
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromadendrin: a dual amyloid promoter to accelerate fibrillization and reduce cytotoxicity of both amyloid-β and hIAPP
Yanxian
Zhang
 a,
Dong
Zhang
a,
Yijing
Tang
a,
Baiping
Ren
a,
Fufeng
Liu
b,
Lijian
Xu
c,
Yung
Chang
d and
Jie
Zheng
*a
a,
Dong
Zhang
a,
Yijing
Tang
a,
Baiping
Ren
a,
Fufeng
Liu
b,
Lijian
Xu
c,
Yung
Chang
d and
Jie
Zheng
*a
aDepartment of Chemical, Biomolecular, and Corrosion Engineering, The University of Akron, Ohio, USA. E-mail: zhengj@uakron.edu
bKey Laboratory of Industrial Fermentation Microbiology, College of Biotechnology, Tianjin University of Science & Technology, Tianjin 300457, China
cHunan Key Laboratory of Biomedical Nanomaterials and Devices, College of Life Science and Chemistry, Hunan University of Technology, Zhuzhou 412007, China
dDepartment of Chemical Engineering and R&D Center for Membrane Technology, Chung Yuan Christian University, Taoyuan 320, Taiwan
First published on 30th June 2020
Abstract
Abnormal aggregation of misfolded amyloid proteins into amyloid fibrils is associated with many neurodegenerative diseases, including Alzheimer's disease (AD) and Type II diabetes (T2D). In principle, any strategy to alter the amyloid aggregation process is considered as a potential therapeutic treatment for these diseases. Significant efforts and progress have been made to develop amyloid inhibitors that can slow down and prevent the amyloid aggregation process. However, much less research has been reported to discover general amyloid promoters to accelerate amyloid progress and remodel toxic amyloids. Here, for the first time we report a repurposing of the drug of aromadendrin as a dual amyloid promoter to be effective in accelerating amyloid aggregation/fibrillization and reducing neuroblastoma/insulinoma toxicity of both Aβ42 (associated with AD) and hIAPP37 (associated with T2D). ThT, AFM, and CD results showed that addition of aromadendrin to amyloid solutions with 1 to 5 molar ratios caused significant acceleration in Aβ42 fibrillization by 86–114% and hIAPP fibrillization by 20–68% as evidenced by shortening or bypassing of the lag phase, promoting the growth phase, and rapidly converting the amyloid species towards the higher ordered β-structure-rich aggregates. Seeding experiments further revealed that aromadendrin is more effective in accelerating the aggregation and structural conversion of the amyloid species at the lag and early growth phases, but unable to escalate the fibrillization of higher order protofibrils. Moreover, MTT and LDH cell assays showed that aromadendrin-treated cell samples enabled the rescue of cells from both Aβ- and hIAPP-induced toxicity by increasing cell viability by 12–15% (Aβ) and 10–49% (hIAPP) and reducing cell apoptosis by 45–67% (Aβ) and 10–30% (hIAPP). LUV assays explained the protective role of aromadendrin in cell toxicity due to the suppression of toxic oligomer formation and thus the decrease of membrane leakage. This work offers a new strategy for repurposing aromadendrin as an amyloid promoter, not as an amyloid inhibitor, to accelerate amyloid formation and remedy the amyloid-mediated toxicity for both Aβ and hIAPP, and the sequence-independent promotion effect of aromadendrin could also be applied to other amyloid proteins.
1. Introduction
The pathological fibrillogenesis of amyloid peptides is believed to be closely associated with different protein-misfolding diseases (PMDs), including Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and Type II diabetes (T2D).1–3 Despite different sequences, native structures, and biological functions of amyloid peptides, they can all transform from unstructured monomers to β-structure-presented oligomers to β-strand-rich fibrils via a nucleation–polymerization process.4 It is generally accepted that the intermediate oligomers are the most cytotoxic species to neuronal cells.5,6 Therefore, in principle, any biomedical and clinical strategy to alter amyloid aggregation pathways and/or conformational changes is considered as a potential therapeutic method for PMD treatment. Significant efforts have been made to develop a wide variety of amyloid inhibitors, including antibodies,7–9 short peptides,10–12 organic molecules,13–16 metal compounds,17–19 and nanoparticles,20–22 to delay and prevent toxic amyloids at different aggregation stages. But, none of these inhibitors have been successful in (pre)clinical trials for PMDs.Alternatively, it is also interesting and important, from different fundamental and clinical viewpoints, to discover new compounds that can promote amyloid aggregation and fibrillization by not only quickly converting toxic oligomers formed at the lag and growth stage to non- or less toxic fibrils, but also shortening the exposure time of tissues/cells to toxic aggregates. However, very few studies have been reported on amyloid promoters. Ellagic acid (EA),23 a polyphenolic compound, was identified to reduce Aβ42 neurotoxicity by promoting Aβ42 fibrillization at the expense of oligomer loss. O4,24 as a small molecule derived from orcein, was found to interact with Aβ42 oligomers, which in turn promote subsequent fibrillogenesis and suppress cell toxicity. A commercial synthetic compound 2002-H20 was identified to enhance Aβ42 fibril formation and inhibit Aβ42-induced cytotoxicity in a dose-dependent manner.25 AcPHF6 peptide26 derived from native tau sequence VQIVYK enabled the promotion of both Aβ40 and Aβ42 fibrillization and reduced their Aβ-mediated toxicity. Cellular cationic polyamines (spermine, spermidine, and putrescine) were also found to promote the aggregation of both α-synuclein27 and Aβ,28 but the cell toxicity data is not available for these reported polyamines. Another polyamine of trodusquemine5 showed an ability to promote Aβ42 aggregation and reduce the toxicity of Aβ42 in neuroblastoma cells.
Even less hIAPP promoters have been reported in the literature so far. Bisphenol A29 and free fatty acids30 have been discovered to promote hIAPP fibrillization, but they have failed to protect cells from hIAPP-induced cytotoxicity, possibly due to the formation of on-pathway, toxic hIAPP-molecule complexes to exacerbate cell cytotoxicity.29 DOPC liposomes also promoted the conversion of hIAPP oligomers into mature fibrils to reduce cell cytotoxicity due to hIAPP oligomers.31 A star polymer poly(2-hydroxyl ethyl acrylate) (PHEA) possessed abundant rodlike arms for promoting hIAPP binding and aggregation, which in turn reduced cell toxicity.32 Among these amyloid promoters, not all of them can simultaneously promote amyloid aggregates and prevent cell toxicity. However, these studies still provide alternative strategies to develop amyloid promoters as potential therapeutic methods for PMDs.
However, the challenge still remains. Most of these amyloid promoters show a sequence-dependent promotion ability, i.e. a compound that promotes the aggregation and fibrillization of one amyloid sequence does not necessarily promote those of another amyloid sequence. Desirable amyloid promoters are also expected to alleviate cell cytotoxicity by reducing the exposure time of toxic oligomers to cells and directing the aggregation away from toxic pathways. As shown in Fig. 1, six Aβ promoters and four hIAPP promoters do not show any overlap compound to work for both Aβ and hIAPP. Thus, developing sequence-independent amyloid aggregation promoters is imperative for the better fundamental understanding and potential therapeutics towards different PMDs. Among different PMDs, AD and T2D are connected by some common molecular denominators, including inflammation, defective insulin signaling, and mitochondrial dysfunction.37 A number of molecular dynamics (MD) simulations discovered that the binding of insulin with Aβ and hIAPP aggregates affected the pathological aggregation of Aβ and hIAPP.38,39 MD simulations also revealed that Aβ and hIAPP can cross-seed each other to form enlongated fibrils and laterally associated fibrils, which in turn exacerbate the neurodegenerative process.40–42 To address this issue, we reported a drug repurposing study of aromadendrin (Fig. 2), a flavanonol found in the wood of Pinus sibirica, for examining its new ability as an amyloid promoter for both Aβ42 (associated with AD) and hIAPP37 (associated with T2D). Aromadendrin has long been used for its therapeutic uses as it has antioxidant,43 anti-inflammatory,44,45 antitumor,46 stimulation of glucose uptake, high insulin resistance,47 and inhibition of β-site amyloid precursor protein cleaving enzyme 1 (BACE1) functions.48 A combination of biophysical, imaging and computational techniques showed that aromadendrin was able to interact with both Aβ42 and hIAPP37 to promote their amyloid fibrillization and to reduce Aβ- and hIAPP-induced cell toxicity. This work not only demonstrates aromadendrin as an amyloid promoter (not an inhibitor), but also suggests a new drug repurposing strategy to potentially treat both AD and T2D diseases.
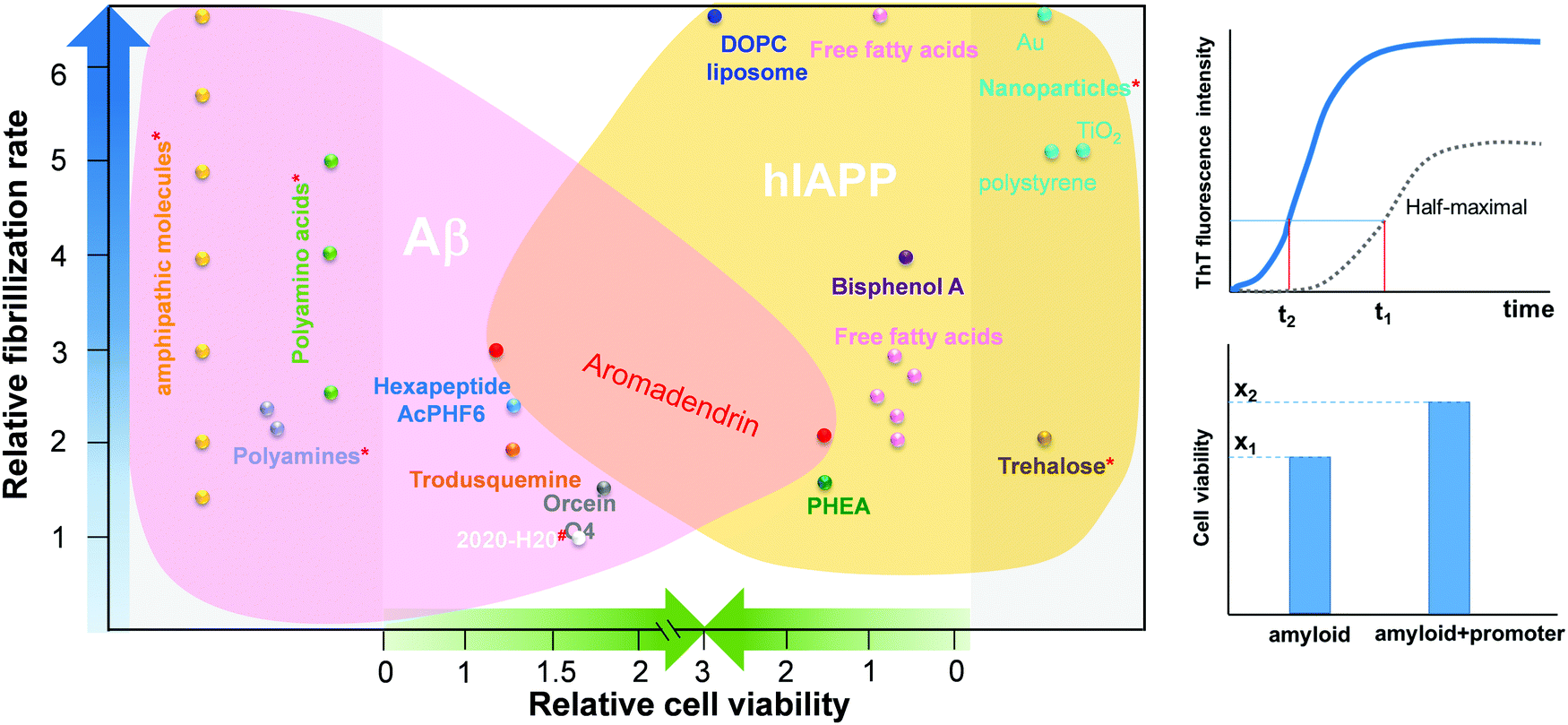 | ||
Fig. 1 Comparison of Aβ and hIAPP promoters in terms of their acceleration of amyloid fibrillization and reduction in cell toxicity.5,24,26,28–30,33–36 The relative fibrillization rate (t1/t2) is defined by a time ratio between amyloid + promoter and pure amyloid (control) systems to achieve the same half-maximal ThT intensity of the control system. The relative cell viability (x2/x1) is defined by a cell viability ratio between amyloid + promoter and pure amyloid (control) systems. *![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) No cell viability data is available in the references. #No fibrillization rate data is available in the references. No cell viability data is available in the references. #No fibrillization rate data is available in the references. | ||
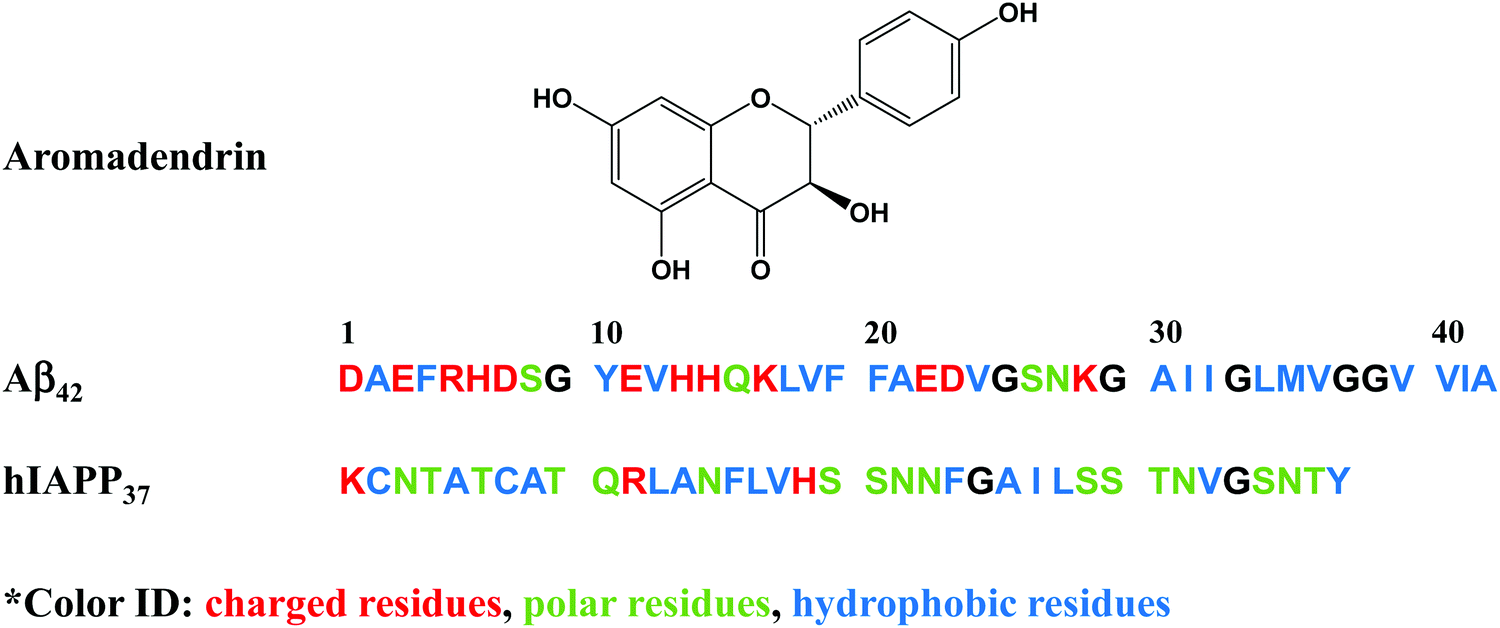 | ||
| Fig. 2 Chemical structure of aromadendrin and the sequences of amyloid-β (Aβ42) and human islet polypeptide (hIAPP37). | ||
2. Results and discussion
2.1. Aromadendrin promotes the aggregation and fibrillization of both Aβ and hIAPP
Aromadendrin, belonging to a flavanonol family, was first discovered in the heartwood of Pinus sibirica27 and was later identified by its anti-inflammatory and antioxidant functions. Further studies found aromadendrin to be effective in inhibiting BACE1 activity in AD28 and in improving glucose uptake and insulin resistance,49 indicating that aromadendrin has a biological function in both AD and T2D. However, there is no evidence about whether aromadendrin can affect the aggregation and toxicity of Aβ and hIAPP.
Fig. 3 shows the ThT aggregation kinetics of Aβ and hIAPP in the presence of aromadendrin at different peptide:aromadendrin molar ratios of 1:1, 1:2, and 1:5 under physiological conditions (37 °C and pH 7.4). As a control, pure aromadendrin (125 μM) did not produce any ThT signal during 12 h incubation, ruling out a possibility of background signal influence from the aromadendrin. For the Aβ–aromadendrin systems (Fig. 3a), freshly prepared Aβ (25 μM) alone showed a typical sigmoidal growth curve, starting with an early lag phase of ∼2 h, followed by a gradual growth phase between 2 and 5 h, and finally ending in a saturation phase with a final ThT intensity of 70 a.u. after 10 h, an indictor of mature fibril formation. When co-incubating aromadendrin with Aβ at three different Aβ:aromadendrin molar ratios (1:1, 1:2 and 1:5), aromadendrin significantly promoted Aβ aggregation and fibrillization, as evidenced by (i) almost eliminating the lag phase, (ii) exhibiting an exponential increase in growth phase, and (iii) achieving almost 2-times higher ThT fluorescence intensity. The aromadendrin-induced promotion displayed a smaller concentration-dependent effect, because three different concentrations of aromadendrin led to a similar final ThT density of 130–150 a.u. On the other hand, a higher concentration (125 μM) of aromadendrin showed a much faster growth rate than the lower ones, as evidenced by its aggregation half-time of 1.7 h, which was much shorter than that of 2.8 h for 25 μM and 2.5 h for 50 μM concentrations.
