
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning protein delivery from different architectures of layer-by-layer assemblies on polymer films†
Giovanne
Delechiave
,
Alliny F.
Naves
,
Elayaraja
Kolanthai
,
Rubens A.
da Silva
,
Raphael C.
Vlasman
,
Denise F. S.
Petri
 ,
Roberto M.
Torresi
and
Luiz H.
Catalani
*
,
Roberto M.
Torresi
and
Luiz H.
Catalani
*
Departamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, CP 26077, 05508-000, São Paulo, Brazil. E-mail: catalani@usp.br; Tel: +55(11)30911811
First published on 27th July 2020
Abstract
In the past two decades, the fabrication of nanolayer coatings on polymeric materials has triggered considerable interest in the tissue-engineering field. Layer-by-Layer (LbL) assembly has attracted substantial attention due to being a convenient and low-cost method to coat materials with biomolecules while keeping their original biofunctionalities. This work aims to access a deeper understanding of the processes of adsorption and desorption of proteins by studying different architectures for the loading and release of model proteins on spin-coated flat surfaces of biodegradable polyesters such as poly(ε-caprolactone) (PCL) and poly(L-lactic acid) (PLLA) by the LbL technique. Surfaces of Si-wafers coated with PCL and PLLA were used as substrates for deposition of model proteins such as bovine serum albumin and lysozyme using heparin and chitosan as a pair of polyelectrolytes for LbL assembly. The analysis of the attributes of these protein reservoirs such as the protein layer position, layer thickness, surface morphology, protein adsorption and desorption quantification and kinetics was performed by ellipsometry, atomic force microscopy, fluorescence microscopy, and quartz-crystal microbalance with enhanced dissipation measurements. A homogenous LbL coating on the surface of the polyester films was achieved by assembling ten layer-pairs of reservoirs. The release profiles were evaluated, and the results showed an intricate dependence on the protein volume charge density and its relation to coacervation/complexation processes with both polyelectrolytes. Our results clearly suggest that the protein delivery kinetics can be fully controlled by precisely positioning the protein within the assembly (bottom vs. top position). They also suggest that the size and total charge of the protein are key factors for controlling its total load in the assembly.
1. Introduction
The combination of bioactive molecules and biodegradable scaffolds gives new possibilities in regenerative medicine. In addition to physical support for cell attachment, these scaffolds can also provide the appropriate mechanical and biological signals to initiate and guide the growth of new tissue. Efforts have been made to include a bioactive proteic material on the surface of scaffolds, as well as to develop the means to control its delivery. Several articles report the use of Layer-by-Layer (LbL) assembly as an efficient architecture to resolve this quest and open perspectives to apply these reservoirs in tissue engineering.1–5LbL assembly, complexation between oppositely charged polyelectrolytes,6 has been applied to a myriad of surfaces (metal,7 glass,8 polymers,9etc.) and shapes (flat,1 porous,9 spherical,10etc.). The main applications include drug delivery devices used to incorporate biomolecules of interest10 or to act as a barrier for sustained release.11 In the case of signaling biomolecules, some examples include the fabrication of poly(lactic-co-glycolic) acid (PLGA) membranes coated with polyelectrolyte multilayers comprised of poly(beta-amino esters) and poly(acrylic acid) for entrapment of bone morphogenetic protein-2 and platelet-derived growth factor, combined or not in the same device. The sustained dual release of growth factors could induce bone repair in a critical-size defect model in rat calvaria and accelerate bone reconstruction.12 In this same trend, the building of nano reservoirs made from chitosan/bone morphogenetic protein-7 (BMP7) multilayers on polycaprolactone (PCL) fibers was able to accelerate in vivo bone mineralization and regeneration.13 The loading of bFGF in PLGA microspheres,14,15 or free-standing films,16 and vascular endothelial growth factor in PLLA17 and PLGA14 electrospun fibers also demonstrates the use of LbL for storage of signaling molecules.
The recent demonstration of the use of heparin (Hep) and chitosan (Chi) solutions loaded with acid or basic fibroblast growth factors (aFGF and bFGF, respectively) opens questions regarding the role of the total charge of the protein in the event. One of the advantages observed using co-solutions of polyelectrolytes and proteins is the long-term storage detected for such LbL reservoirs.18 Even after nine months, the (Hep/Chi) multilayers kept the activity of the embedded growth factors. Also, the preparation of co-solutions with suitable polyelectrolytes creates a mild environment that prevents protein degradation in solution due to the loss of its tertiary structure, enabling the protein to maintain its stability for the prolonged periods required to prepare the reservoirs.
Therefore, the current work aims to create LbL reservoirs of Hep/Chi assembled on polymeric films of PCL and PLLA, using acidic and basic model proteins, namely lysozyme (LYS) and bovine serum albumin (BSA). We have presented a systematic study of reservoir growth onto these polymeric surfaces, including the development of control over the amount of loaded proteins and their release, by elucidating essential parameters in the LbL preparation process. In this way, we intend to gather essential information for the development of reservoirs containing new cell signaling molecules and their application in tissue engineering.
2. Materials and methods
2.1. Materials
PCL (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w 80
w 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Aldrich, USA) and PLLA (intrinsic viscosity 0.32 g dL−1, w 130000 g mol−1, Purasorb PL 32) were used to prepare thin spin-coated films supported on Si-wafers (University Wafers, USA) with a native SiO2 layer. LYS and BSA (pI 10.2 and 5.1, respectively, experimentally obtained on a Zetasizer Nano Zs – Malvern), from Sigma (USA), were chosen as model proteins to assemble the reservoirs. Heparin sodium salt (w 15000 g mol−1) was from CalBiochem (USA). Chitosan (w 83600 g mol−1), from Aldrich (USA), was purified before use.19 Polyethyleneimine (PEI, w 25000 g mol−1) and fluorescein 5-isothiocyanate (FITC), from Aldrich (USA), and chloroform, ethanol, acetic acid, CaCl2, P2O5, and NaOH, from Synth (Brazil), were used as received. Chloroform, from Synth (Brazil), was dried under P2O5 and distilled before use. Ultrapure water of Milli-Q quality (18.2 MΩ cm at 25 °C, Milli-Q Gradient, Millipore, USA) was used for all the experiments and rinsing steps.
000 g mol−1, Aldrich, USA) and PLLA (intrinsic viscosity 0.32 g dL−1, w 130000 g mol−1, Purasorb PL 32) were used to prepare thin spin-coated films supported on Si-wafers (University Wafers, USA) with a native SiO2 layer. LYS and BSA (pI 10.2 and 5.1, respectively, experimentally obtained on a Zetasizer Nano Zs – Malvern), from Sigma (USA), were chosen as model proteins to assemble the reservoirs. Heparin sodium salt (w 15000 g mol−1) was from CalBiochem (USA). Chitosan (w 83600 g mol−1), from Aldrich (USA), was purified before use.19 Polyethyleneimine (PEI, w 25000 g mol−1) and fluorescein 5-isothiocyanate (FITC), from Aldrich (USA), and chloroform, ethanol, acetic acid, CaCl2, P2O5, and NaOH, from Synth (Brazil), were used as received. Chloroform, from Synth (Brazil), was dried under P2O5 and distilled before use. Ultrapure water of Milli-Q quality (18.2 MΩ cm at 25 °C, Milli-Q Gradient, Millipore, USA) was used for all the experiments and rinsing steps.
2.2. Methods
:protein mass ratios were prepared from stock protein solutions of LYS and BSA (1 mg mL−1 in water) and polyelectrolyte stock solutions of Hep (1 mg mL−1 in 0.15 M saline solution) and Chi (1 mg mL−1 in NaCl 0.15 M and 2% acetic acid). The initial protein concentration was 0.3 mg mL−1. The solutions were left under room temperature at rest for 15 min (same conditions as the LbL assembly), followed by soft stirring (homogenization) before the assay.
| Samples | Thickness (nm) | |
|---|---|---|
| Ellipsometry | AFM | |
| (Si/SiO2/PEI)(Hep/Chi)20 | 82 ± 1 | 75 ± 17 |
| (Si/SiO2/PEI)(Hep + BSA/Chi)20 | 80 ± 1 | 93 ± 11 |
| (Si/SiO2/PEI)(Hep/Chi + LYS)20 | 70 ± 1 | 83 ± 6 |
| (Si/SiO2/PEI)(Hep/Chi)10 | 34 ± 5 | 34 ± 2 |
| (Si/SiO2/PEI)(Hep + BSA/Chi)10 | 29 ± 3 | 40 ± 2 |
| (Si/SiO2/PEI)(Hep/Chi + LYS)10 | 39 ± 6 | 45 ± 5 |
| (Si/SiO2/PEI)(Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3 | 29 ± 1 | n.d. |
| (Si/SiO2/PEI)(Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi)3 | 25 ± 2 | n.d. |
The solutions for LbL assembly were prepared according to a methodology described previously.18 PEI 2.5 mg mL−1 was prepared in water (pH = 10.7). Stock solutions of Hep 1 mg mL−1 and Chi 1 mg mL−1 were prepared in NaCl 150 mmol L−1 (physiological conditions), and 2% acetic acid (v/v) was added to the chitosan solution. The dipping solutions were dilutions Hep 0.1 mg mL−1 and Chi 0.1 mg mL−1 in NaCl 150 mmol L−1 (pH = 6.1 and pH = 3.7, respectively). A co-solution of Hep 0.1 mg mL−1 with the model protein BSA 0.1 μg mL−1 (Hep + BSA, pH = 6.7) and a co-solution of Chi 0.1 mg mL−1 with the model protein LYS 0.1 μg mL−1 (Chi + LYS, pH = 3.0) were used as dipping solutions to build the reservoirs described in Table 1. PEI was adsorbed only in the primer layer to enable uniform LbL assembly. PEI deposition was performed by dipping the Si-wafers for 15 minutes, while for the spin-coated polyester films it was carried out overnight, followed by three rinsing steps and drying under an air stream. After the deposition of PEI, the Hep/Chi multilayers as well as the Hep + BSA/Chi or Hep/Chi + LYS reservoirs were assembled in an LbL manner by dipping the substrates (Si-wafers covered or not with polyester spin-coated films) alternately into polyelectrolyte solutions for 10 min, followed by three rinsing steps of one minute each. After each adsorption step, the substrates were dried under an air stream. The multilayers (Hep/Chi)10, (Hep + BSA/Chi)10, and (Hep/Chi + LYS)10, and the hybrid (Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3 and (Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi)3 were assembled on top of PCL or PLLA spin-coated films to measure the protein release according to the position of the reservoir in the multilayers. In addition, the reservoirs (Hep + BSA/Chi)10 and (Hep/Chi + LYS)10 were built on polyester films using co-solutions in which the concentration of BSA or LYS was raised to 1, 10 or 100 μg mL−1 (by keeping the polyelectrolytes at 0.1 mg mL−1) to evaluate the stability of the LbL reservoirs according to the increase of protein concentration in the co-solution.
| eiΔtanΨ = Rp/Rs = f(nk,dk,λ,φ) | (1) |
For calculation purposes, the refractive indexes of the components are considered as constants, assuming the films being uniform and isotropic. The refractive indexes were assumed as 3.88 for Si, 1.462 for SiO2, and 1 for air. Due to the small thickness of the LbL multilayers, their refractive index was assumed to be constant at 1.51 (generic values for proteins due to the presence of model proteins), and despite their slightly heterogeneous morphology, they were assumed to be uniform and isotropic films. Although this procedure gives slightly incorrect values for the absolute film thickness, it allows quick and precise determination of the relative film thicknesses. Each data point in the graphs (thickness versus layer number) is obtained from random measurements at two different points of the substrate surface. Samples were prepared in duplicate.
The adsorption of PEI, using a solution with concentration 2.5 mg mL−1, was measured overnight in flow on Au/PCL. PEI became a surface suitable for heparin immobilization, which is warranted by the formation of hydrogen bonds between the amine group residues of the PEI and the hydroxyl groups of the disaccharide. Due to the difference in charge densities between the polyelectrolytes, chitosan (positively charged) was immobilized on heparin (negatively charged). The adsorption and washing tests were performed in flow with the aid of a peristaltic pump. The interaction time of the polyelectrolytes (concentration at 0.1 mg mL−1) for the adsorption assays was 20 minutes, followed by 8 minutes of washing in NaCl solution (0.15 M). The BSA and LYS proteins were adsorbed on the multilayers using co-solutions with the polyelectrolytes (BSA in the solution of Hep and LYS in the Chi solution), where the concentrations of both proteins were 100 μg mL−1. This procedure was repeated 10 times. The following systems were coded as (Au/PCL/PEI)(Hep/Chi)10; (Au/PCL/PEI)(Hep + BSA/Chi)10 and (Au/PCL/PEI)(Hep/Chi + LYS)10, respectively.
A QCM-D experiment was performed for each modification step on the substrate using a Q-Sense E4 (Q-Sense – Sweden) in a flow cell module. For these measurements an AT-cut quartz crystal covered with Au (d = 14 mm) was used, with a fundamental frequency of 5 MHz. All steps were performed at a flow rate of (320 ± 20) μL min−1 at (25.00 ± 0.05) °C. The changes in resonance frequency (Δfn) and dissipation (ΔDn), where n denotes the fundamental tone recorded from the 5th to the 13th overtones, were simultaneously monitored as a function of time. For acoustically rigid films, the mass variation can be calculated from the resonance frequency variation, as described in Sauerbrey's equation (eqn (2)):21,22
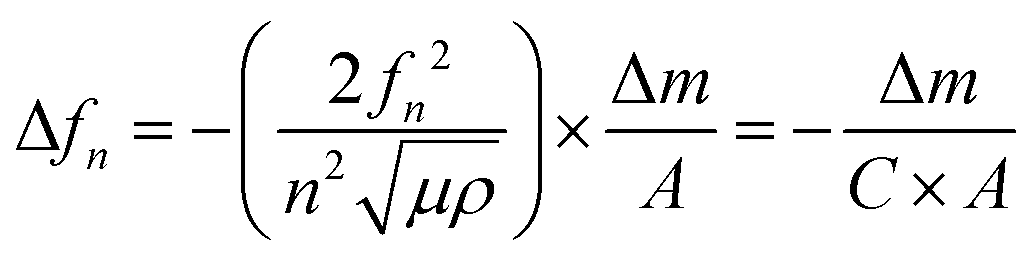 | (2) |
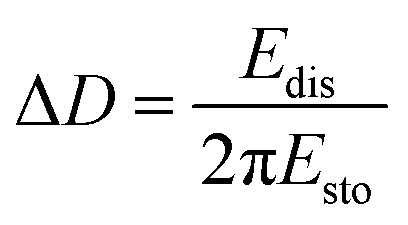 | (3) |
3. Results and discussion
Layer-by-layer self-assembly is a simple construct made from alternate adsorption of a positive and a negative polyelectrolyte on a substrate material such as glass, metal, polymers, etc., where its stability is controlled by electrostatic interactions, hydrogen bonding or other secondary interactions. In this technique, the substrate also plays an important role based on its wettability and surface charge. We chose PCL and PLLA as two substrates showing different wettability and degradability rate, to test the storage of two model proteins of different charges – BSA and LYS – in between heparin and chitosan multilayers. Since BSA presents a pI of 5.1 (experimentally obtained, Zetasizer Nano Zs – Malvern), it is, therefore, negatively charged at neutral pHs; its inclusion in the reservoir was provided by the use of a co-solution with Hep. Likewise, the pI value for LYS is 11.5 (experimentally obtained, Zetasizer Nano Zs – Malvern) and it was included by the use of a co-solution with Chi. The inclusion of both proteins was tested in assemblies of 20 and 10 layer-pairs. Also, the inclusion of proteins only in the inner layers was examined to test eventual barrier effects on the release profile.3.1. Analyses of protein–polyelectrolyte interactions
Protein inclusion on the LbL assemblies was provided by the use of a co-solution with a polyelectrolyte of the same bulk charge. The central hypothesis is that the protein will interact with the superficial layer of the polyelectrolyte of opposite charge. It is, therefore, significant to understand beforehand the interaction of the opposed charge pairs Chi–BSA and Hep–LYS.A turbidimetric titration assay was used to access the formation of insoluble protein–polyelectrolyte complexes, in the form of coacervates. The experiment consisted of UV-vis analysis of progressive addition of polyelectrolytes to a fixed concentration of protein solution, qualifying the phase separation of the complexes LYS–Hep or BSA–Chi by the change in the transmittance values. Fig. 1A shows the progressive attenuation of the transmittance at 500 nm of a LYS solution with an increasing concentration of Hep. At a molar ratio of 0.25 for the Hep:LYS co-solution, a reduction of 25% in the transmittance is observed, showing that a coacervate is readily formed. With increasing molar ratio, the attenuation of transmittance levels up as a result of saturation of the interaction, evidencing the strong interaction between Hep and LYS. Contrarily, Fig. 1A shows a minimal change in transmittance for the Chi:BSA solution, probably due to a much weaker interaction between Chi and BSA, despite the opposite charge of the components at the working pH.
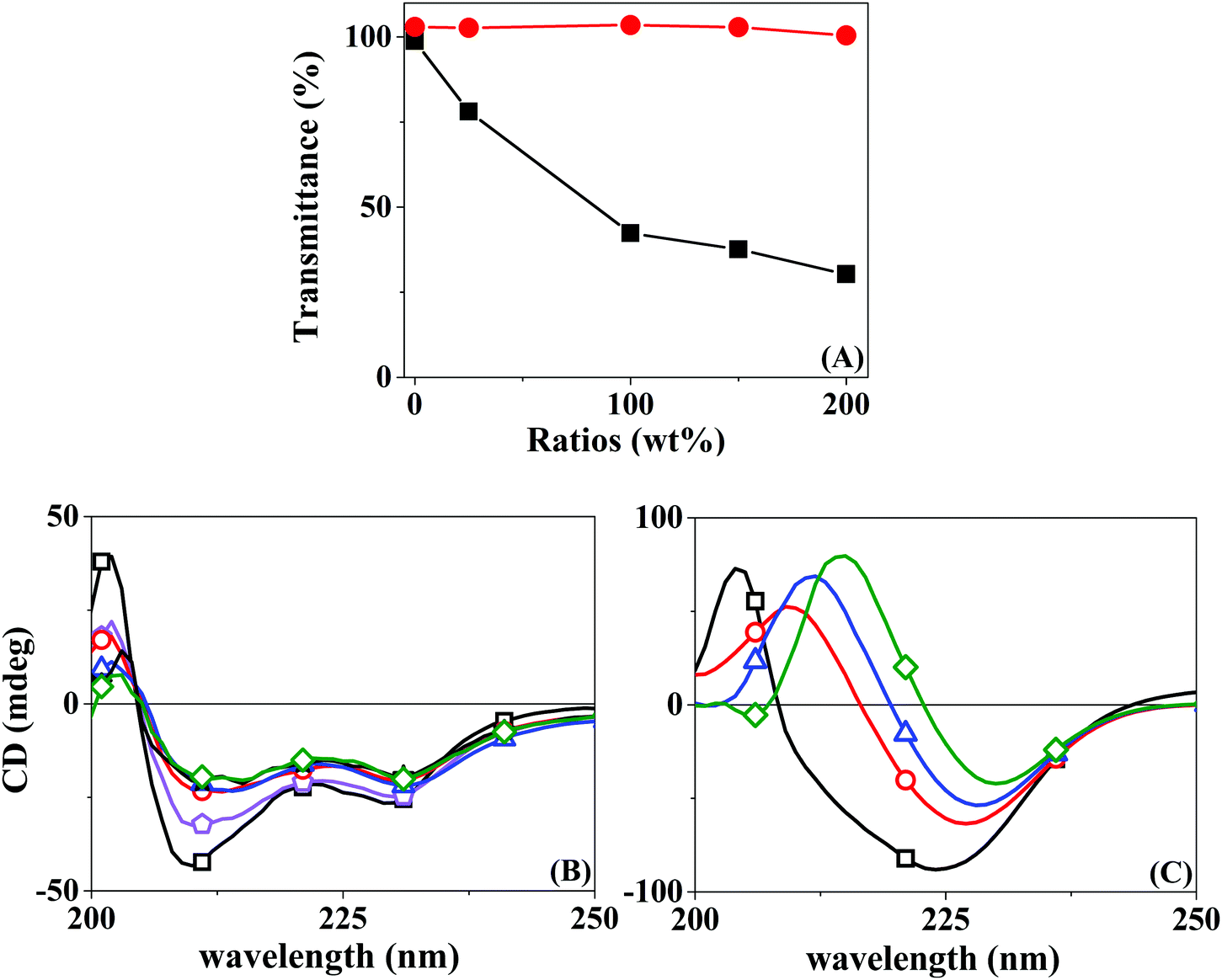 | ||
Fig. 1 (A) Turbidimetry assay transmittance at 500 nm of Hep:LYS (—■—) and Chi:BSA ( ) at different polyelectrolyte:protein ratios, from a fixed protein concentration of 0.3 mg mL−1; (B) CD spectra of Hep:LYS and (C) Chi:BSA with a polyelectrolyte:protein ratio of 0 (—□—), 0.25 ( ) at different polyelectrolyte:protein ratios, from a fixed protein concentration of 0.3 mg mL−1; (B) CD spectra of Hep:LYS and (C) Chi:BSA with a polyelectrolyte:protein ratio of 0 (—□—), 0.25 ( ), 1.0 ( ), 1.0 ( ), 1.5 ( ), 1.5 ( ), and 2.0 ( ), and 2.0 ( ), from a fixed protein concentration of 0.06 mg mL−1. ), from a fixed protein concentration of 0.06 mg mL−1. | ||
Circular dichroism can also be used to confirm the complex formation by observing changes in the secondary structure of the protein. Likewise, CD spectra were acquired for the protein solution under progressive increments of the polyelectrolyte, in the region of 200–250 nm. Fig. 1B shows a progressive change in the absorption spectra when Hep is added to a LYS solution. This result seems to relate to the partial loss of the α-helix structure, with stabilization at a higher Hep:LYS ratio. Similar results were observed by Billsten et al.27 for LYS adsorption over silica nanoparticles. For the pair Chi:BSA, the changes are more prominent, with a continuous alteration at higher mass ratios, suggesting denaturation of the protein structure. Pristine BSA contains 67% helical structures and 23% extended chain configurations.28 The changes observed in Fig. 1C can be correlated to the loss of α-helices and an increase of extended chains.29 The impact on the protein structure is mostly due to the acidity of the polyelectrolyte solution since Chi is solubilized by the addition of 2% acetic acid, as shown by the control experiment. Although Takeda et al.30 showed a significant decrease in the helical content of a BSA solution at pH 2.8 in the range of 2–65 °C, the changes observed were shown to be fully reversible.
The differences between the two pairs are evident. While by turbidimetry, a test for macrophase separation, Hep and LYS seem to form a complex readily, BSA is less sensitive to the presence of Chi. On the other hand, the CD results show a finite effect of Hep over LYS, but a softer and rising effect of Chi over BSA. In a recent review, Obermeyer et al.31 discuss the design parameters for an effective protein–polyelectrolyte complex. The authors claim that “protein charge patchiness” is a crucial factor for coacervate formation. Indeed, the total protein charge is not the only parameter for complex formation. Although the two proteins used here are of opposite charges, as indicated by their pI values, the sizes are rather different. While LYS is a protein of only 14 kD in size, the BSA molecular weight is 66 kD.
To quantify the protein–polyelectrolyte interactions studied here, the energy of the complexation process was obtained by using Isothermal Titration Calorimetry (ITC). By applying a one-site model to the obtained results (Fig. 2), the complexation of Hep:LYS was shown to be an exothermic and spontaneous process, reaching a ΔH value of −69.1 ± 0.7 kcal mol−1 (ΔS = −205 cal mol−1 deg−1, ΔG = 35.3 kcal mol−1 K−1, K = 1.7 ± 0.1 × 106 M−1) with 0.136 ± 0.001 ligation sites for Hep per LYS molecule (Fig. 2A).
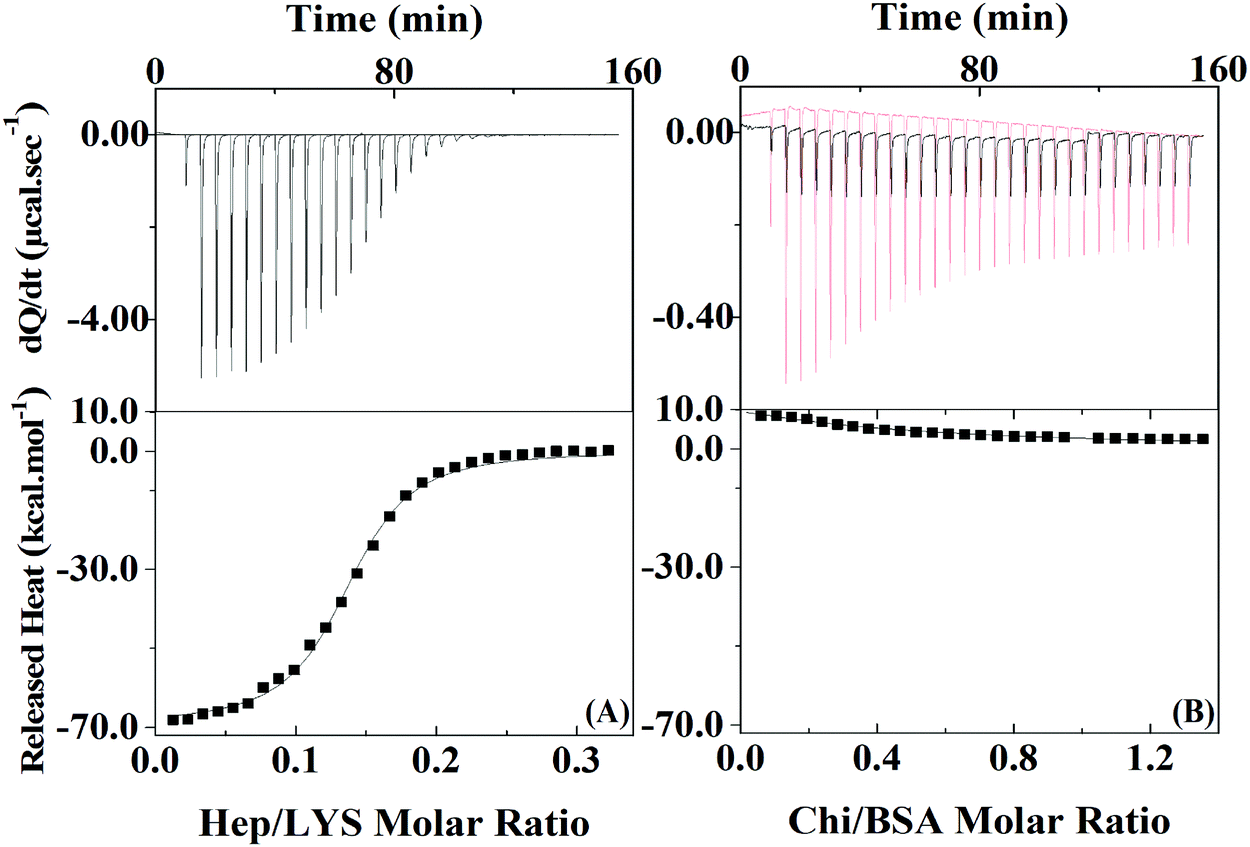 | ||
| Fig. 2 ITC measurements for Hep:LYS (left) and Chi:BSA (right) at 23 °C analyzed by the one-site model. Injections of polyelectrolyte were added to an initial protein concentration of 152 μM for LYS and 20 μM for BSA. | ||
On the other hand, the ITC results for the complexation between Chi and BSA (Fig. 2B) showed it to be slightly endothermic. The one site model is not valid in this case since the errors of each parameter are on the same magnitude or higher than the mean value. Therefore, it is possible to extrapolate the heat of interactions to the first injection of polyelectrolyte to calculate ΔH,32 which is 8.5 kcal mol−1, as it is reasonable to assume that approximately 100% of the polyelectrolyte at this point is bonded to at least one protein of the initial solution. Since the system with BSA and Chi does not correlate with the one-site model, it is likely that no or just a small quantity of polyelectrolyte is complexing with the protein under the conditions of the LbL solutions.
3.2. Analyses of reservoir assembly by AFM
LbL assembly on PCL or PLLA coated silicon substrates did not give a clear-cut layer thickness measurement since the polymer material absorbs the energy of the incident laser beam. Hence, multilayers of Hep and Chi were built on bare Si-wafers to have a proper characterization of the LbL assembly by ellipsometry. Fig. 3 shows that the Hep/Chi multilayers or Hep + BSA/Chi and Hep/Chi + LYS reservoirs present an increased thickness profile based on exponential growth (Table 1) when depositing 20 (Fig. 3A) or 10 (Fig. 3B) layer-pairs. In neither case did the inclusion of BSA or LYS influence the growth profile.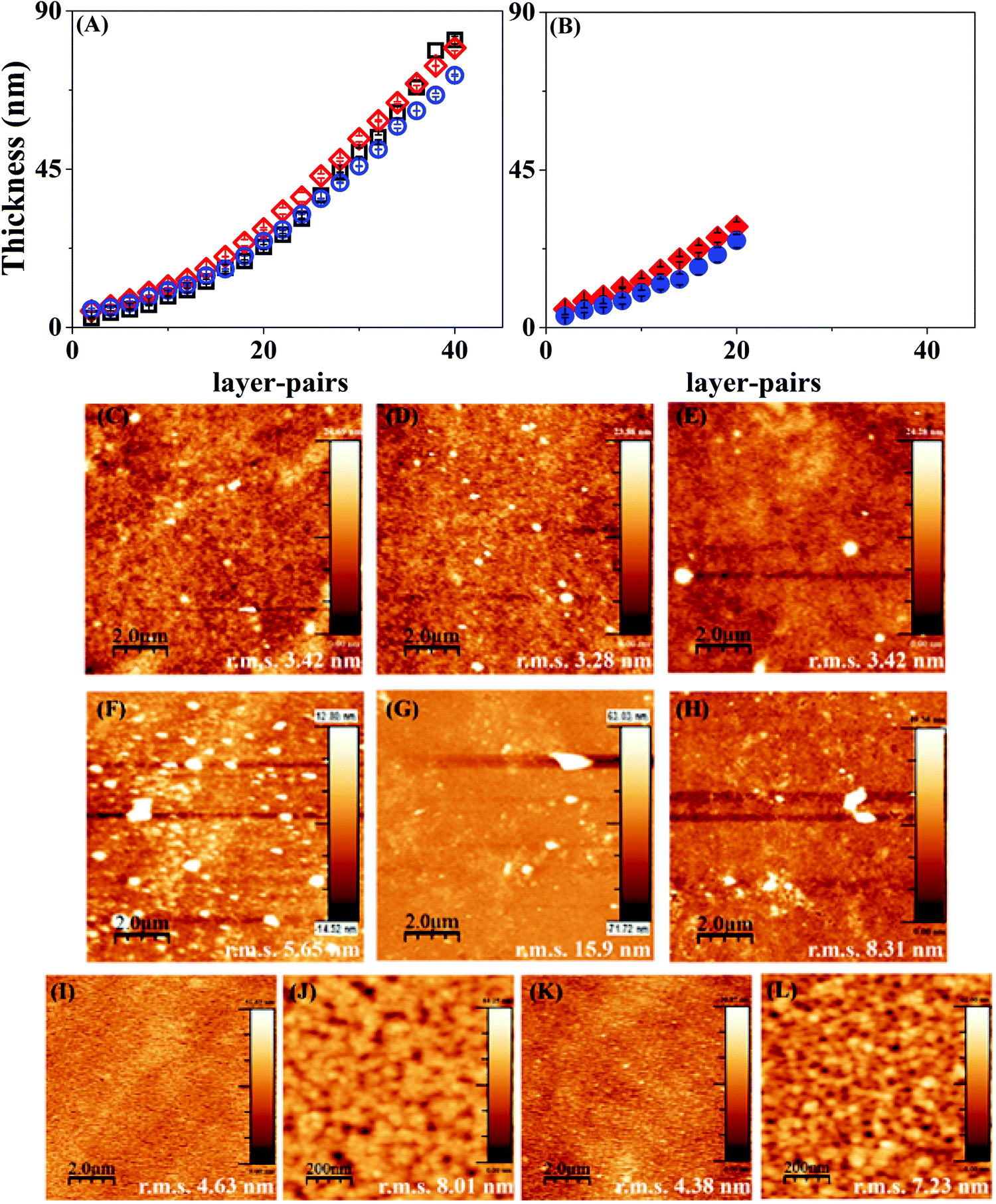 | ||
Fig. 3 Ellipsometry measurements for Hep/Chi multilayers with inclusion of BSA and LYS. (A) (Si/SiO2/PEI)(Hep/Chi)20 (—□—), (Si/SiO2/PEI)(Hep + BSA/Chi)20 ( ), (Si/SiO2/PEI)(Hep/Chi + LYS)20 ( ), (Si/SiO2/PEI)(Hep/Chi + LYS)20 ( ); (B) (Si/SiO2/PEI)(Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3 ( ); (B) (Si/SiO2/PEI)(Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3 ( ), (Si/SiO2/PEI)(Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi) ( ), (Si/SiO2/PEI)(Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi) ( ). AFM images for various multilayer configurations. (C) (Si/SiO2/PEI)(Hep/Chi)20, (D) (Si/SiO2/PEI)(Hep + BSA/Chi)20, (E) (Si/SiO2/PEI)(Hep/Chi + LYS)20, (F) (Si/SiO2/PEI)(Hep/Chi)10, (G) (Si/SiO2/PEI)(Hep + BSA/Chi)10, (H) (Si/SiO2/PEI)(Hep/Chi + LYS)10, (I and J) (Si/SiO2/PEI)(Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3, (K and L) (Si/SiO2/PEI)(Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi)3. The scanning areas are 10 μm × 10 μm, except for (J) and (L) which are 1 μm × 1 μm. ). AFM images for various multilayer configurations. (C) (Si/SiO2/PEI)(Hep/Chi)20, (D) (Si/SiO2/PEI)(Hep + BSA/Chi)20, (E) (Si/SiO2/PEI)(Hep/Chi + LYS)20, (F) (Si/SiO2/PEI)(Hep/Chi)10, (G) (Si/SiO2/PEI)(Hep + BSA/Chi)10, (H) (Si/SiO2/PEI)(Hep/Chi + LYS)10, (I and J) (Si/SiO2/PEI)(Hep/Chi)4(Hep + BSA/Chi)3(Hep/Chi)3, (K and L) (Si/SiO2/PEI)(Hep/Chi)4(Hep/Chi + LYS)3(Hep/Chi)3. The scanning areas are 10 μm × 10 μm, except for (J) and (L) which are 1 μm × 1 μm. | ||
The multilayers Hep/Chi, Hep + BSA/Chi, and Hep/Chi + LYS assembled with 20 or 10 layer-pairs on bare Si-wafers were also characterized by AFM measurements. Fig. 3(C–E) show that multilayers with 20 LP (samples ![[1 with combining low line]](https://www.rsc.org/images/entities/b_char_0031_0332.gif) –
–![[3 with combining low line]](https://www.rsc.org/images/entities/b_char_0033_0332.gif) ) have more homogenous topography than multilayers with 10 LP (Fig. 3(F–H); samples
) have more homogenous topography than multilayers with 10 LP (Fig. 3(F–H); samples ![[4 with combining low line]](https://www.rsc.org/images/entities/b_char_0034_0332.gif) –
–![[6 with combining low line]](https://www.rsc.org/images/entities/b_char_0036_0332.gif) ). Smaller r.m.s. values, around 3 nm for 20 LP multilayers, confirm their reduced roughness when compared to 10 LP multilayers as a consequence of the deposition process. The differences in thickness values obtained by ellipsometry or blade cutting measured by AFM (Table 1) suggest an anisotropic LbL structure due to the introduction of model proteins of BSA or LYS. Moreover, the thickness values determined by AFM show that the reservoirs containing BSA or LYS are thicker than the pristine Hep/Chi multilayers due to the presence of the proteins. BSA inclusion led to a somewhat higher increment of thickness compared to LYS, even when the proteins are included in inner layer pairs, as seen in Table 1 and Fig. 3B.
). Smaller r.m.s. values, around 3 nm for 20 LP multilayers, confirm their reduced roughness when compared to 10 LP multilayers as a consequence of the deposition process. The differences in thickness values obtained by ellipsometry or blade cutting measured by AFM (Table 1) suggest an anisotropic LbL structure due to the introduction of model proteins of BSA or LYS. Moreover, the thickness values determined by AFM show that the reservoirs containing BSA or LYS are thicker than the pristine Hep/Chi multilayers due to the presence of the proteins. BSA inclusion led to a somewhat higher increment of thickness compared to LYS, even when the proteins are included in inner layer pairs, as seen in Table 1 and Fig. 3B.
The AFM images of mixtures containing the proteins in inner layers (Fig. 3(I–L); samples ![[7 with combining low line]](https://www.rsc.org/images/entities/b_char_0037_0332.gif) and
and ![[8 with combining low line]](https://www.rsc.org/images/entities/b_char_0038_0332.gif) ) show that the additional 3 LP of Hep/Chi is enough to render a homogeneous coating on the top of the reservoirs. A similar roughness measured for both samples, around 4 nm (Fig. 3I and K), supports this observation. Higher magnification scanning (Fig. 3J and L) evidenced a homogeneous topography of the Hep/Chi top layers.
) show that the additional 3 LP of Hep/Chi is enough to render a homogeneous coating on the top of the reservoirs. A similar roughness measured for both samples, around 4 nm (Fig. 3I and K), supports this observation. Higher magnification scanning (Fig. 3J and L) evidenced a homogeneous topography of the Hep/Chi top layers.
When PCL and PLLA spin-coated films were used as substrates for LbL, the deposition of the PEI first layer was ineffective as compared to using standard Si-wafer conditions. The lack of stability of the LbL assembly thus formed was evident even after increasing tenfold the concentration of PEI (to a 2.5 mg mL−1 solution during 1 h), and Hep/Chi multilayers started desorbing from the surface of the spin-coated film after three layer-pairs. The AFM images for the PEI first layer after one hour of dipping show aggregates dispersed in the surface of both the PCL (Fig. 4A) and PLLA (Fig. 4C) spin-coated films. The low PEI first layer coating renders a non-homogeneous LbL assembly, and, as a consequence, the multilayers end up desorbing from the polymeric surface.
 | ||
| Fig. 4 AFM images for the first PEI layer adsorbed on PCL (1 h: (A), overnight: (B)) or PLLA (1 h: (C), overnight: (D)); scanning areas: 2.5 μm × 2.5 μm. AFM images for (PCL/PEI)(Hep/Chi)10 multilayers prepared with polyelectrolyte solutions at 1 mg mL−1 (E) and 0.1 mg mL−1 (F); (PLLA/PEI)(Hep/Chi)10 multilayers prepared with polyelectrolyte solutions at 1 mg mL−1 (G) and 0.1 mg mL−1 (H); scanning areas: 10 μm × 10 μm. | ||
The production of a stable LbL assembly on top of spin-coated films was possible by increasing the PEI dipping time to about 16 hours and using a higher concentration (2.5 mg mL−1) solution. Such observations can be explained by the lower interaction of PEI with polymeric films compared to Si-wafers. The electrostatic interactions are replaced by hydrophobic ones, which take more time to be established in non-charged polymeric surfaces. For the samples prepared by overnight dipping in PEI, AFM images evidence a more homogenous PEI layer and a surface of lower roughness (decrease in r.m.s. values) for both PCL (Fig. 4B) and PLLA (Fig. 4D). Based on these observations, the preparation of the PEI first layer from PEI 2.5 mg mL−1 dipping solution overnight was defined as most efficient to build the subsequent LbL multilayers on top of the polymeric films.
Another parameter to control the assembly of Hep/Chi multilayers on top of the polymeric films was the concentration of the polyelectrolytes, here tested at 1 and 0.1 mg mL−1. AFM images evidenced that multilayers prepared from higher concentrations (Fig. 4E and G, for PCL and PLLA, respectively) presented rougher surfaces (higher r.m.s. values) than the ones prepared with diluted solutions (Fig. 4F and H, for PCL and PLLA, respectively). The LbL assembly from diluted polyelectrolyte solutions showed a more homogeneous coating of the polymeric surfaces.
Also, the smaller r.m.s. values for multilayers built on PLLA films evidenced a more homogeneous LbL assembly as compared to PCL. This fact is the result of differences in the topography of pure PCL and PLLA after spin-coating. Thin films made of PCL are rougher and display topographic domains due to their higher crystallinity structure (Fig. 5A). Contrarily, PLLA films show flatter surfaces (Fig. 5D) and lower roughness, similar to materials like glass and mica surfaces (r.m.s. values around 0.18 nm and 0.05 nm, respectively33). The building of Hep + BSA/Chi or Hep/Chi + LYS reservoirs on top of the polymeric films showed a homogeneous topography after 10 LP (Fig. 5). By comparing the topographic profiles, it is possible to confirm that the BSA and LYS reservoirs were adsorbed on top of PCL (Fig. 5B and C, respectively) and PLLA (Fig. 5E and F, respectively).
 | ||
| Fig. 5 AFM images for the (A) PCL spin-coated film, (B) (PCL/PEI)(Hep + BSA/Chi)10, (C) (PCL/PEI)/(Hep/Chi + LYS)10, (D) the PLLA spin-coated film, (E) (PLLA/PEI)(Hep + BSA/Chi)10, (F) (PLLA/PEI)(Hep/Chi + LYS)10. Scanning areas: 2.5 μm × 2.5 μm. | ||
The stability of the reservoir assemblies was investigated by increasing the protein concentration in the dipping co-solution. No changes in roughness and topography were observed when comparing the r.m.s. values for the same scanning areas (2.5 μm × 2.5 μm). Reservoirs built on top of PCL films (Fig. S1, ESI†) present rougher surfaces than PLLA (Fig. S2, ESI†). However, this seems to be an effect of the PCL rugosity, as evidenced in Fig. 5A. As controls, the Hep/Chi multilayers assembled on top of PCL and PLLA (Fig. S3(A–D) and S3(E–H), respectively, ESI†) showed smaller r.m.s. values on average than the reservoirs with higher amounts of protein. The detailed AFM images from inside the holes that appear in PLLA – as a consequence of the dewetting process – confirm the assembly of both reservoirs with BSA and LYS (Fig. S3(I–J) and S3(K–L), respectively, ESI†). The protein position in the reservoir – bottom, middle, or top of the LbL assembly – did not influence the topography of the multilayers adsorbed on PCL and PLLA (Fig. S4 and S5, respectively, ESI†).
Fluorescence images were obtained for LbL reservoirs prepared with model proteins (Fig. 6(A–D)) or FITC-bound model proteins (Fig. 6(E–H)). LbL assemblies with non-bound proteins did not present fluorescence, while the fluorescent greenish color distributed in the surface, due to the presence of BSA–FITC or LYS–FITC, is evidence of the loading of the proteins on the LbL assembly.
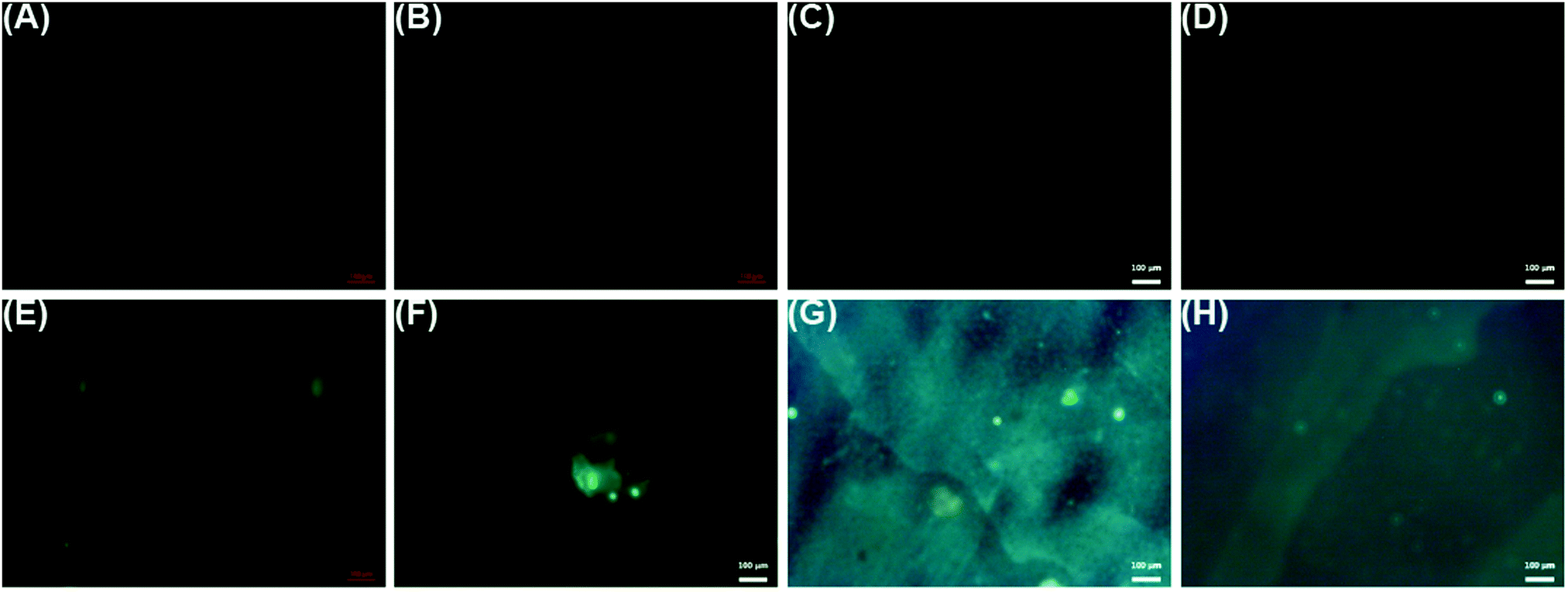 | ||
| Fig. 6 LbL reservoirs assembled with bare proteins: (A) (PCL(PEI)(Hep + BSA/Chi)10), (B) (PLLA/PEI)(Hep + BSA/Chi)10, (C) (PCL/PEI)(Hep/Chi + LYS)10, (D) (PLLA/PEI)(Hep/Chi + LYS)10; LbL reservoirs assembled with FITC-bound proteins: (E) (PCL/PEI)(Hep + BSA–FITC/Chi)10, (F) (PLLA/PEI)(Hep + BSA–FITC/Chi)10, (G) (PCL/PEI)(Hep/Chi + LYS–FITC)10, (H) (PLLA/PEI)(Hep/Chi + LYS–FITC)10. Scales: 100 μm. | ||
3.3. Analyses of reservoir assembly by QCM-D
The multilayers and reservoirs were assembled onto a Au sensor modified by adsorption of PCL and PEI (Au/PCL/PEI). Fig. 7 shows the amount of adsorbed mass (Δm) as a function of the layer-pair, for the experimental control (Hep/Chi) and the BSA reservoir (Hep + BSA/Chi) and LYS reservoir (Hep/Chi + LYS). The different assemblies onto Au/PCL/PEI feature rigidity with low dissipation shifts (Fig. S6, ESI†), allowing one to apply the Sauerbrey equation (eqn (2)).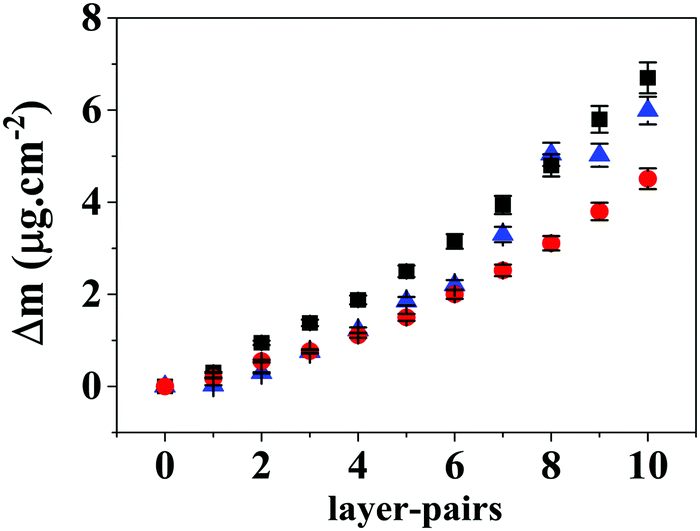 | ||
Fig. 7 Amount of deposited mass (Δm) as a function of the number of layer-pairs onto Au/PCL/PEI determined at (25.00 ± 0.05) °C for Hep/Chi ( ), Hep + BSA/Chi (■) and Hep/Chi + LYS ( ), Hep + BSA/Chi (■) and Hep/Chi + LYS ( ). Each point is the average of triplicates ± SD. ). Each point is the average of triplicates ± SD. | ||
The reported values are the average of triplicates of the data for different electrodes with a deviation of <5%. The adsorption of polyelectrolytes in multilayers (control experiment, blue triangles) and reservoir systems (Hep + BSA/Chi, black squares; Hep/Chi + LYS, red circles) was carried out in 150 mM NaCl at 25.00 ± 0.05 °C. The total amount of material adsorbed onto Au/PCL/PEI increases regardless of the protein (BSA or LYS) added to each layer-pair. The multilayers do not present a tendency of an adsorption plateau. At the tenth layer pair, the total mass deposited was 6.0 ± 0.3 μg cm−2, 6.7 ± 0.3 μg cm−2 and 4.5 ± 0.2 μg× cm−2 for (Au/PCL/PEI)(Hep/Chi)10, (Au/PCL/PEI)(Hep + BSA/Chi)10 and (Au/PCL/PEI)(Hep/Chi + LYS)10, respectively.
Fig. 8 shows the resonance frequency normalized by the overtone (Δf/n) and dissipation for each overtone (ΔDn) as a function of time, for the control experiment (Au/PCL/PEI)(Hep/Chi) (Fig. 8(A and B)), and BSA and LYS reservoirs (Au/PCL/PEI)(Hep + BSA/Chi)2 (Fig. 8(C and D)) and (Au/PCL/PEI)(Hep/Chi + LYS)2 (Fig. 8(E and F)), respectively. Here, two polyelectrolyte layer-pairs are assembled with a long time of adsorption and rinsing, to evaluate the multilayer stabilization and structure changes during each polyelectrolyte adsorption.
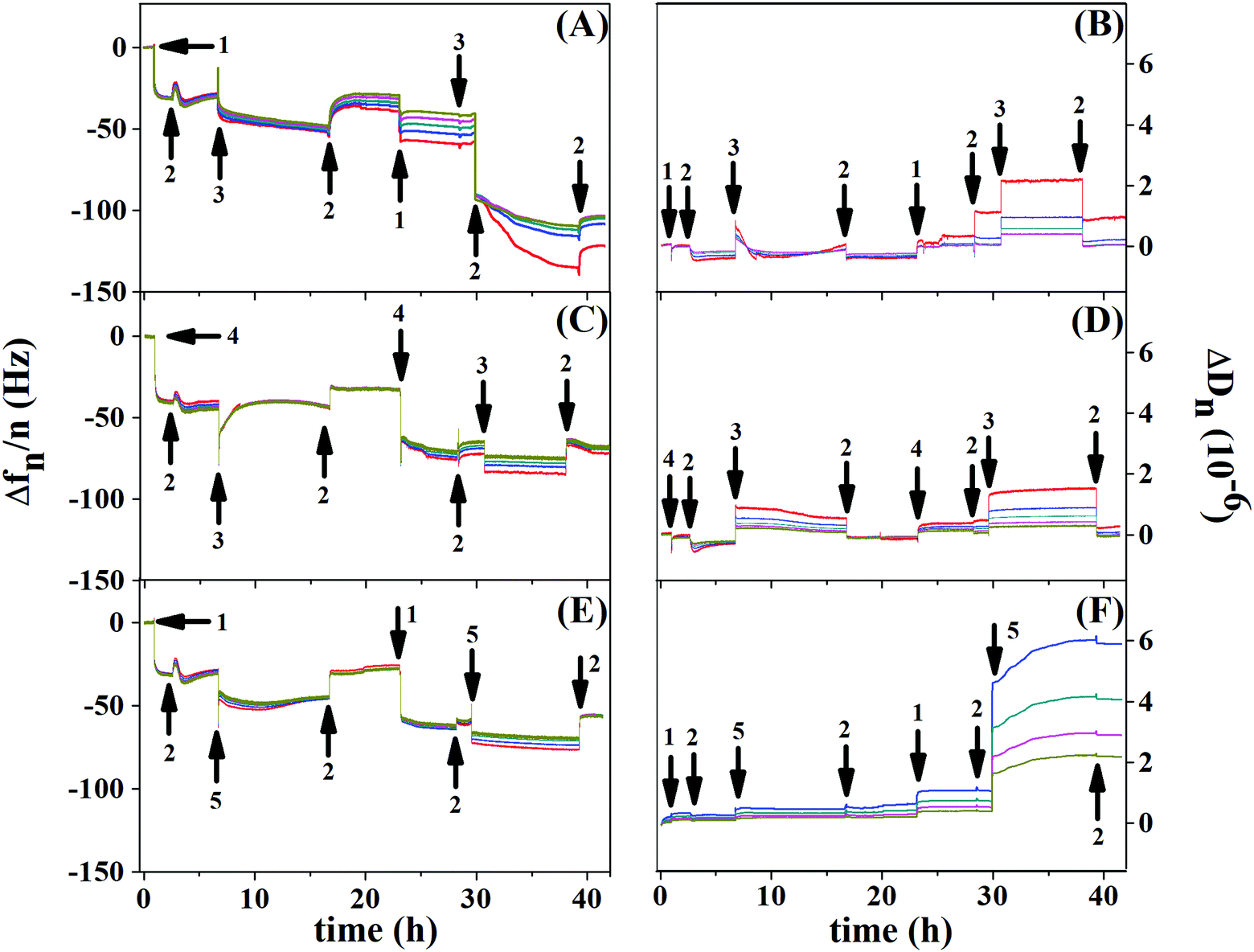 | ||
| Fig. 8 QCM-D in situ response given by Δfn/n (A, C and E) and ΔDn (B, D, and F) as a function of time for two layer-pairs for (Au/PCL/PEI)(Hep/Chi)2 (A and B), (Au/PCL/PEI)(Hep + BSA/Chi)2 (C and D) and (Au/PCL/PEI)(Hep/Chi + LYS)2 (E and F), respectively. The overtones (n) are 5 (red), 7 (blue), 9 (dark cyan), 11 (magenta), and 13 (dark yellow) for the eight steps involved in the modification of the gold substrates. Arrows 1, 2, 3, 4, and 5 indicate the addition of Hep, saline solution, Chi, Hep + BSA, and Chi + LYS, respectively. | ||
The first layer of Hep (Fig. 8A and E, arrow 1) and Hep + BSA (Fig. 8C, arrow 4) adsorbs over Au/PCL/PEI with a Δf decrease of 30 Hz for the control experiment, 40 Hz for the BSA reservoir and 44 Hz for the LYS reservoir. Rinsing with saline solution retains the frequency value for all systems (first arrow 2). However, exchange with Chi (Fig. 8A and C, first arrow 3) or Chi + LYS (Fig. 8E, first arrow 5) causes removal of the Hep layer previously adsorbed, as shown by the second rinse with saline solution (second arrow 2). Despite that, the building of the second layer-pair presents a frequency shift, which indicates new adsorption of Hep or HEP + BSA.
The second adsorption of Chi (Fig. 8A and C, second arrow 3) or Chi + LYS (Fig. 8E, second arrow 5) addition causes a frequency shift until stabilization, with a split in the overtones. This situation is changed during rinsing, likely due to the removal of the superficial Hep or HEP + BSA, leading to a frequency shift of −104, −70, and −74 Hz for the control experiment, and BSA and LYS reservoirs, respectively. Nevertheless, the rinsing can remove part of the polyanions previously adsorbed, a situation caused by strong complexation between Hep and Chi.34–36
The addition of Hep or HEP + BSA in the second bilayer removes the overtone splitting for both reservoirs (Fig. 8C, second arrow 1, and Fig. 8E, second arrow 4). The addition of Chi caused a split in the overtones for all the systems. Similar behavior was observed by Lundin and Kovacevic in layer-by-layer assemblies built with different polyelectrolytes.23,25 They attributed that phenomenon to the redissolution of complexes induced by strong complexation between the polyanion and polycation, likely due to hydrogen bonding between the carboxyl residue group of Hep and amino residue group of Chi. However, after the removal of the shallow Hep layer, Chi might diffuse into the thin film matrix of the deposited polyanion to find an electrostatic cross-linking partner. A more refined discussion on additional aspects shown by the results of Fig. 8 is presented in the ESI.†
Richert et al. reported similar phenomena in QCM-D responses for Au/(Chi/HA)36.24 Through the confocal laser microscopy technique, the authors showed fluorescently labeled Chi migrating inside the multilayers during their formation, while hyaluronan labeled (HA) remains fixed without migration.24 In our study, the diffusion of Chi into Hep fixed layers might cause a decrease in stiffness for the first layer-pair of the control system. Nevertheless, during the deposition of the following polyelectrolyte, a recovery of stiffness was observed, perhaps justified by associated events, such as an increase of density,23,25 demonstrated by dissipation frequencies smaller than 10 Hz (Fig. 8B, D, and F).
A similar QCM-D procedure to that applied to PCL was tried for PLLA; however, the PLLA film detached from the Au sensor because it swelled by absorbing water during the deposition of PEI, preventing the analysis.
3.4. Protein release assays
Considering that the Hep layers stay fixed while the Chi shifts through the multilayer (Section 3.3), therefore, keeping levels of intermixing of the layers and stratification, we propose here a control of the release dynamics by changing the relative position at which the protein is placed inside the reservoir. Three different configurations were designed using an assembly of 10 layer-pairs (Scheme 1): (a) (Hep/Chi)5(Hep + BSA/Chi)5 and (Hep/Chi)5(Hep/(Chi + LYS–FITC)5) where both proteins were loaded on the top 5 layer-pairs; (b) [(Hep/Chi)(Hep + BSA/Chi)]5 and [(Hep/Chi)(Hep/(Chi + LYS–FITC))]5 when 5 layers of proteins were evenly distributed within the 10 layer-pair assembly; and (c) (Hep + BSA/Chi)5(Hep/Chi)5 and (Hep/(Chi + LYS–FITC)5(Hep/Chi)5), where the proteins were loaded on the bottom 5 layer-pairs. The reservoirs for the release assays were assembled using dipping co-solutions of the polyelectrolytes containing protein concentrations of 100 μg mL−1 and release assays were performed in PBS at 37 °C in order to simulate physiological conditions.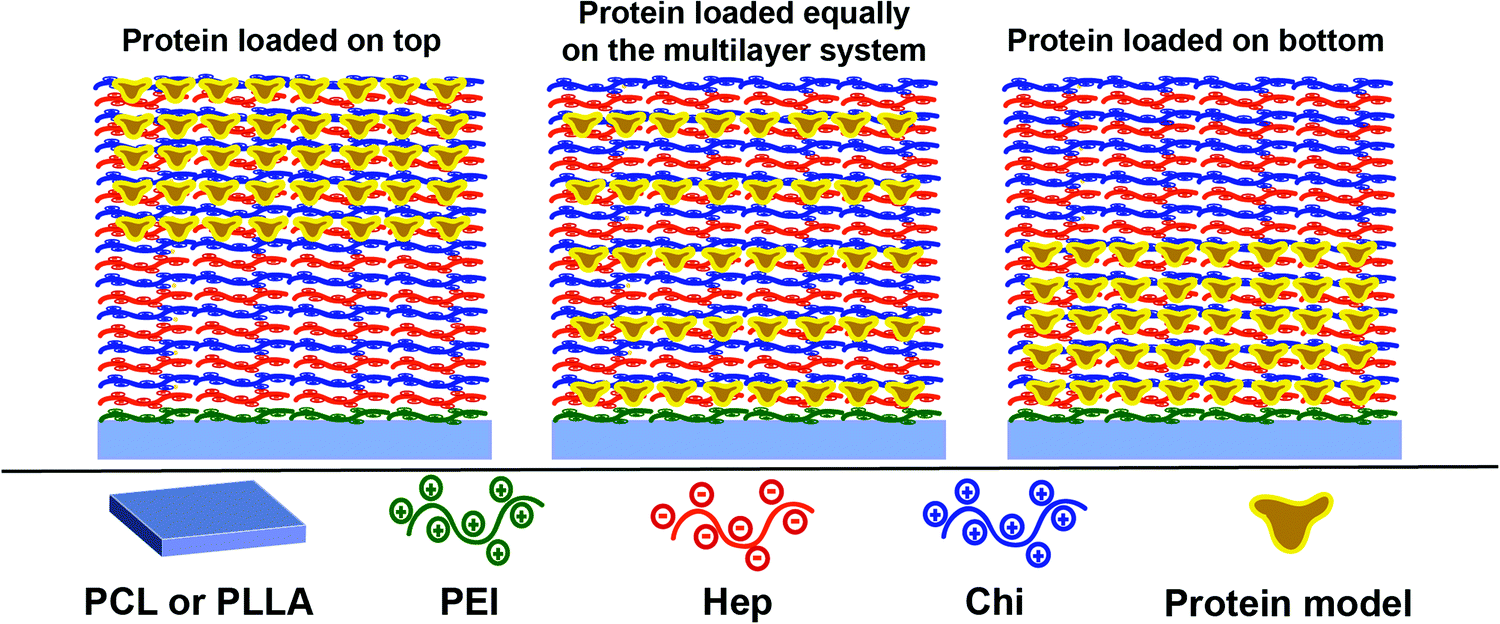 | ||
| Scheme 1 LbL reservoirs applied for the BSA and LYS release studies. | ||
The release of LYS was monitored by fluorescence using an FITC-labeled protein, since the functionalization process promoted only a light change of its pI, from 10.3 to 10.5. The release of BSA was monitored by immunofluorescence using BSA ELISA since the functionalization promoted an expressive drop of 0.8 of its isoelectric point (from 5.1 to 4.3), which risks a divergent situation of protein distribution.
Fig. 9A and B show the BSA release profiles from PCL and PLLA films. All profiles are similar and display an initial burst of around 0.1 to 0.3 ng cm−2 released after 3 h, followed by sustained release that reaches a plateau at about 0.8 ng cm−2 of BSA after 15 days, for both polymeric surfaces. Reservoirs assembled on PCL films with the protein loaded in the top 5 layer-pairs exhibited release slightly faster than the other two configurations. No significant differences were observed between BSA loaded on the bottom layers or evenly distributed. When the PLLA film was the support, no differences can be pinpointed among the three configurations. The amount of BSA released from PLLA reservoirs is somewhat lower than from PCL reservoirs. This result suggested that the surface of the base substrate (polymer) also plays a central role in the LbL self-assembly.
LYS (pI 11.5) was labeled with FITC (LYS–FITC, pI 11.3), a fluorescent probe, to enable the monitoring of its release by fluorescence techniques. Fig. 9C and D show LYS–FITC release profiles from the different multilayer architectures built on PCL and PLLA films, respectively. Similar to BSA, all reservoirs initially showed burst release followed by sustained release of LYS–FITC, reaching a plateau over 15 days. The final amount of LYS–FITC released from the PCL supports gradually decreased in the following order of reservoirs: protein in the top layers, protein evenly distributed, and protein in the bottom layers. This result indicated that a higher amount of LYS protein could be stored in the top layers. Moreover, the reservoir on the PCL substrate showed a significantly higher amount of LYS–FITC released compared to the PLLA film, somewhat repeating the observation from the BSA loaded reservoirs.
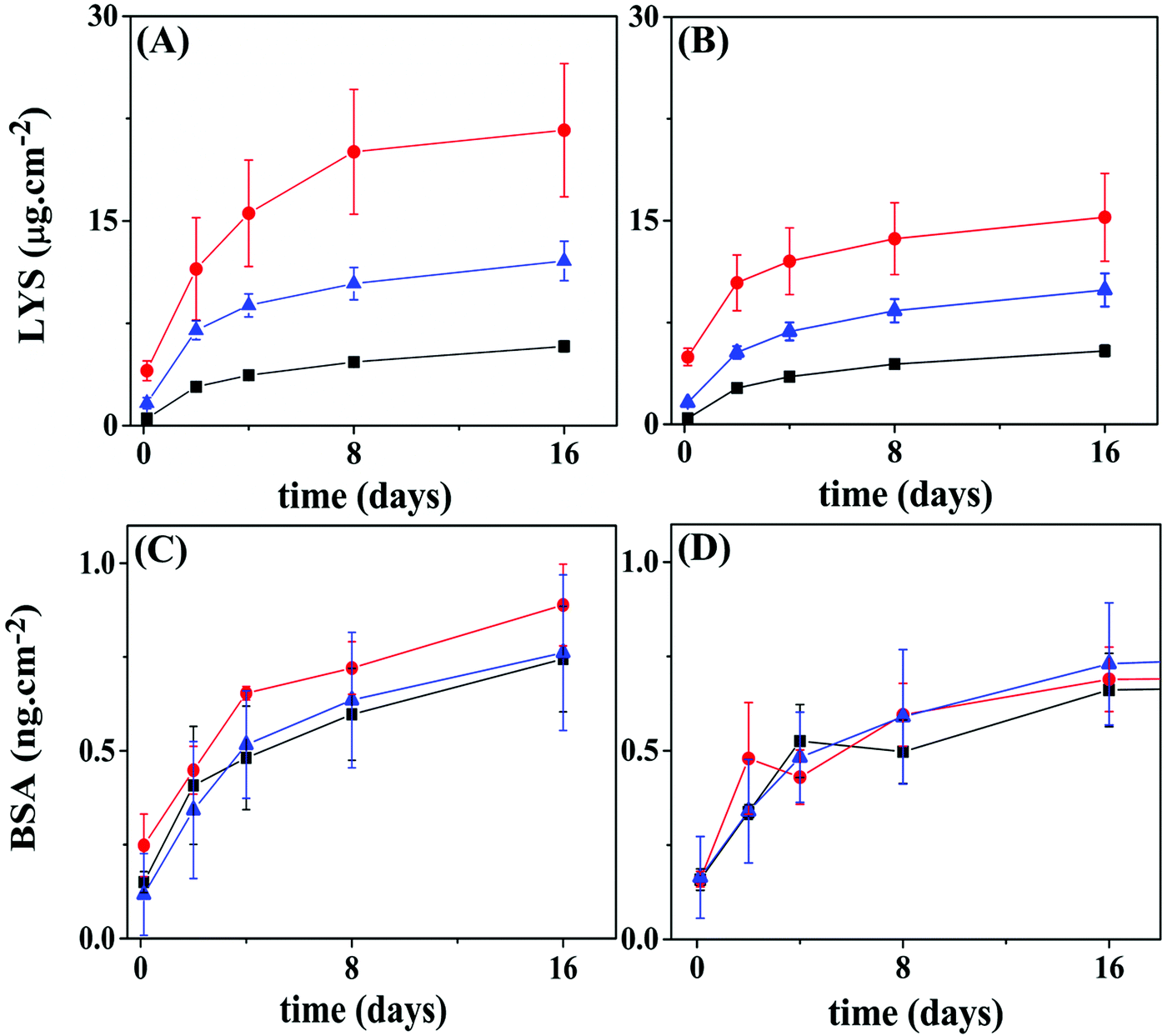 | ||
| Fig. 9 Release profiles for BSA or LYS from 10 layer pairs of Hep/Chi LbL reservoirs built onto PCL or PLLA films. Proteins were placed within the 5 top layers (red circles), evenly distributed (blue triangles), or within the 5 bottom layers (black squares) of the reservoirs. (A) LYS–FITC reservoir on a PCL substrate; (B) LYS–FITC reservoir on a PLLA substrate; (C) BSA reservoir on a PCL substrate; (D) BSA reservoir on a PLLA substrate. Statistical information is provided in Fig. S7 (ESI†). | ||
The amount of BSA or LYS released from the PLLA reservoirs is somewhat lower than from the PCL reservoirs. Here, the surface of the polymer substrate seems to play a role by showing different specific surface area. Although using the same spin-coating conditions, the PCL film presented high rugosity (r.m.s. = 13 nm), likely due to the presence of spherulite-like crystals, not present in PLLA (r.m.s. = 0.46 nm) as observed by AFM (Fig. 5A and D). According to Wu et al.,37 the strong interaction between PLLA and CHCl3 precludes crystal formation.37,38
As shown in Fig. 9, LYS achieved a total release of four orders of magnitude higher than BSA for the same concentration (100 μg mL−1) of the dipping solution. Generally, the rinsing process removes the unbound polyelectrolyte or protein molecules. Alternatively, a polyelectrolyte of opposite charge also operates to remove free or loose charged molecules from the surface, as confirmed here by the QCM-D results. Once inside of the inner layers of the reservoir, the protein would – in theory – be protected from ejection, unless diffusion to the outer layers takes place. A diffusion/redistribution process within the ten layer-assembly, leading to loss of protein during the construction process, can also explain the results shown in Fig. 9. Assuming that the same amount of protein is included during a single loading cycle – five protein-loading cycles in all the configurations – the number of rinsing cycles which each load would be submitted to, i.e., “leaking cycles”, would differentiate the three configurations. For example, the last protein-loading cycle of the “5-bottom” would be submitted to 6 extra rinsing cycles, while the last protein-loading cycle of the “5-top” has only one rinsing cycle ahead. This picture is essentially the same for PLLA as the polymeric substrate.
A shallow intermixing of either polyelectrolyte diffusing into the matrix was detected by QCM-D experiments (Fig. 8), an event observed during the deposition of the 1st and 2nd layers. As the assembly grows to a higher number of layers, the intermixing should grow further in distance to the original positioning. The same behavior is expected for the proteins; however, with different amplitudes since they interact dissimilarly with the LbL assemblies. The turbidimetry (Fig. 1) and ITC (Fig. 2) results show a rather distinct complexing capacity of each protein. The release kinetics (Fig. 9) can be viewed as a representation of this distinct intermixing event: (i) highly precluded for LYS (a tightly bound protein), resulting in different kinetics for different positioning (Fig. 9A and B), and (ii) more prominent for BSA (a loose and free molecule within the LbL assembly), when intermixing equalizes the “bottom vs. top” positioning approach (Fig. 9C and D).
When considering these profiles, the release mechanism is based on the dismantling of LbL multilayers following a physical erosion model, where the gradual disruption of electrostatic interactions can lead to the gradual release of both polyelectrolytes and proteins embedded in the reservoirs,7 combined with a diffusion/redistribution process. In our previous work, we observed thickness loss by ellipsometry measurements for bare Hep/Chi multilayers and FGF reservoirs after seven days of incubation,18 which indicates erosion-controlled release. Other studies also reported similar release behavior for multilayers comprised of different polyamines,7–9 where the release occurs by gradual hydrolysis of weak polyacids and subsequent gradual disruption of the physical electrostatic interactions in the multilayer.
Assessing the volume charge densities of both proteins at the loading pH also shines some light on the differences observed. While BSA is assumed to present a prolate ellipsoid shape with 14.1 and 4.2 nm for the c and a axes,39 LYS shows a spheroid shape with a hydrodynamic radius of 1.89 nm.40 By using PROPKA software,41 pKa predictor software, a total charge of −29.3 and +23.4 was calculated for BSA and LYS at the loading pH (6.7 and 3.0, respectively). These numbers lead to total volume charge densities of 35.9 and 132.3 C cm−3 (in modulus) for BSA and LYS, respectively, demonstrating that LYS is 3.7 times more densely charged, hence explaining the higher loading of LYS.
4. Conclusions
An LbL framework made from a multilayer of alternating charged polyelectrolytes heparin and chitosan was used to store BSA and LYS on biodegradable polyester films like PCL and PLLA. The proteins were chosen based on their interactions with the layer of opposite charge, considering its total charge (positive or negative) at the dipping solution pH. The results observed for the two pairs of Hep/LYS and Chi/BSA are strikingly different. Higher binding activity was observed for Hep and LYS, in clear opposition to that between BSA and Chi. Here, the total size and total charge of the protein must be at play. Indeed, LYS presents a volume charge density almost four-fold higher than BSA. The release of protein from the reservoir was controlled by building different architectures of the protein layer in between multilayers on top of PCL and PLLA substrates, demonstrating a novel key-approach to produce LbL-based protein reservoirs. The choice of positioning the same amount of protein within the bottom five layers or in the top five layers can result in clearly different release profiles.The analysis of the in vitro loading/release profile suggests that the protein delivery can be adjusted by precise positioning within the LbL assembly, demonstrating the importance of planning the material, as well as its architecture. An essential feature of this study was to show the significance of the total charge density of the protein and its interaction with the polyelectrolyte pairs. The total volume charge density of the protein (size versus total protein charge at the loading solution pH) is a crucial factor in controlling the total load, stability, and release kinetics. These findings will help in planning a device based on the size and charge of the protein, as well as the choice of the polyelectrolyte, in constructing LbL reservoirs for protein release.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), Brazil (Grants 2011/21442-6, 2015/10009-0, 2015/19694-8, 2017/06394-1, 2018/13492-2, 2018/14532-8), and Conselho Nacional para o Desenvolvimento Científico e Tecnológico (CNPq), Brazil (Grants 4001145/2014-6, 302152/2017-2), for financial support.References
- J. Hong, B.-S. Kim, K. Char and P. T. Hammond, Biomacromolecules, 2011, 12, 2975–2981 CrossRef CAS.
- R. C. Smith, A. Leung, B.-S. Kim and P. T. Hammond, Chem. Mater., 2009, 21, 1108–1115 CrossRef CAS.
- B.-S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS.
- L. Gu, Z. J. Deng, S. Roy, P. T. Hammond, C. A. Paula, T. Hammond and D. H. Koch, Clin. Cancer Res., 2017, 23, 7312–7323 CrossRef CAS.
- A. vander Straeten, A. Bratek-Skicki, A. M. Jonas, C.-A. Fustin and C. Dupont-Gillain, ACS Nano, 2018, 12, 8372–8381 CrossRef CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- N. E. Vrana, A. Dupret-Bories, C. Chaubaroux, E. Rieger, C. Debry, D. Vautier, M.-H. Metz-Boutigue and P. Lavalle, J. Visualized Exp., 2013, 1–9 Search PubMed.
- W. Li, P. Zhao, C. Lin, X. Wen, E. Katsanevakis, D. Gero, O. Félix and Y. Liu, Biomacromolecules, 2013, 14, 2647–2656 CrossRef CAS.
- X. He, Y. Wang and G. Wu, Appl. Surf. Sci., 2012, 258, 9918–9925 CrossRef CAS.
- R. Hashide, K. Yoshida, Y. Hasebe, M. Seno, S. Takahashi, K. Sato and J.-I. Anzai, J. Nanosci. Nanotechnol., 2014, 14, 3100–3105 CrossRef CAS.
- R. Luo, S. S. Venkatraman and B. Neu, Biomacromolecules, 2013, 14, 2262–2271 CrossRef CAS.
- N. J. Shah, M. N. Hyder, M. A. Quadir, N.-M. Dorval Courchesne, H. J. Seeherman, M. Nevins, M. Spector and P. T. Hammond, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12847–12852 CrossRef CAS.
- L. Keller, S. Eap, J. Schiavi, O. Huck, L. Jacomine, C. Gauthier, V. Sebastian, P. Schwinté, N. Jessel and F. Fioretti, Int. J. Nanomed., 2015, 1061 CrossRef.
- C. D. Easton, A. J. Bullock, G. Gigliobianco, S. L. McArthur and S. Macneil, J. Mater. Chem. B, 2014, 2, 5558–5568 RSC.
- D. P. Go, J. A. Palmer, G. M. Mitchell, S. L. Gras and A. J. O’Connor, J. Biomed. Mater. Res., Part A, 2015, 103, 1849–1863 CrossRef.
- D. Chen, M. Wu, J. Chen, C. Zhang, T. Pan, B. Zhang, H. Tian, X. Chen and J. Sun, Langmuir, 2014, 30, 13898–13906 CrossRef CAS.
- G. Gigliobianco, C. K. Chong and S. MacNeil, J. Biomater. Appl., 2015, 30, 50–60 CrossRef CAS.
- A. F. Naves, M. Motay, R. Mérindol, C. P. Davi, O. Felix, L. H. Catalani and G. Decher, Colloids Surf., B, 2016, 139, 79–86 CrossRef CAS.
- R. Signini and S. P. C. Filho, On the preparation and characterization of chitosan hydrochloride, 1999, vol. 42 Search PubMed.
- R. M. A. Azzam and N. M. Bashara, Ellipsometry and polarized light, North-Holland, 1987 Search PubMed.
- L. E. Valenti, V. L. Martins, E. Herrera, R. M. Torresi and C. E. Giacomelli, J. Mater. Chem. B, 2013, 1, 4921–4931 RSC.
- A. H. B. Dourado, R. A. Silva, R. M. Torresi, P. T. A. Sumodjo, M. Arenz and S. I. Cordoba de Torresi, ChemPhysChem, 2018, 19, 2340–2348 CrossRef CAS.
- M. Lundin, F. Solaqa, E. Thormann, L. Macakova and E. Blomberg, Langmuir, 2011, 27, 7537–7548 CrossRef CAS.
- L. Richert, P. Lavalle, E. Payan, X. Z. Shu, G. D. Prestwich, J.-F. Stoltz, P. Schaaf, J.-C. Voegel and C. Picart, Langmuir, 2004, 20, 448–458 CrossRef CAS.
- D. Kovacevic, S. van der Burgh, A. de Keizer and M. A. Cohen Stuart, Langmuir, 2002, 18, 5607–5612 CrossRef CAS.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS.
- P. Billsten, M. Wahlgren, T. Arnebrant, J. McGuire and H. Elwing, J. Colloid Interface Sci., 1995, 175, 77–82 CrossRef CAS.
- T. Peters, R. G. Reed, R. C. Feldhoff and O. L. Clute, Biochemistry, 1975, 14, 4578–4583 CrossRef.
- B. Ranjbar and P. Gill, Chem. Biol. Drug Des., 2009, 74, 101–120 CrossRef CAS.
- K. Takeda, A. Wada, K. Yamamoto, Y. Moriyama and K. Aoki, J. Protein Chem., 1989, 8, 653–659 CrossRef CAS.
- J. Horn, R. Kapelner and A. Obermeyer, Polymers, 2019, 11, 578 CrossRef.
- X. Wang, K. Zheng, Y. Si, X. Guo and Y. Xu, Polymers, 2019, 11, 1–18 CAS.
- M. J. Sherratt, D. F. Holmes, C. Adrian Shuttleworth and C. M. Kielty, Biophys. J., 2004, 86, 3211–3222 CrossRef CAS.
- T. Hattori, K. Kimura, E. Seyrek and P. L. Dubin, Anal. Biochem., 2001, 295, 158–167 CrossRef CAS.
- M. Rodahl, F. Höök, A. Krozer, P. Brzezinski and B. Kasemo, Rev. Sci. Instrum., 1995, 66, 3924–3930 CrossRef CAS.
- M. D. Levi, N. Shpigel, S. Sigalov, V. Dargel, L. Daikhin and D. Aurbach, Electrochim. Acta, 2017, 232, 271–284 CrossRef CAS.
- N. Wu, S. Lang, H. Zhang, M. Ding and J. Zhang, J. Phys. Chem. B, 2014, 118, 12652–12659 CrossRef CAS.
- M. Sun, P. J. Kingham, A. J. Reid, S. J. Armstrong, G. Terenghi and S. Downes, J. Biomed. Mater. Res., 2009, 93, 1470–1481 Search PubMed.
- A. K. Wright and M. R. Thompson, Biophys. J., 1975, 15, 137–141 CrossRef CAS.
- A. S. Parmar and M. Muschol, Biophys. J., 2009, 97, 590–598 CrossRef CAS.
- M. H. M. Olsson, C. R. SØndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00432d |
| This journal is © The Royal Society of Chemistry 2020 |