delay) at 490 nm (2.85 eV) which is similar to the PMMA films (Fig. S25, ESI†). The DPEPO film studies further ensure that three excited states (3CTPPN, 1CTCzPN, 3CTCzPN) are involved in the emission, which is in line with the results obtained in PMMA except for the change in the nature of the higher-lying triplet state (Fig. 8). The energy of the singlet states (1CTCzPN) was calculated to be 2.98 eV (CPPN) and 2.94 eV (CPPNF) whereas the triplet state energy (3CTCzPN, TADF) is found to be 2.85 eV for both molecules (Fig. S25 and Table S3, ESI†). The ΔEST (TADF) is calculated to be 0.13 eV (CPPN) and 0.09 eV (CPPNF). Based on the observations of alteration of the 3LE state to the 3CT state along with high ΔEST and low PLQY values, we reason that mixing of the excited triplet states (RTP, TADF) through vibronic-coupling is substantially reduced in polar DPEPO films as compared to that observed in PMMA due to which substantial reduction in the fluorescence quantum yield was observed. In spite of this, mixing of the intermediate state (3CTPPN) contributes to the rISC process which further leads to the TADF emission in DPEPO films as reported by Noda et al.15 Comparing photophysical analyses both in PMMA and DPEPO films, we conclude that the higher-energy triplet state originating from the PPN/PPNF part of the molecules acts as an intermediate state that contributes independently as BRTP and BTADF via spin–vibronic coupling. Our studies further confirm that the nature of the higher-lying triplet state plays a decisive role in observing highly efficient simultaneous BRTP and BTADF in D2D2′–A conjugates. Besides, the selection of suitable host materials is a requisite to observe such photophysical properties of the molecule.
3.5. Emission characteristics in PVA films and crystals
In order to understand the effect of hydrogen bonding (H-B) on the photophysical properties, CPPNF was studied in polyvinyl alcohol (PVA) (0.1%, w/w). The steady state and phosphorescence emission bands were observed at 480 nm (τPF = 9.85 ns and τDF = 4.25 μs) and 470 nm (τP = 39.51 ms, 269.21 ms), respectively (Fig. S26, ESI†). TRES measurements of the PVA films without detector delay and with a delay of 3.4 μs showed similar emission behaviour as compared to both in PMMA and DPEPO films (Fig. 9), which further confirms TADF associated in the PVA matrix. However, when the delay was increased to 1.75 ms (Fig. S27, ESI†), a broad emission band was observed at 470 nm (3.06 eV, 2.2 ms) caused by the PPNF part of the molecule. On a later time scale (10.6 ms), the emission band was further red shifted to 525 nm (2.79 eV) with a reduction in intensity of the higher-lying emission band (3.06 eV), which is inconsistent with the emission behaviour observed in both PMMA and DPEPO films where no such red shifted emission band was observed. However, comparing this observation with the phosphorescence spectra (50 μs detector delay) of both powder (λem, 510 nm; τP = 71.83 ms, 187.39 ms) and crystals (λem, 515 nm; τP = 71.98 ms, 173.97 ms) of CPPNF recorded under ambient conditions (Fig. 9c and Fig. S26d, ESI†), we found that the TRES spectra (recorded at 66.9 ms in PVA films) closely overlaps with the phosphorescence spectra recorded for powder and crystals (Fig. S27d, ESI†). Now, it is clear that the further bathochromic shift of the lower energy emission band at 2.79 eV is due to aggregation of the molecules, which leads to the formation of an additional lower energy triplet state. With a 1.75 ms detector delay in films, we observed an isoemissive point in the TRANES spectra, suggesting that two emissive triplet states are involved. The higher energy RTP band originates due to the PPNF part of the molecule while the lower energy emission is due to the new triplet state formed via aggregation (see the ESI† for details). Therefore, three triplet states (TPPNF, TCzPN, Tagg) are responsible for the emission. Furthermore, the persistent-RTP obtained in films can be explained by the stabilization of the triplet states caused by both aggregation of CPPNF, and H-B interactions between the F atoms of CPPNF and –OH groups present in PVA.30,33
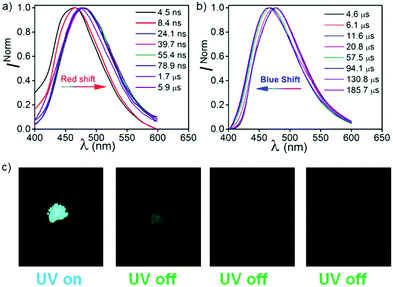 |
| | Fig. 9 TRES of CPPN in PVA (0.1%) at (a) without detector delay except the spectrum recorded at time 1.7–5.9 μs and (b) 3.4 μs detector delay under ambient conditions. (c) Images showing PRTP in CPPNF crystals (λex = 370 nm). | |
Considering the PL studies of all the films (0.1%) (PMMA, DPEPO, PVA), we believe that the phenoxy groups covalently connected at the 4,5-positions of the phthalonitrile acceptor cause blue-RTP, while blue-TADF is observed due to the presence of carbazolyl groups attached to the 3,6-positions of the acceptor core. Moreover, the precise substitution of the phenoxy donors can lead to persistent RTP in different host materials and crystals.
4. Conclusions
In summary, we have demonstrated simultaneous emission via BRTP and BTADF from carbazolyl-phenoxy-phthalonitrile based donor (D2D2′)–acceptor (A) conjugates. Experimental results provide unequivocal evidence of BRTP via radiative decay of the higher-lying triplet state from the phenoxy-phthalonitrile (PPN) part, while TADF occurs from the carbazolyl-phthalonitrile (CzPN) part utilizing the triplet state of PPN via spin–vibronic coupling. Moreover, both blue-RTP and blue-TADF characteristics can be tuned using different host materials and chemical modification of the phenoxy rings. This design strategy may lead to the enhancement of our understanding of highly efficient intrinsic simultaneous emission via RTP and TADF. These systems with asymmetric donors can be further developed to understand the complex exciton dynamics of both singlet and triplet states essential for security applications, imaging and sensors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
D. R. is grateful to the Board of Research and Nuclear Science (BRNS) (File No. 37(3)/14/08-2018-BRNS/37130), DAE; and Shiv Nadar University (SNU) for generous support. H. B. thanks SNU for fellowship.
Notes and references
- I. Bhattacharjee, N. Acharya, S. Karmakar and D. Ray, J. Phys. Chem. C, 2018, 122, 21589–21597 CrossRef CAS.
- C. A. DeRosa, S. Hiroto and C. L. Fraser, J. Phys. Chem. C, 2019, 123, 20488–20496 CrossRef CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS.
- A. Pigliucci, P. Nikolov, A. Rehaman, L. Gagliardi, C. J. Cramer and E. Vauthey, J. Phys. Chem. A, 2006, 110, 9988–9994 CrossRef CAS PubMed.
-
N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry, University Science Books, Sausalito, CA, 2009 Search PubMed.
- I. Bhattacharjee, N. Acharya, H. Bhatia and D. Ray, J. Phys. Chem. Lett., 2018, 9, 2733–2738 CrossRef CAS PubMed.
- Y. Xu, C. Wang, X. Zhou, J. Zhou, X. Guo, X. Liang, D. Hu, F. Li, D. Ma and Y. Ma, J. Phys. Chem. Lett., 2019, 10, 6878–6884 CrossRef CAS PubMed.
- J. S. Ward, N. A. Kukhta, P. L. dos Santos, D. G. Congrave, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem. Mater., 2019, 31, 6684–6695 CrossRef CAS PubMed.
- M. Li, Y.-F. Wang, D. Zhang, L. Duan and C.-F. Chen, Angew. Chem., Int. Ed., 2020, 59, 3500–3504 CrossRef CAS PubMed.
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen and S.-J. Su, Angew. Chem., Int. Ed., 2019, 58, 11301–11305 CrossRef CAS PubMed.
- P. Stachelek, J. S. Ward, P. L. dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125–27133 CrossRef CAS PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, ChemPhysChem, 2016, 17, 2956–2961 CrossRef CAS PubMed.
- H. Noda, X.-K. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J.-L. Brédas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- Y. Olivier, M. Moral, L. Muccioli and J.-C. Sancho-García, J. Mater. Chem. C, 2017, 5, 5718–5729 RSC.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439–6469 RSC.
- A. Fermi, G. Bergamini, M. Roy, M. Gingras and P. Ceroni, J. Am. Chem. Soc., 2014, 136, 6395–6400 CrossRef CAS PubMed.
- M. Shimizu, R. Shigitani, M. Nakatani, K. Kuwabara, Y. Miyake, K. Tajima, H. Sakai and T. Hasobe, J. Phys. Chem. C, 2016, 120, 11631–11639 CrossRef CAS.
- H. Zhao, L. Zang, H. Zhao, F. Qin, Z. Li, Z. Zhang and W. Cao, J. Phys. Chem. C, 2015, 119, 10558–10563 CrossRef CAS.
- G. Chen, H. Feng, F. Feng, P. Xu, J. Xu, S. Pan and Z. Qian, J. Phys. Chem. Lett., 2018, 9, 6305–6311 CrossRef CAS PubMed.
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS.
- S. Hirata, J. Mater. Chem. C, 2018, 6, 11785–11794 RSC.
- S. Hirata and M. Vacha, Adv. Opt. Mater., 2017, 5, 1600996 CrossRef.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Angew. Chem. Int. Ed., 2014, 53, 11177–11181 CrossRef CAS PubMed.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- Y. Xu, W. Yang, D. Yao, K. Bian, W. Zeng, K. Liu, D. Wang and B. Zhang, Chem. Sci., 2020, 11, 419–428 RSC.
- H. Bhatia, I. Bhattacharjee and D. Ray, J. Phys. Chem. Lett., 2018, 9, 3808–3813 CrossRef CAS PubMed.
- H. Bhatia and D. Ray, J. Phys. Chem. C, 2019, 123, 22104–22113 CrossRef CAS.
- G. Baryshnikov, B. Minaev and H. Ågren, Chem. Rev., 2017, 117, 6500–6537 CrossRef CAS PubMed.
- A. Köhler and H. Bässler, Mater. Sci. Eng., R, 2009, 66, 71–109 CrossRef.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199–241 CrossRef CAS.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7025 CrossRef CAS PubMed.
- S. Hirata, J. Phys. Chem. Lett., 2018, 9, 4251–4259 CrossRef CAS PubMed.
- C. S. Bilen, N. Harrison and D. J. Morantz, Nature, 1978, 271, 235–237 CrossRef CAS.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Pavanello, A. Previtali, S. Righetto and E. Cariati, Angew. Chem., Int. Ed., 2017, 56, 16302–16307 CrossRef CAS PubMed.
- Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed.
- J. Zhang, E. Sharman, L. Yang, J. Jiang and G. Zhang, J. Phys. Chem. C, 2018, 122, 25796–25803 CrossRef CAS.
- Y. Lei, W. Dai, Y. Tian, J. Yang, P. Li, J. Shi, B. Tong, Z. Cai and Y. Dong, J. Phys. Chem. Lett., 2019, 10, 6019–6025 CrossRef CAS PubMed.
- R. Kabe, N. Notsuka, K. Yoshida and C. Adachi, Adv. Mater., 2016, 28, 655–660 CrossRef CAS PubMed.
- R. Huang, J. Avó, T. Northey, E. Chaning-Pearce, P. L. dos Santos, J. S. Ward, P. Data, M. K. Etherington, M. A. Fox, T. J. Penfold, M. N. Berberan-Santos, J. C. Lima, M. R. Bryce and F. B. Dias, J. Mater. Chem. C, 2017, 5, 6269–6280 RSC.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612–2615 RSC.
- I. Bhattacharjee, N. Acharya and D. Ray, Chem. Commun., 2019, 55, 1899–1902 RSC.
- L. Zhan, Z. Chen, S. Gong, Y. Xiang, F. Ni, X. Zeng, G. Xie and C. Yang, Angew. Chem. Int. Ed., 2019, 58, 17651–17655 CrossRef CAS PubMed.
- P. Data, M. Okazaki, S. Minakata and Y. Takeda, J. Mater. Chem. C, 2019, 7, 6616–6621 RSC.
- T. Serevičius, T. Bučiūnas, J. Bucevičius, J. Dodonova, S. Tumkevičius, K. Kazlauskas and S. Juršėnas, J. Mater. Chem. C, 2018, 6, 11128–11136 RSC.
- Y. Takeda, T. Kaihara, M. Okazaki, H. Higginbotham, P. Data, N. Tohnai and S. Minakata, Chem. Commun., 2018, 54, 6847–6850 RSC.
- A. Tomkeviciene, T. Matulaitis, M. Guzauskas, V. Andruleviciene, D. Volyniuk and J. V. Grazulevicius, Org. Electron., 2019, 70, 227–239 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR spectra, additional photophysical analysis and single-crystal X-ray data. CCDC 1990136 and 1990137. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ma00382d |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*