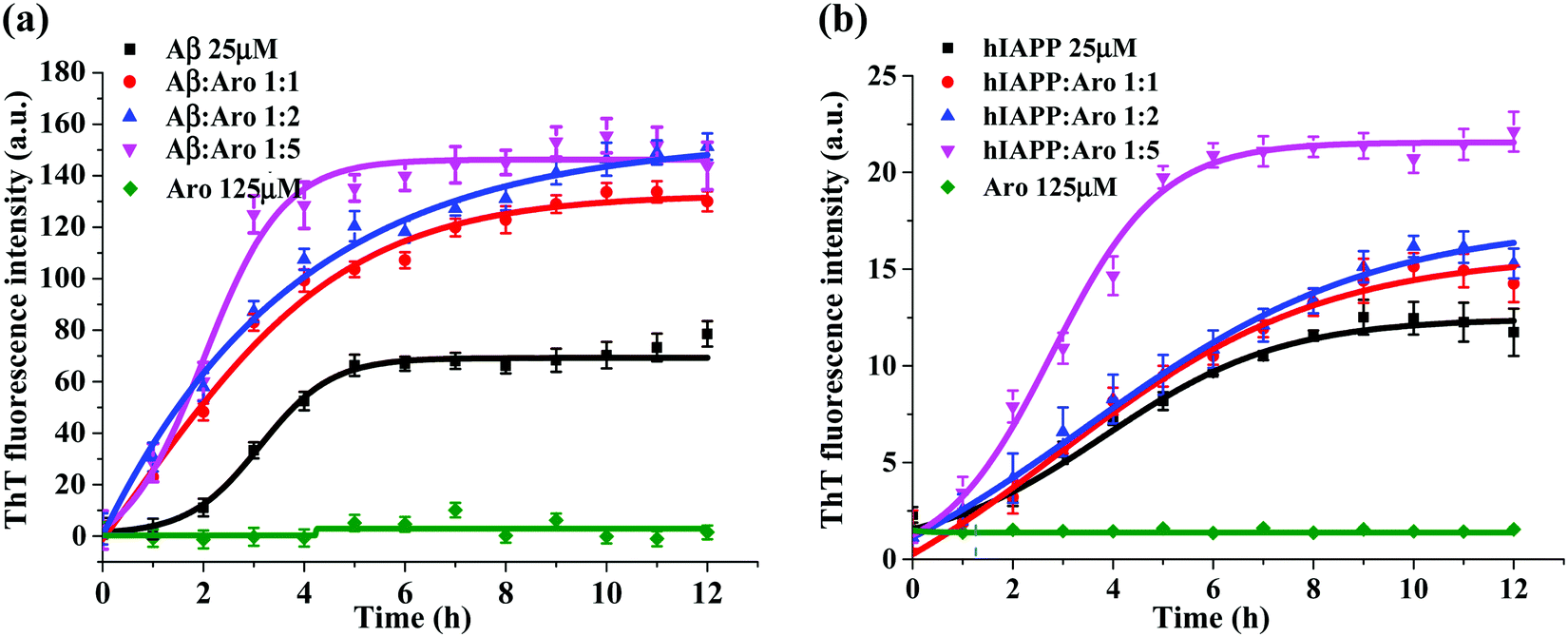 | ||
| Fig. 3 Time-dependent ThT fluorescence profiles for monitoring the aggregation kinetics of (a) Aβ42 (25 μM) and (b) hIAPP37 (25 μM) in the absence and presence of aromadendrin with different molar ratios of 1:1, 1:2, and 1:5 at 37 °C and at a pH of 7.4. | ||
Similarly, in the case of hIAPP-aromadendrin systems, Fig. 3b showed that with the addition of aromadendrin, hIAPP fibril formation was accelerated. Specifically, after 12 h incubation, the final fluorescence intensity increased to 120%, 130%, and 168% with the addition of 25, 50, and 125 μM concentrations of aromadendrin, respectively. This indicates that aromadendrin displays a more obvious concentration-dependent promotion effect on hIAPP fibrillization than on Aβ fibrillization. The aggregation kinetics in the presence of a higher aromadendrin concentration of 125 μM was markedly different and became more prominent in accelerating hIAPP fibrillization. Overall, the aromadendrin-accelerated aggregation kinetics reveal that aromadendrin is able to dramatically increase the fibrillogenesis of both Aβ and hIAPP at an amyloid:aromadendrin ratio of 1:5.
2.2. Aromadendrin modifies the morphology and conformation of Aβ and hIAPP aggregates
To support the aggregation kinetics of aromadendrin-accelerated Aβ and hIAPP fibrillization, AFM images were used to monitor the progressive morphology changes of Aβ and hIAPP aggregates in the absence and presence of aromadendrin. In Fig. 4a, pure Aβ self-assembled into small spherical oligomers in 2 h. In the presence of 25 μM aromadendrin, Aβ aggregates did not show obvious differences in size and shape from pure Aβ aggregates. As Aβ:aromadendrin molar ratios were further increased to 1:2 and 1:5, a few protofibrils of different lengths of 400–2000 nm were observed and co-existed with many globular clusters. At 4 h incubation, the morphological differences between different Aβ cases with and without aromadendrin became more obvious. Addition of aromadendrin already yielded a large number of thin, smooth, and isolated fibrils, in sharp contrast to large and dominant amorphous aggregates of pure Aβ. This indicates that aromadendrin can promote Aβ fibrillogenesis by quickly converting spherical Aβ oligomers to linear-like Aβ fibrils. After 12 h, while AFM images from all four Aβ cases with and without aromadendrin displayed the formation of amyloid fibrils, aromadendrin-promoted fibrils were more densely packed and entangled with a longer length than those of pure Aβ fibrils. A similar promotion effect of aromadendrin on hIAPP aggregation was also observed in Fig. 4b. Clearly, the addition of aromadendrin (25, 50, and 125 μM) led to an increase in the amount of hIAPP aggregates at three different time points of 2, 4, and 12 h. It can be seen from the AFM images that, in particular between 4 h and 12 h, hIAPP aggregates without aromadendrin exhibited considerably fewer, shorter, thinner, and less dense fibrils compared to those of the other three samples with aromadendrin. Such promotion effect became more pronounced at a high aromadendrin concentration of 125 μM. AFM images support the data from the ThT fluorescence assay that aromadendrin promotes the amyloid aggregation and fibrillization of both Aβ and hIAPP.
 | ||
| Fig. 4 AFM images for quantifying the morphological changes of (a) Aβ42 (25 μM) and (b) hIAPP37 (25 μM) aggregates in the absence and presence of aromadendrin at different amyloid:aromadendrin molar ratios of 1:1, 1:2, and 1:5 after 2 h, 4 h, and 12 h. Scale bar = 1 μm. | ||
From a structural viewpoint, it is interesting and important to measure how aromadendrin affects the structural transition of Aβ and hIAPP aggregates and to understand how such structural transition links to the accelerated amyloid fibrillization. The CD spectra in Fig. 5 show the conformational changes of Aβ and hIAPP in the presence and absence of aromadendrin at different molar ratios. For Aβ-aromadendrin systems (Fig. 5a), at the beginning of aggregation (t = 0 h), Aβ alone and Aβ with different concentrations of aromadendrin presented a single negative peak at ∼198 nm, a characteristic spectrum of random coil structures, indicating that Aβ in all the samples adopts an initial random coil conformation. As the aggregation proceeded to 2 h, Aβ co-incubated with aromadendrin experienced a much larger structural transition than Aβ only, as evidenced by the larger spectral shift that arises from the negative region of ∼198 nm towards the positive regions and the formation of another negative peak at 218 nm. This indicates the occurrence of a β-sheet structure with assistance of aromadendrin. At the end of aggregation (12 h), all the Aβ samples with and without aromadendrin showed both a characteristic positive peak at 195 nm and a negative peak at 218 nm, a signature of the β-sheet structure; however, Aβ incubated with aromadendrin at all molar ratios only showed higher peak intensities than Aβ. Quantitative analysis of the secondary structure contents in Fig. 5a2 showed that all freshly prepared samples had almost identical secondary structure contents (i.e., 1.2–4.5% α-helix, 25% β-sheet, and 70% random coils), but after 12 h of incubation, the final β-sheet content showed an increasing order of Aβ only (47.9%) < 1:1 of Aβ:aromadendrin (58.4%) < 1:2 of Aβ:aromadendrin (59.9%) < 1:5 of Aβ:aromadendrin (61.6%). Clearly, the increase of aromadendrin concentration facilitates the structural transition of Aβ towards the β-sheet formation by 22–29%, providing structure-based evidence about the accelerating effect of aromadendrin on Aβ aggregation. In parallel, we also recorded and analyzed the CD spectra of hIAPP in the absence and presence of aromadendrin (Fig. 5b). As a control, hIAPP alone increased its β-sheet structure content from 29.7% to 41.8% during the 12 h incubation. Co-incubating of hIAPP with 25, 50, and 125 μM aromadendrin increased its β-sheet structure content to 46.4%, 46.9%, and 50.9%, respectively, showing the concentration-dependent effect of aromadendrin on the secondary structure of hIAPP aggregates. Taken together, when comparing the aromadendrin-induced second structure changes between Aβ and hIAPP, aromadendrin at the same concentration and incubation time appears to be more efficient at accelerating the β-sheet formation of Aβ than that of hIAPP. Specifically, aromadendrin of 25, 50, and 125 μM concentrations promoted the β-sheet content of Aβ by 22%, 25%, and 29% and hIAPP by 11%, 12%, and 22%, respectively. Acceleration of β-sheet formation by aromadendrin also explains the promotion of Aβ and hIAPP fibrillogenesis as shown in the ThT and AFM data. Such amyloidosis promotion effect induced by aromadendrin may be due to the interaction of hydroxyphenyl and chromane rings of aromadendrin with aromatic residues of Aβ and hIAPP, which could act as an anchor to associate amyloid peptides and to stabilize the β-hairpin structure, thus promoting amyloid aggregation.
