
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBulk and interfacial decomposition of formamidinium iodide (HC(NH2)2I) in contact with metal oxide†
Sampreetha
Thampy‡
 a,
Boya
Zhang‡
a,
Jong-Goo
Park
b,
Ki-Ha
Hong
*b and
Julia W. P.
Hsu
*a
a,
Boya
Zhang‡
a,
Jong-Goo
Park
b,
Ki-Ha
Hong
*b and
Julia W. P.
Hsu
*a
aDepartment of Materials Science and Engineering, University of Texas at Dallas, Richardson, TX 75080, USA. E-mail: jwhsu@utdallas.edu
bDepartment of Materials Science and Engineering, Hanbat National University, Yuseong-Gu, Daejeon, 34158, Republic of Korea. E-mail: kiha.hong@hanbat.ac.kr
First published on 16th October 2020
Abstract
The thermal stability and decomposition pathway of formamidinium iodide (FAI, HC(NH2)2I) in contact with NiO and TiO2 are investigated by combined experimental studies and density functional theory (DFT) calculations. Based on the decomposition temperature, we find that the stability decreases as FAI ∼ FAI + TiO2 > FAI + NiO. Moreover, FAPbI3 in contact with NiO and TiO2 shows similar thermal stability behaviour to FAI. The bulk decomposition of FAI occurs via the formation of sym-triazine, and can also produce HCN, and NH4I at ∼280 °C, which further decomposes to NH3 and HI above 300 °C. When FAI comes into contact with NiO, the interfacial reaction triggers decomposition at a much lower temperature (∼200 °C), resulting in the formation of NiI2 as the solid product while releasing NH3 and H2O into the gas phase; sym-triazine and HCN are observed near the FAI bulk decomposition temperature. In contrast, when FAI comes into contact with TiO2, the decomposition temperature is similar to bulk FAI; however, HCN is released at a lower temperature (∼260 °C) compared to sym-triazine. The difference in the degradation behavior of FAI with NiO and TiO2 is elucidated using DFT calculations. Our results show that the interfacial reaction between the organic component of perovskite material and NiO occurs similarly for MA and FA, which thereby can induce device instability.
Introduction
Compositional engineering of organic–inorganic halide perovskite materials using mixed cations and/or mixed halides shows potential to achieve stable photovoltaic and optoelectronic devices with high efficiency and tunable energy levels.1–4 Mixed cations consisting of methylammonium (MA), formamidinium (FA), cesium (Cs), or rubidium (Rb) are often used in perovskite solar cells (PSCs), with FA being the major component—with the molar fraction varying from 0.75 to 0.85—due to its desired bandgap, photo stability, and reproducibility.1,4–7 Although high performance has been achieved in these mixed-cation halide PSCs, their long-term operational stability still presents a critical challenge.8,9 Even after eliminating environmental factors, e.g. humidity and oxygen, through encapsulation, the inherent chemical reactivity and volatility of organic cations remain major factors in halide perovskite material degradation under light and heat.8,10–13 The degradation of MAPbI3 has been widely studied,14–19 and the comprehensive understanding of its instability and decomposition mechanisms results in efforts to eliminate MA from halide perovskite compounds.6,7,20 In PSCs, the perovskite, irrespective of composition, has been reported to degrade through interfacial reactions with neighbouring materials,7,12,21–29 yielding lower device stability and performance. Although recently a few studies have been performed on the decomposition of FAPbI3 by themselves,10,18,30–34 there is little to no understanding of the interface-induced degradation in FA-based perovskites. Thus, to evaluate the stability of this material as a potential absorber in PSCs, it is imperative to identify possible interfacial reactions between the FA cation and contact layer materials.While previous work focused on the thermal stability and degradation mechanism in formamidinium iodide (FAI, HC(NH2)2I) and FAPbI3 by themselves, here we investigate the thermal stability and decomposition pathway of FAI in contact with NiO, a commonly used hole transport layer material,27,35 or TiO2, a commonly used electron transport layer material.36 In our previous work, we showed that the inorganic component of the perovskite materials—PbI2—does not undergo any change in this temperature range (<400 °C), both by itself or in contact with metal oxides.22 The instability of halide perovskites primarily arises from the decomposition of the organic component. Hence, studying FAI degradation will provide an understanding of FAPbI3 stability. The thermal stability and degradation reactions are studied using thermogravimetric analysis complemented with differential scanning calorimetry (TGA-DSC), as well as the temperature-programmed desorption technique combined with mass spectrometry (MS) and Fourier transform infrared spectroscopy (TPD-MS-FTIR) for simultaneous detection and unequivocal identification of gas-phase decomposition products. The solid decomposition products are examined by X-ray diffraction (XRD). Density functional theory (DFT) modelling is employed to explain the experimental results. This combination allows us to construct an accurate delineation of the decomposition pathways.
Experimental
Materials
All chemicals in this study were used as received: FAI (>99.5%, Greatcell Solar Materials), NiO (<50 nm, 99.8%, Sigma-Aldrich), TiO2 (50 nm, 99.9+%, US Research Nanomaterials, Inc.), and NH4I (99.999%, Alfa Aesar). The NiO and TiO2 powders were dried in a vacuum oven at 250 °C and 150 °C, respectively, prior to mixing with FAI.Thermal analysis
The TGA-DSC analysis was carried out in an SDT Q600 (TA Instruments). The powder samples of FAI and FAPbI3 in contact with metal oxides were prepared by mixing FAI/FAPbI3 with NiO and TiO2 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio using a vortex mixer (Vortex 3, IKA Works, Inc.) for 1 min. The NH4I in contact with NiO was also prepared in a 1:1 molar ratio. From the prepared samples, ∼8–9 mg of the powder was placed in an alumina pan and heated from 25 °C to 450 °C at a rate of 10 °C min−1 under 100 sccm of N2 gas.
:1 molar ratio using a vortex mixer (Vortex 3, IKA Works, Inc.) for 1 min. The NH4I in contact with NiO was also prepared in a 1:1 molar ratio. From the prepared samples, ∼8–9 mg of the powder was placed in an alumina pan and heated from 25 °C to 450 °C at a rate of 10 °C min−1 under 100 sccm of N2 gas.
Gas phase thermal degradation studies
The thermal decomposition studies were performed using the TPD-MS-FTIR technique described in our previous work.22 The samples were prepared as above. For TPD-MS-FTIR experiments, the weight of FAI in all samples was kept constant at 40 mg. For the 1:1 molar ratio FAI + oxide experiments, 57 mg FAI + NiO and 59 mg FAI + TiO2 powders were used. For the 1:4 molar ratio experiments, 110 mg FAI + NiO and 114 mg FAI + TiO2 powders were used. In 1:1 molar ratio NH4I + NiO experiments, 61 mg was used. The samples were placed in a quartz tube, sandwiched between quartz wool. Prior to the analysis, the sample cell, heated gas lines, MS (Vision 1000-C, MKS Instruments, Spectra Products), and FTIR (Thermo Nicolet Nexus 670) were thoroughly purged using He gas at a flow rate of 30 sccm for 30 min to minimize environmental contributions such as H2O, O2, and CO2. Then the samples were heated from 25 °C to 400 °C at 10 °C min−1 under 30 sccm He flow at atmospheric pressure. The gaseous products were carried to FTIR and MS instruments for simultaneous in situ gas-phase analysis. The FTIR spectra were recorded with a resolution of 4 cm−1 in the range of 650–4000 cm−1 at 5 s intervals. The mass analysis was carried out by scanning sequentially from m/z = 2 to 300 and detected with a Faraday cup.
Solid decomposition product analysis
To identify the phase and composition of decomposed solid products, we mimicked the decomposition reactions by heating powders of FAI, FAI + NiO, or FAI + TiO2, to 100 °C, 150 °C, 200 °C, 250 °C, and 300 °C, sequentially—holding for 10 min at each temperature—on a hot plate (Thermo Scientific) inside a N2 purged glove box (Plas-Labs, Inc.) to prevent environmental contributions. The powder XRD data were collected on samples at each temperature using a Rigaku Ultima III diffractometer (40 kV/44 mA) equipped with Cu Kα radiation (λ = 1.5406 Å) over a 2θ range from 10° to 50° with a step size of 0.02° and a scan speed of 2° min−1. The crystalline phases were determined by comparing the experimental XRD patterns with the powder diffraction files (PDFs).Computational methodology
DFT modelling was employed to calculate adsorption and reaction energies using the VASP program package.37,38 We used plane wave basis expansions with an energy cutoff of 400 eV, and the Perdew–Burke–Ernzerhof (PBE) type generalized gradient approximation (GGA) for the exchange–correlation.39 The core-valence interaction was considered by selecting the projector-augmented wave (PAW) method.40 All atomic positions and lattice were relaxed until residual forces and the energy change were less than 0.01 eV Å−1 and 10−6 eV, respectively, to obtain unit cell configurations. Spin-polarized DFT and Hubbard U (U = 6.2 eV for Ni 3d electrons) correction was used to calculate NiO systems.41,42 The Tkatchenko–Scheffler method with iterative Hirshfeld partitioning was employed to reflect van der Waals interactions.43,44 NiO surface structures were made by multiplying converged unit cell lattice structures. Monkhorst–Pack sampling using 2 × 2 × 1 and 3 × 3 × 1 Γ-centered grids was used to calculate the NiO and TiO2 surfaces, respectively. NiO/TiO2 surfaces consisted of 128/135 atoms, and the vacuum layer was set to be larger than 12 Å. The positions of atoms below half of the slabs were fixed to mimic the surface structure. 3p3d4s and 3d4s were considered as valence states of Ti and Ni, respectively.Adsorption energy calculations
The adsorption energies were estimated by subtracting the surface and the adsorbed molecule energies from the total system energy. Therefore, the more negative the adsorption energies, the stronger the binding on the oxide surface.| Eads = Et(molecule adsorbed surface) − E(surface) − E(molecule) | (1) |
| ΔE = E(HCN* + NiO) + E(HI* + NiO) + E(NH3* + NiO) − Et((HC(NH2)2I)* + NiO) − 2E(NiO) | (2) |
Results
We performed TGA-DSC analysis to determine the thermal stability of neat FAI powders, which is compared to when FAI is mixed with dried NiO or TiO2 nanoparticles. For neat FAI (Fig. 1a), the onset of weight loss (black) occurs at ∼250 °C (blue dashed line) with almost complete weight loss (99.5%) by ∼320 °C. Two endothermic features are observed in the DSC data (Fig. 1a, red) during the FAI decomposition when it is by itself. Our neat FAI TGA-DSC results are in agreement with previous reports.18,29 When FAI is in contact with NiO (Fig. 1b), a more complex weight-loss transition (black) and corresponding multiple endothermic peaks (red) are observed in the TGA-DSC data. Most noticeably, the onset of decomposition shifts lower to ∼200 °C (Fig. 1b, blue dashed line). The 20% weight loss by 350 °C agrees with the amount of FA, not FAI, in the FAI + NiO sample. In contrast, when FAI is in contact with TiO2 (Fig. 1c), TGA-DSC data show no obvious difference from the neat FAI; the onset of decomposition occurs at ∼250 °C (Fig. 1c, blue dashed line) and completes by 300 °C. The 68% weight loss corresponds to the weight of FAI in the FAI + TiO2 sample. It is clear that contact with metal oxide affects the thermal stability of FAI while the weight loss percentage indicates that volatile products come from only FA, not FAI. In order to confirm that the thermal degradation of the FAPbI3 perovskite is largely determined by FAI, we performed TGA-DSC of neat FAPbI3, FAPbI3 + NiO, and FAPbI3 + TiO2 (Fig. S3, ESI†). The neat FAPbI3 is found to be stable up to 330 °C. However, when FAPbI3 contacts NiO, the thermal stability lowers to 220 °C while in contact with TiO2, the onset of decomposition occurs at 260 °C. Note that the decomposition temperatures of FAPbI3 with NiO and TiO2 are similar to FAI + NiO (200 °C) and FAI + TiO2 (250 °C). Although interactions between FA cations and inorganic Pb–I matrices are thought to be stronger, resulting in higher structural stability in FAPbI3 perovskite,18 our results explicitly show that the physical contact between perovskite and metal oxides does induce intrinsic instability in these materials. In particular, contact with NiO substantially lowers the thermal stability of FAI and FAPbI3 alike. Thus, the similar thermal stability behaviour between FAI and FAPbI3 validates our rational to perform further degradation studies using FAI.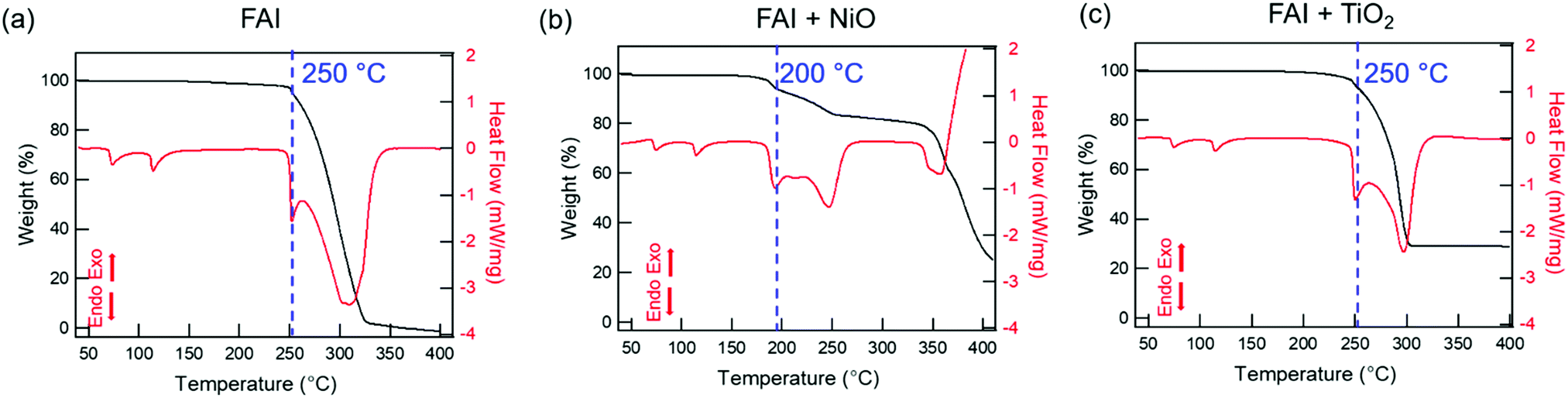 | ||
| Fig. 1 TGA (black, left y-axis) and DSC (red, right y-axis) curves for (a) FAI, (b) FAI + NiO (1:1), and (c) FAI + TiO2 (1:1) heated from 25 °C to 400 °C at 10 °C min−1 under 100 sccm N2 flow. The blue dashed lines represent the onset of thermal decomposition in each case. | ||
To identify the volatile decomposition products associated with thermal events observed in TGA-DSC, we performed TPD-MS-FTIR experiments with neat FAI, FAI + NiO, and FAI + TiO2 powders. Such simultaneous detections of evolved gases by FTIR and MS help to accurately identify molecular species as ionization probability and fragmentation into smaller ions in MS complicate the analysis while many organic moieties have overlapping vibrational frequencies in FTIR. The top panel in Fig. 2 shows FTIR temperature profiles representing infrared absorption intensity versus temperature for evolved gases. Comparing the observed IR spectra of the gas species released from the decomposition to NIST database,45 we assign the evolved gases at 967 cm−1, 3600–3800 cm−1, 739 cm−1, and 1551 cm−1 to ammonia (NH3, black), water (H2O, blue), hydrogen cyanide (HCN, red), and sym-triazine ((HCN)3, green), respectively. Note that the higher wavenumber region is used for H2O to avoid the overlap with NH3 signals in the 1500–1600 cm−1 region. The full FTIR line spectra at different temperatures for the three samples are shown in Fig. S4 (ESI†). In MS, a molecule can have several fragments with different mass to charge ratio (m/z) values. By comparing the intensity ratios at different m/z of all detected ions to NIST database,46 we identify the released gases to be NH3, H2O, HCN, and sym-triazine (Fig. S5, ESI†). Using the m/z of the parent ions, NH3+ (m/z = 17), H2O+ (m/z = 18), HCN+ (m/z = 27), and sym-triazine ((HCN)3+, m/z = 81), the MS temperature profiles are shown in the bottom panel of Fig. 2. For neat FAI (Fig. 2a and d), the FTIR and MS results show the decomposition temperature (Td), at which gases begin to evolve, to be ∼280 °C. While Td of the neat FAI observed here is closer to Perez et al.'s work (260 °C),30 Ma et al. reported it to be at 245 °C.18 The Td differences in these three works could arise from the different ramping rates used in heating the samples and the geometry of the experimental apparatus.17 In contrast, when FAI is in contact with NiO (Fig. 2b and e), gases begin to evolve at ∼200 °C, 100 °C lower than Td of neat FAI. When FAI is in contact with TiO2 (Fig. 2c and f), HCN starts to appear at ∼260 °C.
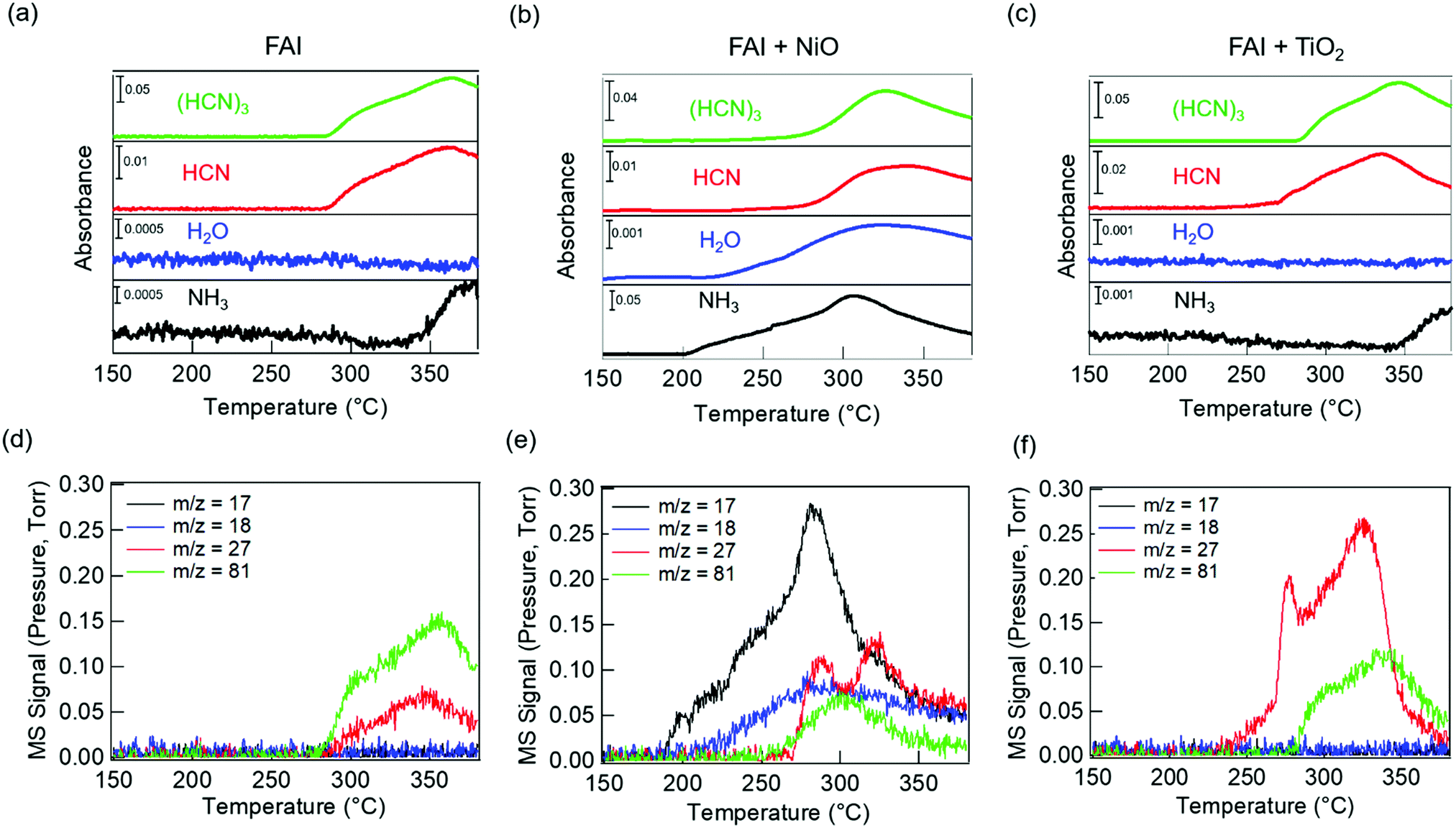 | ||
| Fig. 2 TPD-MS-FTIR results: (a–c) FTIR temperature profiles (absorbance versus temperature) and (d–f) MS signals of evolved gases for FAI (left), FAI + NiO (middle), and FAI + TiO2 (right). Signals at the characteristic vibrational frequencies of NH3 (967 cm−1), H2O (3600–3800 cm−1), HCN (739 cm−1), and sym-triazine (1551 cm−1) gases are used in FTIR profiles and represented with black, blue, red, and green lines, respectively, in (a–c). MS signals of m/z = 17 (black), m/z = 18 (blue), m/z = 27 (red), and m/z = 81 (green) represent NH3+, H2O+, HCN+, and sym-triazine parent ions, respectively. All assignments are based on the NIST database.45,46 | ||
Correlating the TPD results with TGA-DSC results, we can attribute the first endothermic peak in DSC (Fig. 1a, blue dashed line) to bulk decomposition of neat FAI, releasing sym-triazine and HCN gases simultaneously at Td ∼ 280 °C as detected by both FTIR and MS (Fig. 2a and d). In addition, FTIR (Fig. 2a) also shows NH3 evolution at a higher temperature ∼340 °C, indicating that bulk FAI decomposition does not produce NH3 directly; this temperature corresponds to the second endothermic peak in DSC. From these results, we can infer that the bulk decomposition of neat FAI occurs via a two-step process. Previous work on neat FAI thermal decomposition also reported sym-triazine, HCN, and NH3 as the gaseous products, but no detection of HI or I2.18,30 We also did not observe HI in FTIR or MS (Fig. 2a and d) as HI is known to adhere to cold surfaces in the apparatus.10 The fact that NH3 is not detected by MS (Fig. 2d) can be attributed to its low concentration, i.e. below the MS detection limit; FTIR is sensitive to the N–H symmetric deformation mode, but the intensities of these peaks are extremely low in this case (Fig. S4a, ESI,† black dotted rectangle in the zoomed-in view).
In the case of FAI + NiO, we observe two distinct degradation processes, with gaseous products of NH3, H2O, sym-triazine, and HCN (Fig. 2b and e). At 200 °C, NH3 and H2O are released simultaneously, corresponding to the first endothermic peak in DSC (Fig. 1b, blue dashed line), while sym-triazine and HCN only begin to evolve at ∼270 °C, which aligns well with the second endothermic peak in DSC. Because the high-temperature evolved gases are the same as neat FAI and also occur at a similar temperature, we attribute this process to bulk decomposition of FAI. The low-temperature event, during which NH3 and H2O are released, is a new degradation pathway that is not previously known. To accentuate the interfacial effects, we increased the molar ratio of FAI to NiO to 1:4. With excess NiO, both FTIR (Fig. S6a, ESI†) and MS (Fig. S6c, ESI†) results are dominated by the NH3 and H2O evolution at 200 °C, confirming that the low-temperature process arises from the interaction of FAI with NiO. At the same time, the sym-triazine signal is low in FTIR and not observed in MS, indicating that a very small amount is produced whereas HCN is detected using both techniques. It is noteworthy that the Td ∼200 °C and the released NH3 and H2O gas products are the same as observed for the decomposition of MAI in contact with NiO.22
For FAI + TiO2 samples, although there is no significant change in Td from neat FAI, HCN is released first at a lower temperature of ∼260 °C while sym-triazine is released at ∼280 °C (Fig. 2c and f). Similar to neat FAI, FTIR shows evidence of NH3 released at ∼340 °C (Fig. 2c and Fig. S4c, ESI†), in agreement with the DSC result (Fig. 1c) where we observed a high temperature endothermic peak at ≥300 °C. With the increased molar ratio of FAI:TiO2 to 1:4, Td remains the same as 1:1 FAI:TiO2, but much more HCN is produced compared to sym-triazine (Fig. S6b and d, ESI†).
Further insight into the degradation process can be gained by examining the solid decomposed products using XRD. The XRD patterns taken before heating and after heating at each temperature are shown in Fig. S7 (ESI†). The XRD patterns of neat FAI, FAI + NiO, and FAI + TiO2 measured after heating to 250 °C are shown in Fig. 3. For neat FAI (top panel), XRD shows strong NH4I peaks (maroon squares) and weak sym-triazine signals (green inverted triangles), indicating that neat FAI decomposition results in the formation of NH4I as the solid product. The XRD patterns of FAI + NiO (Fig. 3, middle panel) show predominantly NiI2 peaks (violet diamonds) along with weak NH4I signals (maroon squares). The formation of NiI2 coincides with the observed weight loss of FA only in the TGA of FAI + NiO (Fig. 1b), further substantiating the reaction between FAI and NiO, i.e. the interfacial reaction. NiI2 has been identified previously as the reaction product of halide perovskite and NiO.12,21,22 On the other hand, in the XRD pattern of FAI + TiO2 (Fig. 3, bottom panel), no peaks of TiI4, only TiO2 reflections (blue dumbbells), are observed at 250 °C. Thus, the absence of TiI4 is consistent with the weight loss in TGA of FAI + TiO2 (Fig. 1c), which corresponds to the total weight of FAI in the sample and supports the fact that no reaction occurs between TiO2 and FAI.
 | ||
| Fig. 3 XRD patterns of neat FAI (top panel), FAI + NiO (middle panel), and FAI + TiO2 (bottom panel) after heating to 250 °C (brown) for 10 min. Peaks associated with crystalline NH4I, sym-triazine, NiI2, and TiO2 are marked by maroon squares, green inverted triangles, violet diamonds, and blue dumbbells, respectively. | ||
Discussion
Unlike MAI, the decomposition pathway and products of FAI are currently still not well understood. Although there are broad agreements on two overall reactions,18,30,32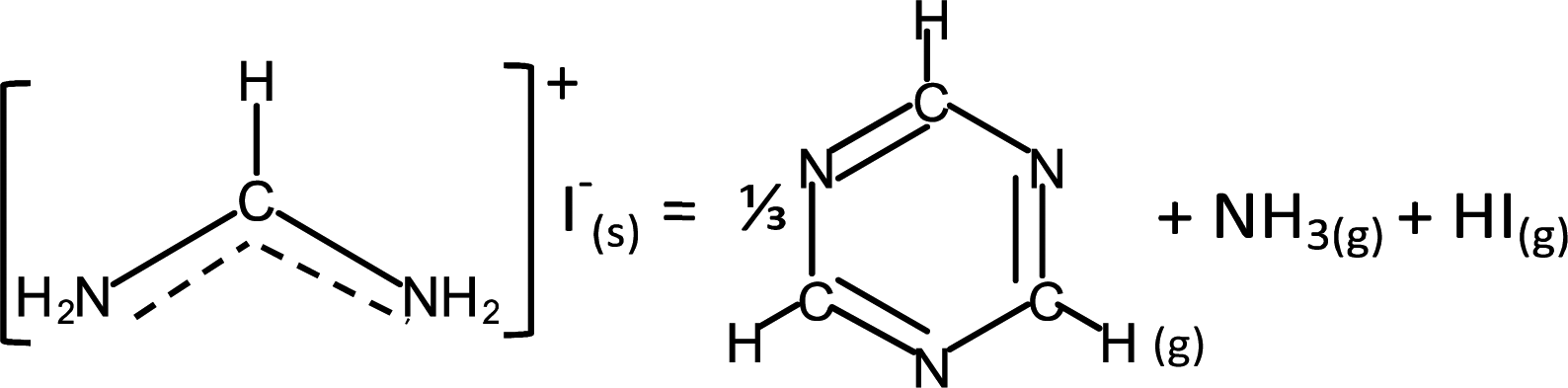 | (3a) |
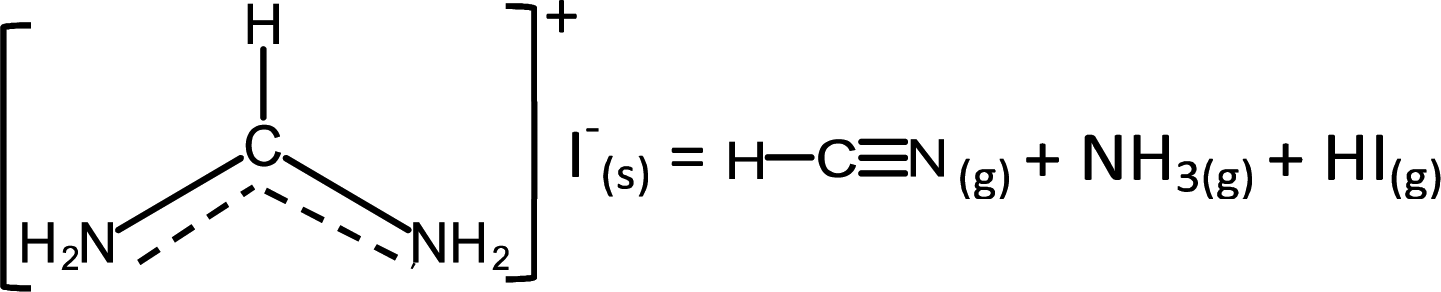 | (3b) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N while sym-triazine does not. FTIR results by both Ma et al. and this work show no evidence of the spectroscopic signature of CN in malononitrile at ∼2190 cm−1.47 Moreover, aminomalononitrile is unstable; it would have reacted quickly with HCN to produce diaminomaleonitrile.48 The relation between FA and sym-triazine was also studied previously under the context of sym-triazine synthesis from FACl; the authors reported that high reactivity of FA arising from its negligible steric effect from H atom favours the sym-triazine route while nitrile formation occurs when H is replaced by neutral groups.49 Therefore, based on these reports and our combined FTIR and MS results, the assignment of the gas species to sym-triazine is valid.
N while sym-triazine does not. FTIR results by both Ma et al. and this work show no evidence of the spectroscopic signature of CN in malononitrile at ∼2190 cm−1.47 Moreover, aminomalononitrile is unstable; it would have reacted quickly with HCN to produce diaminomaleonitrile.48 The relation between FA and sym-triazine was also studied previously under the context of sym-triazine synthesis from FACl; the authors reported that high reactivity of FA arising from its negligible steric effect from H atom favours the sym-triazine route while nitrile formation occurs when H is replaced by neutral groups.49 Therefore, based on these reports and our combined FTIR and MS results, the assignment of the gas species to sym-triazine is valid.
To explain our experimental observations and to elucidate the degradation pathways in neat FAI and when FAI contacts NiO or TiO2, we employed DFT calculations. The energy changes for decomposition reactions in the gas phase and on oxide surfaces are calculated with adsorption energy data sets and are represented in Fig. 4 and Table S1 (ESI†). Here our calculation results shed light on the conflicting reports of whether sym-triazine or HCN is formed first. Fig. 4 shows that, for a given sample, neat FAI, FAI + NiO, or FAI + TiO2, the reactions that produce sym-triazine (green dashed lines) have lower energies than equivalent reactions that produce HCN (red dashed lines). Thus, sym-triazine is a thermodynamically favored product over HCN, i.e.reaction (3a) dominates over (3b). The middle panel in Fig. 4 shows that contact with NiO (pink pentagons) and TiO2 (blue dumbbells) lowers the energies of both reaction (3a) and (3b) compared to neat FAI (black circles). The reaction energies decrease as FAI > FAI + TiO2 > FAI + NiO, consistent with the decrease in Td observed in TPD-FTIR-MS (Fig. 2), i.e. 280 °C for FAI > 260 °C for FAI + TiO2 > 200 °C for FAI + NiO. Note that our calculations present the thermodynamic energy changes for selected reactions, but not the activation energy barrier heights. Thus, the negative energy of reaction (3a) on NiO indicates that the reaction is thermodynamically favoured but does not mean that it will occur spontaneously.
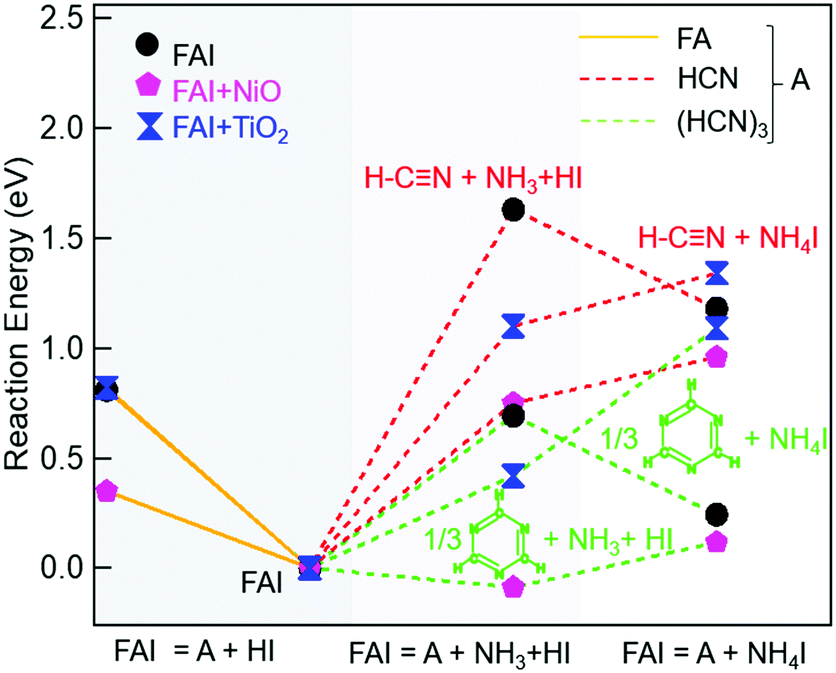 | ||
| Fig. 4 Decomposition energy diagram of FAI. The energy of FAI is set to 0 as a reference. Left column represents decomposition to FA + HI (orange solid lines). The middle column represents decomposition into sym-triazine (green dashed lines)/HCN (red dashed lines) + NH3 + HI and is considered to be the first degradation pathway. The right column represents the reaction energies of NH4I formation, the difference from the previous step represents the energy needed for NH4I formation from NH3 + HI. The black circles, pink pentagons, and blue dumbbells represent FAI, FAI + NiO, and FAI + TiO2, respectively. | ||
While Perez et al. and Akbulatov et al. proposed NH4I in FAI or FAPbI3 decomposition,30,32 our experimental results are the first to unambiguously identify its presence in the decomposition products. The XRD results of partially decomposed FAI after 250 °C heat treatment (Fig. 3, top panel) show the existence of sym-triazine (green inverted triangles) and NH4I (maroon squares). Our DFT calculations (Table S1, ESI†) show that when FAI decomposes, the formation of NH4I from NH3 and HI is energetically favorable (−0.45 eV). In this case (black circle), the lowest energy reaction produces sym-triazine + NH4I (0.24 eV, green dashed line, right column), while the reaction that produces sym-triazine + NH3 + HI, i.e.reaction (3a), has a higher energy of 0.69 eV (green dashed line, middle column). Moreover, the sym-triazine + NH4I reaction requires less energy than the equivalent reaction that produces HCN + NH4I (1.2 eV, red dashed line, right column). We therefore propose that FAI first transforms to sym-triazine and NH4I. sym-triazine and HCN readily desorb at ∼280 °C in the TPD experiment (Fig. 2a and d), while NH4I is still a solid at this temperature. As the temperature further rises, NH4I undergoes complete decomposition to NH3 + HI by 340 °C with NH3 being detected (Fig. 2a). The TGA-DSC of neat NH4I shown in Fig. S8 (ESI†) provides further evidence that NH4I decomposes ≥300 °C. Comparing the DSC curves (red) of NH4I with FAI (Fig. 1a), the endothermic peak at ∼330 °C matches well with the second endothermic peak in FAI, supporting our hypothesis that FAI decomposition occurs in two steps with the release of NH3 (Fig. 2a) as the result of NH4I decomposition. Since the reaction to form NH4I is highly exothermic, the reformation of solid NH4I on the colder parts of the experimental apparatus can occur, which has been suggested previously.32 In neat FAI and FAI + TiO2 TPD experiments, we observed white deposits lining the exhaust capillary of the cell. Comparing the XRD pattern of this white deposit (Fig. S9, ESI,† orange) to neat NH4I (Fig. S9, ESI,† maroon), it is identified as NH4I. Furthermore, NH4I TPD results show no NH3 or HI (m/z = 128) gases (Fig. S10a, ESI†) and a large amount of white deposits in the exhaust capillary of the sample cell (Fig. S10b, ESI†). Thus, based on these results, we determine that the neat FAI undergoes decomposition via sym-triazine + NH4I first and NH4I further decomposes to NH3 + HI at higher temperature, with the possibility of NH4I reformation on colder surfaces.
A notable difference in neat FAI vs. FAI + oxides is the decomposition via NH4I vs. NH3 + HI. Since solid NH4I is not stable on either NiO (0.21 eV) or TiO2 (0.67 eV) surfaces (Table S1, ESI†), it dissociatively adsorbs as NH3* and HI* on the oxide surfaces instead of as NH4I*. This difference in the adsorption characteristics on oxide surfaces explains the presence of NH4I (maroon squares) in the XRD of neat FAI (Fig. 3, top panel), while a lower amount is found in that of FAI + NiO (Fig. 3, middle panel) and none in that of FAI + TiO2 (Fig. 3, bottom panel). Hence, the decomposition of FAI on NiO or TiO2 follows decomposition reaction (3a). We next analyse the effects of oxide surfaces on FAI decomposition pathways based on the adsorption energies of molecules summarized in Table 1. As shown in Fig. 4, FAI decomposition into sym-triazine, NH3, and HI (reaction (3a)) on NiO has a significantly lower reaction energy (−0.09 eV, green dashed lines, middle column) compared to neat FAI decomposing into sym-triazine and NH4I (0.24 eV, green dashed lines, right column). The low energy of reaction (3a) for FAI + NiO is in good agreement with the experimental results, where we also observed NH3 (not NH4I) and H2O at 200 °C, and sym-triazine and HCN at 270 °C (Fig. 2b and e). The low desorption temperature of NH3 (200 °C) on the NiO surface is attributed to its low binding energy (−0.77 eV, Table 1) and hence it can be readily desorbed from the surface. On the other hand, the HI gas that must be formed as a decomposition product and adsorbed on the surface as HI* reacts further with NiO producing H2O and NiI2. H2O desorbs from the surface as water vapor and is detected in both FTIR and MS (Fig. 2b and e, blue), and NiI2 remains as a solid decomposed product as observed in XRD (Fig. 3, middle panel, violet diamonds). Thus, the detection of NiI2, NH3, and H2O at low temperature (∼200 °C) points to the prevalence of the interfacial reaction of FAI with NiO. To further substantiate these findings, we performed TGA-DSC and TPD-FTIR-MS experiments on NH4I + NiO (1:1 molar ratio). We also observed the decomposition of this sample starting at 220 °C and a clear endothermic peak at 250 °C (Fig. S11, ESI†), while both FTIR (Fig. S12a, ESI†) and MS (Fig. S12b, ESI†) in the TPD experiment detect evolution of NH3 (black) and H2O (blue) gases starting at 220 °C. Thus, the similar behaviours of FAI + NiO and NH4I + NiO unambiguously show that FAI reacting with NiO at the interface results in FAI decomposing prematurely. In addition, this result also validates the simulation that NH4I is unstable on the NiO surface. As pointed out earlier, FAI and MAI when in contact with NiO show similar thermal stability, with both undergoing interfacial decomposition at ∼200 °C. These results suggest that the intrinsic stability is dictated by the oxide, rather than the perovskite.
| NiO(001) (eV) | TiO2(001) (eV) | |
|---|---|---|
| NH3* | −0.77 | −1.43 |
| HI* | −1.28 | −2.15 |
| sym-triazine* | −0.87 | −1.42 |
| HCN* | −0.39 | −0.83 |
| NH4I* | −1.39 | −2.46 |
| CH(NH2)2I* | −1.56 | −3.78 |
| NH–CH–NH2* | −0.74 | −1.62 |
While interfacial decomposition is evident for FAI + NiO with lower Td, the released gases at 200 °C are only NH3 and H2O. The reason why sym-triazine is not detected at 200 °C along with NH3 is due to its higher adsorption energy than NH3* (−0.87 eV vs. −0.77 eV, Table 1). Thus, we observe sym-triazine from both interfacial and bulk decomposition starting at 270 °C as a result of its strong adsorption energy on the NiO surface. While HCN has a lower adsorption energy (Table 1), the reason that HCN is not observed at low temperature is because on the NiO surface, the reaction energy for FAI decomposing to produce sym-triazine is lower than HCN (−0.09 vs. 0.75 eV, Table S1, ESI†). Therefore sym-triazine, not HCN, is the decomposition product at 200 °C. At higher temperatures, configurational entropy favors HCN over sym-triazine. The free energies of HCN and sym-triazine can be estimated by considering entropy contribution using the values from JANAF table: −TΔS @227 °C = −1.15 eV.50 The entropy of sym-triazine is assumed to be 1/3 of the entropy of HCN. The free energy difference then becomes 0.07 eV at 227 °C and −0.11 eV at 327 °C. This implies that HCN can be generated from sym-triazine between 227 °C and 327 °C, without revoking the attack of hydrogen radicals from HI previously proposed.18 However, the formation energy for HCN from sym-triazine is higher by 0.84 eV on NiO (Table S1, ESI†) compared to 0.58 eV for FAI + TiO2. Therefore, HCN evolves at 270 °C along with sym-triazine on the NiO surface. The lower reaction energy along with lower Td supports the dominance of the interfacial reaction when FAI is in contact with NiO. Based on these results, the interfacial decomposition reaction of FAI + NiO can be written as:
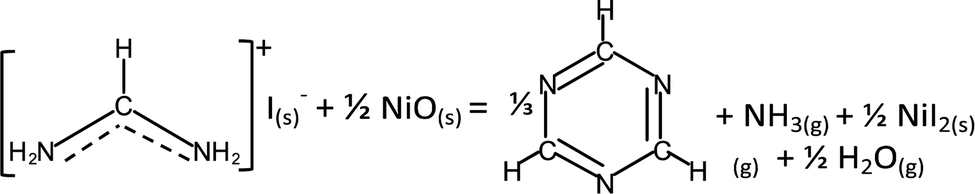 | (4) |
In contrast to NiO, the energy for FAI decomposition on the TiO2 surface according to reaction (3a) is 0.42 eV (green dashed lines, middle column) which is slightly higher than neat FAI decomposing into sym-triazine and NH4I (0.24 eV, green dashed lines, right column), so FAI + TiO2 mostly follow the bulk FAI decomposition pathway. Experimentally, we do not observe a significantly different Td (Fig. 1c, 2c and f). Similar to neat FAI, sym-triazine, HCN, and NH3 are the decomposed gaseous products. The decomposition of FAI + TiO2 should also produce HI. However, there is no reaction between TiO2 and HI* as the formation enthalpy of TiI4 is less negative compared to NiI2 (−0.9 eV vs. −2.4 eV),51,52 and is consistent with no TiI4 in the XRD (Fig. 3, bottom panel). Moreover, sym-triazine and NH3 are released at higher temperatures of 280 °C and 340 °C, respectively, while HCN is detected at a lower temperature of 260 °C. This is because the adsorption energies of sym-triazine* (−1.42 eV), NH3* (−1.43 eV), and HI* (−2.15 eV) are quite strong on TiO2 compared to the NiO surface, which explains why we cannot detect them under 280 °C. On the other hand, the HCN* binding energy (−0.83 eV) is significantly lower than the other three molecules and HCN formation energy from sym-triazine is also lower (−0.58 eV), and readily desorbs from the TiO2 surface.
It is noteworthy that the HCN formation is also affected by the oxide surface. We see that a higher amount of HCN is formed on both NiO and TiO2 surfaces compared to neat FAI. This is because the formation energy of HCN from sym-triazine on NiO (−0.84 eV) and TiO2 (−0.58 eV) surfaces is lower compared to neat FAI (−0.94 eV). In addition, the adsorption energies of HCN on both NiO and TiO2 surfaces are lower than sym-triazine (Table 1). Thus, the lower formation energy compared to FAI coupled with lower adsorption energy of HCN on oxide surfaces explains the larger amount of HCN detection on the FAI + TiO2 (Fig. 2f) and FAI + NiO samples (Fig. 2e), in particular when FAI is mixed with excess oxides (1:4 molar ratios, Fig. S6, ESI†).
Finally, the decomposition of FAI to gas-phase FA and HI (Fig. 4, orange solid lines and Table S1, ESI†) for all three cases is unfavourable, consistent with TPD results where we did not observe FA (m/z = 44) in any samples.
Conclusions
In conclusion, we have shown that interfacial interaction between the perovskite and metal oxide contact layer can trigger degradation and induce instability in PSCs. The bulk decomposition of FAI occurs at 250 °C via a two-step decomposition process: FAI first decomposes to sym-triazine and NH4I at ∼280 °C, and then NH4I further decomposes to NH3 and HI ≥ 300 °C. Among the two oxides, NiO is much more reactive and Td is lowered to 200 °C compared to the bulk Td of neat FAI. The interfacial reaction between FAI and NiO releases NH3 and H2O at 200 °C while producing NiI2 as a solid decomposed product; on the other hand, sym-triazine, from both interfacial and bulk decomposition of FAI, is released at 270 °C due to its strong adsorption energy on the NiO surface. The interfacial decomposition temperature reported here is similar to that of MAI in contact with NiO,22 indicating the fundamental importance of oxide transport layer materials on perovskite device stability. FAI adsorbed on the TiO2 surface slightly lowers Td, but the stability of TiO2, relative to NiO, prevents chemical reactions from taking place. The similar thermal stability behaviors of FAI and FAPbI3 further emphasize that the degradation studies should not be performed on the perovskite material alone, but should also consider the chemical reactivity of perovskites with materials that they might come into contact with, so that strategies to propel PSCs towards commercialization can be developed.Full legal disclaimer
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank K. Cho for insightful discussions. This material is based on work partially supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Solar Energy Technology Office Award Number DE-EE0008544. S. T. acknowledges support from VPR Post-doctoral Accelerator Award from UT Dallas. B. Z. acknowledges support from the National Science Foundation (CBET-1916612). J.-G. P. and K.-H. H. acknowledges the support from the National R&D Program through the National Research Foundation of Korea (NRF) (NRF-2015M1A2A2055836, NRF-2018R1A2B6007888, and NRF-2017M3A7B4041698) and the New & Renewable Energy Core Technology Program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) (No. 20183010013820). J. W. P. H. acknowledges support from the Texas Instruments Distinguished Chair in Nanoelectronics.Notes and references
- M. Saliba, J. P. Correa-Baena, C. M. Wolff, M. Stolterfoht, N. Phung, S. Albrecht, D. Neher and A. Abate, Chem. Mater., 2018, 30, 4193–4201 CrossRef CAS.
- F. Xu, T. Zhang, G. Li and Y. Zhao, J. Mater. Chem. A, 2017, 5, 11450–11461 RSC.
- L. K. Ono, E. J. Juarez-Perez and Y. Qi, ACS Appl. Mater. Interfaces, 2017, 9, 30197–30246 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- M. Saliba, T. Matsui, K. Domanski, J.-Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J.-P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Science, 2016, 354, 206–209 CrossRef CAS.
- S.-H. Turren-Cruz, A. Hagfeldt and M. Saliba, Science, 2018, 362, 449–453 CrossRef CAS.
- J.-W. Lee, D.-H. Kim, H.-S. Kim, S.-W. Seo, S. M. Cho and N.-G. Park, Adv. Energy Mater., 2015, 5, 1501310 CrossRef.
- R. Wang, M. Mujahid, Y. Duan, Z. K. Wang, J. Xue and Y. Yang, Adv. Funct. Mater., 2019, 29, 1–25 Search PubMed.
- S. He, L. Qiu, L. K. Ono and Y. Qi, Mater. Sci. Eng., R, 2020, 140, 100545 CrossRef.
- Z. Song, C. Wang, A. B. Phillips, C. R. Grice, D. Zhao, Y. Yu, C. Chen, C. Li, X. Yin, R. J. Ellingson, M. J. Heben and Y. Yan, Sustainable Energy Fuels, 2018, 2, 2460–2467 RSC.
- E. J. Juarez-Perez, L. K. Ono, M. Maeda, Y. Jiang, Z. Hawash and Y. Qi, J. Mater. Chem. A, 2018, 6, 9604–9612 RSC.
- A. G. Boldyreva, I. S. Zhidkov, S. Tsarev, A. F. Akbulatov, M. M. Tepliakova, Y. S. Fedotov, S. I. Bredikhin, E. Y. Postnova, S. Y. Luchkin, E. Z. Kurmaev, K. J. Stevenson and P. A. Troshin, ACS Appl. Mater. Interfaces, 2020, 12, 19161–19173 CrossRef CAS.
- A. F. Akbulatov, L. A. Frolova, N. N. Dremova, I. Zhidkov, V. M. Martynenko, S. A. Tsarev, S. Y. Luchkin, E. Z. Kurmaev, S. M. Aldoshin, K. J. Stevenson and P. A. Troshin, J. Phys. Chem. Lett., 2020, 11, 333–339 CrossRef CAS.
- A. E. Williams, P. J. Holliman, M. J. Carnie, M. L. Davies, D. A. Worsley and T. M. Watson, J. Mater. Chem. A, 2014, 2, 19338–19346 RSC.
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC.
- J. A. McLeod and L. Liu, J. Phys. Chem. Lett., 2018, 9, 2411–2417 CrossRef CAS.
- A. Ciccioli and A. Latini, J. Phys. Chem. Lett., 2018, 9, 3756–3765 CrossRef CAS.
- L. Ma, D. Guo, M. Li, C. Wang, Z. Zhou, X. Zhao, F. Zhang, Z. Ao and Z. Nie, Chem. Mater., 2019, 31, 8515–8522 CrossRef CAS.
- A. Latini, G. Gigli and A. Ciccioli, Sustainable Energy Fuels, 2017, 1, 1351–1357 RSC.
- X. X. Gao, W. Luo, Y. Zhang, R. Hu, B. Zhang, A. Züttel, Y. Feng and M. K. Nazeeruddin, Adv. Mater., 2020, 32, 1–9 CrossRef.
- W. A. Dunlap-Shohl, T. Li and D. B. Mitzi, ACS Appl. Energy Mater., 2019, 2, 5083–5093 CrossRef.
- S. Thampy, B. Zhang, K.-H. Hong, K. Cho and J. W. P. Hsu, ACS Energy Lett., 2020, 5, 1147–1152 CrossRef CAS.
- T. H. Schloemer, J. A. Raiford, T. S. Gehan, T. Moot, S. Nanayakkara, S. P. Harvey, R. C. Bramante, S. Dunfield, A. E. Louks, A. E. Maughan, L. Bliss, M. D. McGehee, M. F. A. M. van Hest, M. O. Reese, S. F. Bent, J. J. Berry, J. M. Luther and A. Sellinger, ACS Energy Lett., 2020, 5, 2349–2360 CrossRef CAS.
- R. A. Kerner and B. P. Rand, J. Phys. Chem. Lett., 2017, 8, 2298–2303 CrossRef CAS.
- W. A. Dunlap-Shohl, R. Younts, B. Gautam, K. Gundogdu and D. B. Mitzi, J. Phys. Chem. C, 2016, 120, 16437–16445 CrossRef CAS.
- I. S. Zhidkov, D. W. Boukhvalov, A. I. Kukharenko, L. D. Finkelstein, S. O. Cholakh, A. F. Akbulatov, E. J. Juárez-Pérez, P. A. Troshin and E. Z. Kurmaev, J. Phys. Chem. C, 2020, 124, 14928–14934 CrossRef CAS.
- D. Di Girolamo, F. Di Giacomo, F. Matteocci, A. G. Marrani, D. Dini and A. Abate, Chem. Sci., 2020, 11, 1–26 RSC.
- J. Byeon, J. Kim, J.-Y. Kim, G. Lee, K. Bang, N. Ahn and M. Choi, ACS Energy Lett., 2020, 5, 2580–2589 CrossRef CAS.
- C. C. Boyd, R. C. Shallcross, T. Moot, R. Kerner, L. Bertoluzzi, A. Onno, S. Kavadiya, C. Chosy, E. J. Wolf, J. Werner, J. A. Raiford, C. Paula, A. F. Palmstrom, Z. J. Yu, J. J. Berry, S. F. Bent, Z. C. Holman, J. M. Luther, E. L. Ratcliff, N. R. Armstrong and M. D. McGehee, Joule, 2020, 4, 1759–1775 CrossRef CAS.
- E. J. Juarez-Perez, L. K. Ono and Y. Qi, J. Mater. Chem. A, 2019, 7, 16912–16919 RSC.
- W. T. M. Van Gompel, R. Herckens, G. Reekmans, B. Ruttens, J. D’Haen, P. Adriaensens, L. Lutsen and D. Vanderzande, J. Phys. Chem. C, 2018, 122, 4117–4124 CrossRef CAS.
- A. F. Akbulatov, V. M. Martynenko, L. A. Frolova, N. N. Dremova, I. Zhidkov, S. A. Tsarev, S. Y. Luchkin, E. Z. Kurmaev, S. M. Aldoshin, K. J. Stevenson and P. A. Troshin, Sol. Energy Mater. Sol. Cells, 2020, 213, 110559 CrossRef CAS.
- P. E. Marchezi, E. M. Therézio, R. Szostak, H. C. Loureiro, K. Bruening, A. Gold-Parker, M. A. Melo, C. J. Tassone, H. C. N. Tolentino, M. F. Toney and A. F. Nogueira, J. Mater. Chem. A, 2020, 8, 9302–9312 RSC.
- H. Wei, S. Chen, J. Zhao, Z. Yu and J. Huang, Chem. Mater., 2020, 32, 2501–2507 CrossRef CAS.
- M. B. Islam, M. Yanagida, Y. Shirai, Y. Nabetani and K. Miyano, ACS Omega, 2017, 2, 2291–2299 CrossRef CAS.
- K. Wang, S. Olthof, W. S. Subhani, X. Jiang, Y. Cao, L. Duan, H. Wang, M. Du and S. Liu, Nano Energy, 2020, 68, 104289 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- Q. Xu, S. Cheah and Y. Zhao, J. Chem. Phys., 2013, 139, 024704 CrossRef.
- T. Yu, Z. Li, H. Zheng, L. Chen, W. Song, Z. Zhao, J. Li and J. Liu, Mol. Catal., 2019, 474, 110417 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- P. Bultinck, C. Van Alsenoy, P. W. Ayers and R. Carbó-Dorca, J. Chem. Phys., 2007, 126, 144111 CrossRef.
- W. E. Wallace, ‘Infrared Spectra’ in NIST Chemistry WebBook NIST Standard Reference Database Number 69, 2017.
- W. E. Wallace, ‘Mass Spectra’ in NIST Chemistry WebBook NIST Standard Reference Database Number 69, 2017.
- L. De Vries, J. Org. Chem., 1971, 36, 3442–3450 CrossRef CAS.
- D. S. Donald and O. W. Webster, Adv. Heterocycl. Chem., 1987, 41, 1–40 CrossRef CAS.
- F. C. Schaefer, I. Hechenbleikner, G. A. Peters and V. P. Wystrach, J. Am. Chem. Soc., 1959, 81, 1466–1470 CrossRef CAS.
- M. Chase, J. Phys. Chem. Ref. Data, Monogr., 1998, 9, 1–1951 Search PubMed.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- A. Jain, G. Hautier, S. P. Ong, C. J. Moore, C. C. Fischer, K. A. Persson and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1–10 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: FAPbI3 synthesis and XRD, adsorption configurations, TGA-DSC, FTIR line spectra, TPD-MS bar charts, TPD-MS-FTIR of 1:4 FAI + NiO and FAI + TiO2, XRD patterns, reaction energy table, TGA-DSC of NH4I, XRD of FAI after TPD, TPD-MS of NH4I, TGA-DSC and TPD-MS-FTIR of 1:1 NH4I + NiO are provided. See DOI: 10.1039/d0ma00624f |
| ‡ These two authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |