
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering TiO2 nanosheets with exposed (001) facets via the incorporation of Au clusters for boosted photocatalytic hydrogen production†
Xiaobin
Liu
a,
Huaqiang
Zhuang
 *b,
Jiale
Huang
c,
Wentao
Xu
b,
Liqin
Lin
b,
Yanmei
Zheng
*c and
Qingbiao
Li
*ac
*b,
Jiale
Huang
c,
Wentao
Xu
b,
Liqin
Lin
b,
Yanmei
Zheng
*c and
Qingbiao
Li
*ac
aCollege of the Environment & Ecology, Xiamen University, Xiamen 361005, P. R. China. E-mail: kelqb@xmu.edu.cn
bCollege of Chemical Engineering and Materials Science, Quanzhou Normal University, Quanzhou 362000, P. R. China. E-mail: huaqiangz@163.com
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: zym@xmu.edu.cn
First published on 12th August 2020
Abstract
Au cluster-incorporated TiO2 nanosheets with exposed (001) facets were designed and fabricated using a facile self-assembly strategy. The glutathione-protected Au clusters act as both a co-catalyst and a photosensitizer and significantly enhance the light absorption efficiency and photocatalytic hydrogen production simultaneously.
Conversion of solar energy into storable fuels via photocatalytic technology is considered as a feasible approach to tackle the rising energy crisis with minimal environmental impact.1–10 For instance, harvesting hydrogen gas as a clean energy source through photocatalytic water splitting is one of the most promising strategies.11–18 Inspired by Fujishima and Honda's first discovery of photoelectrochemical water splitting on a TiO2 photoanode,19 significant efforts have been devoted to taking TiO2-based photocatalysts to the next level, owing to their low cost and remarkable stability.20–26 With proven catalytic activity, TiO2 is the model material in photocatalysis and photoelectrochemistry.27–30 Yang et al.31 first reported TiO2 nanosheets with exposed (001) facets that show superior photoreactivity, and further studies uncovered that its photocatalytic performance is improved by the high surface energy and efficient chemisorption capacity.32–34 Although the extensive utilization of TiO2 in practical applications is limited by its wide band gap, different strategies have been adopted to tailor its absorption edge into the visible-light region to overcome the limitations.35–41 Inorganic sensitization, owing to its convenience and flexibility, is a common and effective technique to tailor the light absorption capability of wide-band gap materials.
Novel metal clusters protected by thiolate ligands have been used as a new-type photosensitizer to enhance the photocatalytic performance of the wide bandgap semiconductors, by broadening their photo-absorption range.42–44 Compared to conventional bulk Au nanoparticles, Au clusters composed of a precise number of metal atoms in the core show more favorable physico-chemical properties, including strong electron energy quantization induced by the ultra-small cluster size, tunable band-gap, and controllable catalytic properties.45,46 In particular, recent studies unveiled that photo-generated electrons in glutathione-protected Au (Au-GSH) clusters can be transferred to adjacent TiO2 under simulated solar light or visible light irradiation.47,48 The Au-GSH clusters generally exhibit nonmetallic properties with energy quantization manifested in their HOMO–LUMO gap due to the small scale at the nanometer level, in which electrons can be transferred from the HOMO to the LUMO and the generated electron–hole pairs can participate in certain photocatalytic reaction processes.46,49 Liu's investigation50 showed that the Au GSH-cluster–TiO2 composites have excellent visible-light activity for the degradation of RhB and reduction of Cr(VI). In addition, the electronic property and energy level of Au GSH-clusters can be easily designed and tuned by altering the size distribution, ligand selection and cluster composition.51,52 Therefore, optimized Au clusters can be applied as a promising photosensitizer to accelerate the development progress of photocatalysis and photoelectrochemistry. In this work, the heterostructure of Au GSH-clusters and TiO2 nanosheets with exposed (001) facets has been proposed to study the enhancement mechanisms in photocatalytic hydrogen evolution, and a clear and consensual understanding is necessary to disclose the differences between TiO2 with exposed (001) facets and TiO2 with exposed (101) facets after Au clusters are introduced.
With these motivations, Au-cluster/TiO2 nanosheets with an exposed (001) facet (Au/TiO2-001) heterostructure were designed and constructed by a facile self-assembly strategy, where the specially-made Au clusters can be uniformly dispersed onto TiO2 surface. The XRD patterns of the TiO2-001 and Au/TiO2-001 nanosheets are presented in Fig. S1 (see ESI†). As expected, the characteristic diffraction peaks of anatase TiO2 (JCPDS no. 21-1272) can be clearly observed for the TiO2-001 nanosheets.53 Meanwhile, the Au/TiO2-001 nanosheets display only the characteristic diffraction peaks of anatase TiO2, in which no additional diffraction peak ascribed to Au can be observed. This result is attributed to the low loading amount or high dispersion of Au clusters on the surface of TiO2 nanosheets. The micro-structure and morphologies of the TiO2-001 and Au/TiO2-001 nanosheets were further investigated by transmission electron microscopy (TEM) measurement, as displayed in Fig. 1. Obviously, the TEM and HRTEM images of TiO2 nanosheets with exposed (001) facets are clearly displayed in Fig. 1a and b. The uniform TiO2 nanosheets have an average side length of ca. 40 nm. Meanwhile, the lattice fringes with a d-spacing of about 0.235 nm can be obtained, which is indexed towards the {001} direction of the vertical TiO2 nanosheets.54 Compared with nude TiO2 nanosheets, many nanodots with sizes in the range of 1.6 ± 0.5 nm on the surface of TiO2 nanosheets can be distinctly observed in Fig. S3 (ESI†). Furthermore, a magnified HRTEM image of Au/TiO2-001 nanosheets is shown in Fig. 1d, in which an interplanar spacing of 0.23 nm that is in good agreement with the {111} crystal plane of face-centered cubic gold can be clearly observed.55 Simultaneously, the corresponding energy-dispersive spectroscopy (EDS) of Au/TiO2-001 nanosheets further confirms the existence of Au clusters as demonstrated by the fluorescence signal in Fig. S2 (ESI†). In addition, according to the EDS parameters of the Au/TiO2-001 sample listed in Table S1 (ESI†), the Ti/Au atomic ratio is 0.019, and thus the loading amount of Au can be calculated as 4.8 wt%. Au clusters are rather crowded in this test area. Overall, the above results distinctly demonstrate that heterostructures between Au clusters and TiO2 nanosheets are successfully constructed using this facile strategy.
 | ||
| Fig. 1 TEM and HRTEM images of (a and b) TiO2 nanosheets and (c and d) Au/TiO2-001 nanosheets. | ||
Interestingly, the optical absorption properties of TiO2-001 and Au/TiO2-001 nanosheets display considerable variation due to the introduction of Au-GSH clusters, as depicted in Fig. 2. Therein, TiO2-001 nanosheets show a typical absorption band gap of anatase TiO2, and its absorption band edge is around 390 nm, which is in good agreement with the band-gap energy of ca. 3.1 eV in the inset graph.27 However, compared to nude TiO2-001 nanosheets, Au/TiO2-001 nanosheets corresponding to the band-gap energy of ca. 2.7 eV display a gratifying visible-light absorption region from 400 nm to 800 nm, owing to the formation of the heterostructure between TiO2 and Au-GSH clusters. It is mainly because of the Au-GSH clusters exhibiting a non-metallic semiconductor property of photosensitizer to extend the absorption edge of TiO2 into the visible light region and improve the light absorption capacity. As shown in Fig. 2b, the subtracted DRS spectrum between nude TiO2-001 and Au/TiO2-001 nanosheets further demonstrates the function of Au-GSH clusters, which can be excited via the HOMO–LUMO gap to inject photo-generated electrons into TiO2.49 Furthermore, in order to compare the differences between the photosensitization effect of Au-GSH clusters and surface plasmon resonance effect, the DRS spectrum of Au particles/TiO2-001 prepared via a photodeposition method is presented in Fig. S4 (ESI†). It depicts an evident surface plasmon resonance peak due to a larger size of Au particles obtained via this strategy. When the size of the metal nanoparticles is reduced to around 2 nm or less, their optical absorption cross section becomes very small and plasmonic effects become negligible.46,56–58 Therefore, it can be seen that the visible light absorption of Au/TiO2-001 is enhanced by the photosensitization effect of the Au-GSH clusters, instead of its surface plasmon resonance effect.
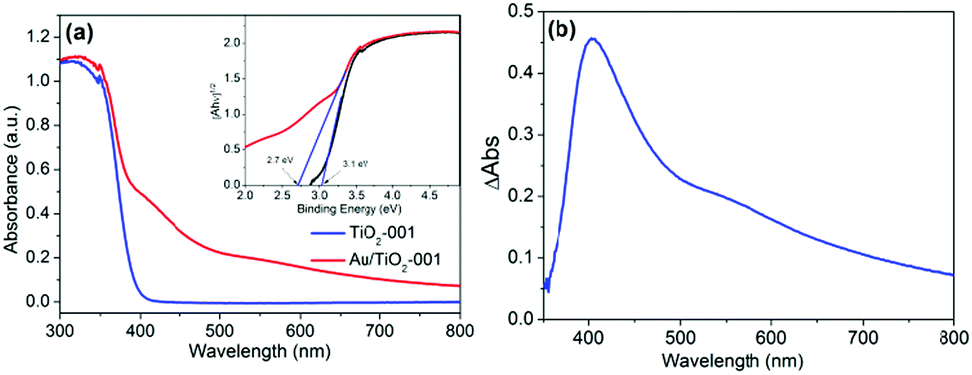 | ||
| Fig. 2 (a) UV-vis DRS spectra of TiO2-001 and Au/TiO2-001 nanosheets. The inset graph shows the plots of [Ahν]1/2versus photon energy (hν). (b) The subtracted DRS spectrum between TiO2-001 and Au/TiO2-001 nanosheets. | ||
The surface composition and chemical bonding state of Au, Ti and O elements are investigated by X-ray photoelectron spectroscopy (XPS) analysis. Fig. 3a presents the XPS survey spectra of TiO2-001 and Au/TiO2-001 nanosheets, indicating that the Au/TiO2-001 sample contains Ti, O and Au elements. The high resolution XPS spectra for Ti 2p, Au 4f and O 1s are presented in Fig. 3b, c and d, respectively. The binding energy of Ti 2p3/2 in TiO2-001 nanosheets is ca. 458.9 eV, which is assigned to the intrinsic Ti4+ oxidation state in TiO2.59 Nevertheless, after Au clusters are introduced, the binding energy of Ti 2p appears slightly shifted towards the negative binding energy direction. In addition, the O 1s XPS spectrum presents two characteristic peaks, as shown in Fig. 3d. The peak at low binding energy belongs to lattice oxygen in the Au/TiO2-001 nanosheets, while the other peak pertains to surface hydroxyls. Simultaneously, the binding energy of O 1s also appears shifted to the negative direction after introduction of the Au clusters. This phenomenon suggests that there is an intimate interfacial interaction between TiO2-001 nanosheets and Au clusters.46 In the meantime, the existence of Au clusters is further affirmed by the high resolution XPS spectrum for Au 4f, as presented in Fig. 3c, which appears with two core peaks at ca. 87.8 and 84.1 eV in the Au/TiO2-001 nanosheets, belonging to the Au 4f7/2 and Au 4f5/2 peaks of metallic Au0 species, respectively.60 These results clearly confirm that the Au clusters are successfully introduced on TiO2-001 nanosheets.
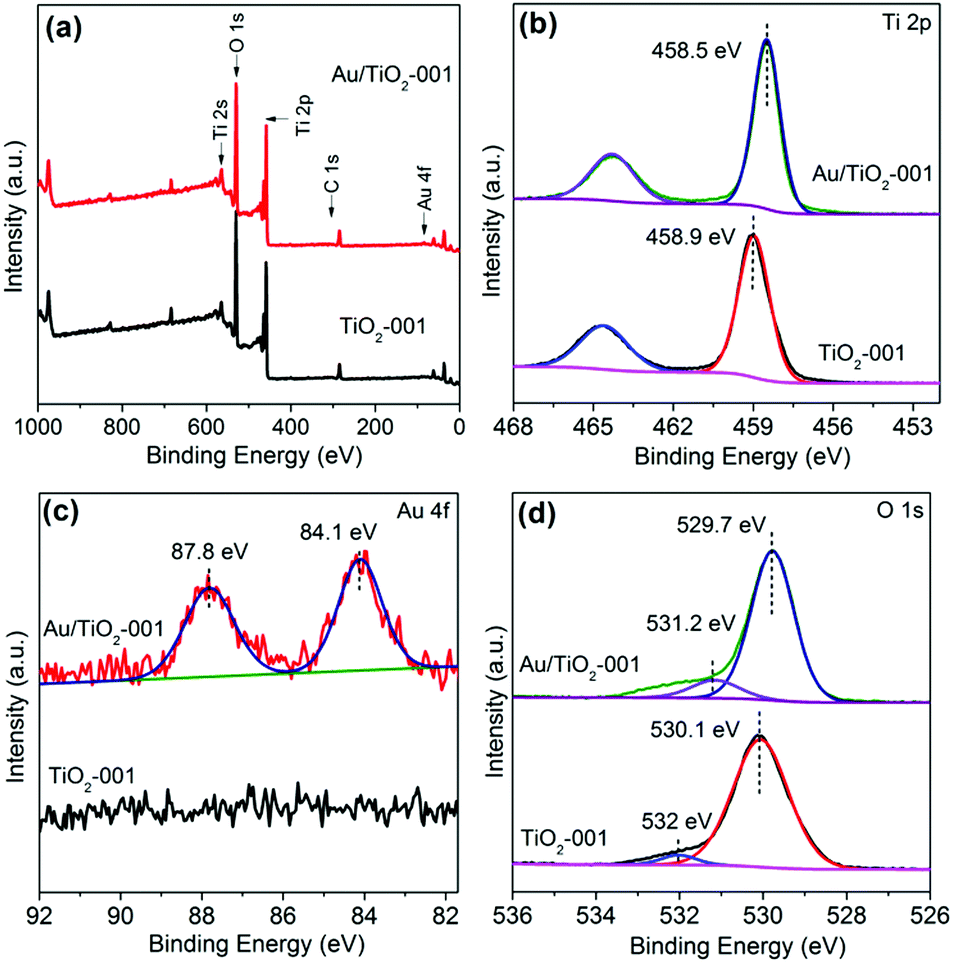 | ||
| Fig. 3 (a) Typical XPS survey spectra of TiO2-001 and Au/TiO2-001 nanosheets. The high resolution XPS spectra of Ti 2p, Au 4f and O 1s (b, c and d, respectively) for the TiO2-001 and Au/TiO2-001 nanosheets. | ||
The photocatalytic hydrogen evolution is selected as a model reaction to evaluate the photocatalytic performance of TiO2-101, Au/TiO2-101, TiO2-001 and Au/TiO2-001 samples under visible light or simulated solar light irradiation, as depicted in Fig. 4. In order to evaluate the advantages of TiO2-001 and Au/TiO2-001 nanosheets, the TiO2-101 and Au/TiO2-101 samples are chosen as reference samples. It can be found that TiO2-101 and TiO2-001 samples show a low and similar photocatalytic activity under simulated solar light irradiation, as displayed in Fig. 4a. Nevertheless, after coupling with Au clusters, the Au/TiO2-101 and Au/TiO2-001 samples display a sharp improvement in photocatalytic hydrogen production, indicative of the co-catalyst property of Au clusters. This figure also reveals that the H2 evolution rates of the Au/TiO2-101 and Au/TiO2-001 samples are as high as 178 and 338 μmol g−1 h−1, respectively. In the other words, the photocatalytic hydrogen production rate of Au/TiO2-001 nanosheets is almost double that of the Au/TiO2-101 sample. This is because TiO2 with exposed (001) facets has higher surface energy, which is more beneficial to the dissociative adsorption of reactant molecules, in comparison with thermodynamically stable (101) facets.32,61 In addition, these as-prepared samples were further examined under visible light irradiation, as shown in Fig. 4b. As expected, Au/TiO2-001 nanosheets show a higher hydrogen evolution rate than that of the Au/TiO2-101 sample under visible light irradiation, which agrees with the former changing regularity under simulated solar light irradiation. As a contrast, TiO2-101 and TiO2-001 samples are disabled to catalyze the hydrogen production under visible light irradiation, which is attributed to the wide band gap of pure TiO2. The Au/TiO2-101 and Au/TiO2-001 samples are endowed with photocatalytic hydrogen generation ability under visible-light via forming a heterostructure between TiO2 and Au clusters. Furthermore, it can be concluded that the glutathione-protected Au clusters not only act as an efficient co-catalyst, but also take on the function of a photosensitizer. Furthermore, in order to explore the merit of Au clusters/TiO2-001, the Au particles/TiO2-001 sample was chosen as a reference material to investigate its photocatalytic hydrogen production, as shown in Fig. S5 (ESI†). The Au particles/TiO2-001 sample exhibited a weak photocatalytic performance under visible light irradiation, suggesting that Au-clusters/TiO2-001 nanosheets possessed great potential in the field of photocatalytic water splitting. Moreover, the results of cycling experiments show that Au/TiO2-001 nanosheets have high catalytic activity and stability, as indicated in Fig. S6 (ESI†). The photocatalytic performance of Au/TiO2-001 nanosheets can continuously produce hydrogen, with no obvious activity loss after three cycles, attesting to the excellent stability. In order to further investigate the situation of the photocatalyst after the photocatalytic reaction, the SEM and TEM measurements were carried out. No significant changes can be observed from the SEM graph (Fig. S7, ESI†), which is due to the small size of the TiO2 nanosheets and Au clusters. But in Fig. S8 (ESI†) the TEM image of the Au/TiO2-001 sample after photocatalytic reaction reveals that the Au clusters got larger after the photocatalytic reaction, suggesting that Au/TiO2-001 nanosheets undergo a certain change during the photocatalytic reaction process.
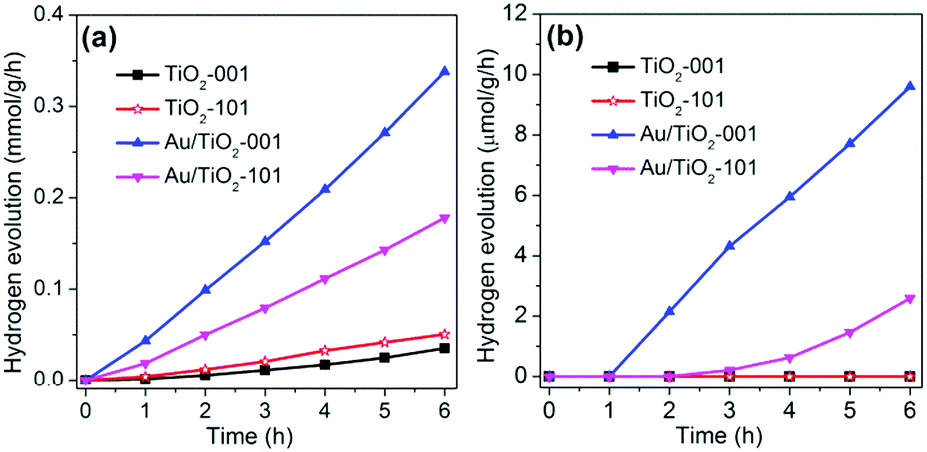 | ||
| Fig. 4 The photocatalytic activity of hydrogen production rate for TiO2-101, Au/TiO2-101, TiO2-001 and Au/TiO2-001 samples under (a) simulated solar light or (b) visible light (λ ≥ 420 nm) irradiation. | ||
In order to fully elucidate the role of Au clusters in our visual light photocatalytic hydrogen production, the transient photocurrent response curves of TiO2-101, Au/TiO2-101, TiO2-001 and Au/TiO2-001 samples are displayed in Fig. S9 (ESI†). The photocurrent response measurement can be employed to elucidate the mechanism, in which the generation and transfer of photoexcited charge carriers in the photocatalytic process can be indirectly monitored by the photo-generated current.27 Among them, the TiO2-101 and TiO2-001 samples show a lower photocurrent response, suggesting a poor generation and transfer efficiency of photoexcited electron–hole pairs, which is consistent with the photocatalytic activity for hydrogen production. In contrast, the Au/TiO2-001 and Au/TiO2-101 samples present a stronger visible light response. This can be ascribed to the integration of the Au-GSH clusters, which intensifies the visible light absorption of nude TiO2. In addition, Au/TiO2-001 nanosheets display the highest photocurrent density, far more than that of the Au/TiO2-101 sample, indicating that the Au/TiO2-001 nanosheets own a much higher separation and transfer efficiency of the photo-excited electron–hole pairs. As mentioned above, the Au/TiO2-001 nanosheets possess higher photocatalytic activity due to more efficient separation and transfer efficiency of the photo-generated charge carriers. It clearly demonstrates that the glutathione-protected Au clusters present the properties of a co-catalyst and a photosensitizer in TiO2 nanosheets, and the as-obtained Au/TiO2-001 is a promising photocatalytic material in comparison to the Au/TiO2-101 sample. Hence, a visible-light-induced photocatalytic reaction mechanism can be proposed in Fig. 5. Given the optical absorption and semiconductor property of the glutathione-protected Au clusters, the Au clusters can be inspired to generate electron–hole pairs under visible light irradiation. In addition, the photoexcited process for the glutathione-protected Au clusters has been clearly demonstrated by many precursors.46,56–58 Consequently, the photo-generated electrons can be rapidly injected into the conduction band (CB) position of the TiO2 nanosheets owing to the appropriate LUMO potential of Au clusters, in which these electrons can participate in the hydrogen evolution reaction to reduce H+ into H2. Meanwhile, the holes on the HOMO of the Au cluster react with the sacrificial agent (EDTA-2Na) to produce related oxidation products.
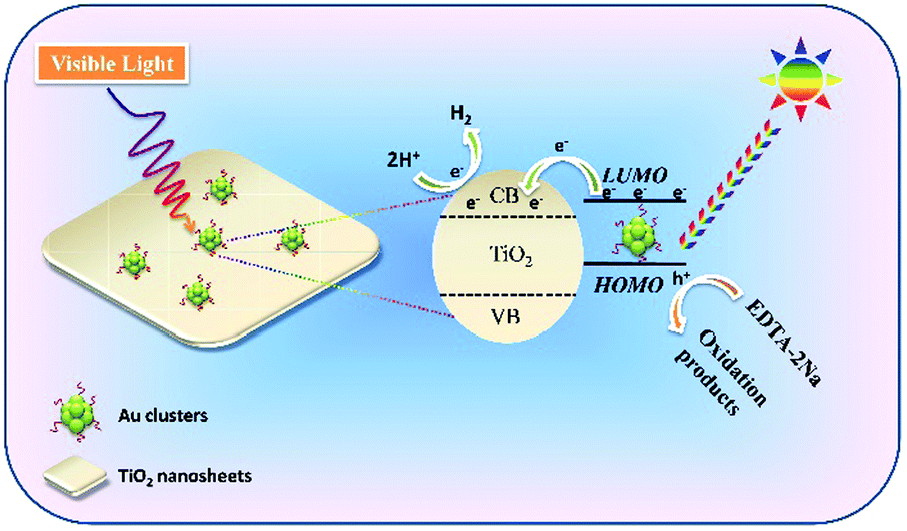 | ||
| Fig. 5 Proposed mechanism of Au/TiO2-001 nanosheets for visible-light-induced photocatalytic hydrogen production. | ||
Conclusions
In this work, TiO2 nanosheets with exposed (001) facets were fabricated with Au clusters by a facile self-assembly strategy. These catalysts display a noteworthy H2 generation rate as high as 338 μmol g−1 h−1 under simulated solar light irradiation, which is far more than that of TiO2-001 nanosheets and the Au/TiO2-101 sample. Its high efficiency and stable performance reveal that the Au/TiO2-001 material is an efficient and potential photocatalyst for hydrogen evolution. More significantly, the visible light photocatalytic activity also reflects that Au clusters are a promising photosensitizer. The in-depth understanding of the glutathione-protected Au clusters and the different properties between the TiO2-001 and TiO2-101 materials makes a contribution to exploitation of the glorious photocatalytic water splitting system and provides a strategy to delicately design and fabricate composite materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Quanzhou Science and Technology Project (2017Z031) and Open Project Program of Provincial Key Laboratory of Green Energy and Environment Catalysis (Grant No. FJ-GEEC201907), Ningde Normal University.Notes and references
- R. Shi, H.-F. Ye, F. Liang, Z. Wang, K. Li, Y. Weng, Z. Lin, W.-F. Fu, C.-M. Che and Y. Chen, Adv. Mater., 2018, 30, 1705941 CrossRef PubMed
.
- Y.-X. Pan, H.-Q. Zhuang, H. Ma, J. Cheng and J. Song, Chem. Eng. Sci., 2019, 194, 71–77 CrossRef CAS
- H. Zhuang, Z. Cai, W. Xu, X. Zhang, M. Huang and X. Wang, Catal. Commun., 2019, 120, 51–54 CrossRef CAS
- H. Zhang, G. Liu, L. Shi, H. Liu, T. Wang and J. Ye, Nano Energy, 2016, 22, 149–168 CrossRef CAS
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef CAS PubMed
- W. Wei, P. Sun, Z. Li, K. Song, W. Su, B. Wang, Y. Liu and J. Zhao, Sci. Adv., 2018, 4, eaap9253 CrossRef PubMed
- H. Zhuang, W. Xu, L. Lin, M. Huang, M. Xu, S. Chen and Z. Cai, J. Mater. Sci. Technol., 2019, 35, 2312–2318 CrossRef
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, M. E. A. Warwick, K. Kaunisto, C. Sada, S. Turner, Y. Gönüllü, T.-P. Ruoko, L. Borgese, E. Bontempi, G. Van Tendeloo, H. Lemmetyinen and S. Mathur, Adv. Mater. Interfaces, 2015, 2, 1500313 CrossRef
- D. Barreca, G. Carraro, M. E. A. Warwick, K. Kaunisto, A. Gasparotto, V. Gombac, C. Sada, S. Turner, G. Van Tendeloo, C. Maccato and P. Fornasiero, CrystEngComm, 2015, 17, 6219–6226 RSC
- A. Lepcha, C. Maccato, A. Mettenbörger, T. Andreu, L. Mayrhofer, M. Walter, S. Olthof, T. P. Ruoko, A. Klein, M. Moseler, K. Meerholz, J. R. Morante, D. Barreca and S. Mathur, J. Phys. Chem. C, 2015, 119, 18835–18842 CrossRef CAS
- H. Zhang, P. Zhang, M. Qiu, J. Dong, Y. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1804883 Search PubMed
- D. Barreca, G. Carraro, A. Gasparotto, C. Maccato, M. E. A. Warwick, E. Toniato, V. Gombac, C. Sada, S. Turner, G. Van Tendeloo and P. Fornasiero, Adv. Mater. Interfaces, 2016, 3, 1600348 CrossRef
- H. Han, F. Riboni, F. Karlicky, S. Kment, A. Goswami, P. Sudhagar, J. Yoo, L. Wang, O. Tomanec, M. Petr, O. Haderka, C. Terashima, A. Fujishima, P. Schmuki and R. Zboril, Nanoscale, 2017, 9, 134–142 RSC
- S. J. Kim, K. Xu, H. Parala, R. Beranek, M. Bledowski, K. Sliozberg, H.-W. Becker, D. Rogalla, D. Barreca, C. Maccato, C. Sada, W. Schuhmann, R. A. Fischer and A. Devi, Chem. Vap. Deposition, 2013, 19, 45–52 CrossRef CAS
- H. Zhuang, W. Chen, W. Xu and X. Liu, Int. J. Energy Res., 2020, 44, 3224–3230 CrossRef CAS
- H. Lim, J. L. Young, J. F. Geisz, D. J. Friedman, T. G. Deutsch and J. Yoon, Nat. Commun., 2019, 10, 3388 CrossRef PubMed
- C. D. Windle, H. Kumagai, M. Higashi, R. Brisse, S. Bold, B. Jousselme, M. Chavarot-Kerlidou, K. Maeda, R. Abe, O. Ishitani and V. Artero, J. Am. Chem. Soc., 2019, 141, 9593–9602 CrossRef CAS PubMed
- X. Tao, Y. Zhao, L. Mu, S. Wang, R. Li and C. Li, Adv. Energy Mater., 2018, 8, 1701392 CrossRef
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CrossRef CAS
- M. Xie, X. Fu, L. Jing, P. Luan, Y. Feng and H. Fu, Adv. Energy Mater., 2014, 4, 1300995 CrossRef
- R. Shi, Z. Li, H. Yu, L. Shang, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu and T. Zhang, ChemSusChem, 2017, 10, 4650–4656 CrossRef CAS PubMed
- H. Zhuang, J. Miao, H. Huang, J. Long, Y. Zhang, H. Yang, S. He, Y. Yang, X. Wang and B. Liu, ChemPhysChem, 2015, 16, 1352–1355 CrossRef CAS PubMed
- G. Carraro, C. Maccato, A. Gasparotto, M. E. A. Warwick, C. Sada, S. Turner, A. Bazzo, T. Andreu, O. Pliekhova, D. Korte, U. Lavrenčič Štangar, G. Van Tendeloo, J. R. Morante and D. Barreca, Sol. Energy Mater. Sol. Cells, 2017, 159, 456–466 CrossRef CAS
- D. Primc, M. Bärtsch, D. Barreca, G. Carraro, C. Maccato, C. Sada and M. Niederberger, Sustainable Energy Fuels, 2017, 1, 199–206 RSC
- C. Fàbrega, D. Monllor-Satoca, S. Ampudia, A. Parra, T. Andreu and J. R. Morante, J. Phys. Chem. C, 2013, 117, 20517–20524 CrossRef
- H. Zhuang, Y. Zhang, Z. Chu, J. Long, X. An, H. Zhang, H. Lin, Z. Zhang and X. Wang, Phys. Chem. Chem. Phys., 2016, 18, 9636–9644 RSC
- J. Wang, Z. Wang, P. Qu, Q. Xu, J. Zheng, S. Jia, J. Chen and Z. Zhu, Int. J. Hydrogen Energy, 2018, 43, 7388–7396 CrossRef CAS
- Z. U. Rahman, N. Wei, M. Feng and D. Wang, Int. J. Hydrogen Energy, 2019, 44, 13221–13231 CrossRef CAS
- C. Peng, X. Yang, Y. Li, H. Yu, H. Wang and F. Peng, ACS Appl. Mater. Interfaces, 2016, 8, 6051–6060 CrossRef CAS PubMed
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed
- X. Hu, S. Lu, J. Tian, N. Wei, X. Song, X. Wang and H. Cui, Appl. Catal., B, 2019, 241, 329–337 CrossRef CAS
- H. Zhang, J. Cai, Y. Wang, M. Wu, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng, Z. Jiang and J. Gong, Appl. Catal., B, 2018, 220, 126–136 CrossRef CAS
- Z.-W. Yin, S. B. Betzler, T. Sheng, Q. Zhang, X. Peng, J. Shangguan, K. C. Bustillo, J.-T. Li, S.-G. Sun and H. Zheng, Nano Energy, 2019, 62, 507–512 CrossRef CAS
- J.-C. Wang, H.-H. Lou, Z.-H. Xu, C.-X. Cui, Z.-J. Li, K. Jiang, Y.-P. Zhang, L.-B. Qu and W. Shi, J. Hazard. Mater., 2018, 360, 356–363 CrossRef CAS PubMed
- F. Xu, K. Meng, B. Cheng, J. Yu and W. Ho, ChemCatChem, 2019, 11, 465–472 CrossRef CAS
- H. Zhao, X. Zheng, X. Feng and Y. Li, J. Phys. Chem. C, 2018, 122, 18949–18956 CrossRef CAS
- H. Xu, S. Ouyang, L. Liu, P. Reunchan, N. Umezawa and J. Ye, J. Mater. Chem. A, 2014, 2, 12642–12661 RSC
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed
- H. Zhuang, X. Liu, F. Li, W. Xu, L. Lin and Z. Cai, Int. J. Energy Res., 2019, 43, 7197–7205 CrossRef CAS
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, 1802981 CrossRef PubMed
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC
- F. F. Schweinberger, M. J. Berr, M. Döblinger, C. Wolff, K. E. Sanwald, A. S. Crampton, C. J. Ridge, F. Jäckel, J. Feldmann, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2013, 135, 13262–13265 CrossRef CAS PubMed
- F.-X. Xiao, Z. Zeng and B. Liu, J. Am. Chem. Soc., 2015, 137, 10735–10744 CrossRef CAS
- B. Weng, K.-Q. Lu, Z. Tang, H. M. Chen and Y.-J. Xu, Nat. Commun., 2018, 9, 1543 CrossRef PubMed
- F.-X. Xiao, S.-F. Hung, J. Miao, H.-Y. Wang, H. Yang and B. Liu, Small, 2015, 11, 554–567 CrossRef CAS PubMed
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed
- Y.-S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822–8825 CrossRef CAS PubMed
- Y.-S. Chen and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 6075–6082 CrossRef CAS PubMed
- S. Liu and Y.-J. Xu, Sci. Rep., 2016, 6, 22742 CrossRef CAS PubMed
- A. Sreedhar, I. N. Reddy, J. H. Kwon, J. Yi, Y. Sohn, J. S. Gwag and J.-S. Noh, Ceram. Int., 2018, 44, 18978–18986 CrossRef CAS
- G. Li and R. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed
- H. Zhuang, Q. Gu, J. Long, H. Lin, H. Lin and X. Wang, RSC Adv., 2014, 4, 34315–34324 RSC
- J. Long, H. Chang, Q. Gu, J. Xu, L. Fan, S. Wang, Y. Zhou, W. Wei, L. Huang, X. Wang, P. Liu and W. Huang, Energy Environ. Sci., 2014, 7, 973–977 RSC
- S.-I. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251–1254 CrossRef CAS PubMed
- M. A. Abbas, P. V. Kamat and J. H. Bang, ACS Energy Lett., 2018, 3, 840–854 CrossRef CAS
- W. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS
- F. Xu, J. Chen, S. Kalytchuk, L. Chu, Y. Shao, D. Kong, K.-H. Chu, P. H. L. Sit and W. Y. Teoh, J. Catal., 2017, 354, 1–12 CrossRef CAS
- S. Yang, Y. Li, J. Sun and B. Cao, J. Power Sources, 2019, 431, 220–225 CrossRef CAS
- H. Song, L. Wei, C. Chen, C. Wen and F. Han, J. Catal., 2019, 376, 198–208 CrossRef CAS
- S. Kenmoe, O. Lisovski, S. Piskunov, D. Bocharov, Y. F. Zhukovskii and E. Spohr, J. Phys. Chem. B, 2018, 122, 5432–5440 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00479k |
| This journal is © The Royal Society of Chemistry 2020 |