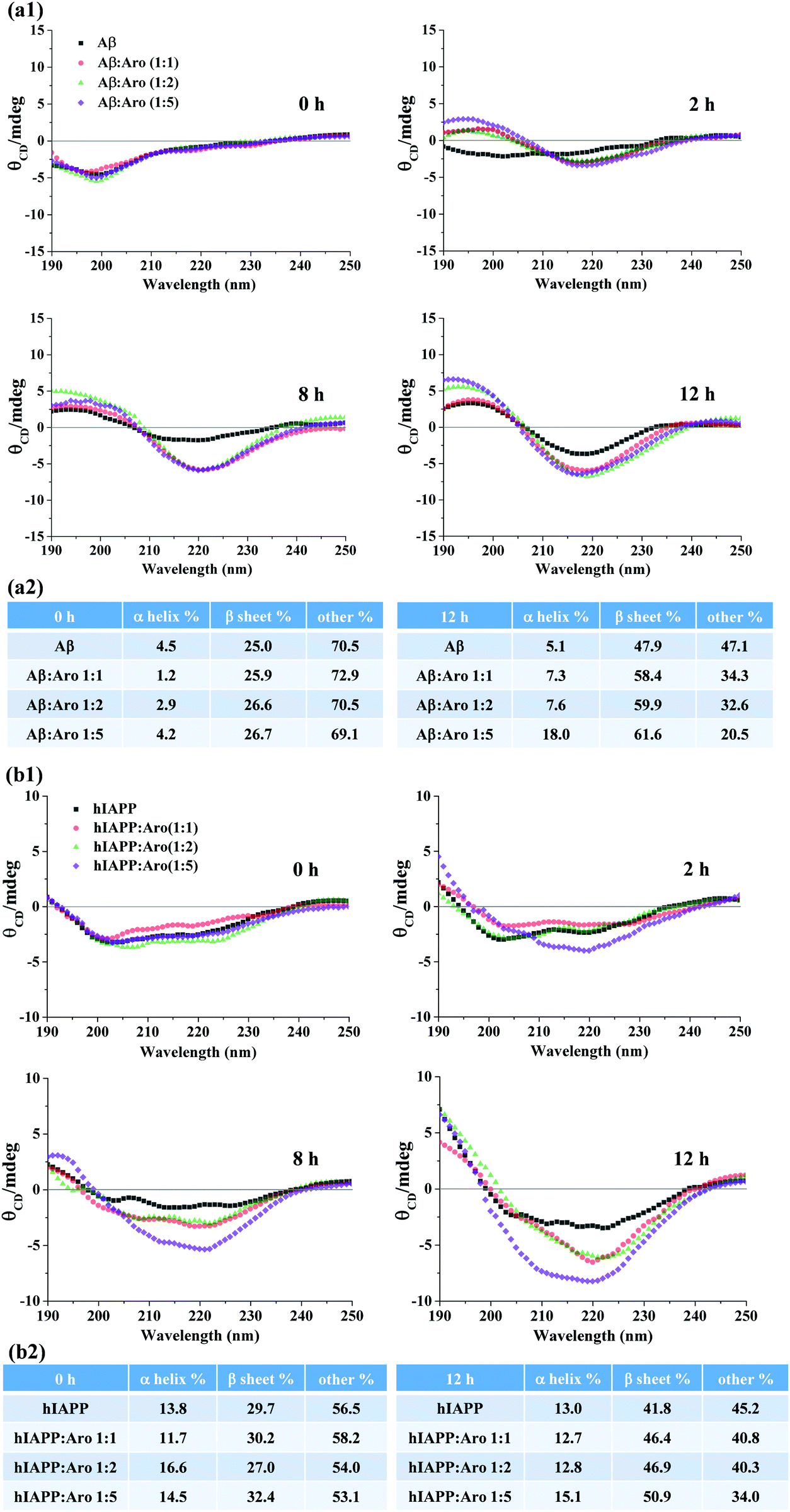 | ||
| Fig. 5 Time-dependent far-UV CD spectra for (a1) Aβ42 (25 μM) and (b1) hIAPP37 (25 μM) in the absence and presence of aromadendrin at different amyloid:aromadendrin molar ratios of 1:1, 1:2, and 1:5, with quantitative analysis of the secondary structure contents of the (a2) Aβ42 and (b2) hIAPP37 systems. | ||
2.3. Aromadendrin is more effective in converting early amyloid aggregates to amyloid fibrils
The acceleration effect of aromadendrin on Aβ42 and hIAPP37 aggregations as shown in Fig. 3 was observed only when adding aromadendrin to freshly prepared Aβ or hIAPP monomer solutions. But little is known about which amyloid species can more effectively interact with aromadendrin and be converted into a higher-order species. To address this issue, we designed a new experimental protocol by adding one equivalent aromadendrin to Aβ or hIAPP solutions seeded at different times (i.e., 1, 3, 5, 7, and 8 h) in 37 °C, then monitored the changes of the aggregation kinetics before and after adding aromadendrin by recording ThT signals. Different amyloid seed solutions preformed at 1, 3, 5, 7 and 8 h presented different amyloid species at the lag, growth, and equilibrium phases. In Fig. 6a, the addition of aromadendrin to the 1 h- or 3 h-seeded Aβ solutions displayed almost the same ThT aggregation kinetics curve as the addition of aromadendrin to freshly prepared Aβ solution, indicating that aromadendrin can interact with both Aβ monomers and small oligomers efficiently to accelerate their conversion towards mature fibrils. When adding aromadendrin to the 5 h-seeded Aβ solutions, on one hand, the aggregation rate was still accelerated and the final fibril amount was increased by 14% as compared to those of pure Aβ; on the other hand, this promotion effect by aromadendrin decreased as compared to the 25–32% increment of final fibril amount in the above-mentioned cases. Finally, when adding aromadendrin to the preformed Aβ fibrils at 8 h, no obvious promotion effect was observed, as indicated by the lack of growth rate increment and final fibril amounts in the ThT curve.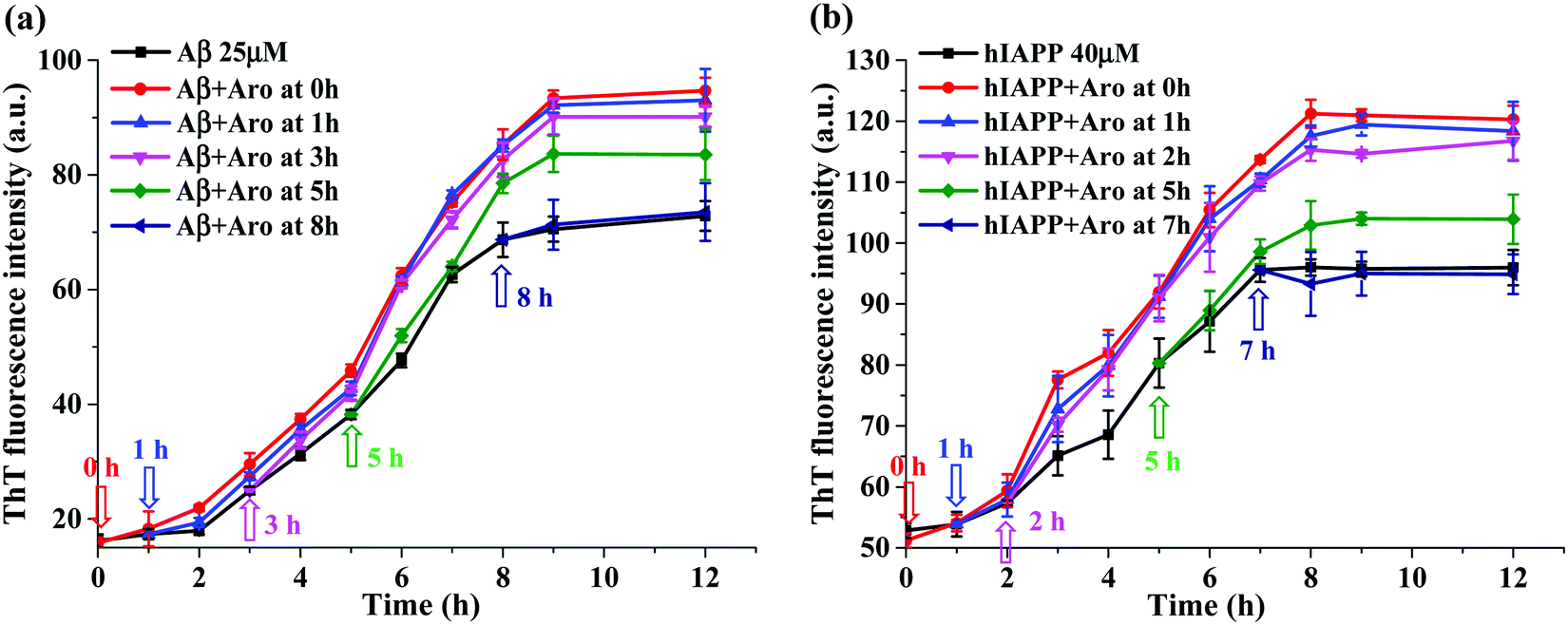 | ||
| Fig. 6 Time-dependent ThT fluorescence profiles by addition of equimolar aromadendrin to (a) Aβ42 (25 μM)- and (b) hIAPP37 (40 μM)-seeded solutions at different time points. Arrows indicate the time points of addition of aromadendrin to the amyloid-seeded solutions. Error bars represent the average of three replicate experiments. | ||
A similar amyloid seeding-dependent promotion effect was also observed for the aromadendrin-hIAPP cases. Due to the overall small fluorescence signal that comes from the aggregation of 25 μM hIAPP37, the concentration of hIAPP37 was increased (40 μM) in this study to improve the fluorescence intensity for better comparison. As shown in Fig. 6b, aromadendrin accelerated the aggregation of hIAPP monomers (0 h-seeded), early oligomers (1 h-and 2 h-seeded), and larger oligomers (5 h-seeded), as compared to that of pure hIAPP aggregation without aromadendrin. But, the reduced acceleration effect of aromadendrin on the 5 h-seeded hIAPP was observed and evidenced by ThT curves, in which the increment of final fibril amount was 10.5%, less than the 26.3% increment of hIAPP fibrils as promoted from —1- and 2h-seeding by aromadendrin. Finally, this promotion effect of aromadendrin on 7 h-seeded hIAPP seeds disappeared. All these seeding results confirm that aromadendrin is more effective to promote the aggregation and conversion of amyloid species formed at the early lag and growth phase, but once the higher-ordered amyloid species with more β-structure are formed, aromadendrin becomes less effective to transform them into more β-structure-rich aggregates and to catalyze them into mature fibrils.
