
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGliadin-coated gold nanoparticles for rapid colorimetric test for celiac disease†
Anantdeep
Kaur
ab,
Ying
Wang
a,
Michael
Wallach
*b and
Olga
Shimoni
 *ac
*ac
aInstitute for Biomedical Materials and Devices, Faculty of Science, The University of Technology Sydney, 15 Broadway, Ultimo, Sydney, New South Wales 2007, Australia. E-mail: olga.shimoni@uts.edu.au
bSchool of Life Sciences, Faculty of Science, The University of Technology Sydney, 15 Broadway, Ultimo, Sydney, New South Wales 2007, Australia
cARC Research Hub for Integrated Device for End-user Analysis at Low-levels (ARC IDEAL Hub), Faculty of Science, University of Technology Sydney, 15 Broadway, Ultimo, NSW 2007, Australia
First published on 22nd September 2020
Abstract
Celiac disease (CD) is an immune-mediated disorder affecting the small intestine in genetically predisposed individuals. Despite the significant progress made in research on CD, there is still an urgent need for accurate detection or a point-of-care test for the rapid diagnosis of the disease. In this paper, we present a novel detection test for CD based on the coating of gold nanoparticles with gliadin, the highly antigenic protein that induces CD. Although the protein is cheap and stable, its hydrophobic nature prevented its efficient use in diagnostics. In our work, we successfully demonstrated the binding of the whole hydrophobic gliadin protein to the surface of the gold nanoparticles that we further translated into a simple assay for the detection of CD specific biomarkers in serum as well as saliva. Finally, we compared the diagnostic accuracy of the novel test on 30 previously tested patient serum samples and saliva from 35 untested participants in a blinded assessment that were further compared to the previous serological and pathological tests on those patients. The data showed that the developed test using gold nanoparticles had an overall accuracy of over 96% in detecting celiac disease. Our developed test method offers a substantial advantage over the existing blood tests by eliminating multiple steps associated with routine serological testing, providing results within 15–30 minutes that can be determined by eye without any specialized equipment for signal reading. Finally, the developed assay is valuable for testing CD patients using saliva as a non-invasive, point of care test.
Introduction
The field of diagnostics has experienced explosive growth over the last decade.1,2 Portable diagnostic kits are highly sought after as they offer a relatively fast, easy and simple technology to detect a large number of biomarkers. Despite these advancements, there are still diseases that would greatly benefit from developing a lab-based or point-of-care diagnostic test. One such condition is celiac disease.Celiac disease (CD) is a chronic illness that affects the small intestine due to an auto-immune reaction induced by the consumption of gluten, a protein found in wheat and other cereal grains.3 Epidemiological studies have shown that there is a worldwide distribution of celiac disease with a high proportion of CD sufferers going undiagnosed.4 Early diagnosis is vital in order to prevent the progression of CD that can often lead to severe complications.
Symptoms of CD vary significantly in different individuals and include anemia, anorexia, weight loss, abdominal pain, diarrhea, chronic fatigue, constipation, joint pain as well as an increased level of liver enzymes.5 Due to the wide variability in symptoms and similarity to other gastrointestinal disorders, such as Crohn's disease and Inflammatory Bowel Disease (IBD), it has proven difficult to correctly diagnose CD at an early stage by a medical practitioner.
The first step for CD diagnosis in clinical practice is serologic testing for gliadin-induced autoantibodies; the two key ones are an anti-gliadin antibody (AGA) and a tissue anti-transglutaminase (tTG) antibody. The most reliable method for diagnosing CD, however, is gastrointestinal endoscopy, which is invasive, costly, and time-consuming. This method relies on the clinician to acquire a tissue sample for a mucosal biopsy for visualization of varying degrees of intestinal damage, from mild abnormalities to completely flat mucosa.3,6 In addition, genetic testing for individuals with a high probability of developing CD, i.e., those showing the presence of the main susceptibility genes HLA-DQ2 (specifically HLA-DQ2.5 and HLA-DQ2.2) and HLA-DQ8, is also used in combination with the other serological approaches for diagnosis.7 Typically, CD patients present higher serum titers of AGA and anti-tTG antibodies that are detectable using a lab-based, enzyme-linked immunosorbent assay (ELISA) test.8,9 In non-CD patients, these two types of antibodies are generally undetectable in serum.
Several commercial kits based on the serological testing of antibodies against tTG and DGP have been introduced and used for large-scale screening of the general population. However, the existing point-of-care methods for identifying CD are unable to provide the required diagnostic accuracy that might be affected by the higher variability in the characteristics of patients, such as their age, family history, or other clinical conditions associated with auto-immune diseases.10
Over the last decade, nanoparticle-based technology has been applied to the early detection of multiple diseases, such as HIV,11,12 other pathogenic infections,13,14 pregnancy testing in women,15 and many other applications.16–20 This technology has proven to be highly sensitive, accurate, and easy to perform. Nanotechnology-based approaches have been employed to deliver a point-of-care test for CD detection, involving the coating of DGP on a carrier protein as the antigen and using colloidal gold anti-human antibodies as the signal detector to identify anti-DGP antibodies in serum samples. Although it is a plausible approach, the test was found to be less specific than expected, which limited its potential as a population screening tool due to the functional complexity of the method.21
In this paper, we demonstrate a simple, cheap and accurate colorimetric immunoassay to detect celiac disease-specific anti-gliadin antibodies in bodily fluids. To exhibit it, (1) we assembled a stable suspension (without any significant level of aggregation) of gold nanoparticles (AuNPs) coated with the hydrophobic whole gliadin protein; (2) we adapted and validated it to develop a novel screening test for rapid detection of celiac disease using simulated bodily fluids (Fig. 1). Interestingly, we found that the use of the whole gliadin protein with AuNPs showed a better binding towards antibodies than in typical flat surface tests (i.e., ELISA), and we predict that the curvature of AuNPs' increased the exposure of hidden epitopes in gliadin towards antibody recognition. (3) Importantly, we tested our invention on 30 patient serum and 35 saliva samples and found that the assay achieved a high level of accuracy (>95%) in distinguishing CD from non-CD patients. We contemplate that this immunoassay represents a simple, straightforward nanoparticle-based assay for the detection of celiac disease that can be easily translated into clinical practice.
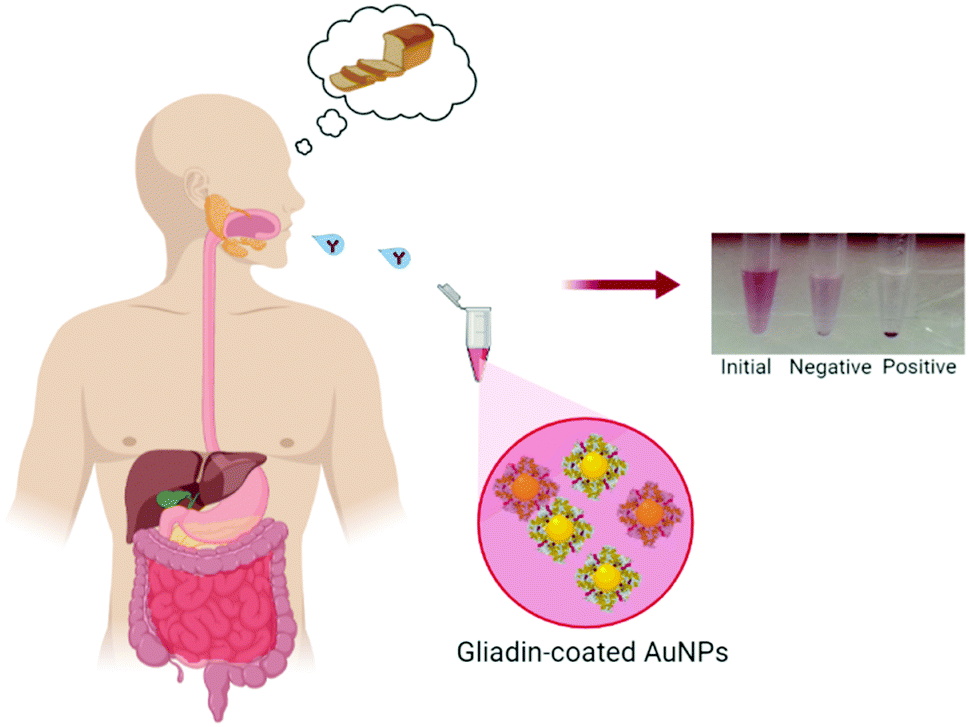 | ||
| Fig. 1 Rapid screening test for celiac disease. Schematic representation of the gliadin-coated immunoassay for fast and accurate detection of the celiac disease biomarker in saliva. The colorimetric output can be observed by a naked eye, where AGA binding to gliadin-coated AuNPs induces nanoparticle aggregation, leading to a change to a change in color. The absence of the AGA causes just a bit of color weakening. | ||
This level of accuracy is in the range required for a serological testing while in the clinic or home we would expect to find only a relatively small proportion of people tested with CD. In addition, these results for the detection of AGA eliminate multiple steps followed in existing serological tests that sometimes lead to user-dependent error. A test enabling early detection would be particularly useful in aiding the large-scale screening of the general population, particularly in the preselection of CD in small children by parents or medical practitioners, which can be then be confirmed by mucosal biopsy.
Results and discussion
Gliadin protein, found in wheat, barley, and rye, is the antigenic protein that induces CD in genetically predisposed people. Gliadin epitopes are recognized by T cells that stimulate the formation of CD4, natural killer (NK)-like cells, as well as the release of pro-inflammatory cytokines such as IFN-γ thereby, activating the adaptive immune response.22,23 Typically, patients with CD present higher serum titers of AGA and anti-tTG antibodies that are detectable using a lab-based, enzyme-linked immunosorbent assay (ELISA) tests.8,9Over the years, tTG antibodies showed higher specificity and sensitivity values as compared to AGA tests, which led to the abandoning of whole gliadin protein-specific AGA as the biomarker for diagnosing CD. More recently, ELISA tests using human recombinant tTG (h-tTG) or deamidated gliadin peptide (DGP) as the antigen source have further improved the sensitivity of CD using the patient's blood serum. However, increased tTG antibody titers are also associated with Type I diabetes as well as some liver disorders, and this can generate an unacceptable number of false-positive results.
One of the reasons that AGA lacks high specificity towards gliadin is that gliadin is a mostly hydrophobic wheat protein that is only slightly soluble in aqueous solution. It is challenging to use this protein as an antigen in typical ELISA testing.
To use gliadin as an antigen and to coat AuNPs for AGA detection, we overcame the problem of aggregation of the coated nanoparticles. In previous studies using coated AuNPs, the vast majority of work used water-soluble molecules, such as DNA/RNA,24–26 peptides,27,28 aptamers29 and albumins,30 to coat the nanoparticles. Because of the hydrophobicity of the used protein, we had to develop a new method to achieve a stable colloidal suspension of gliadin-coated AuNPs under physiological conditions.
We tested a variety of protic and aprotic solvents, such as acetone, chloroform, dichloromethane, dimethylformamide and methanol, where we observed only partial solubility of the protein. We developed a protocol for sufficient solubilization of gliadin using the cationic surfactant CTAB and 70% isopropanol previously used in other studies to extract some plant-based proteins.31 In the aqueous environment, gliadin would have a compact globular shape with polar groups and hydrophilic amino acids oriented outwards as it is considered energetically favourable conformation. Gliadin protein has a considerate amount of glutamine amino acids in its sequence,32 and they can interact with the gold surface due to high affinity for amines, thiols and phosphines.34 The use of CTAB surfactant provides a further stabilization of the nanoparticles by incorporation steric hindrance and positive charge leading to electrostatic repulsion between nanoparticles.
In the present study, 20 nm citrate stabilized gold nanoparticles were used. The citrate layer provides long term stability to the AuNPs and is weakly associated with the nanoparticle surface. This citrate layer can be easily displaced by a range of molecules, including proteins. Adsorption of gliadin on the surface of AuNPs followed by displacement of the citrate layer involved non-covalent processes based on the ionic interactions between the negatively charged nanoparticle and the positively charged amino acids in the protein. Some interactions also occurred due to covalent binding between the AuNPs and the conducting electrons of nitrogen and sulphur atoms present in the protein.33 These interactions help in providing stability to the coated AuNPs in the aqueous solution.
The solubilization of gliadin allowed us to adsorb the protein on the surface of AuNPs, which we monitored using UV-Vis spectrophotometry measurements.34 We found that there was a redshift in the absorbance maximum from 525 nm to 532 nm, indicating a shift in the localized plasmon resonance of AuNPs due to the protein adsorbed onto the AuNPs surface (Fig. 2A). In addition, we did not observe a measurable decrease of absorbance at peak wavelength or an increase of absorbance at longer wavelengths (600–700 nm), suggesting the colloidal dispersion of protein-adsorbed AuNPs was stable, and no strong AuNP-to-AuNP interactions or aggregation occurred.
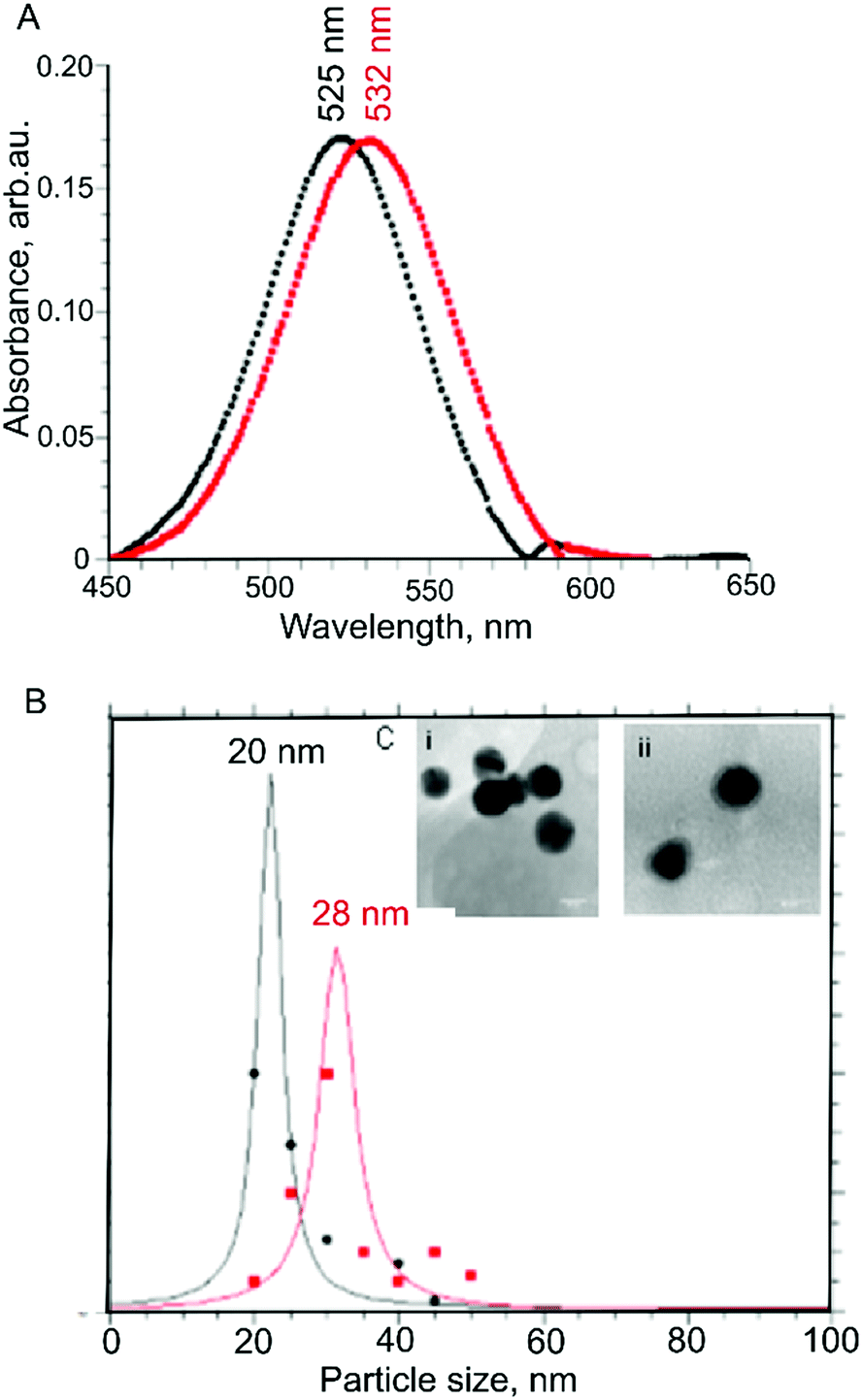 | ||
| Fig. 2 Characterization of gliadin coated AuNPs. (A) Characterization of AuNPs adsorbed with gliadin using a UV-Vis Spectrophotometer show a spectral redshift in wavelength from 525 nm (for 20 nm AuNPs only) to 532 nm (for 20 nm AuNPs adsorbed with gliadin). (B) Characterization of AuNPs adsorbed with gliadin using DLS, showing an increase in the hydrodynamic size of the uncoated vs. coated particles from 20 nm to 28 nm, respectively. (C) High-resolution TEM images of (i) uncoated AuNPs, (ii) gliadin coated AuNPs showing a ‘halo’ layer indicating coating of the gold with the protein had occurred. In contrast, the ‘halo’ effect was not observed on the surface of the un-coated AuNPs. | ||
We further confirmed there was no aggregation using DLS, which showed an increase in hydrodynamic diameter from 20 nm to 28 nm following adsorption with gliadin (Fig. 2B). As the molar mass of the protein is directly proportional to its hydrodynamic radius in solution,35 an increase in the hydrodynamic radius indicates that the coating of AuNPs with gliadin had occurred. We carried out a similar test using control AuNPs absorbed with BSA (molecular weight = 66 kDa) protein, where we found an average hydrodynamic particle diameter of 32 nm, correlating with the larger size of the protein.
We observed zeta potential change of AuNPs as a result of gliadin absorption. The value changed from −27.5 mV for citrate-capped AuNPs to +18.1 mV after absorption of gliadin. This further confirms the assumption that in the aqueous solution gliadin protein present polar amino acids (mostly glutamine) on the surface.
To directly observe gliadin absorbed on AuNPs, high-resolution TEM imaging was used. The presence of a thin layer of material (1–2 nm) (Fig. 2C(ii)) surrounding the nanoparticles, which was not observed on the surface of the uncoated nanoparticles (seen in Fig. 2C(i)) further confirmed the absorption of protein.36
Immunoassay of gliadin-coated AuNPs with AGA
To examine the ability of gliadin-coated AuNPs to detect AGA, we first tested serial dilutions of rabbit anti-gliadin IgG polyclonal antibody (AGA) in a range that exists typically in human serum.37,38 After 30 minutes incubation, we observed a significant reduction in color as well as a decrease in the absorbance peak with a shift from 532 nm to 580 nm (Fig. 3A). These results showed that in the presence of the AGA, increased nanoparticle-to-nanoparticle interaction leads to aggregation and precipitation of the AuNPs. Interestingly, we have not observed color change to blue/purple. While the AuNPs absorb and scatter light intensely at their surface plasmon resonance (SPR) frequency that is sensitive to the inter-particle distance between the nanoparticles. The color change happens due to aggregation of AuNPs that increases electric dipole–dipole interaction and overlapping of plasmons of neighbouring particles. However, in this study when the inter-particle distance is substantially greater than the average particle diameter, the AuNPs appear red. Specifically, if we take into account the size of the protein adsorbed on the surface of the AuNPs (4–5 nm) together with the complex of antibody (∼10 nm), it creates a greater distance between adjacent nanoparticles. Therefore, the aggregates formed in this study precipitate out and appear as red while leaving the suspension translucent. | ||
| Fig. 3 Testing gliadin-coated AuNPs in buffer with spiked AGA (positive test) or IgG from human serum(control). (A) Change in absorbance (filled markers) and colorimetric response (open markers) after incubation with different concentrations of antibodies (Blue markers for AGA and red markers for control IgG antibodies). The initial solution of AuNPs coated with gliadin in water was defined as 100% absorbance. (B) Visual performance: no significant change in color or shift in peak wavelength was observed in gliadin coated AuNPs incubated with control IgG (top)at all dilutions tested (2, 4, 6, 8, 10 μg mL−1) while there is visible precipitation formed in samples after mixing with AGA (bottom). | ||
As a control, we tested normal serum IgG antibody with the gliadin-coated AuNPs. We found that there was no significant change in color or shift in the wavelength or size of the absorbance peak after taking the dilution factor into account (Fig. 3A and B). We found that at all the tested concentrations, the absorbance was significantly lower using AGA as compared to normal IgG, reaching its minimum value in the range of 4–8 μg mL−1 (p < 0.005, Fig. 3A). These results demonstrate the specificity and sensitivity of the assay.
We tested uncoated AuNPs and those coated with an irrelevant antigen, such as BSA, incubated with AGA as controls for specificity. Our results did not show any significant change in absorbance or aggregation (Fig. S3 and S4, ESI†), confirming the specificity of the assay for whole gliadin.
To assess the specificity of the AGA toward gliadin coated AuNPs, we calculated a colorimetric response using our assay (Fig. 2C and Fig. S5, ESI†). We found that the response reaches a maximum value at a concentration of 6 μg mL−1 of AGA. The near-constant colorimetric response curve obtained for the control antibody as compared to the response curve obtained for AGA further demonstrates the specificity of the assay.
Testing AGA in spiked serum and saliva
Human serum is a complex fluid containing various proteins, peptides, exosomes as well as nucleic acids. To reduce background binding, we used 1% BSA as a blocking agent to lower the non-specific interaction with the gliadin-coated AuNPs. Spiked human serum containing 2–10 μg mL−1 AGA was incubated with gliadin-coated AuNPs. We observed a reduction in the color of the solution from red to translucent coupled with the formation of aggregation and precipitation, which was easily detectable by eye (Fig. 4A). This change was further supported by UV-Vis measurements, where the maximum change was observed at 8 μg mL−1 of AGA (Fig. S6 and S7, ESI†). In contrast, when we added normal IgG to the human serum, no precipitate formation or change in color was observed. The normal serum itself did not show any change in absorbance, and the solutions preserved a pink color over a long period of time (Fig. 4A).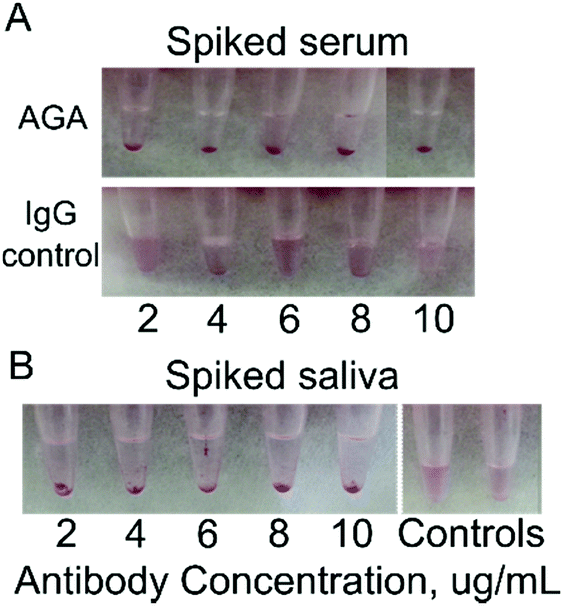 | ||
| Fig. 4 Detection of AGA in spiked human serum and saliva samples using gliadin-coated AuNPs. (A) AuNPs adsorbed with gliadin showed intense precipitation in the presence of AGA and serum at different concentration. The precipitation can be observed by the naked eye within 20 min after incubation. No reduction in color from red to translucent or precipitate formation was observed in AuNPs adsorbed with gliadin in the presence of control IgG and serum. All serum samples were diluted 1:20 before the addition of antibodies. (B). Testing gliadin-coated AuNPs with AGA in saliva at different dilutions. Reduction in color from red to translucent and formation of precipitation was clearly observed in AuNPs coated with gliadin incubated with AGA at various dilutions. As human saliva contains multiple antibodies, no spiking of control antibodies was required. Saliva samples were only slightly diluted. | ||
Human saliva is another complex fluid that contains multiple molecular species. Developing a test that would be able to detect biomarkers in the saliva is of utmost interest for truly non-invasive detection. In this work, we first tested the ability of gliadin-coated AuNPs to detect AGA in spiked saliva from healthy volunteers. The antibody concentrations used in saliva were similar to those typically observed in the saliva of patients with CD.39,40
Human saliva spiked at various concentrations of AGA was mixed with gliadin coated AuNPs, and the results we observed after 20 min by the naked eye. We confirmed that the gliadin-AuNPs were able to interact with AGA in saliva samples (Fig. 4B). AGA-spiked saliva showed a visible precipitate while control samples remained pink in color. Human saliva did not require spiking with control antibodies as it already contains large amounts of IgG and IgA endogenous antibodies, peptides, nucleic acids and more. When observed by UV-Vis spectrometry, a decrease in the absorption wavelength was detected when the gliadin-coated AuNPs were added to saliva samples spiked with AGA. Normal saliva itself did not show any absorbance change. These readings confirmed that the AGA could trigger aggregation in saliva, and their specificity is not affected by other constituents in saliva. A change in color from red to translucent was clearly observed (Fig. 4B).
Clinical samples testing
To assess the clinical relevance of the developed method, we next used the gliadin-coated AuNPs to test a selected set of human serum and saliva samples obtained from patients with CD or healthy controls. The goal of the small clinical study was to evaluate the ability of the assay to distinguish between previously diagnosed CD patients from non-CD individuals. The testing of clinical samples has been performed blinded without prior knowledge of the clinical outcome for the tested patient's serum and saliva.The results for the 30 clinical serum samples and 35 saliva samples were recorded after the visual examination of precipitate formation and the determination of a shift or change in absorbance values using a UV-Vis spectrophotometer (Fig. 5). Based on the results observed by the eye, the samples have been divided into three categories: evident precipitation, aggregation and colloidal suspension.
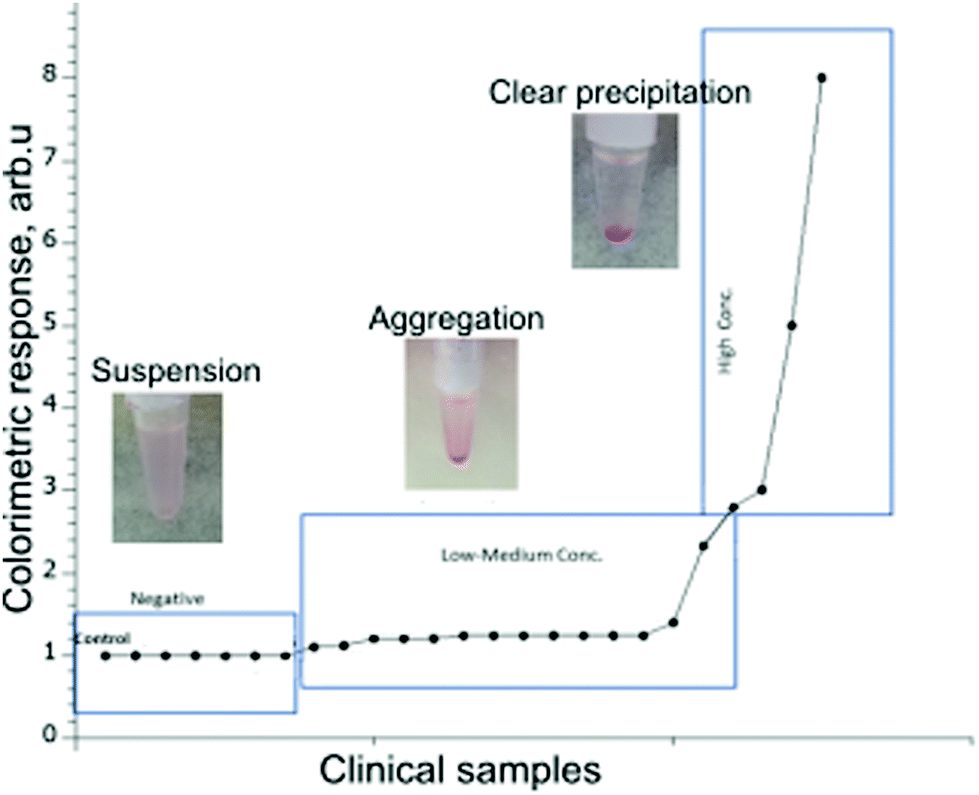 | ||
| Fig. 5 Testing gliadin-coated AuNPs with patients serum samples: colorimetric response curve of different clinical samples as a result of AGA titre concentrations. Inset images: Representative images of tubes after incubation with gliadin-coated AuNPs and serum. CD negative samples presented a pink colloidal suspension, low and medium concentration samples formed a visible aggregation, and the samples with high AGA titre showed clear precipitation at the bottom of the tube. | ||
Based on eye observation and UV-Vis spectra performance, the outcomes are summarized in Tables 1 and 2 after comparing the results with those reported using other serological methods, obtained through biopsy and histology.
Out of the 30 clinical serum samples tested, 19 samples showed evident precipitation within 15 minutes and formed a pellet indicating they were positive for CD. Seven samples showed the formation of aggregated particles (without clear pellet) within 15 minutes, and those were observed for another 15 minutes for further confirmation as CD positive. In four samples, there was no precipitation or formation of aggregates within 15 minutes, and remained as a colloidal suspension, and were classified as negative for CD.
In the cohort of tested serum samples, there were two cases where the patients had previously been diagnosed with CD and therefore followed a gluten-free diet (GFD). While one person had been on a GFD for more than 8 weeks, the other person had been on a GFD for less than 2 weeks. The AuNPs-AGA based method distinguished the person following GFD for more than 8 weeks as negative for CD, while serum from the person who has been on GFD for less than 2 weeks formed a suspension. Interestingly, the conventional serology test identified those two patients as negative, suggesting that our test can detect actual biopsy-confirmed cases even when traditional serology is negative. The analysis of these two exceptional patients using the AuNPs-AGA method illustrates that this method can potentially also be used for monitoring patients on a GFD over time. Further work using sera from more CD patients on GFD must be tested in the future to confirm further the efficacy of the test for monitoring patients on GFD.
Another encouraging result was the correct identification of an individual diagnosed with Type 1 diabetes mellitus (T1DM) as positive for CD using the AuNPs-AGA test method that essentially matched with the previously conducted biopsy and serological profile of the patient. For patients with T1DM, the prevalence of CD varies from 3% to 16%,41 due to the common genetic background with multiple environmental and immunological factors.42 Also, most T1DM patients, particularly in children, present with a silent or latent form of the disease characterized by the absence of both gastrointestinal and extra-intestinal signs and are often regarded as asymptomatic. Therefore, the CD diagnosis of diabetic patients is difficult and requires continuing careful clinical and serological follow-up.43 This, however, is a single test result and needs to be further explored in more extensive clinical studies.
Three serum samples were classified as latent celiac sufferers that have positive CD serology and display mild mucosal atrophy initially, which then developed with typical atrophy of small intestine mucosa.41 In addition, four cases have been identified as “potential” CD positives; they are characterized by a normal villous architecture but demonstrate pathological findings such as increased γδ+ intraepithelial lymphocytes as well as the presence of gliadin specific antibodies, usually at low titres (<1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40).44 Although histology, in general, is considered the most reliable method of testing for CD, it has shown lowered predictive value in recognizing “potential” CD cases. This situation requires an evaluation of serological markers for the correct diagnosis of potential CD cases, which are then monitored for the appearance of pathological symptoms.
:40).44 Although histology, in general, is considered the most reliable method of testing for CD, it has shown lowered predictive value in recognizing “potential” CD cases. This situation requires an evaluation of serological markers for the correct diagnosis of potential CD cases, which are then monitored for the appearance of pathological symptoms.
The cohort included four samples identified as negative for CD based on biopsy. Out of the four samples, three samples were correctly identified as CD negative by AuNPs-AGA based method, while one sample showed the formation of aggregates and was identified as positive. The DGP-IgG serology titers for that sample, however, indicate positive CD necessitating a clinical follow-up on the patient to confirm CD status. As intestinal biopsy has been used as a “gold standard” for CD confirmation, that one sample has been referred to as a false-positive result.
Overall, the results for the clinical samples tested showed that 22 of the 23 samples gave the correct result based on previous biopsy and serology. Furthermore, 7 samples were either potential or latent celiac that all tested positive for CD (Fig. S8 and S9, ESI†). Finally, there was one sample that was referred to as a false positive, giving an overall accuracy for this cohort of 96.7% (Table 1).
In the saliva test cohort from volunteers, out of the 35 clinical saliva samples tested, four samples showed evident precipitation within 15 minutes and formed a pellet indicating they were positive for CD. The results of the 4 positive samples matched the previously conducted biopsy and serology investigations. Out of the four samples, one individual was a latent celiac sufferer and was not following a GFD when tested using the AuNPs-AGA test method. The remaining three individuals had not been following GFD strictly, and cross-contamination in the diet might have led to an increase in the titers of the AGA antibodies in the saliva.
In the other 30 samples, there was no precipitation or formation of aggregates within 15 minutes, and remained as a colloidal suspension, and were classified as negative for CD. One sample showed the formation of aggregates and identified as positive using the AuNPs-AGA test method. The intestinal biopsy, used as a “gold standard” for CD confirmation, confirmed non-CD diagnosis (but had another related auto-immune condition) and that one sample had been referred to as a false positive result in this study. Finally, the saliva analysis showed that the test method has an overall accuracy for this cohort of at least 96% (Table 2).
In future work, AGA in serum and saliva as a biomarker for CD needs further validation in larger patient cohorts, particularly in patients suffering from other auto-immune diseases. This is necessary not only to verify the diagnostic accuracy but also to confirm specificity for the diagnosis of CD.
Our developed test method offers a substantial advantage over the existing blood tests by eliminating multiple steps associated with routine serological testing, providing results within 15–30 minutes that can be determined by eye without any specialized equipment for signal reading. Finally, the developed assay is valuable for testing CD patients using saliva as a non-invasive, point of care test.
Experimental
Materials and methods
600 μL of 20 nm AuNPs were added dropwise to the tube containing 2.6 mL of the filtered solubilized gliadin while vortexing. The dispersion was mixed for 60 minutes with repeated vortexing every 10 minutes, followed by centrifugation at 4500 × g for 30 minutes. The supernatant was discarded, and the pellet was re-dissolved in 150 μL of MilliQ water. The 20 nm AuNPs coated with gliadin were stored at 4 °C for up to 4 weeks.
The hydrodynamic diameter of nanoparticle was measured using Zetasizer Nano (Malvern Technologies, Inc.). Measurements were carried out at 25 °C in disposable cuvettes using a sample volume of 500 μL. Each sample was measured in duplicates, followed by mean value calculation.
High-resolution transmission electron microscopy (TEM) micrographs were obtained using a FEI Tecnai TEM 200 V fitted with a Gatan (Pleasantville, CA) CCD camera. Samples were prepared by placing 2 μL of AuNPs adsorbed with gliadin onto a carbon-coated TEM grid (Agar Scientific, UK) and the film allowed to air dry for 15 minutes.
UV-Vis measurements were carried out using a Cary Series UV-Vis spectrophotometer (Agilent Technologies) using a standard 1 cm path-length quartz cuvette. Spectra were obtained from 200 nm to 800 nm. MilliQ water was used as the blank.
:10, 1:20 and 1:50 using 10 mM HEPES buffer. 75 μL of serum from each of the dilutions was spiked with AGA at various dilutions comparable to that seen in celiac patients.
To prevent non-specific binding, 1 μL of 20% BSA dissolved in MilliQ water and was added to 150 μL of 20 nm AuNPs adsorbed with gliadin. The tubes were incubated for 30 minutes at room temperature. 75 μL of normal serum spiked with AGA at increasing dilutions of 2 μg mL−1, 4 μg mL−1, 6 μg mL−1, 8 μg mL−1 and 10 μg mL−1 was then added to 20 nm AuNPs adsorbed with gliadin. The tubes were then incubated for 30 minutes at room temperature.
Before testing, each human serum sample was diluted to 1:10, 1:20 and 1:50 using 10 mM HEPES buffer. 1 μL of 20% BSA dissolved in MilliQ water was added to 20 nm AuNPs adsorbed with gliadin to prevent non-specific binding. 75 μL of serum from each of the dilutions was then added to 20 nm AuNPs adsorbed with gliadin. The tubes were incubated for 15 minutes at room temperature before the absorbance was measured using a UV-Vis spectrophotometer.
Zeba™ Spin desalting columns (ThermoFisher Scientific) were used for the desalting of the immunoglobulins from the concentrated serum solution according to the manufacturer's instructions. The final concentration of the total immunoglobulins was measured using the NanoDrop and stored at −20 °C until further use.
Prior to testing, each human saliva sample was diluted to 1:2 using 10 mM HEPES buffer. 1 μL of 20% BSA dissolved in MilliQ water was added to 20 nm AuNPs adsorbed with gliadin to prevent non-specific binding. 75 μL of saliva from the dilution was then added to 20 nm AuNPs adsorbed with gliadin. 1 μL of protease inhibitor cocktail (P2714) was added to the saliva sample to prevent protein degradation. The tubes were incubated for 15 minutes at room temperature before the absorbance was measured using a UV-Vis spectrophotometer.
Conclusions
In the present study, we demonstrated that gliadin-coated AuNPs could be used as an efficient tool to detect a biomarker for CD from serum and saliva. We developed a methodology to adsorb the highly hydrophobic gliadin protein on the surface of AuNPs without causing aggregation. The addition of AGA to gliadin adsorbed AuNPs at levels associated with CD resulted in color reduction and absorbance peak shift due to the aggregation of AuNPs. We confirmed that the developed assay could detect AGA not only in quantitatively spiked samples but also in a small-scale study on real CD patient's samples. The analysis of the clinical samples demonstrated not only the ease of the test procedure but also its high accuracy. This study demonstrates the immense potential of the AuNPs-AGA based assay in for pre-selecting CD sufferers for mucosal biopsy for CD as well as for monitoring the effectiveness of gluten-free diet treatment. The method can be adapted as a point-of-care test as well as in resource-limited laboratory settings for the screening to aid in early identification of CD. Future work will include more comprehensive clinical trials on a large cohort of people suffering from a variety of gastrointestinal and auto-immune conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank A/Prof Jason Tye-Din, MBBS, PhD, FRACP, Immunology Division, The Walter and Eliza Hall Institute of Medical Research, Parkville, Victoria, Australia, for providing clinical samples as well for his critical review and feedback on the manuscript. This research is supported by an Australian Government Research Training Program Scholarship. OS acknowledges the Australian Research Council, National Health and Medical Research Council (APP1101258, IH150100028) and AMP Foundation for financial support. Table of content figure and Fig. 1 were created with BioRender.com.Notes and references
- A. A. Kumar, J. W. Hennek, B. S. Smith, S. Kumar, P. Beattie, S. Jain, J. P. Rolland, T. P. Stossel, C. Chunda-Liyoka and G. M. Whitesides, Angew. Chem., Int. Ed., 2015, 54, 5836–5853 CrossRef CAS.
- A. Malekjahani, S. Sindhwani, A. M. Syed and W. C. W. Chan, Acc. Chem. Res., 2019, 52, 2406–2414 CrossRef CAS.
- J. F. Ludvigsson, D. A. Leffler, J. C. Bai, F. Biagi, A. Fasano, P. H. R. Green, M. Hadjivassiliou, K. Kaukinen, C. P. Kelly, J. N. Leonard, K. E. A. Lundin, J. A. Murray, D. S. Sanders, M. M. Walker, F. Zingone and C. Ciacci, Gut, 2013, 62, 43 CrossRef.
- M. N. Marsh, V. Villanacci and A. Srivastava, Gastroenterol. Hepatol. Bed Bench, 2015, 8, 171–177 Search PubMed.
- E. Tonutti and N. Bizzaro, Autoimmun. Rev., 2014, 13, 472–476 CrossRef CAS.
- G. Oberhuber, G. Granditsch and H. Vogelsang, Eur. J. Gastroenterol. Hepatol., 1999, 11, 1185 CrossRef CAS.
- J. A. Tye-Din, D. J. Cameron, A. J. Daveson, A. S. Day, P. Dellsperger, C. Hogan, E. D. Newnham, S. J. Shepherd, R. H. Steele, L. Wienholt and M. D. Varney, Intern. Med. J., 2015, 45, 441–450 CrossRef CAS.
- I. Dahlbom, B. I. Nyberg, L. Berntson and T. Hansson, Scand. J. Clin. Lab. Invest., 2016, 76, 208–216 CrossRef CAS.
- F. Benkebil, C. Combescure, S. I. Anghel, C. Besson Duvanel and M. G. Schäppi, World J. Gastroenterol., 2013, 19, 5111–5117 CrossRef.
- A. Kaur, O. Shimoni and M. Wallach, J. Gastroenterol., 2017, 52, 1001–1012 CrossRef CAS.
- R. Banerjee and A. Jaiswal, Analyst, 2018, 143, 1970–1996 RSC.
- B. A. Rohrman, V. Leautaud, E. Molyneux and R. R. Richards-Kortum, PLoS One, 2012, 7, e45611 CrossRef CAS.
- E. I. Laderman, E. Whitworth, E. Dumaual, M. Jones, A. Hudak, W. Hogrefe, J. Carney and J. Groen, Clin. Vaccine Immunol., 2008, 15, 159–163 CrossRef CAS.
- A. L. Tomás, M. P. de Almeida, F. Cardoso, M. Pinto, E. Pereira, R. Franco and O. Matos, Front. Microbiol., 2019, 10, 2917, DOI:10.3389/fmicb.2019.02917.
- J. Su, Z. Zhou, H. Li and S. Liu, Anal. Methods, 2014, 6, 450–455 RSC.
- R. E. Biagini, D. L. Sammons, J. P. Smith, B. A. MacKenzie, C. A. F. Striley, J. E. Snawder, S. A. Robertson and C. P. Quinn, Clin. Vaccine Immunol., 2006, 13, 541–546 CrossRef CAS.
- N. Tippkötter, H. Stückmann, S. Kroll, G. Winkelmann, U. Noack, T. Scheper and R. Ulber, Anal. Bioanal. Chem., 2009, 394, 863–869 CrossRef.
- Q. Li, L. Liu, W. Chen, C. Peng, L. Wang and C. Xu, Int. J. Environ. Anal. Chem., 2009, 89, 261–268 CrossRef CAS.
- S. Gupta, H. Andresen, J. E. Ghadiali and M. M. Stevens, Small, 2010, 6, 1509–1513 CrossRef CAS.
- R. Chandrawati and M. M. Stevens, Chem. Commun., 2014, 50, 5431–5434 RSC.
- F. Bienvenu, C. Besson Duvanel, C. Seignovert, P. Rouzaire, A. Lachaux and J. Bienvenu, Eur. J. Gastroenterol. Hepatol., 2012, 24, 1418–1423 CrossRef CAS.
- V. Abadie, L. M. Sollid, L. B. Barreiro and B. Jabri, Annu. Rev. Immunol., 2011, 29, 493–525 CrossRef CAS.
- G. Malamut, R. El Machhour, N. Montcuquet, S. Martin-Lannerée, I. Dusanter-Fourt, V. Verkarre, J. J. Mention, G. Rahmi, H. Kiyono, E. A. Butz, N. Brousse, C. Cellier, N. Cerf-Bensussan and B. Meresse, J. Clin. Invest., 2010, 120, 2131–2143 CrossRef CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078 CrossRef CAS.
- J.-S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 4093–4096 CrossRef CAS.
- Q. Wang, R. Liu, X. Yang, K. Wang, J. Zhu, L. He and Q. Li, Sens. Actuators, B, 2016, 223, 613–620 CrossRef CAS.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS.
- C.-C. Huang, Y.-F. Huang, Z. Cao, W. Tan and H.-T. Chang, Anal. Chem., 2005, 77, 5735–5741 CrossRef CAS.
- S. H. Brewer, W. R. Glomm, M. C. Johnson, M. K. Knag and S. Franzen, Langmuir, 2005, 21, 9303–9307 CrossRef CAS.
- J. C. Watson and W. F. Thompson, Methods in Enzymology, Academic Press, 1986, pp. 57–75, vol. 118 Search PubMed.
- Y. Li, R. Xin, D. Zhang and S. Li, Crop J., 2014, 2, 10–21 CrossRef.
- D. Tsai, F. W. DelRio, A. M. Keene, K. M. Tyner, R. I. MacCuspie, T. J. Cho, M. R. Zachariah and V. A. Hackley, Langmuir, 2011, 27, 2464–2477 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- H. Jans, X. Liu, L. Austin, G. Maes and Q. Huo, Anal. Chem., 2009, 81, 9425–9432 CrossRef CAS.
- L. Jürgens, A. Nichtl and U. Werner, Cytometry, 1999, 37, 87–92 CrossRef.
- C. O’Farrelly, J. Kelly, W. Hekkens, B. Bradley, A. Thompson, C. Feighery and D. Weir, Br. Med. J., 1983, 286, 2007–2010 CrossRef.
- H. F. al-Bayaty, M. J. Aldred, D. M. Walker, R. G. Newcombe, G. Swift, P. M. Smith and P. J. Ciclitira, J. Oral Pathol. Med., 1989, 18, 578–581 CrossRef CAS.
- M. Lenander-Lumikari, R. Ihalin and H. Lähteenoja, Arch. Oral Biol., 2000, 45, 347–354 CrossRef CAS.
- N. T. Tucker, F. S. Barghuthy, T. J. Prihoda, V. Kumar, A. Lerner and E. Lebenthal, J. Pediatr., 1988, 113, 286–289 CrossRef CAS.
- U. Volta, F. Tovoli and G. Caio, Expert Rev. Gastroenterol. Hepatol., 2011, 5, 479–487 CrossRef CAS.
- G. K. Holmes, Diabetic Med., 2001, 18, 169–177 CrossRef CAS.
- K. Kaukinen, M. Mäki, J. Partanen, H. Sievänen and P. Collin, Dig. Dis. Sci., 2001, 46, 879–887 CrossRef CAS.
- U. Volta and V. Villanacci, Cell. Mol. Immunol., 2011, 8, 96–102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00495b |
| This journal is © The Royal Society of Chemistry 2020 |