2.4. Aromadendrin reduces the Aβ- and hIAPP-induced cell toxicity
Our ThT and AFM data show that the introduction of aromadendrin shortened the lag phase of both Aβ and hIAPP aggregation, which may provide another strategy to suppress the cell toxicity of Aβ and hIAPP by quickly bypassing their toxic oligomers. To test this hypothesis, we studied the effect of aromadendrin molecules on the cell toxicity of Aβ and hIAPP aggregates using MTT and LDH assays, where SH-SY5Y and RIN-m5F cell lines were used separately to study the toxicity of Aβ and hIAPP. As a control, we first measured the signal intensity of the separately cultured SH-SY5Y and RIN-m5F cells and then set it as a baseline corresponding to 100% cell viability. Then, aromadendrin (125 μM)-treated SH-SY5Y cells exhibited 8.4% (24 h)/11.7% (48 h) cell apoptosis (Fig. 7a1, LDH assay) and 88.7% (24 h)/77.2% (48 h) cell viability (Fig. 7a2, MTT assay), while aromadendrin (125 μM)-treated RIN-m5F cells exhibited 3.9% (24 h)/8.4% (48 h) cell apoptosis (Fig. 7b1, LDH assay) and 96.5% (24 h)/90.2% (48 h) cell viability (Fig. 7b2, MTT assay), indicating that aromadendrin presents very low cytotoxicity to both SH-SY5Y and RIN-m5F cells. In all cases, the increase in incubation time always led to an increase in cell apoptosis and a decrease in cell viability. Therefore, below we mainly discuss cell toxicity results after 48 h incubation.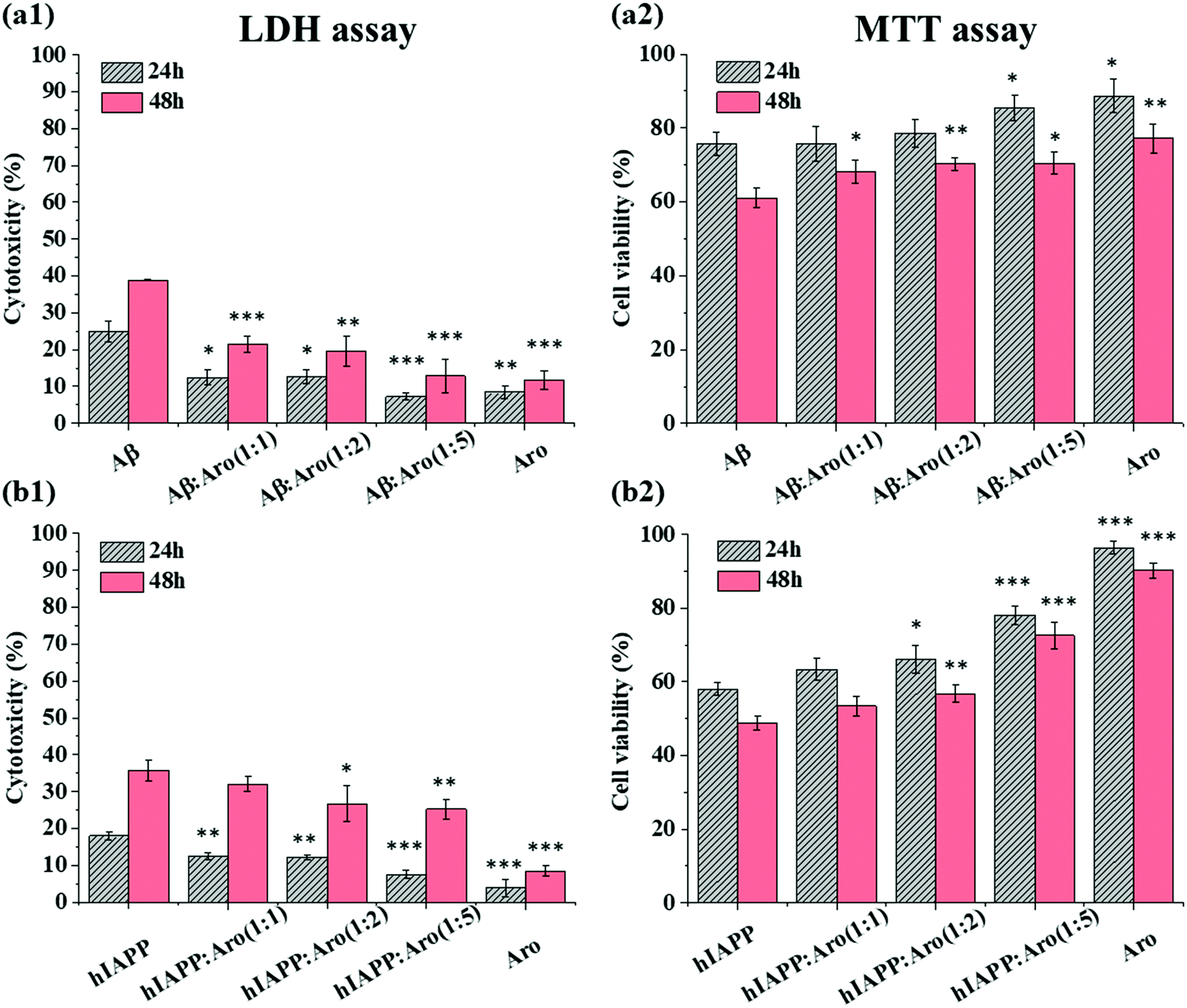 | ||
| Fig. 7 (a1 and b1) LDH cell apoptosis assay and (a2 and b2) MTT cell viability assay for (a1 and a2) Aβ42 (25 μM) and (b1 and b2) hIAPP37 (25 μM)-incubated without and with aromadendrin at different amyloid:aromadendrin molar ratios of 1:1, 1:2, and 1:5. Cells treated with aromadendrin at a concentration of 125 μM alone were tested as a control. In all the figures, the data represent mean ± s.d. of three independent experiments (n = 3). Cells treated with aromadendrin were analyzed using a t-test relative to cells only treated with amyloids (*P < 0.05, **P < 0.01, ***P < 0.001). | ||
In the case of Aβ-aromadendrin, when incubating 25 μM Aβ alone with SH-SY5Y for 48 h, cell apoptosis (LDH assay, Fig. 7a1) and viability (MTT assay, Fig. 7a2) were 38.9% and 61.1%, respectively, showing the high toxicity of Aβ to neuron cells. For comparison, co-incubation of Aβ with aromadendrin of all concentrations increased viability and decreased apoptosis in a concentration-dependent manner, as compared to Aβ-treated cells. Specifically, upon 48 h incubation, aromadendrin of 25, 50, and 125 μM increased the respective viabilities to 68.3% (P < 0.05), 70.3% (P < 0.01), and 70.5% (P < 0.05) and decreased the respective apoptosis to 21.5% (P < 0.001), 19.6% (P < 0.01), and 12.8% (P < 0.001). A similar protective effect of aromadendrin on hIAPP-treated RIN-m5F cells was observed in Fig. 7b. When co-incubating aromadendrin with hIAPP for 48 h, aromadendrin at all concentrations protected the cells from the hIAPP-induced toxicity, as evidenced by the reduction of cell apoptosis from 35.8% to 32.1% (25 μM), 26.7% (50 μM, P < 0.05), and 25.2% (125 μM, P < 0.01) and the increase of cell viability from 48.7% to 53.4% (25 μM), 56.7% (50 μM, P < 0.01), and 72.6% (125 μM, P < 0.001). Taken together, aromadendrin can rescue SH-SY5Y cells from Aβ-induced MTT reduction by 12–15% and apoptosis by 45–67%, as well as RIN-m5F cells from hIAPP-induced MTT reduction by 10–49% and apoptosis by 10–30%. Comparison of amyloid aggregation kinetics with amyloid-induced cell toxicity in the presence of aromadendrin reveals a mechanistic possibility that aromadendrin-promoted amyloid aggregation is likely to sequester toxic oligomers by quickly converting them to less-toxic amyloid fibrils and off-pathway non-toxic aggregates, so that they can no longer effectively engage in the toxic pathways.
Numerous studies have shown that amyloid-induced cell toxicity is likely attributed to the disruption and leakage of cell membranes.50,51 To further explore the underlying protection mechanism of aromadendrin against Aβ- and hIAPP-induced cell apoptosis, we examined the effect of aromadendrin (125 μM) on modulating the Aβ- and hIAPP-induced membrane leakage using LUVs composed of DOPC and DOPS as model cell membranes. LUVs were loaded with 5(6)-carboxyfluorescein that is self-quenched in the interior of LUVs. Any membrane leakage allows the 5(6)-carboxyfluorescein to release into the solution, whose dilution effect leads to relief of self-quenching and enhancement in observable fluorescence. So, the percentage of fluorescence change of the dye is positively related to the percentage of membrane leakage. First, we fabricated LUVs pre-loaded with 5(6)-carboxyfluorescein, showing a uniform size distribution of ∼228 nm by DLS (Fig. 8a). Then, we measured the time-dependent membrane leakage of LUVs when incubating either Aβ or hIAPP (25 μM) in the absence and presence of aromadendrin (125 μM) and the results are presented in Fig. 8b and c. As a control, aromadendrin (125 μM) alone did not cause obvious membrane leakage. On the other hand, both Aβ- and hIAPP-treated LUVs experienced a rapid membrane leakage of 60–70% (i.e. fluorescence intensity increase) at the first ∼4 h and finally reached their maximum levels of membrane leakage of 80–90% at 12 h. Side-by-side comparison of ThT fluorescence curves (Fig. 3a and b) with membrane leakage curves (Fig. 8b and c) revealed that the rapid oligomer formation and the initial burst membrane leakage occur at the same period of 0–4 h, and this indicates that amyloid oligomers formed at the early lag and growth stages are the main species to induce the membrane leakage. Moreover, the addition of aromadendrin (125 μM) led to a significant decrease (P < 0.01) in membrane leakage as induced by Aβ (25 μM) and hIAPP (25 μM), i.e., aromadendrin reduced Aβ-induced membrane leakage by 23% and hIAPP-induced membrane leakage by 24%, relative to aromadendrin-untreated controls. Collectively, cell assay results demonstrate that aromadendrin reduces amyloid-induced cytotoxicity via two possible pathways, (i) by accelerating the aggregation process to quickly transform toxic amyloid oligomers to less-toxic amyloid fibrils and (ii) by reducing membrane leakage caused by membrane penetrating oligomers.
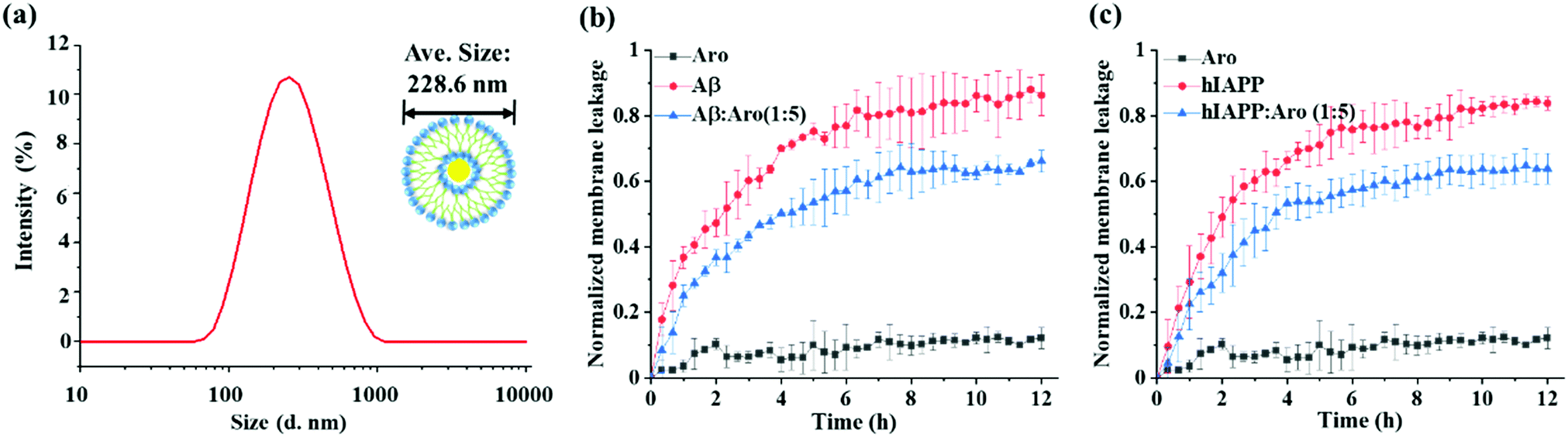 | ||
| Fig. 8 Effect of aromadendrin on membrane leakage. (a) Size distribution of as-prepared large unilamellar vesicles (LUVs). Comparison of (b) Aβ42- and (c) hIAPP37-induced membrane leakage in the absence and presence of aromadendrin at an amyloid:aromadendrin molar ratio of 1:5 (25 μM:125 μM). Data represent mean ± s.d. of three independent experiments (n = 3). The time-dependent membrane leakage in the presence of amyloid or amyloid:aromadendrin (1:5) was analyzed by two-way ANOVA. Comparison of membrane leakage between amyloid:aromadendrin (1:5) and pure amyloid samples revealed a Pvalue <0.01. | ||
3. Conclusions
Amyloid formation, particularly early stages of amyloid aggregation, has been implicated in different cell dysfunctions and death responsible for many neurodegenerative diseases. Since amyloid oligomers are well recognized as the main toxic species causing cell cytotoxicity, development of different drugs/compounds to reduce or avoid the formation of amyloid oligomers is considered as a more effective interfering strategy for neurodegenerative diseases. In this work, we discovered aromadendrin, a natural flavonoid that exists in various medicinal herbs, as an amyloid promoter (not amyloid inhibitor) to dramatically accelerate the amyloid aggregation and fibrillization of both Aβ42 and hIAPP37 by shortening the lag phase and promoting the growth phase to quickly transform the toxic oligomers into less-toxic amyloid fibrils and off-pathway aggregates. Aromadendrin showed the concentration-dependent promotion effect on the aggregation kinetics of both Aβ42 and hIAPP37, leading to 1.86–2.14 fold increase in Aβ fibrils and 1.20–1.68 fold increase in hIAPP fibrils at amyloid:aromadendrin molar ratios of 1:1, 1:2, and 1:5. Moreover, addition of aromadendrin into the preformed amyloid seed solutions incubated at different time points indicated that aromadendrin was very effective in converting early amyloid aggregates to amyloid fibrils, but was unable to promote the growth of preformed amyloid protofibrils. Consequently, both MTT and LDH assays confirmed that aromadendrin reduced cellular toxicity mediated by Aβ42 in SH-SY5Y cells and hIAPP37 in RIN-m5F cells. At an optimal amyloid:aromadendrin molar ratio of 1:5 and 48 h co-incubation with cells, aromadendrin rescued SH-SY5Y cells from Aβ-induced toxicity by 12–15% and apoptosis by 45–67%, and RIN-m5F cells from hIAPP-induced toxicity by 10–49% and apoptosis by 10–30%. LUV assays further showed that the introduction of aromadendrin can largely reduce 23% Aβ-induced membrane leakage and 24% hIAPP-induced membrane leakage. The timelines for amyloid-induced membrane leakage and the formation of amyloid oligomers are well aligned to each other. Such correlation suggests that the introduction of aromadendrin can effectively sequester toxic amyloid oligomers, thus remodeling their aggregation pathways and disruptive interactions with cell membranes. This work provides a different strategy by repurposing old drugs/compounds as a general amyloid promoter to alleviate the devastating aggregation process and toxic amyloid species. More importantly, the sequence-independent promotion effect of aromadendrin on Aβ and hIAPP could also be extended to other amyloid peptides for fighting against different amyloid diseases. Given that (i) this work has demonstrated the promotion effect of aromadendrin on both Aβ and hIAPP aggregation and (ii) our and other previous works have demonstrated the cross-seeding between Aβ and hIAPP,37,52–54 it is of great interest to study the effect of aromadendrin or other dual inhibitors/promoters on the cross-seeding of Aβ and hIAPP in the future, which may provide alternative pharmaceutical strategies to block the cross-seeding between different amyloid peptides so as to prevent their pathological transmission between cells and tissues. In addition, given the limited progress on developing amyloid promoters, it is still challenging to discover therapeutic promoters capable of (i) strong specific binding affinity towards monomeric or oligomeric amyloids, (ii) rapid structural conversion into β-sheet-rich structures, or (iii) producing off-pathway promoter-amyloid aggregates for minimal cytotoxicity.
4. Materials and methods
4.1. Materials
Amyloid β peptide (1–42), Aβ42, and human islet amylin, hIAPP37, with more than 95% purity were purchased from AnaSpec (Fremont, CA). Aromadendrin (≥98%) was purchased from AdipoGen Life Sciences (San Diego, CA). 1,1,1,3,3,3-Hexafluoro-2-propanol (HFIP, ≥99.9%), 10 mM PBS buffer (pH = 7.4), and thioflavin T (ThT, ≥98%) were purchased from Sigma (St Louis, MO). Dimethyl sulfoxide (DMSO, ≥99.9%), and human neuroblastoma SH-SY5Y cell line, and rat insulinoma cells (RIN-m5F) were purchased from ATCC (Manassas, VA). 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC, ≥97.0%) was purchased from TCI America (Portland, OR). 1,2-Dioleoyl-sn-glycero-3-phospho-L-serine (DOPS, ≥99.0%) was purchased from Avanti Polar Lipids (Alabaster, AL).4.2. Aβ and hIAPP peptide monomer preparation
The as-received 1 mg of Aβ42 and hIAPP37 peptides were dissolved in 1 mL of HFIP and kept at ambient temperature for 2 h to eliminate the pre-existing protofibrils or seeds. To further remove the aggregates, the peptide/HFIP solution was sonicated in an ice bath for 30 min, then centrifuged at 14000 rpm, and 4 °C for 30 min. The supernatant was divided into several aliquots and lyophilized for 24 h to obtain the ready-to-use peptide monomers.
4.3. Large unilamellar vesicle (LUV) preparation
The DOPC and DOPS were dissolved in chloroform and mixed in a 7:3 molar ratio. After evaporating the mixture under a nitrogen atmosphere and further drying in a high vacuum for at least 30 min, a lipid film was obtained, which was then hydrated in Tris buffer (10 mM Tris–HCl and 100 mM NaCl at pH 7.4) at a concentration of 10 mM for 1 h. The lipid solution was then subjected to 10 freeze–thaw cycles and extruded 10 times through 200 nm pore size filters to obtain a uniform LUV size. For the membrane leakage experiment, LUVs containing 5(6)-carboxyfluorescein were prepared similarly, except that the lipid film was hydrated in 5(6)-carboxyfluorescein containing Tris buffer. The free 5(6)-carboxyfluorescein was removed from the 5(6)-carboxyfluorescein-filled LUVs using a PD 10 desalting column (GE Healthcare Life Science) by carefully eluting with Tris buffer (10 mM Tris–HCl, 100 mM NaCl, and pH 7.4). Dynamic light scattering was used to ensure the proper diameter and polydispersity and the vesicle solution should be used right after being prepared.
4.4. Thioflavin T (ThT) fluorescence assay
ThT fluorescence was monitored by using the kinetic bottom-read mode of a SpectraMax M3 microplate reader (Molecular Devices, San Jose, CA) with an excitation wavelength of 450 nm and emission wavelength in the range from 470 nm to 500 nm. Samples were prepared by dissolving the peptide in the absence or presence of aromadendrin in 10 μM ThT–Tris buffer solution. The ThT fluorescence measurement was conducted in triplicate per sample, and data were recorded consistently at 37 °C.4.5. Atomic force microscopy (AFM)
During the aggregation process, the morphology changes of the peptides were imaged by using a Nanoscope III multimode AFM instrument in a tapping mode. The peptides were incubated in PBS buffer with or without aromadendrin at 37 °C, and aliquots of the solution (∼20 μL) at different time intervals were deposited on mica for 2 min at ambient temperature. The AFM samples were washed with DI water to eliminate salts, dried using airflow and stored in sealed containers, scanned in the tapping mode with a scan rate of 1.0 Hz and silicon AFM probe with 300 kHz resonant frequency.4.6. Circular dichroism (CD) spectroscopy
The secondary structure of the peptide can be monitored by far-UV CD spectroscopy using a J-1500 spectropolarimeter (Jasco Inc., Japan). The peptide samples were incubated at 37 °C in 10 mM phosphate-buffered saline (PBS, pH 7.4) in the presence or absence of aromadendrin. Aliquots (∼150 μL) were continuously added into the 1 mm optical path length cuvette, and the spectra were scanned between 190 and 250 nm with a step size of 0.5 nm and 50 nm min−1 scan rate. The spectra were analyzed after subtracting the background signal of pure PBS buffer, and taking an average of three scans per sample. The secondary structures were predicted by using the Beta Structure Selection (BeStSel) algorithm52 (http://bestsel.elte.hu/).4.7. Membrane permeability assays
The leakage experiment was conducted to evaluate the peptide affiliation and induced damage to the membrane. All of the samples were incubated in triplicate in a 96-well quartz microplate at 37 °C. The peptide-added 10 mM 5(6)-carboxyfluorescein encapsulated LUV (in 10 mM Tris·HCl, and 100 mM NaCl at pH 7.4) solution was the control group, and the peptide with aromadendrin was added into the LUV solution as the test group. Additionally, maximum leakage should be determined by adding Triton X-100 to a final concentration of 0.1% (v/v) to disrupt membranes thoroughly. The leakage was monitored through the 5(6)-carboxyfluorescein fluorescence signal, and the signal was continuously measured using a SpectraMax M3 microplate reader with excitation and emission wavelengths of 485 nm and 535 nm, respectively. The release of the fluorescent dye was calculated as the percent fluorescence change L(t) = (Ft − F0)/(F100 − F0), where L(t) represents the normalized membrane leakage, Ft is the measured fluorescence intensity at time t, F0 is the initial fluorescence intensity at time t = 0 and F100 is the intensity after addition of Triton X-100.4.8. Cell cultures
Human SH-SY5Y neuroblastoma cells were cultured in sterile-filtered Eagle's minimum essential medium (EMEM, ATCC, Manassas, VA) and Ham's F-12 medium (1:1) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. RIN-m5F were incubated in sterile-filtered RPMI-1640 medium mixed with 10% fetal bovine serum and 1% penicillin/streptomycin. Cell cultures were maintained in a 5% CO2 humidified incubator at 37 °C.
4.9. Cell toxicity assay
The cell toxicity was assessed by lactate dehydrogenase (LDH) and the cell viability was determined by 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT). SH-SY5Y/RIN-m5F cells were incubated in a 96-well plate at 104 cells per well density for 12 h, and the peptide, aromadendrin and peptide–aromadendrin solutions were added into wells in sextuplicate. Following 24/48 h incubation, the supernatant medium was removed from each well, incubation of the cells was continued with 0.5 mg mL−1 MTT at 37 °C. After 4 h incubation, the supernatant was extracted and dimethyl sulfoxide was added to dissolve the formazan crystals. The absorbance values of the purple formazan were determined at 590 nm using a microplate reader, which reflects the number of viable cells present. LDH, a cytosolic enzyme that will be released into the cell culture medium after the plasma membrane is damaged, and the released LDH can be quantified by a coupled enzymatic reaction. To a 96-well plate incubated with 104 cell density, 10 μLs of peptide, aromadendrin, and peptide–aromadendrin solution were added to the test group, 10 μL of sterile and ultrapure water was added to the spontaneous LDH activity control group, and 10 μL of lysis buffer (10×) was added to the maximum LDH activity control group. The leaked LDH in the medium was evaluated using the Pierce™ LDH Cytotoxicity Assay Kit (Thermo, USA), and the absorbance was read at the wavelengths of 490 nm and 680 nm using a microplate reader.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We highly appreciate the previous financial supports from NSF grants (1510099, 1158447 and 0952624) over the past decade to move forward our amyloid works from fundamental research to translational research.References
- J.-C. Rochet and P. T. Lansbury Jr, Curr. Opin. Struct. Biol., 2000, 10, 60–68 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef CAS PubMed.
- W.-F. Xue, S. W. Homans and S. E. Radford, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8926–8931 CrossRef CAS PubMed.
- R. Limbocker, S. Chia, F. S. Ruggeri, M. Perni, R. Cascella, G. T. Heller, G. Meisl, B. Mannini, J. Habchi and T. C. Michaels, Nat. Commun., 2019, 10, 225 CrossRef CAS PubMed.
- H. J. Jung, H. A. Jung, B.-S. Min and J. S. Choi, Chem. Pharm. Bull., 2015, 63, 955–960 CrossRef CAS PubMed.
- B. Solomon, R. Koppel, E. Hanan and T. Katzav, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 452–455 CrossRef CAS PubMed.
- J. McLaurin, R. Cecal, M. Kierstead, X. Tian, A. L. Phinney, M. Manea, J. French, M. H. Lambermon, A. A. Darabie and M. E. Brown, Nat. Med., 2002, 8, 1263–1269 CrossRef CAS PubMed.
- Y. Du, X. Wei, R. Dodel, N. Sommer, H. Hampel, F. Gao, Z. Ma, L. Zhao, W. H. Oertel and M. Farlow, Brain, 2003, 126, 1935–1939 CrossRef PubMed.
- T. Takahashi and H. Mihara, Acc. Chem. Res., 2008, 41, 1309–1318 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Castaño and B. Frangione, Nat. Med., 1998, 4, 822–866 CrossRef CAS PubMed.
- W. Hoyer, C. Grönwall, A. Jonsson, S. Ståhl and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5099–5104 CrossRef CAS PubMed.
- B. Ren, B. Jiang, R. Hu, M. Zhang, H. Chen, J. Ma, Y. Sun, L. Jia and J. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 20476–20485 RSC.
- B. Ren, Y. Liu, Y. Zhang, M. Zhang, Y. Sun, G. Liang, J. Xu and J. Zheng, J. Mater. Chem. B, 2018, 6, 56–67 RSC.
- B. Ren, Y. Liu, Y. Zhang, Y. Cai, X. Gong, Y. Chang, L. Xu and J. Zheng, ACS Chem. Neurosci., 2018, 9, 1215–1224 CrossRef CAS PubMed.
- Q. Wang, X. Yu, K. Patal, R. Hu, S. Chuang, G. Zhang and J. Zheng, ACS Chem. Neurosci., 2013, 4, 1004–1015 CrossRef CAS.
- A. I. Bush, Neurobiol. Aging, 2002, 23, 1031–1038 CrossRef CAS.
- D. J. Hayne, S. Lim and P. S. Donnelly, Chem. Soc. Rev., 2014, 43, 6701–6715 RSC.
- D. T. Meier, L.-H. Tu, S. Zraika, M. F. Hogan, A. T. Templin, R. L. Hull, D. P. Raleigh and S. E. Kahn, J. Biol. Chem., 2015, 290, 30475–30485 CrossRef CAS PubMed.
- Y. H. Liao, Y. J. Chang, Y. Yoshiike, Y. C. Chang and Y. R. Chen, Small, 2012, 8, 3631–3639 CrossRef CAS.
- S. I. Yoo, M. Yang, J. R. Brender, V. Subramanian, K. Sun, N. E. Joo, S. H. Jeong, A. Ramamoorthy and N. A. Kotov, Angew. Chem., Int. Ed., 2011, 50, 5110–5115 CrossRef CAS PubMed.
- N. Xiong, X.-Y. Dong, J. Zheng, F.-F. Liu and Y. Sun, ACS Appl. Mater. Interfaces, 2015, 7, 5650–5662 CrossRef CAS PubMed.
- Y. Feng, S.-G. Yang, X.-T. Du, X. Zhang, X.-X. Sun, M. Zhao, G.-Y. Sun and R.-T. Liu, Biochem. Biophys. Res. Commun., 2009, 390, 1250–1254 CrossRef CAS PubMed.
- J. Bieschke, M. Herbst, T. Wiglenda, R. P. Friedrich, A. Boeddrich, F. Schiele, D. Kleckers, J. M. Lopez del Amo, B. A. Grüning, Q. Wang, M. R. Schmidt, R. Lurz, R. Anwyl, S. Schnoegl, M. Fändrich, R. F. Frank, B. Reif, S. Günther, D. M. Walsh and E. E. Wanker, Nat. Chem. Biol., 2012, 8, 93–101 CrossRef CAS PubMed.
- J. Chen, A. H. Armstrong, A. N. Koehler and M. H. Hecht, J. Am. Chem. Soc., 2010, 132, 17015–17022 CrossRef CAS PubMed.
- T. Mohamed, S. S. Gujral and P. P. N. Rao, ACS Chem. Neurosci., 2018, 9, 773–782 CrossRef CAS PubMed.
- T. Antony, W. Hoyer, D. Cherny, G. Heim, T. M. Jovin and V. Subramaniam, J. Biol. Chem., 2003, 278, 3235–3240 CrossRef CAS PubMed.
- J. Luo, C.-H. Yu, H. Yu, R. Borstnar, S. C. Kamerlin, A. Gräslund, J. P. Abrahams and S. K. Wärmländer, ACS Chem. Neurosci., 2013, 4, 454–462 CrossRef CAS PubMed.
- H. Gong, X. Zhang, B. Cheng, Y. Sun, C. Li, T. Li, L. Zheng and K. Huang, PLoS One, 2013, 8, e54198 CrossRef CAS PubMed.
- L.-P. Gao, H.-C. Chen, Z.-L. Ma, A.-D. Chen, H.-L. Du, J. Yin and Y.-H. Jing, Biochim. Biophys. Acta, Gen. Subj., 2020, 1864, 129422 CrossRef.
- L. Zhang, Q. Chen, P. Li, L. Yuan, Y. Feng, J. Wang, X. Mao and L. Liu, Chem. Commun., 2019, 55, 14359–14362 RSC.
- E. H. Pilkington, M. Lai, X. Ge, W. J. Stanley, B. Wang, M. Wang, A. Kakinen, M.-A. Sani, M. R. Whittaker, E. N. Gurzov, F. Ding, J. F. Quinn, T. P. Davis and P. C. Ke, Biomacromolecules, 2017, 18, 4249–4260 CrossRef CAS PubMed.
- T. M. Ryan, A. Friedhuber, M. Lind, G. J. Howlett, C. Masters and B. R. Roberts, J. Biol. Chem., 2012, 287, 16947–16954 CrossRef CAS PubMed.
- A. Assarsson, S. Linse and C. Cabaleiro-Lago, Langmuir, 2014, 30, 8812–8818 CrossRef CAS PubMed.
- Q. Fan, Y. Liu, X. Wang, Z. Zhang, Y. Fu, L. Liu, P. Wang, H. Ma, H. Ma, N. P. Seeram, J. Zheng and F. Zhou, ACS Chem. Neurosci., 2020, 11, 638–647 CrossRef CAS.
- Y. Peretz, R. Malishev, S. Kolusheva and R. Jelinek, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1810–1817 CrossRef CAS PubMed.
- B. Ren, Y. Zhang, M. Zhang, Y. Liu, D. Zhang, X. Gong, Z. Feng, J. Tang, Y. Chang and J. Zheng, J. Mater. Chem. B, 2019, 7, 7267–7282 RSC.
- M. Baram and Y. Miller, ACS Chem. Neurosci., 2020, 11, 445–452 CrossRef CAS PubMed.
- M. Baram, S. Gilead, E. Gazit and Y. Miller, Chem. Sci., 2018, 9, 4244–4252 RSC.
- M. Zhang, R. Hu, H. Chen, X. Gong, F. Zhou, L. Zhang and J. Zheng, J. Chem. Inf. Model., 2015, 55, 1628–1639 CrossRef CAS PubMed.
- M. Zhang, R. Hu, H. Chen, Y. Chang, J. Ma, G. Liang, J. Mi, Y. Wang and J. Zheng, Phys. Chem. Chem. Phys., 2015, 17, 23245–23256 RSC.
- M. Zhang, R. Hu, B. Ren, H. Chen, B. Jiang, J. Ma and J. Zheng, ACS Chem. Neurosci., 2017, 8, 524–537 CrossRef CAS PubMed.
- A. Muhammad, G. Tel-Cayan, M. Öztürk, S. Nadeem, M. E. Duru, I. Anis, S. W. Ng and M. R. Shah, Ind. Crops Prod., 2015, 78, 66–72 CrossRef CAS.
- X. Zhang, T. M. Hung, P. T. Phuong, T. M. Ngoc, B.-S. Min, K.-S. Song, Y. H. Seong and K. Bae, Arch. Pharmacal Res., 2006, 29, 1102–1108 CrossRef CAS PubMed.
- J.-W. Lee, N. H. Kim, J.-Y. Kim, J.-H. Park, S.-Y. Shin, Y.-S. Kwon, H. J. Lee, S.-S. Kim and W. Chun, Biomol. Ther., 2013, 21, 216 CrossRef CAS PubMed.
- J. H. Kwak, M. W. Kang, J. H. Roh, S. U. Choi and O. P. Zee, Arch. Pharmacal Res., 2009, 32, 1681–1687 CrossRef CAS PubMed.
- W. Y. Zhang, J.-J. Lee, I.-S. Kim, Y. Kim and C.-S. Myung, Pharmacology, 2011, 88, 266–274 CrossRef CAS PubMed.
- P. Arosio, T. P. Knowles and S. Linse, Phys. Chem. Chem. Phys., 2015, 17, 7606–7618 RSC.
- Y. Liu, B. Ren, Y. Zhang, Y. Sun, Y. Chang, G. Liang, L. Xu and J. Zheng, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1906–1916 CrossRef CAS PubMed.
- J. R. Brender, S. Salamekh and A. Ramamoorthy, Acc. Chem. Res., 2011, 45, 454–462 CrossRef PubMed.
- S. Han, M. Kollmer, D. Markx, S. Claus, P. Walther and M. Fändrich, Sci. Rep., 2017, 7, 43577 CrossRef PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci., 2015, 112(24), E3095–E3103 CrossRef CAS PubMed.
- I. Moreno-Gonzalez, G. Edwards III, N. Salvadores, M. Shahnawaz, R. Diaz-Espinoza and C. Soto, Mol. Psychiatry, 2017, 22(9), 1327–1334 CrossRef CAS PubMed.
- R. Hu, M. Zhang, H. Chen, B. Jiang and J. Zheng, ACS Chem. Neurosci., 2015, 6(10), 1759–1768 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |