
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on recent approaches for designing the SEI layer on sodium metal anodes
Jisung
Lee†
 a,
Jinuk
Kim†
a,
Seongseop
Kim
a,
Changshin
Jo
*b and
Jinwoo
Lee
*a
a,
Jinuk
Kim†
a,
Seongseop
Kim
a,
Changshin
Jo
*b and
Jinwoo
Lee
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea. E-mail: jwlee1@kaist.ac.kr
bSchool of Chemical Engineering & Materials Science, Chung-Ang University (CAU), 84 Heukseok-ro, Dongjak-gu, Seoul, 06974, Republic of Korea. E-mail: changshin@cau.ac.kr
First published on 10th November 2020
Abstract
With the ever-increasing demand for high energy density and low-cost energy storage devices, researchers have revitalized the field of alkali metal batteries. One of the emerging candidates are the sodium metal batteries (SMBs) as they have the merits of high theoretical capacity (1166 mA h g−1) and can be easily manufactured owing to the abundance of sodium resources. However, the uncontrollable dendrite growth on sodium metal, mainly attributed to its inhomogeneous and unstable solid–electrolyte interphase (SEI), has severely hindered the practical application of SMBs. In this review, we first summarize the critical challenges faced in the practical application of SMBs based on fundamental studies. We then discuss the recent approaches for designing an SEI layer on sodium metal anodes from two perspectives combined with feasible ideas, i.e., electrolyte modification and artificial interphase engineering. Finally, the practical applications and prospects of SMBs based on the fundamental knowledge and characterization methods are provided. We anticipate that our review will provide essential insights on stable SEI layer formation and also contribute to innovative ideas for developing practical SMBs.
 Jisung Lee | Jisung Lee received his BS degree (2016) from the Department of Chemical and Biological Engineering at Korea University and MS degree (2018) from the School of Chemical Engineering at Pohang University of Science and Technology (POSTECH), Korea. Currently, he is a PhD candidate under the supervision of Prof. Jinwoo Lee in the Department of Chemical and Biomolecular engineering at Korea Advanced Institute of Science and Technology (KAIST). His current research interests focus on the synthesis of advanced materials and their application for energy storage. |
 Jinuk Kim | Jinuk Kim received his BS (2018) and MS (2020) degrees from the Department of Chemical and Biomolecular Engineering at Korea Advanced Institute of Science and Technology (KAIST). Currently, he is a PhD candidate under the supervision of Prof. Jinwoo Lee at the Department of Chemical and Biomolecular Engineering, KAIST. His current research interests focus on protecting alkali metal anodes for next-generation batteries. |
 Changshin Jo | Changshin Jo obtained his BS (2010) and PhD (2016) degrees from Pohang University of Science and Technology (POSTECH) in the field of nanotechnology for energy storage. After postdoctoral research at POSTECH (with Prof. Jinwoo Lee) and University of Cambridge (as Marie Skłodowska-Curie Fellow with Prof. Michael De Volder), he joined the faculty of the School of Chemical Engineering and Materials Science at Chung-Ang University. His current research interests focus on the scalable synthesis of nanostructured materials for energy storage applications. |
 Jinwoo Lee | Jinwoo Lee obtained his BS (1998) and PhD (2003) degrees from the Department of Chemical and Biological Engineering of Seoul National University (SNU), Korea. After postdoctoral research at SNU (with Prof. Taeghwan Hyeon) and Cornell University (with Prof. Ulrich Wiesner), he joined the faculty of the Department of Chemical Engineering at POSTECH (2008–2018). In 2018, he joined the faculty of the Department of Chemical and Biomolecular Engineering at KAIST. His researches focus on the synthesis of designed nano-functional materials for energy conversion and storage devices. |
1. Introduction
Since the continuous consumption of fossil fuels as energy sources would eventually lead to a devastating environmental catastrophe, renewable energy technology is rapidly growing.1–3 Although renewable energy, which comes from natural sources, is low cost and inexhaustible, it needs to be integrated with energy storage devices to complement its intermittent feature.4–9 To date, lithium-ion batteries (LIBs), commercialized in 1991, are the most powerful and fascinating energy storage devices. While LIBs have been developed continuously over the past 30 years, they have approached the limit of their theoretical energy density.10–13 Recently, in a bid to go beyond their stagnant energy density, lithium metal electrodes with extremely high theoretical capacity (3860 mA h g−1) and the most negative standard redox potential (−3.04 V vs. standard hydrogen electrode (SHE)) have revived and are in the limelight again.11,14–21 However, lithium metal is considered as a rare metal owing to its increasing price and limited availability.22–25 Researchers forecast that more than one-third of the global lithium reserves will be exhausted by the year 2050.22 Moreover, the depletion rate would accelerate if the anodes of advanced LIBs are replaced with the lithium metal.The upcoming unstable lithium market has motivated researchers to find alternative secondary ion batteries such as sodium-ion batteries (SIBs), which have electrochemical features similar to those of LIBs.25,26 Generally, sodium can be extracted easily through the electrolytic reduction of sodium chloride, which is naturally abundant in seawater;27 thus, it is unlikely to be affected by limited supply and price fluctuations. In addition, the copper current collector could be replaced with the lighter and cheaper aluminum as sodium does not react with aluminum at a low potential.25,28,29 However, traditional SIBs possess critical obstacles as follows: (1) in typical electrolytes, sodium ions cannot be intercalated into graphite, which has been successfully commercialized as an anode in LIBs because it is thermodynamically unstable for forming binary sodium-intercalated graphite compounds.30,31 Although great efforts have been devoted to searching ultrahigh capacity anode materials such as phosphorus,32 tin,33 and metal oxide/phosphide,32,34 it is necessary to design electrodes via extremely high-cost nano-engineering because they demonstrate extremely poor electrochemical performance in the bulky state. (2) Both the atomic weight and ionic volume of sodium ions are two times larger than those of lithium ions,35,36 requiring the larger and heavier SIBs to deliver the same volumetric and gravimetric energy densities as that of LIBs. Thus, to transcend their intrinsic limitations, sodium metal batteries (SMBs) have been considered as a competitive edge in practical applications.
Sodium metal is regarded as a promising anode considering its high theoretical capacity (1166 mA h g−1), comparatively low standard redox potential (−2.71 V vs. SHE), and low cost, which are beneficial for large-scale energy storage systems.36–39 However, there are key challenges (e.g., the reactivity of sodium metal, severe volume change, dead sodium, and dendrite growth) that hinder further advances in sodium metal batteries. The principle of critical issues faced by SMBs is discussed in Section 2. Most of the critical issues originate from the deteriorated interphase layer. As the highly reactive sodium metal is always in contact with the electrolyte, the interphase layer is inevitably formed by continuous decomposition during cycling. Although the interphase layer serves to prevent further decomposition of the electrolyte in general, it is prone to pulverization due to the drastic volume change in the electrode. Worse, the thick and chemically inhomogeneous layer formed by excessive reaction is attributed to not only low sodium ion conductivity but also rapid sodium dendrite growth at physically vulnerable spots. Accordingly, diverse approaches for designing a homogeneous layer have been proposed.
Several reviews on the comprehensive strategy for sodium metal anodes have been reported, which focusing on (1) modification of salt, solvent, and additives in the electrolyte; (2) engineering of the metal/electrolyte interphase; (3) electroplating on the nanostructured current collector and the sodiophilic host; and (4) developing solid-state electrolytes. We focused on the SEI layer profoundly for effective design. In this review, we first clarify the critical challenges of Na metal anodes that are to be addressed and the fundamental reasons. Second, we summarize the recent progress in tailoring metal/electrolyte interphase films, which are subdivided based on the interphase film-forming process. At the end of each section, we propose research directions and possible candidates for enhancing the performance of sodium metal batteries. Then, we introduce high energy density sodium metal full cells such as Na–S, Na–O2, and Na–CO2 batteries, which are fabricated with sodium metal protected by the film. Finally, the remaining challenges in conclusion and future research directions are presented in the field of tailoring interphase films.
2. Key challenges
Since the lithium and sodium metal anodes possess a highly reductive nature, the electrolyte is spontaneously decomposed to form an organic/inorganic hybrid layer known as SEI layer on the alkali metal anodes as they are directly in contact with the electrolyte.40 Because they belong to group 1 of the periodic table, the principle of formation and morphology, as well as elemental composition, are similar to some extent. The morphology and elemental composition of the formed layer have a crucial impact on the dendritic growth. However, the intrinsically more reactive sodium metal anodes could aggressively attack the electrolyte, resulting in the formation of a thick and inhomogeneous SEI layer, which induces non-uniform sodium ion flux. Besides, unstable organometallic compounds and large proportion of organic species that are not polymerized may cause dissolution of the SEI layer, which is frequently observed in the research of SMBs.41,42 In addition to the inferior SEI layer, SMBs still suffer from an energy density that is far below that of traditional LIBs and the poor cyclability. To mitigate the shortcomings of low energy density, researchers have explored high-capacity cathodes such as sulfur, oxygen, and carbon dioxide, coupled with sodium metal, even though there are still some critical issues that impede the further progress of SMBs. The following sections discuss the key challenges of SMBs along with their fundamentals in detail (Fig. 1).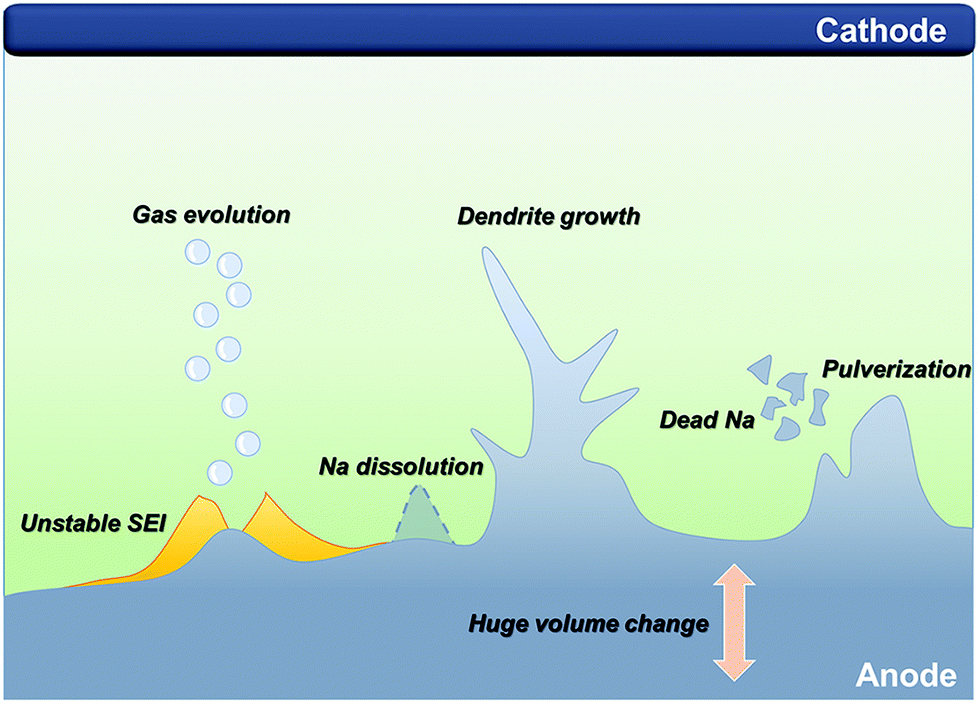 | ||
| Fig. 1 Schematic illustration of the critical challenges in a sodium metal anode for sodium metal batteries. | ||
2.1. Intrinsic properties of Na metal
As sodium is located right below lithium in the alkali metal group, it has one electron in the outermost energy level, which is more reactive than lithium because of its greater atomic radius.36 Accordingly, sodium possesses slightly different physical and electrochemical properties (Table 1). In particular, sodium easily loses its valence electron and has a relatively high negative redox potential, leading to the rapid growth of the unstable SEI layer through electron transfer from the sodium metal to the electrolyte. The unstable SEI layer causes inhomogeneous sodium ion flux through the SEI layer, forming sodium dendrites and leading to the unnecessary consumption of the electrolyte because of the direct contact of fresh sodium metal with the electrolyte, leading to low Coulombic efficiency (CE). The newly formed SEI layer continuously accumulates on the sodium metal, providing a large increase in the interfacial resistance.| Lithium | Sodium | |
|---|---|---|
| Atomic radius (pm) | 152 | 186 |
| Atomic weight (g mol−1) | 6.9 | 23 |
| 1st ionization energy (kJ mol−1) | 520.2 | 495.8 |
| Melting point (K) | 454 | 371 |
| Density (g cm−3) | 0.534 | 0.968 |
| Theoretical gravimetric capacity (mA h g−1) | 3860 | 1166 |
| Standard redox potential (V vs. SHE) | −3.04 | −2.71 |
| Bulk modulus (GPa) | 11 | 6.3 |
Indeed, to verify the effect of highly reactive sodium metal on the detrimental phenomena of SMBs, the optical and electrochemical studies of its behavior have been conducted using sodium metal symmetric cells. Iermakova et al. conducted a comparative study on the stability of the SEI layer on sodium and lithium metal.43 In conventional carbonate electrolytes, they compared solely immersed metals in the electrolyte without the flow of electric current and metals after 30 cyclic voltammetry cycles through infrared spectroscopy. The signals from the lithium metal barely changed, while the signal from the sodium metal was significantly amplified after the electrochemical reaction, indicating the poor stability of the SEI layer on the sodium metal surface. This dissolution phenomenon of the SEI layer was visualized via optical microscopy by Dsoke's group.44 The sodium metal on the polyethylene film was packaged in an air-tight microscopy cell filled with 1 M NaClO4 EC/DMC electrolyte to monitor the surface variation of the sodium metal (Fig. 2A). It is envisaged that the generated film on the interphase between the metal and the electrolyte constantly fell off and dissolved in the electrolyte. When the grown dendrite was in a resting state without electron movement through an external circuit (called a quasi-zero electrochemical field), the sodium dendrite steadily disappeared over time. The low mechanical stability and unstable electrochemical stability could lead to detrimental effects on SMBs, i.e., when vibration force or shear force (by shaking the electrolyte) was applied, the metal dendrite and SEI layer could not withstand the forces and eventually vanished (Fig. 2B).45
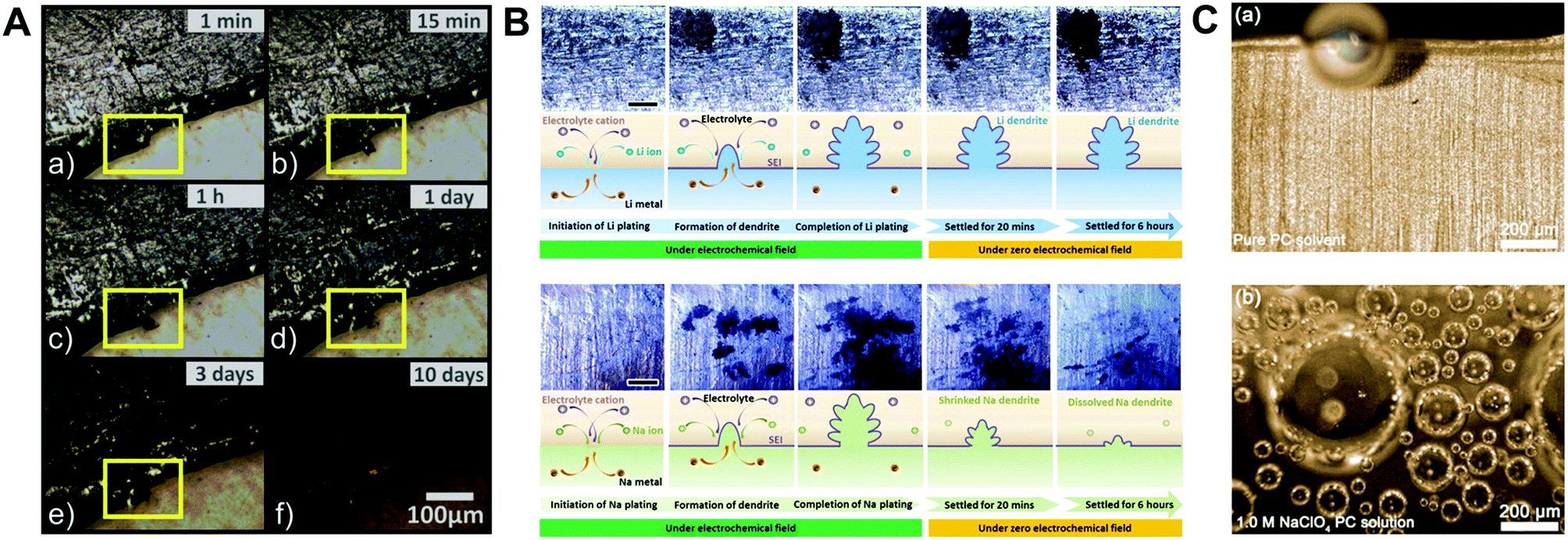 | ||
| Fig. 2 (A) Optical microscopy images of Na metal on polyethylene film, filled with E-NaClO4 in a sealed cell. Pictures were recorded after (a) 1 min, (b) 15 min, (c) 1 h, (d) 1 d, (e) 3 d, and (f) 10 d. The yellow highlighted area shows a time-resolved reaction on the Na surface. Reproduced with permission from ref. 44. Copyright 2019, Wiley. (B) Surface morphology observation: in situ top-view images of the dendritic change on the surface of lithium and sodium anodes during the plating process with galvanostatic current density and subsequent settling without voltage or current. The galvanostatic charge current density was 0.4 mA cm−2 in both the cases. Schemes of the corresponding states are illustrated from cross-sectional views. The scale bars in the optical photographs are all 1 mm. Reproduced with permission from ref. 45. Copyright 2018, Elsevier. (C) In situ optical microscopic images of gas evolution on Na: (a) pure PC solvent. (b) Na+–PC solution. Reproduced with permission from ref. 49. Copyright 2018, Wiley. | ||
In addition, considering that the sodium metal and SEI layer have poor electrochemical/physical stability, gas will evolve constantly in the cell until the electrolyte is entirely decomposed, inducing cell swelling and even explosion.46–48 Several groups have analyzed gas evolution, which is inevitably accompanied by the decomposition of the electrolyte (e.g., carbonate or even ether-based electrolytes) via optical microscopy and DFT calculations. Chen et al. discovered that the lowest unoccupied molecular orbital (LUMO) level of the ion–solvent complex [(Na–X)+, X = PC, TEGDME, DME] is lower than that of the pure solvent, leading to an acceleration in electrolyte decomposition and gassing rate (Fig. 2C).49 In addition, the amount of gas evolution (H2, CH4, CO, and C2H4) obtained by online electrochemical mass spectroscopy varies depending on the type of sodium salt (i.e., NaOTf, NaFSI, NaTFSI, NaClO4, and NaPF6).50 The cycle performance due to the severe side reaction of the electrolyte is deteriorated with an increase in released gas evolution. For most gases excluding CO, the largest gas evolution was observed for NaTFSI. Although using a solution containing only NaTFSI salt could cause poor cyclability, blending NaTFSI with another salt may protect the sodium metal and prevent further decomposition of the electrolyte due to the sodium hydride passivation film formed by hydrogen gas.51 Thus, it is imperative to design a suitable artificial SEI layer to mitigate the inherent drawbacks of sodium metal.
2.2. Dendrite growth
The space charge model typically illustrates the growth of metallic dendrites by electrodeposition in a dilute salt solution under a high electric field. Chazalviel et al. analytically calculated the distribution of electrostatic potential and ionic concentration.52 It was inferred that metallic dendrite growth is initiated when the ionic concentration in the vicinity of the negative electrode is completely depleted, inducing a strong electric field near the interface. In addition to the space charge model, sand's time model can provide the initial time of metallic dendrite growth, which is called sand's time and the variables affecting it are given as follows:where the subscripts a and c correspond to the anion and cation, respectively, τs is the sand's time at which the dendrite growth starts, D is the ambipolar diffusion constant, C0 is the initial cationic concentration of the electrolyte, zc is the cationic charge number, J is the effective electrode current density, and μ is the ionic mobility.53
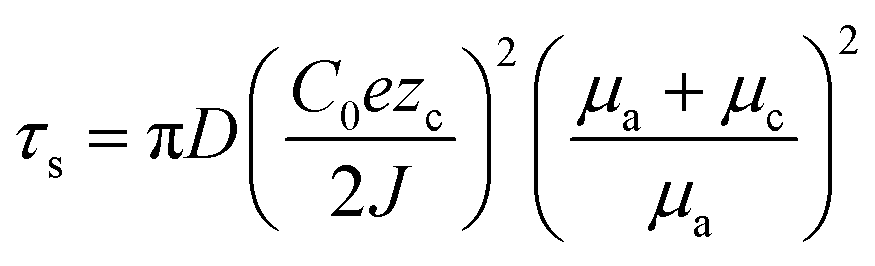
Brissot et al. discovered two different phenomena depending on the effective current density, which were divided into two regimes.54 The critical current density dividing the two regimes can be represented as follows:
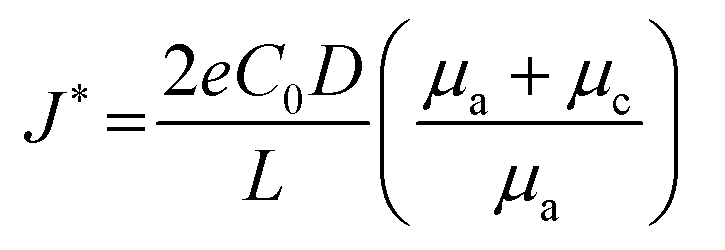
There have been several reports about the formation and morphology transition of sodium metal dendrites.48,51 We divided the formation of sodium metal dendrites into three stages. In stage I, granular sodium particles were observed in the first cathodic process (deposition) and then, the needle-like dendrites grew on granular sodium metal as the deposition proceeded. In stage II, the bottom part of the needle-like sodium dendrites dissolved in the following anodic process (stripping), resulting in the formation of an electrically isolated sodium metal (known as “dead-sodium”), which led to low CE (Fig. 3A). In stage III, the electrolyte was decomposed preferentially at the tip of the sodium metal owing to high local current density, leading to selective accumulation of the SEI layer, thus rendering the non-axial growth of a moss-like sodium metal (Fig. 3B).
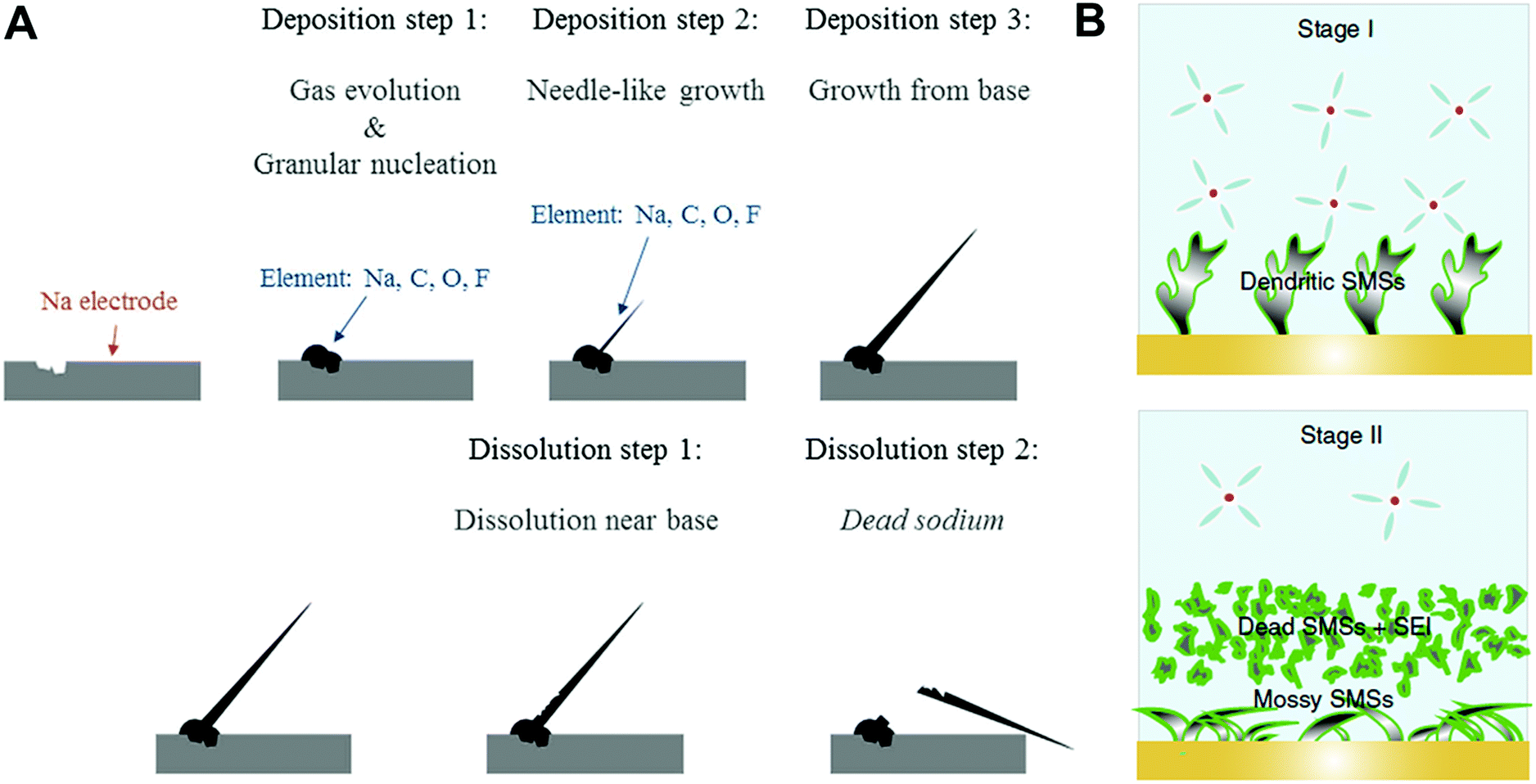 | ||
| Fig. 3 (A) Pattern diagram of electrochemical sodium deposition and dissolution on copper electrode. Reproduced with permission from ref. 48. Copyright 2016, Springer Nature. (B) The SMSs tend to be dendritic below the specific voltage (stage I). When exceeding the specific voltage (stage II), serious electrochemical decomposition of electrolyte is triggered, which results in a change in the surface morphology of the Na metal, which accelerates the depletion of electrolytes. Reproduced with permission from ref. 51. Copyright 2020, Springer Nature. | ||
In addition, as inferred from the sand's time model, various strategies have been proposed to maximize the sand's time. First, the increasing concentration of the electrolyte (i.e., solvent in salt electrolyte), denoted as C0, retards sand's time. Second, substantially increasing the active surface for the electrodeposition of sodium metal is beneficial for significantly reducing the local current density, leading to a dendrite-free sodium metal composite. Third, anchoring anions with fixed structures or close-to-unity transference numbers of cations, which makes sand's time infinity, could effectively restrain the strong electric field induced by anion depletion near the surface of the electrode. Last, introducing an artificial homogeneous SEI layer could promote uniform sodium ion flux, thus suppressing dendritic growth.
Although significant research on the strategies for averting dendritic deposition of sodium metal has been devoted to applying similar technology used in LMBs, research on the fundamental principle remains at its infancy.
2.3. Severe volume change
The problems caused by drastic volume variations have been presented constantly in alkali ion secondary batteries composed of conversion/alloy reaction-based electrodes.55–57 Similarly, owing to the host-less nature of the Na metal anode, the Na metal anode suffers an infinite volume change during repeated Na plating/stripping, resulting in a spatially non-uniform SEI layer, poor CE, and capacity fading towards the end of the battery life.Indeed, a large variation in the volume and shape could lead to particle pulverization and fracture of the SEI layer. Consequently, the pulverized sodium metal particles could be peeled off from the sodium metal anode, causing electrical and ionic isolation of the active materials that are known as “dead sodium” and even capacity fading. In addition, the fracture of the active materials and the SEI layer produces a fresh active surface that spontaneously reacts with the electrolyte to form an additional SEI layer. This phenomenon gives rise to two critical issues, i.e., the thickness fluctuation of the SEI layer, which hinders the homogeneous Na ion flux contribution,58 leading to inhomogeneous electroplating of sodium metal, denoted as sodium dendrite and continuously growing sodium dendrites, which exacerbate the fracture of the SEI layer and, in turn, accelerate the reduction of the electrolyte until the whole electrolyte is consumed.
Recently, diverse micro/nanostructured frameworks have been proposed to complement the host-less nature of sodium metal. The sodiophilic or electrically-conductive frameworks such as metal foam,59,60 doped carbon,61,62 metal oxide,59 and MXene63 could alleviate the volume expansion problem and mitigate the strain to some extent. However, introducing frameworks increases the volume and weight of SMBs, which is a consequence of both the low gravimetric and volumetric energy densities. Although the high mechanical modulus of lithium dendrites cannot be effectively suppressed by engineering the SEI layer, the low mechanical modulus of sodium enables the fabrication of a robust SEI layer, thereby posing a promising technology.64 This review provides a meaningful insight to promote the artificial engineering of the SEI layer for progressive applications in SMBs (Fig. 4).
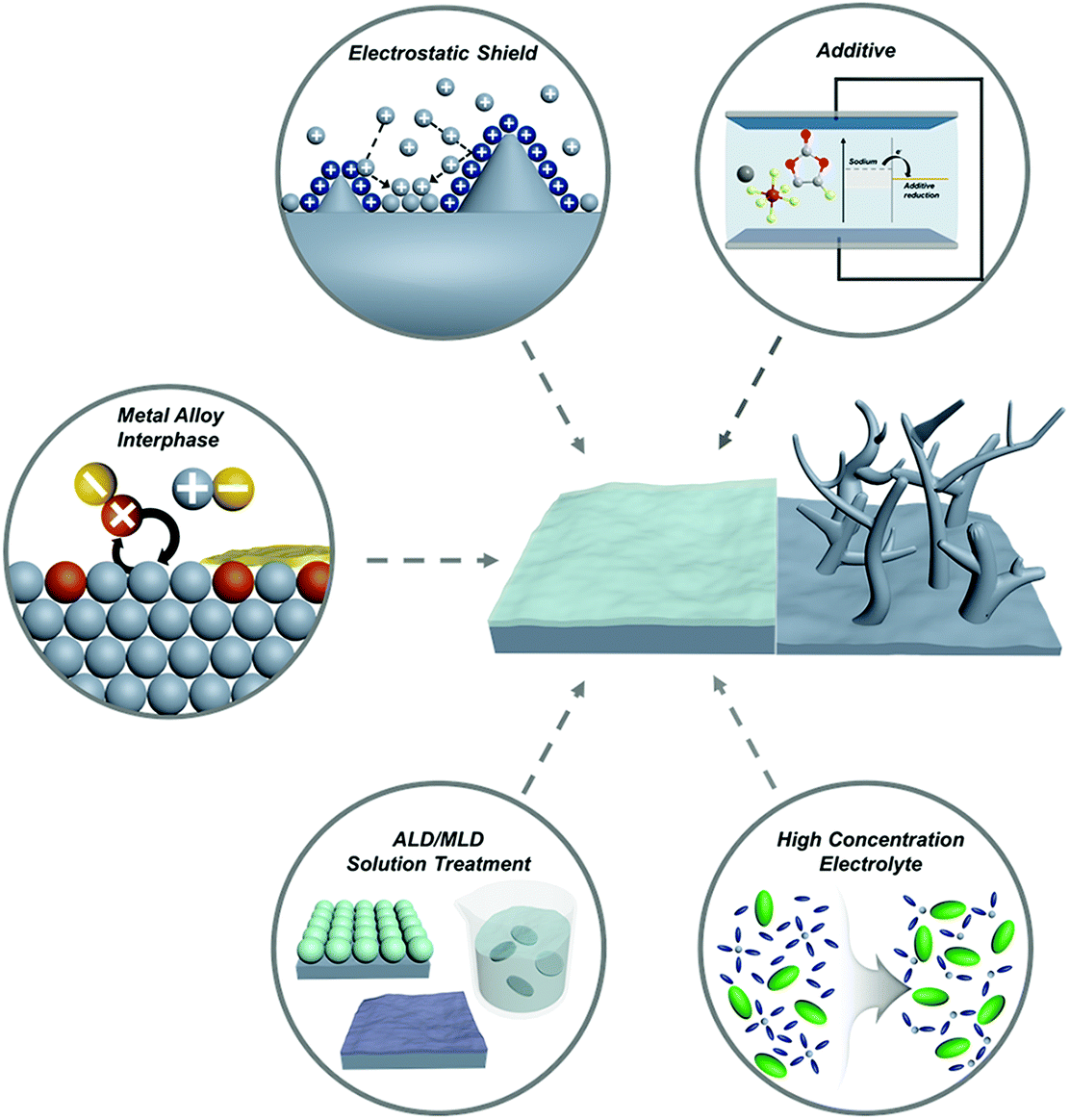 | ||
| Fig. 4 Schematic illustration of the strategies for tailoring the metal/electrolyte interphase film on sodium metal anodes. | ||
3. Strategies for tailoring the metal/electrolyte interphase film
3.1. Additional substances in typical electrolytes
As the electrodes are in direct contact with the electrolyte, interphase films (known as solid electrolyte interphase (SEI) and cathode electrolyte interphase (CEI) on the anode and cathode, respectively) between the electrode and the electrolyte are spontaneously formed by the transfer of electrons from the electrode to the electrolyte (or vice versa) and the oxidative corrosion of the metal.14,65 As represented in Fig. 5A,![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) Anode and Cathode are the electrochemical potentials of the anode and cathode, respectively.66 In the case of Anode > −e[EReduction], sodium metal is prone to transfer electrons to the electrolyte, inducing a rapid reduction of the electrolyte on the surface of the anode. The products of the reduced electrolyte as a passivation film on the metal surface prevent further decomposition of the electrolyte by blocking electron flow to the electrolyte. The interphase films passivate the further decomposition of the electrolyte. However, the SEI on sodium metal is mechanically weak and thermodynamically unstable, which is easily broken and dissolved into the electrolyte.
Anode and Cathode are the electrochemical potentials of the anode and cathode, respectively.66 In the case of Anode > −e[EReduction], sodium metal is prone to transfer electrons to the electrolyte, inducing a rapid reduction of the electrolyte on the surface of the anode. The products of the reduced electrolyte as a passivation film on the metal surface prevent further decomposition of the electrolyte by blocking electron flow to the electrolyte. The interphase films passivate the further decomposition of the electrolyte. However, the SEI on sodium metal is mechanically weak and thermodynamically unstable, which is easily broken and dissolved into the electrolyte.
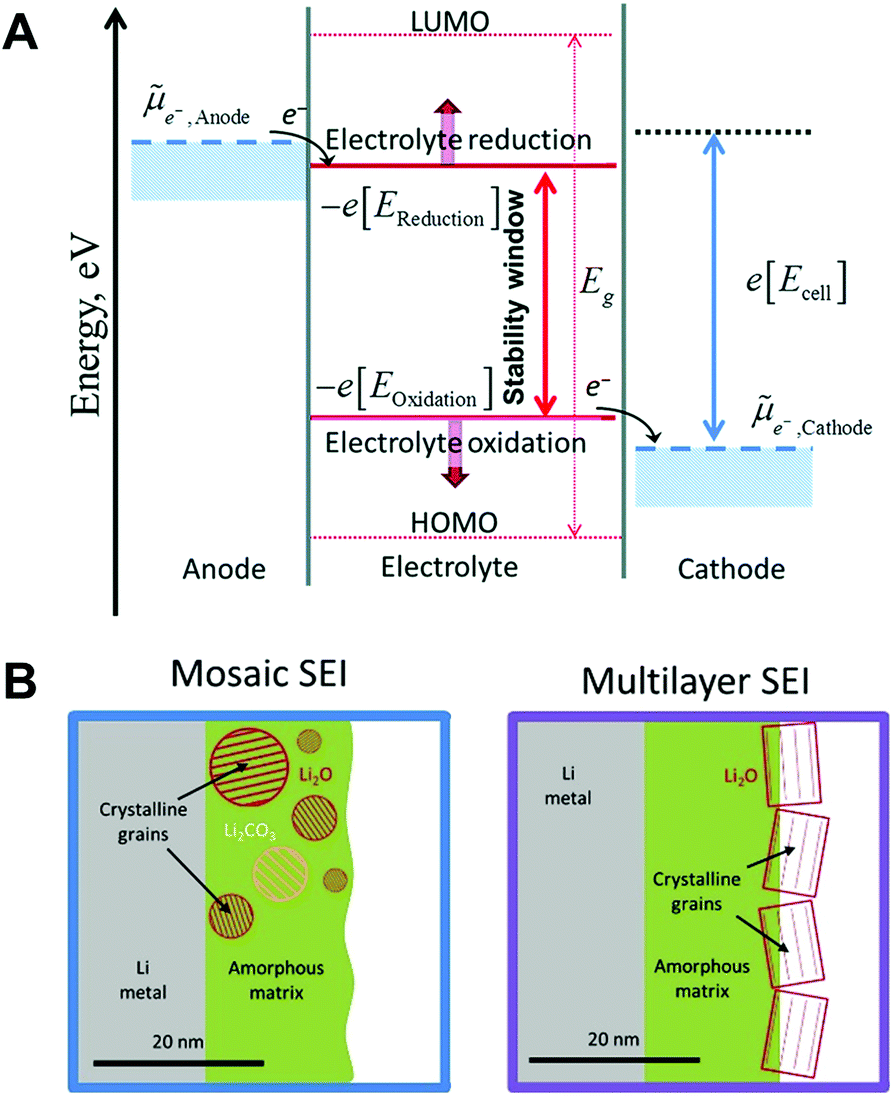 | ||
| Fig. 5 (A) The negative and positive potential limits for the electrolyte stability and the energy levels of HOMO and LUMO. Reproduced with permission from ref. 66. Copyright 2018, The Royal Society of Chemistry. (B) Schematic of the mosaic SEI nanostructure and the multilayer SEI nanostructure on lithium metal. Reproduced with permission from ref. 67. Copyright 2018, Elsevier. | ||
Generally, two representative morphologies of SEI (i.e., mosaic or layered) have been proposed, which consist of organic–inorganic hybrid compounds (Fig. 5B).67 Similar to the SEI layer on lithium metal, the organic components (e.g., ROCO2Na, HCOONa, and polyethylene oxide oligomers) originate from the decomposition of the solvent, while the inorganic components (e.g., NaF, NaCl, and Na2CO3) are generated predominantly from the decomposition of salt anions.68 It is deduced that the composition of the SEI strongly depends on the solvent, salt anion of the electrolyte, additive, and impurities. Since the SEI layer is structurally and chemically heterogeneous, its properties could affect the growth of metallic dendrites, the reversibility, as well as the chemical and mechanical stability of sodium.
A fundamental understanding of the formation and composition of the SEI layer is important for the design of substances that are added into the electrolyte to effectively suppress sodium dendrites. Herein, additive strategies for the protection of sodium metal are classified into 3 categories as follows: (1) formation of a stable SEI film-forming additive during the initial cycle of SMBs, (2) electrostatic shield, which is attributed to the cation of salt, and (3) the alloying reaction between metallic sodium and the cation of the salt.
Fluoroethylene carbonate (FEC) is one of the most studied additives in SMBs. Dugas et al. found that the protective layer induced by FEC can suppress gas evolution originating from the reaction between sodium metal and other components in the electrolyte, despite the increase in the interfacial resistance due to the continuous growth of the protective layer (Fig. 6A).70 Recently, Yang and co-workers quantified SEI components through 23Na magic-angle-spinning NMR spectra.51 By introducing FEC, the proportion of organic species decreased from 62 at% to 39 at%, while the proportion of inorganic species increased from 27 at% to 61 at% (Fig. 6B). Therefore, these phenomena changed the modulus of the SEI and resulted in a superior cycle life for sodium metal batteries. Although adding FEC to carbonate electrolytes can contribute to the enhanced stability of the sodium metal during platting/stripping, leading to an improved CE of 88%, unnecessary electrolyte consumption is still a large part in the aspect of commercialization.
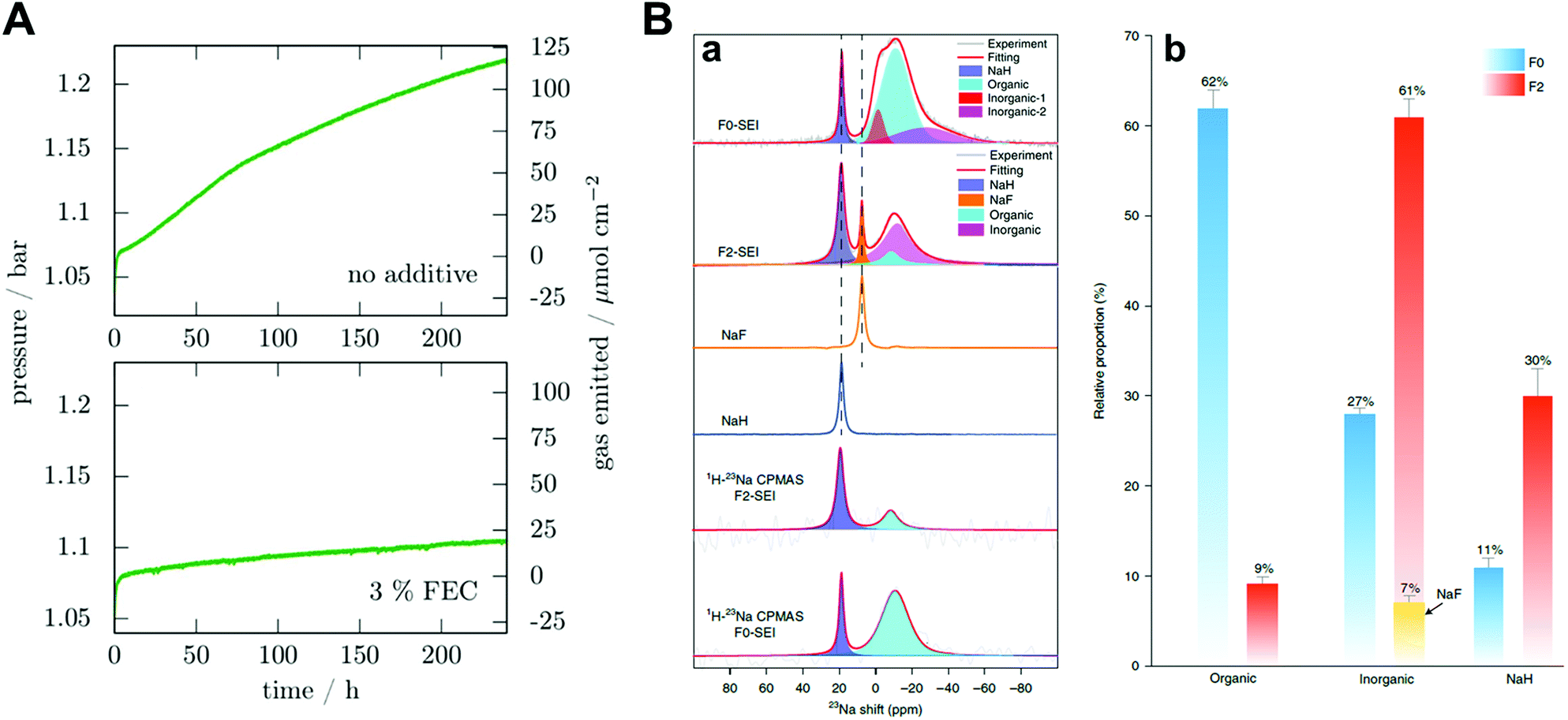 | ||
| Fig. 6 (A) Pressure variation in a 9.2 cm3 cell containing a 0.5 cm2 sodium electrode and 250 μL electrolyte without the additive and with 3% FEC in mass. Reproduced with permission from ref. 70. Copyright 2016, Electrochemical Society. (B) Ex situ NMR characterization of the SEI species. (a) Single-pulse 23Na NMR and 1H → 23Na CPMAS NMR spectra of SEI species harvested from Cu foil after 50 cycles and the reference spectra of NaF and NaH. (b) The relative proportion of organic and inorganic components in the SEI species. Error bars indicate errors when fitting the NMR signals of different SEI components. Reproduced with permission from ref. 51. Copyright 2020, Springer Nature. | ||
Wang et al. proposed sodium hexafluoroarsenate (NaAsF6) as an additive for carbonate electrolytes.71 Through the surface analysis of XPS and Ar ion sputter depth profiling, it was found that both the F atomic ratios and the fraction of NaF in the SEI film were higher than those of the pure electrolyte, which is beneficial for stabilizing the sodium metal and eventually achieving a high Coulombic efficiency. Shi et al. discovered that potassium bis(trifluoromethylsulfonyl)imide (KTFSI) can be used as a bifunctional electrolyte additive with the synergistic effect of electrostatic shielding and generating a stable SEI through K cations and TFSI anions, respectively.72 K+ contributes to electrostatic shielding, hindering the growth of dendrites, while TFSI− is conducive for producing NaN3 and NaNxOy containing a stable SEI film. The symmetric cell with the KTFSI additive demonstrated a long cycle life of more than 40 cycles at an ultrahigh capacity of 10 mA h cm−2 at a current density of 2 mA cm−2. Wang et al. studied the role of sodium polysulfide (Na2S6) and the adverse effects of sodium nitrate (NaNO3) on the formation of a SEI layer (Fig. 7A).73 Unlike the previous report on LMBs that applied polysulfide and LiNO3 as co-additives,74 they discovered that the SEI formed in the diglyme electrolyte with co-additives is unstable, whereas the SEI formed with only Na2S6 consists of Na2O, Na2S2, and Na2S6, which can effectively protect sodium metal. They observed that the morphology of the SEI formed with only the additive was uniform but that formed with the co-additive has a structure with pinholes (Fig. 7B). As inferred from the analysis of the additives, the ideal additive should have the following features:
 | ||
| Fig. 7 (A) (a, d, g and j) Cross-section elemental distribution maps of Na, S, and Cu, and (b, e, h and k) the corresponding SEM images of the Na metal layer plated on the Cu current collector after 40 cycles under 1 mA cm−2–1 mA h cm−2 conditions for the Na II Cu cells with and without the additives in 1 M NaOTf in TEGDME electrolyte. (c, f, h and l) Magnified SEM images of the cross-section of the deposited Na metal layer and the corresponding schematics, showing the stabilization effects of ionic additives for Na metal deposition. Reproduced with permission from ref. 72. Copyright 2018, Wiley. (B) The role of Na2S6 additive and Na2S6–NaNO3 co-additives for affecting the Na stability in diglyme electrolyte. The Na surface morphology difference with (a) PS alone and (b) P–N co-additives. The SEM images of SEI layers formed with (c) 0.033 M PS alone and (b) P–N co-additives, after 10 cycles at 0.5 mA cm−2 and 1 mA h cm−2. Scale bars in (c) and (d) are 10 μm. Reproduced with permission from ref. 73. Copyright 2018, Wiley. | ||
1. The additive should possess lower −e[EReduction] for sacrificing itself to form a stable and dense SEI layer relative to the solvents and sodium salts in the electrolyte in advance.
2. The SEI layer derived from the additive involved in the decomposition process should be thermodynamically and electrochemically stable, thus retaining a dense and uniform film.
3. The additive should be electronically insulating and ionically conductive to further prevent the decomposition of the electrolyte.
4. The additive should have a high elastic modulus to sustain the strain induced by severe volume change during repeated cycles and the growth of sodium dendrites.
Theoretically, Na+ has the highest standard reduction potential among all alkali metal ions (−2.71 V vs. SHE), resulting in wide potential candidates for the SHES mechanism (Table 2). It is worth noting that the SHES mechanism works effectively with decreasing cation radius; thus, Li+ is the most powerful candidate.81,82 Ma et al. demonstrated a strategy by applying Li–Na (Na/Li = 6) bimetallic anode to the O2 battery systems.81 Even though both Li and Na construct anodes, Na only acts as an active material and Li ions, which are supplied during the stripping process, play a key role in the SHES mechanism (Fig. 8A). However, there was huge volume expansion due to the host-less nature of the Li–Na alloy; thus, they introduced 1,3-dioxolane (DOL) as additive for the formation of an elastic SEI layer. The SEI layer formed by the reaction of DOL and Li–Na alloy prevented cracking, which was consistent with the results of little change in the interfacial resistance. Bimetallic anodes showed even better cyclability (137 cycles) under galvanostatic tests with 1000 mA h g−1 capacity compared to the Li (34 cycles) or Na metal (31 cycles) anodes. Furthermore, Zhang et al. designed Li–Na hybrid batteries (LNHBs) consisting of a Na metal anode, LiFePO4 (LFP) cathode, and a Li–Na hybrid electrolyte containing 0.5 M LiPF6 and NaPF6 in DME.83 In this system, Li+ from the electrolyte acts as an active material intercalating into the LFP cathode and generates a self-healing electrostatic shield on the sodium metal anodes. On the other hand, Na+ has a much higher standard reduction potential, resulting in preferential deposition on sodium metal anodes. Through this novel approach, LNHB accomplished highly stable Na metal anodes (99.2% CE, 100 cycles), lower overpotential (42 mV at 2 mA cm−2), and faster charge transfer than LMB (Fig. 8B). As mentioned before, K+ also attempted to take advantage of the SHES mechanism for stabilizing sodium metal anodes, as proposed by Wang and co-workers.72
| Li | Na | K | Ca | Rb | Sr | Cs | Ba | Sm | Eu | |
|---|---|---|---|---|---|---|---|---|---|---|
| Standard reduction potential (vs. SHE) | −3.04 | −2.71 | −2.93 | −2.87 | −2.98 | −2.90 | −3.03 | −2.91 | −2.68 | −2.81 |
| Metal cations concentration/lithium | — | Too low | 0.014 M | Too low | 0.096 M | Too low | 0.578 M | 0.007 M | Too low | Too low |
| Metal cations concentration/sodium | — | — | — | — | — | — | — | — | 0.311 M | — |
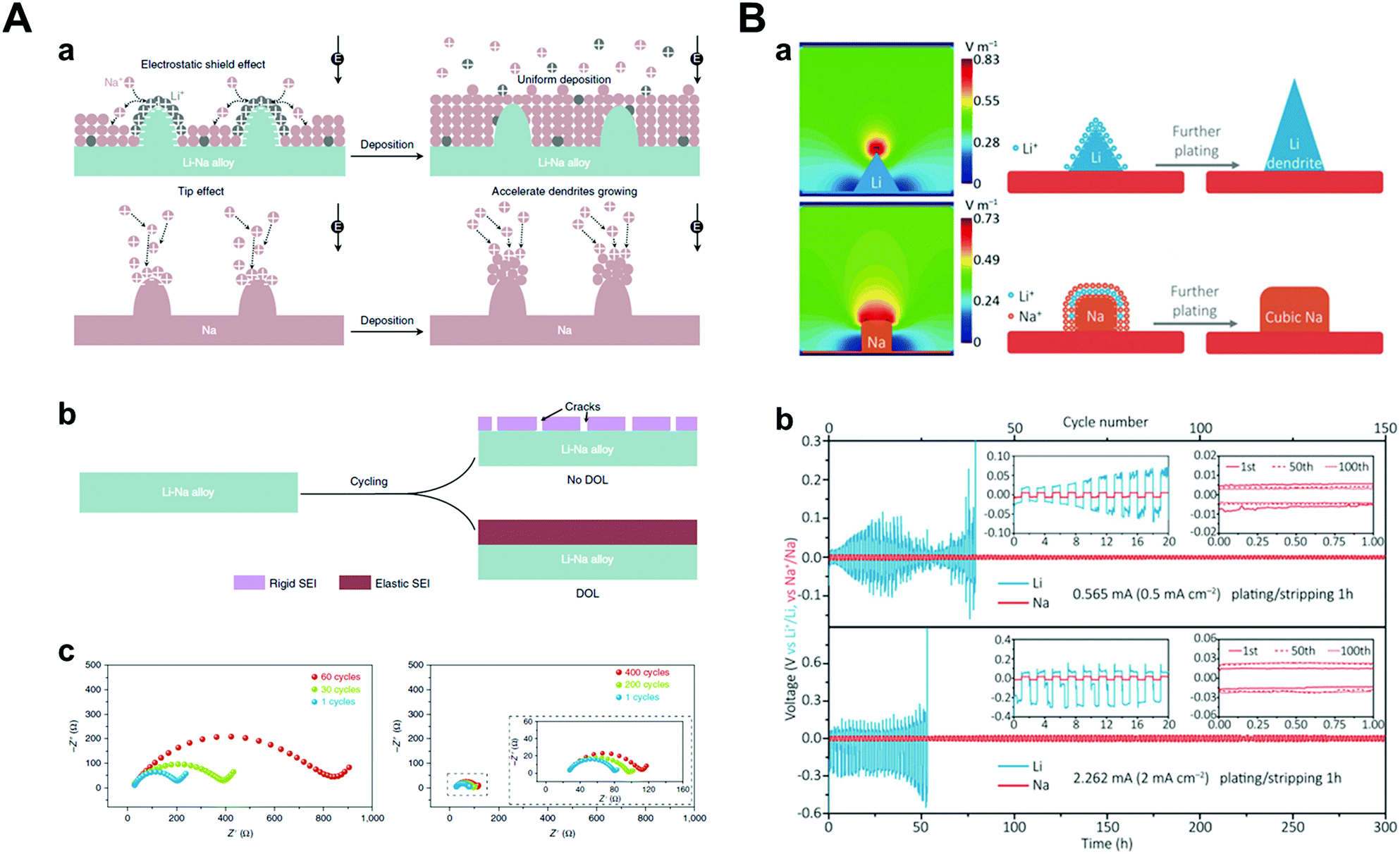 | ||
| Fig. 8 (A) Illustration of dendrite and crack suppression. (a) In the Li–Na alloy electrode, Li+ adsorbs on the tips to repel the incoming Na+ ions and forces Na+ deposition on the adjacent regions away from the tips. At the Na electrode, no Li+ is adsorbed on the tips to repel the incoming Na+; thus, Na+ deposits on the tips. (b) Without DOL, the electrolyte reacts with the alloy to form a rigid SEI that will crack during cycling. After adding DOL, it reacts with the alloy to form an elastic SEI, thus preventing SEI cracking. (c) Electrochemical impedance spectra of symmetric batteries using Li–Na alloy (Na/Li = 6) after the 1st, 30th, and 60th cycles in 0.5 M NaCF3SO3/TEGDME electrolyte and after the 1st, 200th, and 400th cycles in 0.5 M NaCF3SO3/DOL/TEGDME electrolyte. Reproduced with permission from ref. 81. Copyright 2019, Springer Nature. (B) (a) Simulated electric-field intensity distribution and schematic illustration of the Li/Na plating patterns. (b) The electrochemical performance of Li/Na testing in lithium/hybrid electrolyte. Voltage profiles of symmetric cells. Insets of (b) are the detailed voltage profiles. Reproduced with permission from ref. 83. Copyright 2018, Wiley. | ||
Compared to the additives for in situ SEI formation, the electrolyte additives for the SHES mechanism have not been investigated extensively. As a result, we expect studies in this field to start broadening owing to the many potential candidates compared to lithium in sodium metal anodes.
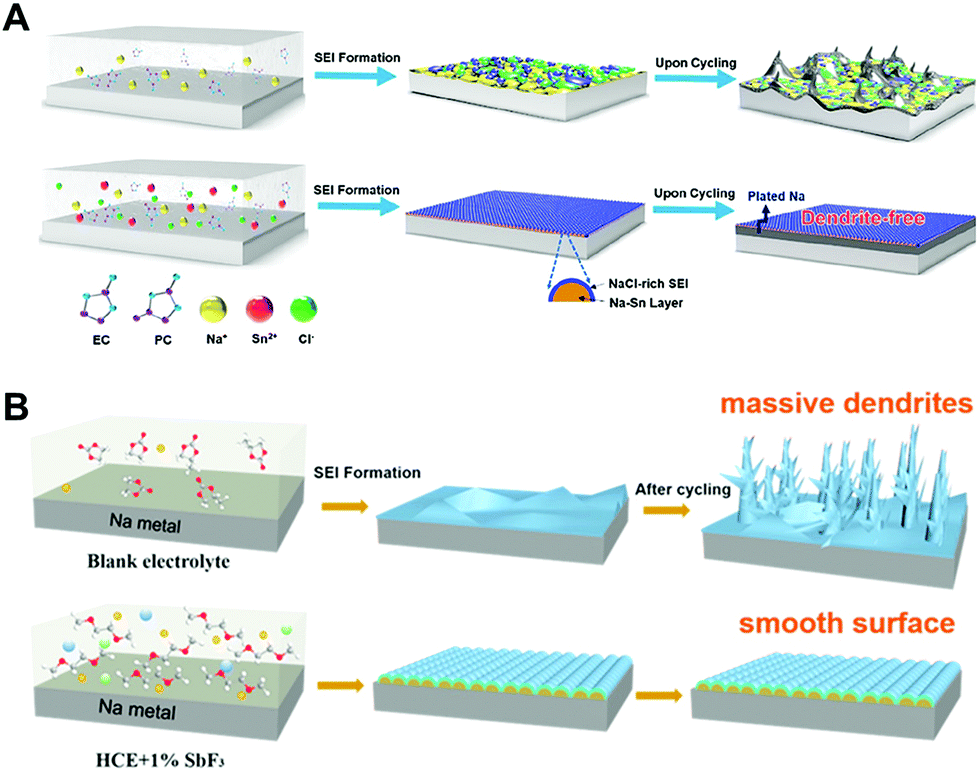 | ||
| Fig. 9 Schematic illustration of (A) the formation of a typical mosaic SEI on the Na metal anode cycled in regular carbonate electrolyte, which drives uneven interfacial ion transfer and Na dendrites (top) as well as fast and uniform ion transport, benefitting from the in situ formed Na–Sn alloy layer plus a NaCl-rich SEI, leading to uniform plating–striping and dendrite-free morphology (bottom). Reproduced with permission from ref. 89. Copyright 2019, American Chemical Society. (B) Formation mechanism illustration of a typical SEI on the Na metal anode using (a) the blank electrolyte, (b) HCE, and (c) HCE + 1% SbF3 electrolytes. Reproduced with permission from ref. 90. Copyright 2020, Elsevier. | ||
It should be noted that there are many candidates, which enable to form an alloy with sodium metal including Sn,91–93 Au,94 Ag,95 Zn,96 Sb,97 and alkaline earth metals.98 In most cases, they have been applied for the synthesis of host materials to reduce the nucleation overpotential during the electrodeposition or synthesis of sodium-alloy anodes using melting infusion. These are also powerful approaches for mitigating dendrite growth but utilizing them as electrolyte additives should also be considered owing to their ease of use.
3.2. High concentration electrolytes
Compared with conventional electrolytes, the high-concentration electrolyte (>3 M) has distinct electrochemical and physicochemical features.99–104 As the concentration of the electrolyte increases, the lowest unoccupied molecular orbital (LUMO) level of the electrolyte shifts from the solvent to the anion.101,105 This indicates that the anion, instead of the solvent, is reduced and decomposed at low potential, forming a firm inorganic SEI layer.106 The inorganic SEI layer derived from the reaction between the anion of the salt and the solvent can facilitate homogeneous stripping/plating of sodium metal to hinder the growth of dendrites. The component of the anion-derived SEI layer was revealed by FT-IR, which clearly shows the participation of the NaFSI salt in SEI formation.107 This anion-derived SEI layer can lead to a stable Coulombic efficiency of 97.7% over 250 cycles compared to typical carbonate electrolytes.108When the cationic concentration is zero at the electrode surface, called “sand's time”, sodium metal dendrites initiate the growth. Notably, the highly concentrated electrolyte leads to the retardation of sand's time owing to the effect of high ionic concentration and large Na ion transference number. In addition, little free-state of the solvent molecule could play an important role in suppressing the side reactions. For example, it is well known that Na stability in DMSO is poor because the bond of DMSO is cleaved.109 However, in the concentrated NaTFSI/DMSO electrolyte, the stability of Na metal significantly improves owing to lesser interaction between the free state of DMSO and Na metal (Fig. 10A). In addition, the corrosion of the Al current collector can be prevented because there is no available free-state solvent molecule that can dissolve Al–Cl or Al–FSI formed on the Al surface (Fig. 10B).107 Finally, the highest occupied molecular orbital (HOMO) level of the coordinated solvent molecule with Na ions can be tuned downwards, enhancing its oxidation stability. Thus, it is feasible to use metal-high voltage class cathode batteries. Although concentrated electrolytes are beneficial for stabilizing the electrochemical reaction of Na metal, critical issues such as high cost and viscosity should be addressed.
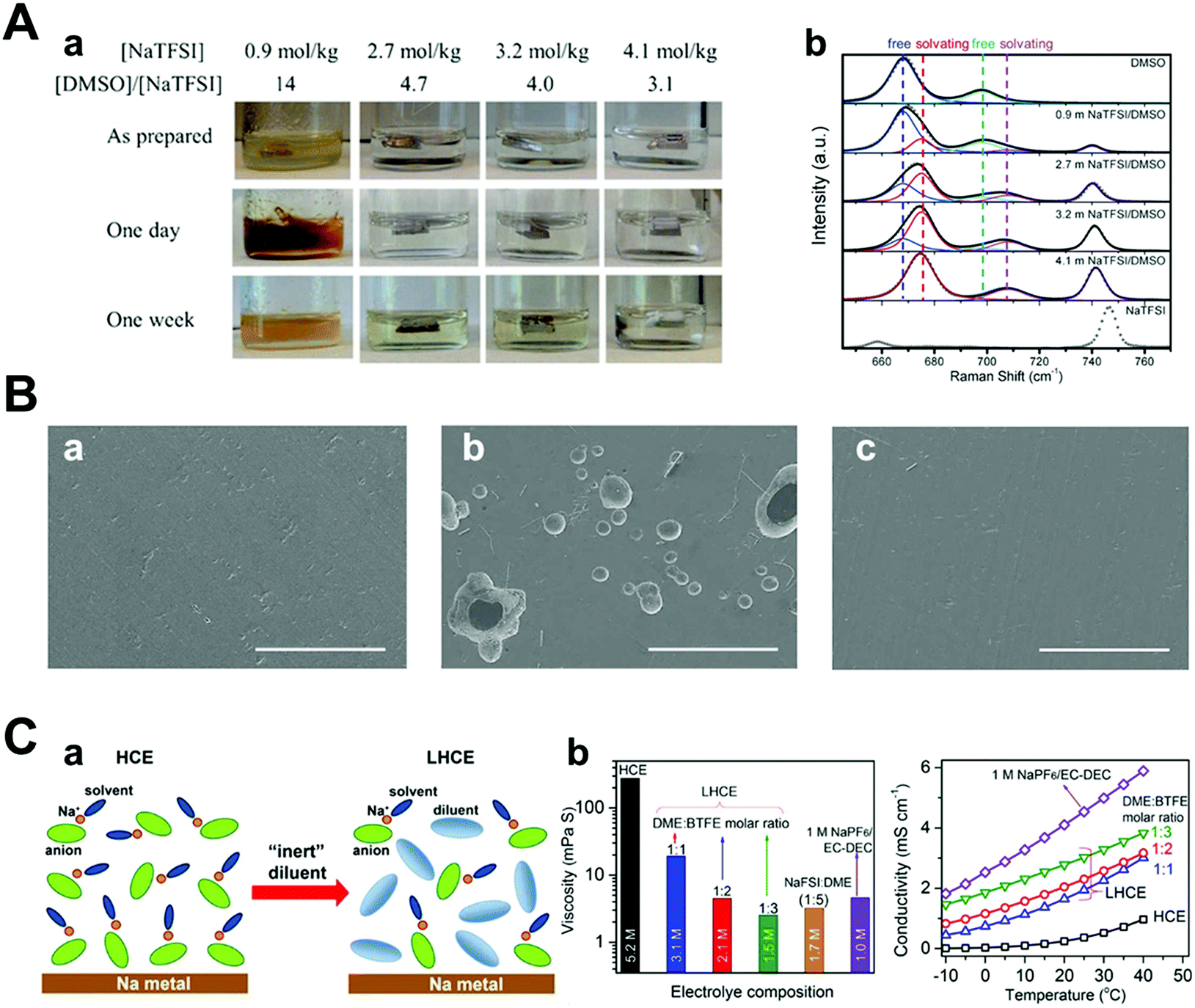 | ||
| Fig. 10 (A) (a) Images of Na in O2-saturated NaTFSI/DMSO solutions as prepared (top), stored for 1 day (middle), and 1 week (bottom). Each solution concentration and its molar ratio of the solvent to the solute are as indicated. (b) Raman spectra of pure DMSO, NaTFSI, and NaTFSI/DMSO solutions with different salt concentrations. Curves in gray dots and color lines are experimental and fitted results, respectively. The spectra were fitted using mixed Gaussian–Lorentzian distribution functions with fixed peak positions at 668, 675, 698, 708, and 741 cm−1, respectively. Reproduced with permission from ref. 109. Copyright 2016, Wiley. (B) SEM images of (a) pristine Al and Al surfaces after LSV measurements in (b) 1 M NaFSI–DME and (c) 5 M NaFSI–DME (NaFSI/DME = 1.06/1 mol ratio) (scale bar: 100 μm). Reproduced with permission from ref. 107. Copyright 2017, American Chemical Society. (C) (a) Schematic illustration of the dilution from HCE to LHCE. (b) Viscosity and ionic conductivity of the dilute electrolyte, HCE, and LHCEs with different NaFSI concentrations. Reproduced with permission from ref. 110. Copyright 2018, American Chemical Society. | ||
Intending to mitigate the drawbacks of concentrated electrolytes, Zheng et al. recently diluted a high-concentration electrolyte using an ‘inert solvent’ (e.g., hydrofluoroether), forming a localized high concentration electrolyte (LHCE), which maintained the aggregated solvation structure of the high-concentration electrolyte (Fig. 10C).110 Notably, this diluent can lower the salt concentration and viscosity of the electrolyte, effectively improving both its ionic conductivity and wettability because of its distinct properties (e.g., low dielectric constant and low donor number).111,112 Indeed, the overpotential of Na metal stripping/plating at a high current of 2 mA cm−2 was small and stable owing to the improved interfacial stability. In this regard, high energy density batteries such as Na–S and Na-high voltage class cathodes can be achieved; thus, researchers have attempted to apply concentrated salt solution strategies to sodium metal batteries.
3.3. Artificial metal/electrolyte interphase film
The solid electrolyte interphase is generally formed by reacting sodium metal and an electrolyte because of its highly reactive nature. Ideally, the SEI layer plays a role in preventing the additional reduction of the electrolyte to protect both the sodium metal and the electrolyte composition but its inhomogeneity and irregularity in chemical compositions and physical characteristics induce dendrite formation.14,113 As a result, researchers have been exploring the development of a stable artificial SEI layer using many different approaches. An artificial SEI layer should satisfy some conditions among them: (1) high ionic conductivity so as not to deplete the ions beneath the SEI layer fast,114 (2) mechanically rigid or elastic so as not to be damaged by dendrite growth easily,115 and (3) homogeneous thickness and chemical composition.116 In this section, we will focus on the strategies for enhancing the cyclability of sodium metal anodes via the generation of a stable and ideal artificial SEI layer before cell assembly.At a relatively early stage in this field, researchers attempted to form artificial SEI layers using deposition processes such as atomic layer deposition (ALD),117 low-temperature plasma-enhanced ALD (PEALD),118 and molecular layer deposition (MLD).119 It is important to conduct the above processes at low temperatures because the melting point of sodium metal is relatively low (98 °C). Zhao et al. carried out the ALD process and obtained an extremely thin Al2O3 layer (3.5 nm), which was controlled by cycles of the ALD process. The symmetric cell with Al2O3-coated sodium metal anode demonstrated a stable voltage profile under a symmetric cell at a high current density (3 mA cm−2, 1 mA h cm−2) for over 500 h (Fig. 11A). The XPS results proved that NaAlOx was generated during cycling, which demonstrated high ionic conductivity. Therefore, it was concluded that high ionic conductivity materials with a thin artificial SEI layer exhibited highly stable electrochemical performances under high current densities owing to facile sodium ion transport.
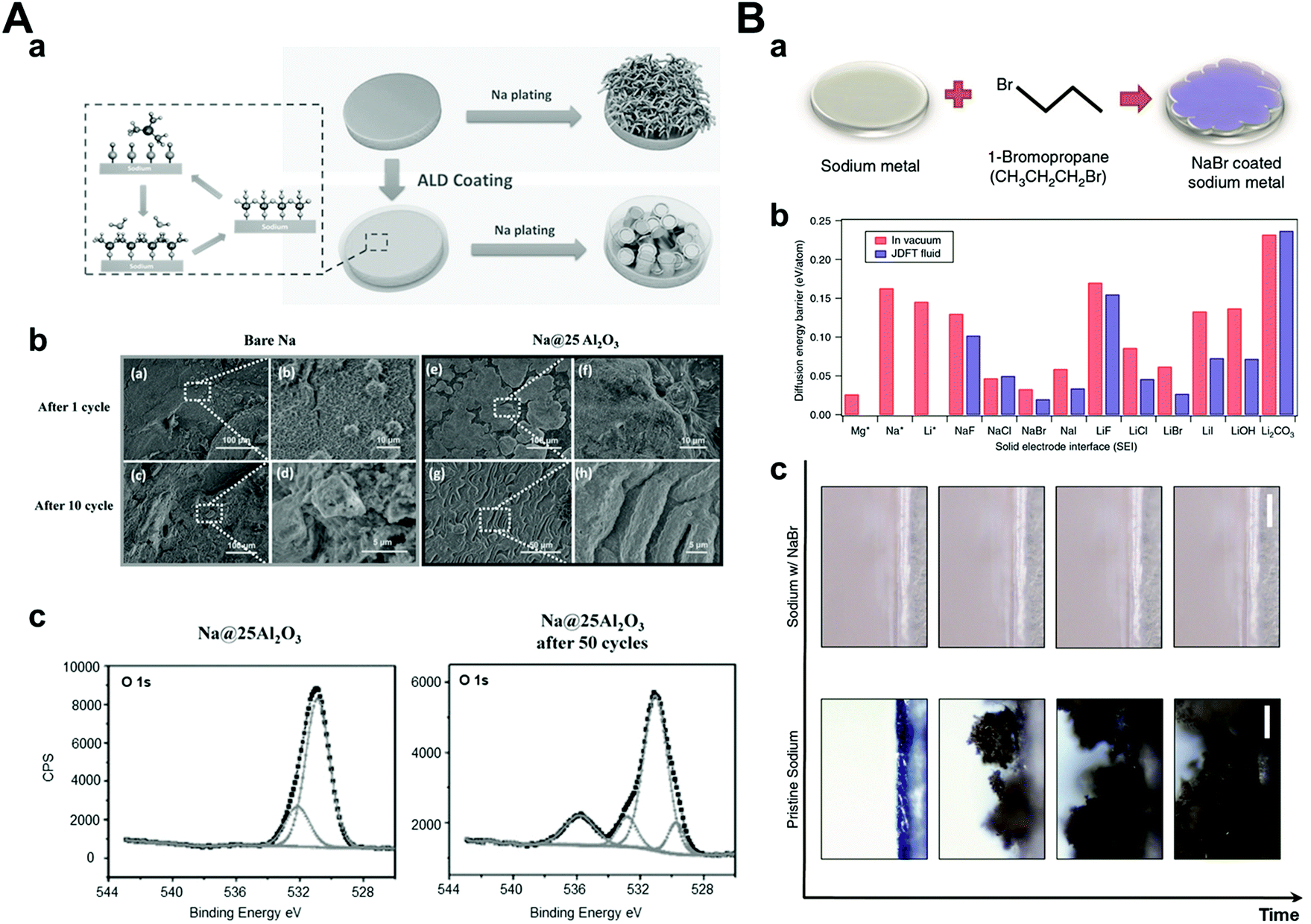 | ||
| Fig. 11 (A) (a) Schematic diagrams of Na stripping/plating on bare Na foil and Na foil with ALD coating. (b) Top-view SEM images of (a and b) bare Na and (e and f) Na@25Al2O3 after 1 cycle of stripping/plating, (c and d) bare Na, and (g and h) Na@25Al2O3 after 10 cycles of stripping/plating. (c) XPS O 1s spectrum of Na–Al2O3 25 and Na–Al2O3 25 after 50 cycles of plating/stripping at the current density of 3 mA cm−2. Reproduced with permission from ref. 117. Copyright 2017, Wiley. (B) (a) Schematic showing the procedure used to coat Na with NaBr. (b) Surface diffusion barriers calculated using joint density functional theory. Diffusion energy barriers computed for Mg, Na, and Li adatoms on surfaces with the noted chemistries. The red bars denote the surface in contact with vacuum and blue bars indicate the same in the presence of acetonitrile. (c) Snapshots from video microscopy of the pristine Na- and NaBr-coated Na electrolyte interface as a function of time. Reproduced with permission from ref. 120. Copyright 2017, Springer Nature. | ||
Another approach for generating an artificial SEI layer is a direct chemical reaction with sodium.120–125 Choudhary et al. conducted joint density functional theory to determine the most effective artificial SEI layer among NaX (X = halogen elements) (Fig. 11B). According to the results, NaBr was chosen considering its lowest diffusion barrier for interfacial ion transport. To synthesize the NaBr artificial SEI layer on sodium metal, they reacted pure sodium metal with 1-bromopropane, which is a well-known Wurtz reaction for the production of symmetric alkanes and sodium halide as a byproduct.120 Depending on the reaction time, NaBr was generated in sub-micron sizes ranging from 2 to 12 μm, which was confirmed by cross-sectional SEM and EDX. The symmetric cuvette-type optical cell was used for the in situ observation of sodium metal and its dendritic growth. It was found that dendritic structures were generated easily on bare sodium metal, whereas NaBr-coated sodium metal was observed without dendrite formation during cycling. In addition, during galvanostatic tests in the coin cell, NaBr-coated sodium showed a stable voltage profile up to 250 cycles at 0.5 mA cm−2 and 0.25 mA h cm−2 using a carbonate-based electrolyte. On the other hand, pristine sodium metal showed an unstable and increasing voltage profile within 50 cycles. This can be attributed to the inhibited extra decomposition of the electrolyte and reduced sodium ion transport barriers by the artificially-formed NaBr SEI layer.
As discussed in Section 3.1.3, alloy-based artificial interphase is a promising approach for protecting sodium anodes and delivering fast ion transport between the electrolyte interphases. This approach in LMBs was reported by Liang et al.126 They introduced 0.167 M metal chloride (In, Zn, Bi, As) dissolved in THF for 20 s to stabilize lithium metal anodes before cell-assembly. After rinsing it with THF and fully evaporating it under vacuum, the alloy SEI layer with LiCl of less than 10 μm thickness was formed, as confirmed by cross-sectional EDS mapping. These alloy metal interphases and LiCl alleviated the volume change of lithium metal during cycling and protected it from damage that could be caused by dendrite growth. Furthermore, the alloy interphase with a high chemical diffusion coefficient facilitated fast lithium diffusion through the interphases. Operando optical microscopy, galvanostatic tests in symmetric cells, and full cell characterization with LTO cathode proved that lithium with alloy interphase has more stable electrochemical performance and also, effectively restrained dendritic growth of lithium metal during cycling. Due to similarity of system, this approach could apply to SMBs. Tu et al. reported the formation of an artificial alloy interphase on an electrochemically active metal (both lithium and sodium) by blending tin bis(trifluoromethanesulfonyl)imide (SnTFSI) with a carbonate electrolyte (Fig. 12A).127 SnTFSI rendered chemical replacement between lithium or sodium to generate Sn–Li or Sn–Na hybrid anodes. They characterized the effect of the concentration of additives by electrochemical impedance spectroscopy for measuring the ionic conductivity. The 500 nm Li–Sn layer formed by an additive concentration of 10 mM had higher ionic conductivity compared to the layer formed by a blank electrolyte and a 2 μm Li–Sn layer formed by 100 mM of the additive. Despite the formation of an inorganic layer, the ionic conductivity of the 2 μm Li–Sn layer was even lower than that of the layer formed by the blank electrolyte. This implied that the high concentration of the additive hindered its original purpose. The hybrid anodes facilitated facile ion transport and physically mitigated the dendrite growth, which was observed from the in situ optical images during cycling. By galvanostatic charging and discharging, it was obvious that these benefits were more pronounced in sodium electrolytes than in lithium electrolytes. The hybrid Sn–Na electrodes were operated for 1700 h at a current density of 0.25 mA cm−2, where pristine sodium showed an extremely high overpotential but only after 300 h under the same conditions. Chen et al. compared the benefits of the electrochemical performances of the Na–Sn alloy made from SnCl2 and SnCl4.128 The two tin chlorides were quite different, especially when comparing their oxidation states and phases at room temperature. SnCl4 is a well-known liquid phase with high reactivity in air and moisture, whereas SnCl2 exists in the solid phase. Thus, SnCl4–Na electrodes are fabricated by simply dropping liquid droplets on sodium, while SnCl2 is dissolved in diglyme to form the SnCl2–Na interphase. According to the SEM images, liquid drops of SnCl4 formed a uniform and dense layer on the surface of sodium. In addition, the components of the artificial SEI layer did not vary depending on its depth, as verified by XPS depth profiling. In comparison, the SnCl2–Na electrode had a heterogeneous structure due to competitive reactions between sodium and SnCl2 or diglyme. As a result, SnCl4–Na showed much longer cyclability than both pristine sodium and SnCl2 in both symmetric cells and full cells.
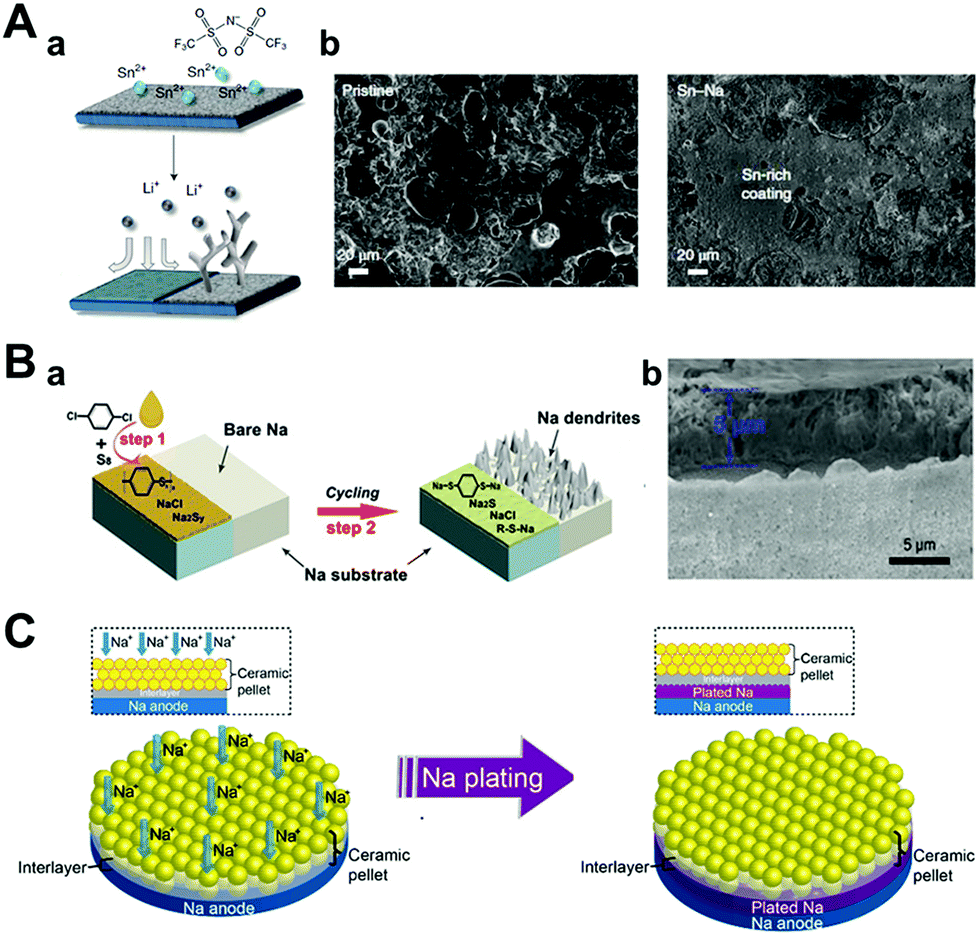 | ||
| Fig. 12 (A) Hybrid anodes based on facile and fast Sn deposition on reactive metals produced by ion exchange. (a) A schematic illustration of the tin protection on the metal electrode. (b) Post-mortem SEM images of the pristine sodium electrode (left) and tin-protected sodium electrode (right) after being cycled at 0.5 mA cm−2 and 0.25 mA h cm−2 for 10 cycles. Reproduced with permission from ref. 127. Copyright 2018, Springer Nature. (B) (a) Diagram for the fabrication of a sodium benzenedithiolate (PhS2Na2)-rich protection layer on sodium metal foil. (b) SEM image of the protected sodium foil. Reproduced with permission from ref. 121. Copyright 2020, Wiley. (C) Contact model of ceramic pellet solid electrolyte and sodium metal with a good wetting ability artificial interlayer during the plating of sodium. Reproduced with permission from ref. 124. Copyright 2017, American Chemical Society. | ||
Zhu et al. introduced two steps of the facile and scalable synthetic route for creating a sodium benzenedithiolate (PhS2Na2)-rich protection layer (Fig. 12B).121 In the first step, sodium metal was reacted with para-dichlorobenzene in THF to form poly(phenylene sulfides) (PPS), Na2Sy, and NaCl. Next, it was converted into PhS2Na2 during cycling. To confirm its protective functionality, galvanostatic tests are performed on symmetric sodium cells under various conditions. Under normal conditions (1 mA cm−2, 1 mA h cm−2), sodium metal with a PhS2Na2 protection layer was operated for over 800 h without any voltage fluctuation, while pristine sodium showed a high overpotential at the initial cycle of approximately 300 mV. Even at a high capacity (3 mA h cm−2) and high current density (5 mA cm−2), it delivered a stable voltage profile of up to 360 h and 280 h, respectively.
Even in an all-solid-state battery, artificial SEI formation can be adopted to suppress dendrite formation. As reported by Monroe and Newman, it is well known that the SEI layer, which has a shear modulus 1.8 times higher than that of lithium metal, can prevent lithium dendritic growth.115 However, in the case of sodium metal, sodium dendrites can penetrate even the elastically much stiffer NASICON solid-state electrolyte by diffusing through the defects of NASICON. For this reason, Zhou et al. carried out a reaction between NASICON and sodium metal at high temperature (380 °C), which produced a black interlayer between liquid sodium metal and NASICON pellets (Fig. 12C). In addition, the wettability of liquid sodium metal on NASICON pellets also dramatically increased at high reaction temperatures.124 By applying heat-treated NASICON (H-NASICON), Na//H-NASICON//Na cells delivered more stable sodium plating–stripping cycles (550 h at 0.15 mA cm−2, 0.15 mA h cm−2, which later increased to 0.25 mA cm−2, 0.25 mA h cm−2). On the contrary, the Na//NASICON//Na cells were short-circuited, lasting only 1 h under the same conditions.
Jin et al. proposed multiple strategies in their report for synthesizing an NaF/SnO2@rGO composite (Fig. 13A).129 To synthesize the composite, SnO2 nanodots were obtained by the hydrothermal method, followed by mixing with a GO suspension to obtain a monolithic SnO2/GO composite film. Next, the film was reduced by hydrazine hydrate steam and magnetron sputtering was conducted to form the NaF thin film. To explain the role of each component, the NaF thin film served as the rigid artificial SEI layer for shielding from damage by dendritic growth and the SnO2 nanodots were converted into Na–Sn alloy and Na2O during the sodiation process, which could lower the nucleation overpotential and produce higher binding anchors, respectively. When Sn nanoparticles were used instead of SnO2 nanoparticles in this composite, the van der Waals interaction between the graphene layer and the Sn nanoparticles were much smaller than that between Na2O and graphene because of the formation of C–O covalent bonds with graphene. The weak interaction between the Sn metal nanoparticles and the graphene host material could induce particle coarsening, which gradually reduced the effect of the designed strategies. Lastly, rGO provided a sandwich-like structure, which physically suppressed dendrite formation by its framework. By comprehensive interfacial engineering as that mentioned above, much more stable sodium metal anodes could be achieved in symmetric cells and full cells. In symmetric cells, the NaF/SnO2@rGO electrodes operated for more than 3500 h at 0.5 mA cm−2, 1 mA h cm−2. The full cell with the FeS2 cathode could deliver a stable capacity of 315.6 mA h g−1 for 100 cycles.
 | ||
| Fig. 13 Schematic illustration of the Na deposition process: (A) SnO2-guided uniform Na plating in NaF/SnO2@rGO and Sn particle coarsening. Reproduced with permission from ref. 129. Copyright 2020, Elsevier. Schematic diagram showing the difference between bare Na and graphene film-protected Na anodes. (B) Illustration of (i–iii) transferring free-standing graphene film onto the Na metal surface and (iv–vi) the high stability of graphene-coated Na anode during stripping/plating without the formation of Na dendrites. Reproduced with permission from ref. 130. Copyright 2017, American Chemical Society. (C) Schematic of the FCPL and FCPL–Na metal electrode integration fabrication. Reproduced with permission from ref. 132. Copyright 2017, American Chemical Society. | ||
In essence, several authors have published their works by laying physically rigid materials on sodium metal, such as graphene.130,131 Wang et al. transferred ultrathin graphene onto a metal substrate, followed by growing graphene on copper foil by chemical vapor deposition (Fig. 13B). Graphene not only provides mechanical strength and flexibility as an artificial SEI layer but also chemical stability against organic electrolytes and facile ion transport through its defects, which perfectly matches the qualifications of an SEI layer. Moreover, they investigated the cycling performance of sodium metal anodes relying on the thickness of the graphene layer. Indeed, even a few nanometer differences showed a pronounced change in the electrochemical performance. As the graphene layer was kept thicker (up to about 15 layers of graphene), the electrochemical stability and rate capability increased. On the other hand, single-layer graphene has similar electrochemical performance as that of pristine sodium. By adopting multilayer graphene on sodium metal in symmetric cells, they obtained more than 300 cycles with a relatively stable voltage profile at 2 mA cm−2 and 3 mA h cm−2.
Kim et al. introduced an inorganic–organic composite protective layer (CPL), which is composed of poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF–HFP) and Al2O3 nano-powder.132 The two components were mixed in an organic solvent (PC) and coated on a glass substrate. After evaporation of the solvents, the dried film was transferred onto the sodium metal by roll pressing (Fig. 13C). They demonstrated that CPL has sufficient mechanical strength including shear modulus (over 6.0 GPa) to endure dendritic growth, which is inversely proportional to the ratio of PC/PVDF. To verify the protective effect of CPL, full cells were fabricated with Na0.6Mn0.65Ni0.25Co0.10O2. Full cells consisting of CPL artificial layer on sodium metal anodes achieved higher capacity retention (91.6% at 60 cycles) compared to pristine sodium (76.7% for the same number of cycles).
There are diverse principles and strategies for generating an artificial SEI layer. Many studies have been carried out in this field, and new materials and synthetic techniques are being updated often. However, creative approaches to overcome challenges, rather than existing theories and strategies, can lead to new ways of creating artificial SEI layers. In additionally, when an SEI layer is directly applied to the metal, its analysis may be interrupted by the strong reductive nature of the metal; thus, an analytical method and solid theoretical proof will be required.
4. High-energy density batteries with stabilized sodium metal anode
Sodium metal batteries are generally composed of sodium metal as the anode and various materials such as metal oxides, metal phosphates, and even organic materials as the cathode.133–137 These cathodes suffer from their low specific capacity, which cannot fulfill the purpose of SMBs. Therefore, researchers have been devoted to developing high specific capacity cathodes such as sulfur, oxygen, and carbon dioxide.38 With these fascinating cathodes, designing a fine SEI layer that can suppress dendrite growth and contamination should be considered because the sodium metal is easily exposed to harsh conditions originating from the cathodes and the operating environment. As a result, we will focus on the strategies for designing the SEI layer in full cells, which could suppress the growth of dendrites and contamination.4.1. Na–S batteries
Since high-temperature Na–S batteries (HT Na–S) were invented in the 1960s, there has been a considerable effort to develop room-temperature Na–S batteries (RT Na–S).138,139 At a high temperature (300–350 °C), sodium and sulfur are both liquid states; thus, the development of a stable solid-state electrolyte was inevitable. The beta-alumina solid electrolyte (BASE) has been applied widely to HT Na–S batteries owing to its high sodium ion conductivity of 0.1 S cm−1.140 However, the HT Na–S batteries pose some shortcomings: (1) a large amount of external energy is required for maintaining the high temperature, (2) high cost for manufacturing complicated systems, (3) safety concerns arising from the use of liquid sodium, and (4) relatively lower theoretical specific capacity because only high-order sodium polysulfides are utilized (557 mA h g−1). As a result, RT Na–S batteries have emerged for utilizing its high specific capacity (1675 mA h g−1) under safe conditions.141,142In RT-Na–S batteries, the dissolution of sodium polysulfides (NaPSs) is a critical issue for realizing long-cycle, high energy density batteries, similar to Li–S batteries.143,144 During the discharge process, sulfur (S8) is electrochemically reduced to NaPSs (NaxSy, x = 1, 2, y = 1–8) at the cathode. High-order NaPSs (Na2S8, Na2S6) are easily dissolved into electrolytes, which then migrate to the sodium metal anodes in ether-based electrolytes.145 These are converted into low order NaPSs and even further to Na2S2 and Na2S due to the highly reactive metallic sodium, which keeps covering the surface of sodium increasing its thickness, resulting in low cyclability and the loss of active material. Furthermore, carbonate-based electrolytes react rapidly with NaPSs via the nucleophilic attack or substitution reaction, which leads to the loss of active sulfur and damage to the electrolyte.146 For these reasons, designing an SEI layer that protects the sodium anode from NaPS shuttling is essential for RT Na–S batteries.
Researchers have focused on optimizing electrolytes as a way of preventing the deterioration of sodium metal anodes such as ionic liquid electrolytes,147,148 solid-polymer electrolytes,149 solid-state electrolytes, and typical organic solvents. Wei et al. introduced a carbonate electrolyte containing the ionic liquid 1-methyl-3-propylimidazolium-chlorate tethered to SiO2 nanoparticles (SiO2–IL–ClO4) as an additive (Fig. 14A).147 Besides, the silica nanoparticles provided an anchor point for ClO4− anions for the supporting electrolyte and reducing the strong electric field through the tethered anion effect. This modified electrolyte played a major role in increasing the cyclability and specific capacity of Na–S batteries owing to ionic conductivity stabilization over a wide temperature range, thus forming a robust and electrochemically stable SEI layer on the sodium metal that effectively prevents side reactions. 600 mA h g−1 of discharge capacity was obtained at 100 cycles with a capacity decay of only 0.31% per cycle at 0.1C rate.
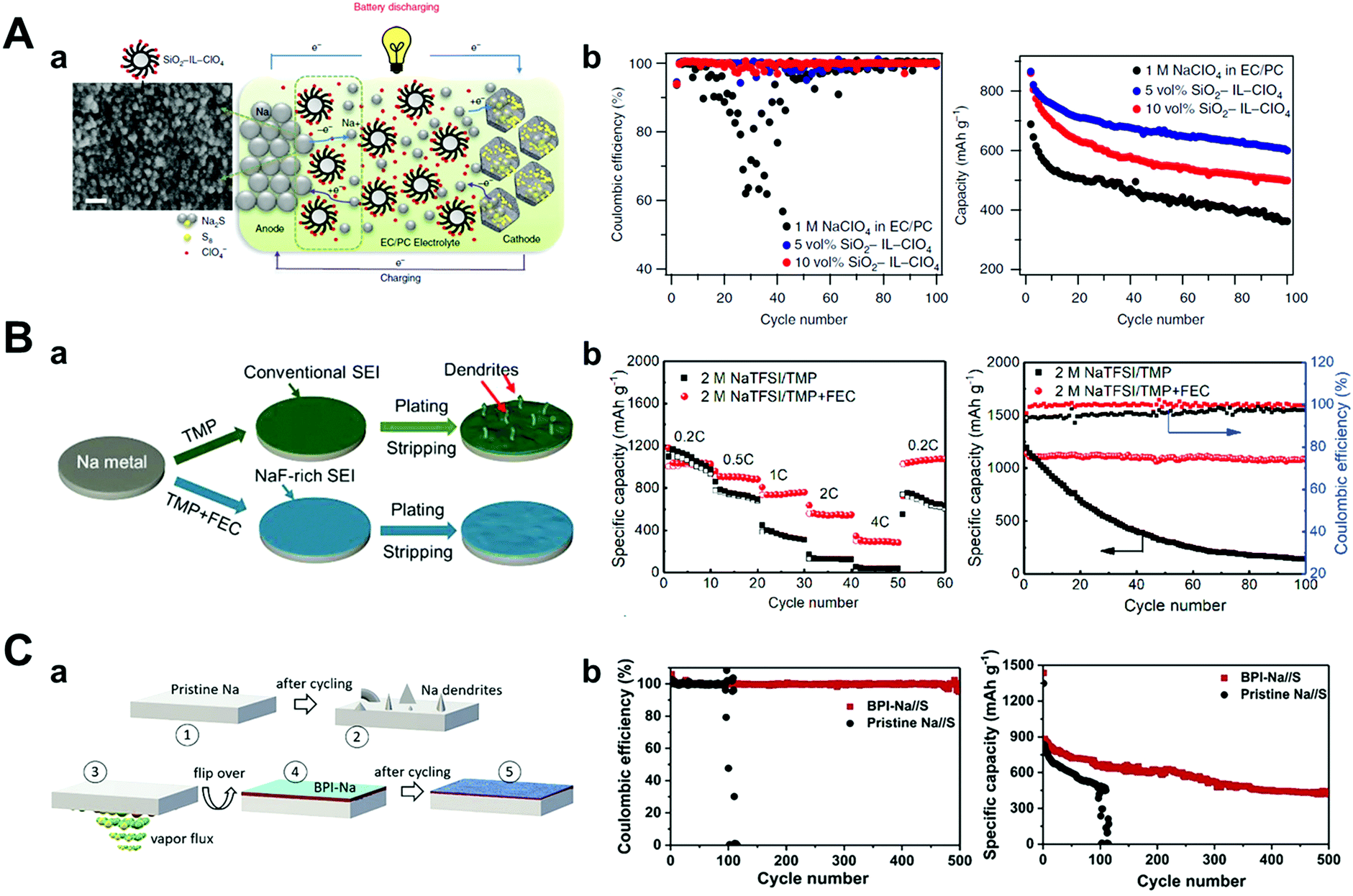 | ||
| Fig. 14 (A) (a) Schematic drawing of the Na–S cell during galvanostatic cycling, using SiO2–IL–ClO4 as additive in 1 M NaClO4 in a mixture of EC/PC. On the anodic side, sodium atom loses an electron to form sodium ion during discharge. Sodium ion diffuses inside the microporous carbon–sulfur composite and reacts with sulfur to form sodium sulfide (Na2S) on the cathodic side, and the reverse reaction takes place during charging, where SiO2–IL–ClO4 helps to stabilize the sodium anode. The SEM image of the sodium metal surface cycled in a cell with 10 vol% of SiO2–IL–ClO4 in the electrolyte shows for the first time that the particles form a conformal layer on the anode surface. Scale bar, 30 nm. (b) Coulombic efficiency and capacity versus cycle number for the cell with different amounts of SiO2–IL–ClO4 in the electrolytes at a current density of 0.5C. Reproduced with permission from ref. 147. Copyright 2016, Springer Nature. (B) (a) Schematic illustration of the impact of NaF-rich SEI on the stability of the Na electrode. Electrochemical performance of the Na/SPAN cells with and without FEC. (b) Rate performance at several current densities and cyclic performance at 0.2C. Reproduced with permission from ref. 150. Copyright 2019, Elsevier. (C) (a) Schematic illustration of the process steps involved in the formation of BPI, (1), (2) pristine sodium before and after cycling, (3) aqueous ammonia vapors react with sodium, (4) sodium with BPI, and (5) sodium with BPI after cycling. (b) Comparison of Coulombic efficiencies between Na//S cells comprising sodium anode with and without BPI at 0.5C, and their corresponding cyclability. Reproduced with permission from ref. 152. Copyright 2020, Elsevier. | ||
Wu et al. developed a novel non-flammable liquid organic electrolyte for RT Na–S batteries. In particular, they applied trimethyl phosphate (TMP) as a solvent owing to its low viscosity (2.3 mPa s), high dielectric constant (21.6), and wide temperature range (−46 to 197 °C).150 However, TMP alone could not generate a stable SEI layer on the sodium metal surface; thus, they blended it with 30 vol% of FEC and highly concentrated salt (2 M NaTFSI) (Fig. 14B). The electrolyte formed fewer dendrites on the sodium surface during galvanostatic cycling owing to the generation of a more NaF-rich SEI layer (when compared to the electrolyte without FEC). Since the FEC's LUMO level is lower than that of TMP, the reduction of FEC occurs before the reduction of TMP, as proven by DFT calculations. Accordingly, the Na–S batteries that adopted a 2 M NaTFSI TMP/FEC electrolyte delivered a discharge capacity of 788 mA h g−1 after 300 cycles with very low capacity decay below 0.04% per cycle at 1C rate.
The use of a metal alloy artificial SEI layer could also be another approach to enhance the chemical stability of sodium metal anodes in Na–S batteries. Kumar et al. suggested an artificial metal-alloy interphase (MAI) synthesized by the facile solid–vapor reaction of metallic sodium with tin tetrachloride vapors, which facilitated the formation of a uniform and thinner (12–15 nm) SEI layer.151 In the case of pristine sodium metal, a short circuit was observed before 100 cycles at 0.5C, while MAI on sodium attained a high specific capacity of 1110 mA h g−1 during the early cycles and maintained 47% of the capacity even after 500 cycles, which meant only a capacity decay of 0.106% per cycle. This long-term stability and high capacity retention originated in MAI, which preserved anodes from any parasitic reaction with NaPSs.
In the same manner, Kumar et al. induced a solid–vapor reaction between sodium metal and ammonia vapor, which generated a stiff biphasic artificial SEI layer consisting of NaOH and NaNH2 with a thickness of 3 μm (Fig. 14C).152 The biphasic SEI layer consisting of NaOH and NaNH2 had high Young's modulus of 31.0 GPa and 16.4 GPa, respectively. In addition, NaNH2 had ductile characteristics as its critical strain (∼56%) is higher than that of NaOH (∼16%), resulting in synergistic effects that hindered sodium dendrite growth. The biphasic interphase on sodium (BPI-Na) delivered high cyclability even at 50 mA cm−2 in BPI-Na//BPI-Na symmetric cells and exhibited the lowest capacity decay of 0.09% per cycle at 0.5C rate for 500 cycles for RT Na–S batteries.
4.2. Na–O2 (CO2) batteries
Na–O2 batteries have also been of interest to many researchers owing to their high gravimetric energy density (1108 W h kg−1). During their discharge process, the oxygen reduction reaction (ORR) occurs at the cathode, resulting in NaO2 or Na2O2 formation, depending on the electrolyte and the catalyst.153–157 Even though most studies have focused on cathode designs, catalysts, and mechanisms for optimizing ORR/OER,158,159 dendrite growth and oxygen crossover, arising from the concentration gradients between the cathode and anode, are also prominent shortcomings of Na–O2 batteries that need to be investigated.For Na–O2 batteries, the migrating O2/O2− react with metallic sodium, leading to the production of an insulating oxide layer on the sodium surface. This insulating thick oxide layer blocks sodium ion transport during cycling, which in turn leads to robust dendrite formation and capacity fading through the formation of an inhomogeneous SEI layer. In this regard, there should be investigations into the development of advanced electrolytes capable of generating homogeneous or stable SEI layers and protective artificial layers that act as barriers to oxygen crossover.
Various electrolytes, such as carbonate-based solvents and ether-based solvents, have been used to enhance the dendritic growth of sodium in Na–O2 batteries. It has been reported that 1.0 M NaPF6 in EC/DMC shows very low reversibility and large overpotential owing to the formation of Na2O2 as a major product.153 In contrast, NaO2 is dominantly formed in the ether-based electrolyte with much lower overpotential and superior cyclability. Some studies have been conducted to reveal the fundamentals of ether-based electrolytes and their interactions with both the cathode and anode. Lutz et al. investigated the influence of four types of salt anions (ClO4−, PF6−, OTf−, and TFSI−) on the performance of Na–O2 batteries.158 Only 0.5 M NaPF6 in DME electrolyte showed long-term stability, while 0.5 M NaClO4 in DME and 0.5 M NaOTf in DME electrolyte is stable, indicating that the SEI layer grows continuously during repeated cycling. In addition, 0.5 M NaTFSI in DME electrolyte destroyed the sodium metal anodes, as confirmed by electrochemical impedance spectroscopy (EIS) analysis and digital photos of sodium metal after galvanostatic cycling. Recently, Vitoriano et al. compared three common ether-based electrolytes (DME, DEGDME, and TEGDME) in Na–O2 batteries with the same NaClO4 sodium salts. DFT calculations and experiments were conducted to estimate their overall properties, including chelating ability, solvation ability, desolvation ability, diffusion coefficient, viscosity, and donor number.160 After a comprehensive evaluation, DME was found to be the fastest ion-transport solvent. However, it was also found to have low stability and required a lot of energy to move the DME molecules during sodium ion solvation/desolvation owing to its small charge screening effect. On the other hand, TEGDME was found to have shortcomings of viscosity, bulkiness, and four chelation sites, resulting in sluggish sodium ion transport and a high desolvation energy barrier. For DEGDME, electrochemical tests demonstrated that the longest cyclability was achieved at various electrolyte concentrations and current densities as it does not have any significant shortcomings as that of the other solvents. Dimethyl sulfoxide (DMSO) was applied in Na–O2 batteries as an electrolyte solvent. DMSO is a well-known solvent in the field of batteries and has many advantages such as low volatility, high conductivity and polarity, and a wide electrochemical window. Nevertheless, DMSO has the fatal shortcoming of high reactivity with sodium metal. To enhance the oxidative and reductive stability of DMSO, the researchers introduced a 3.2 M NaTFSI/DMSO high concentration electrolyte. Na–O2 full cells with this electrolyte showed superior cyclability over 150 cycles at current densities of 0.05 mA cm−2 and 0.15 mA cm−2 with controlled discharge capacity, demonstrating the characteristics of high-concentration electrolytes.109
To prevent the failure of the sodium metal anode by oxygen crossover, Mao et al. introduced fluorinated carbon nanotubes (FCNTs) and mechanically mixed them with sodium metal under high temperature (250 °C) (Fig. 15A).161 A tiny amount of FCNT (1–1.5 wt%) guided the facile formation of an NaF-rich SEI layer, which was confirmed by both experimental and DFT calculations. The strength of the CNT-reinforced SEI layer is due to the high mechanical strength of FCNTs. Na–O2 full cells were composed of Na/FCNTs as the anode and δ-MnO2 on carbon cloth as the cathode. The pristine Na–O2 cell cycled until only 62 cycles but Na/FCNT was cycled up to 112 cycles at a current density of 400 mA g−1 with a limited capacity of 1000 mA h g−1. Wu et al. prepared an NaF-rich artificial SEI layer by galvanostatic cycles using 1 M NaOTf in TEGDME with a 2% FEC electrolyte (Fig. 15B).162 During galvanostatic cycles, FEC was preferentially reduced to form NaF on sodium metal, which effectively hindered the reaction of sodium metal with the chemical species when oxygen crossover occurred. After treatment, the cells were disassembled and reassembled to utilize NaF-rich sodium metal and a new electrolyte, 1 M NaOTf in TEGDME. The Na–O2 full cell, including pristine sodium, experienced cell failure even after 10 cycles, while NaF-rich sodium maintained its stability for 30 cycles under a limited capacity of 1000 mA h g−1 at a current density of 500 mA g−1.
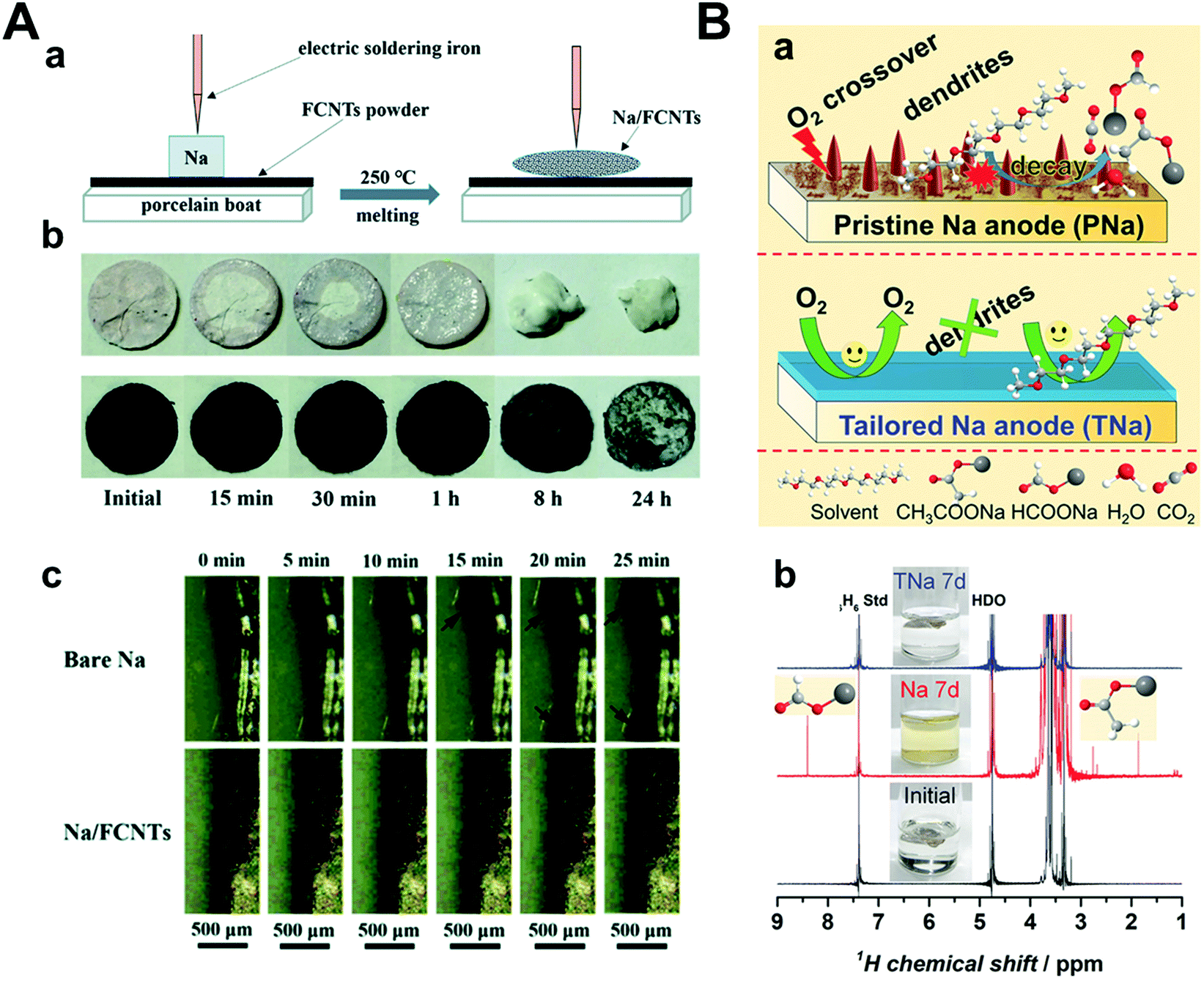 | ||
| Fig. 15 (A) (a) Schematic illustration of the fabrication procedure of the Na/FCNT electrode. (b) Appearance changes of bare Na and Na/FCNT electrodes in open air. (c) In situ optical microscopy images of Na deposition behavior on the bare Na electrode and Na/FCNT electrode after various durations at a current density of 2 mA cm−2. Reproduced with permission from ref. 161. Copyright 2020, The Royal Society of Chemistry. (B) Tailored Na anode (TNa) against side reactions between the electrolyte and the Na anode. (a) Illustration of problems (O2 crossover, dendrite formation, and electrolyte decay) on pristine Na anode (PNa) in Na–O2 battery (top) and improvements on TNa (bottom). (b) 1H nuclear magnetic resonance (NMR) spectra and photos of pristine electrolyte (black line), electrolyte after immersing PNa anode for 7 d (red line), and electrolyte after immersing the TNa for 7 d (blue line), respectively. Reproduced with permission from ref. 162. Copyright 2018, Wiley. | ||
Hu et al. were the first to report Na–CO2 batteries. They used a Ni mesh coated by TEGDME-treated MWCNT as the cathode and 1 M NaClO4 in TEGDME as the electrolyte.163 Since then, several papers have been published on Na–CO2 batteries, most of them aimed at accelerating the sluggish reaction of carbon dioxide reduction/evolution reactions.164,165 As in the case of Na–CO2 batteries, it is important to protect sodium metal anodes from attack by CO2 crossover for long-term stability. To this end, Hu et al. introduced a composite polymer electrolyte (CPE), which is a hemi-crystalline PVDF–HFP-4% SiO2/NaClO4 in TEGDME.166 10–20 nm of SiO2, used as ceramic fillers, enlarged the amorphous region of the CPE matrix to increase the sodium ion conductivity to 1.0 mS cm−1 and improved the mechanical strength to 0.5–0.6 GPa. CPE had a non-flammable and strong electrolyte-locking ability. In addition, for preparing stabilized sodium metal, they contacted the graphene oxide (GO) film with molten sodium metal. The high reactivity of sodium metal reacted with the oxygen functional groups of GO, which converted GO into reduced graphene oxide (rGO) and formed the Na2O or Na2CO3 film. Therefore, the rGO–Na anode delivered non-dendritic sodium ion plating/stripping (5.7 to 16.5 mA cm−2) and operated in full cells for 400 cycles with a high current rate (500 mA g−1) and a limited capacity of 1000 mA h g−1. In the same manner, Wang et al. synthesized a solid polymer electrolyte (SPE) consisting of PEO/NaClO4/3 wt% SiO2 with high ionic conductivity (0.64 mS cm−1) at 70 °C.167 It also had an inhibitory effect on sodium dendrite growth, as confirmed by the SEM images. By applying SPE, the Na–CO2 batteries showed a reversible capacity of 500 mA h g−1 under excellent cycling stability for more than 240 cycles at a current density of 50 mA g−1 and a limited capacity of 500 mA h g−1.
5. Conclusions
In this review, while exploring the various approaches to stabilize Na anodes, we have emphasized on the recent progress in SEI design for sodium metal anode applications. SEI modification is one of the most fascinating approaches for stabilizing the charge–discharge performance of metal anodes. The SEI layer allows ion diffusion and prevents electron transport between the electrode and electrolyte, resulting in stable cycling performance. However, owing to the rapid and random growth and severe volume change of sodium metal, the effective design of the SEI layer is necessary to achieve stable and reversible cycling performance. As a result, we have provided diverse approaches for the design and control of the ideal SEI layer for sodium metal anodes in the following order.First, electrolyte additives have been applied to generate stable in situ SEI films. These additives have lower reductive stability than the electrolyte and react with sodium metal before electrolyte decomposition. FEC is a representative additive for an in situ stable SEI layer and has been studied in different ways. It reduces gas evolution, originating from the reaction between the sodium metal and electrolyte, and guides a high proportion of the inorganic SEI layer, resulting in better mechanical properties of the SEI layer. Furthermore, the SHES mechanism has also been highlighted to induce smooth electrodeposition. Additives including metal cations, with lower reduction potential than sodium ions, are not able to electroplate on the anode but they accumulate adjacent to the tip to form an electrostatic shield. The positively charged barrier of the tip impels the further plating of sodium ions to plate out of the tip. Fortunately, unlike LIBs, there are more potential candidates to generate SHES, such as lithium and potassium ions. Additives for the formation of Na-inorganic alloy interphase have also attracted the attention of researchers. Sn-based electrolytes are typical examples that are capable of galvanic replacement without any external electron transfer. The synthesized Na–Sn SEI layer renders not only facile sodium-ion transportation but also physically inhibits dendrite growth.
Second, a high-concentration electrolyte is also an effective method to stabilize the anode performance as it increases sand's time, which is the time for dendrite formation. Compared to the low-concentration electrolyte, the high-concentration electrolyte shifts its LUMO level from the solvent to the anion, resulting in an increasing preference for salt anion decomposition (inorganic SEI layer) rather than the solvent (organic SEI layer). Recently, in order to reduce its adverse effects such as high cost and low ionic mobility, there have been recent reports of a high-concentration electrolyte with an inert diluent solvent, maintaining the aggregated solvation structure of the high-concentration electrolyte.
Third, the formation of an artificial SEI layer before cell assembly has been proposed in many different methods, including chemical coating processes such as atomic/molecular layer deposition, organic reactions, galvanic replacement, inorganic conversion reactions, and transferring target materials. These artificial SEI layers have at least one of the following merits: (1) materials with high sodium ion conductivity; (2) nanometer-scale thickness for reducing the interfacial resistance; (3) mechanical stiffness or elastic characteristics to endure volume change and dendrite growth; (4) homogeneous thickness and chemical composition.
Finally, various strategies for sodium metal protection in various sodium metal full cells have been summarized. In RT Na–S batteries, polysulfide dissolution should be considered since it converts to low order polysulfides or Na2S when it comes in contact with sodium. In Na–O2 (or CO2) batteries, sodium metal is not only damaged by dendrite growth but also O2 (or CO2) crossover. As sodium is continuously exposed to the gas flowing out from the cathode, a thicker insulating film is formed on the surface of sodium, resulting in lower cyclability and Coulombic efficiency. Therefore, the SEI layer formed by electrolyte additives or an artificially pre-formed SEI layer could be a protective shield for metallic dendrites and the spatial barrier between the sodium metal anode and other species.
6. Perspectives
Although substantial efforts have been devoted to the development of stable, long-life SMBs, so far, there are still numerous challenges to be overcome. In particular, it is necessary to settle both the laboratory and industrial hindrances in SMBs production. In this section, we highlight the comprehensive issues and prospects of SMBs.1. Development of advanced electrolytes
Advanced electrolytes containing sacrificial additives are the most powerful and simplest way for the further development of SMBs. However, these kinds of electrolytes have remained at their infancy due to complex electrochemical reactions and poor compatibility with SMBs. Generally, the conventional electrolytes for SMBs are divided into carbonate-based electrolytes and ether-based electrolytes. Each of these electrolytes has pros and cons, which leads to a complicated dilemma when assembling a full cell. In the case of carbonate-based electrolytes, it is commonly known that these are readily decomposed to generate the thick and non-uniform SEI layer; thus, it delivers very low stability and high overpotential at relatively high current densities (>1 mA cm−2). Therefore, it is essential to apply sacrificial additives, which allow the formation of mechanically robust and high ionically conductive inorganic SEI layer. On the other hand, the carbonate-based electrolyte has prominent oxidation stability at the cathode, resulting in relatively high voltage operation (>4.2 V). Since the available cut-off charging voltage is directly related to the energy density of the battery, it is quite reasonable to use carbonate-based electrolytes to fully utilize the capacity of the cathode materials.Meanwhile, ether-based electrolytes allow relatively uniform and stable SEI layer formation, resulting in long cyclability, unlike carbonate-based electrolyte. Besides, since ether-based electrolytes are operated steadily even at a relatively high current density and capacity, metal anodes are able to withstand it even if the batteries are assembled by high-loading cathode. However, ether-based electrolytes are only available to next-generation cathode materials, which are operated under low upper cut-off voltage, including sulfur, O2, and CO2, because of the low oxidative stability. In addition, sodium metal is easily contaminated and damaged by the crossover of the by-products that inevitably originate from the cathode of these systems, thus hindering the long-term stability.
Both carbonate-based electrolytes and ether-based electrolytes are not sufficient to achieve long-term stable SMBs alone. As a result, it is vital to develop sacrificial additives, which have suitable compatibility with sodium metal, and it would be excellent if the oxidative stability could be improved through the decomposition of the additives on the cathode side as well. Since the development of effective sacrificial additives in the SMBs field has not yet been made, it is necessary to fully understand the electrochemical reactions of various chemical species by computational screening and simulation.
2. Electrochemical performance under realistic conditions
For practical application, high energy density batteries over 250 W h kg−1 should be achieved to meet the social demands. In a full-cell system, a high current density and capacity are inevitably applied to the anode for high energy density with decreasing N/P ratio. However, only a few papers have provided electrochemical data in symmetric cells at high current densities (over 5 mA cm−2) and capacities (over 5 mA h cm−2) in SMBs, whereas in LMBs, electrochemical performance at high current densities and capacities is essential when organic electrolytes are used. Furthermore, under realistic conditions such as low N/P ratio and E/S ratio, it shows much lower lifespan characteristics and overpotential compared to the lab-scale coin cell system. Up to now, most of the papers have shown their cyclability in the short term (below 1000 h) under mild conditions. Future studies need to accommodate a longer lifespan. It might be challenging due to the higher reactivity of sodium metal compared to lithium metal but it should be solved. As a result, designing an SEI layer and advancing the electrolyte development could be a promising approach for stabilizing sodium metal anodes.3. Advanced and accessible characterization methods
Along with the development of the strategies for LMBs, the development of characterization methods can be achieved. Cryo-EM is a representative that enables the observation of both the morphologies of metal electrodes and the thickness and chemical composition of crystalline SEI layers without damage by air and electron beams. XPS depth profiling and Rutherford backscattering spectroscopy (RBS) are also analytical techniques for measuring the relative and absolute thicknesses of the SEI layers, respectively, and determining their chemical composition. Moreover, X-ray tomographic microscopy, AFM, and in situ optical microscopy could be effective characterization methods for 2D and 3D surface investigations of metal deposition processes and dendrite formation. However, most of these techniques are not accessible at the lab scale, which consumes considerable cost and time. Furthermore, these measurements are not able to provide a comprehensive understanding of the interface chemistry between the metal and SEI layers, and the quantitative analysis of both the metal and the SEI layer. As a result, advanced and new accessible characterization methods should be further developed. This will be an attractive research topic in the future.Several challenges remain in the path to achieving high performance for commercially available sodium metal anodes. In addition to Na metal stabilization studies, many researchers have suggested and optimized Na–metal full-cell systems such as Na–S and Na–O2 batteries because of their high theoretical energy density and low price. Many studies on these systems have simultaneously enhanced the cycle life and investigated Na degradation mechanisms under full-cell operation conditions. We believe that the understanding of Na metal and stabilization in both half- and full-cell systems for the broadened commercial use of Na metal anodes in sodium-ion rechargeable batteries is imminent. We expect that this review will provide a general overview of previous SEI designs to researchers in the materials and energy storage fields so that they can design an efficient artificial SEI layer on Na metal anodes.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was supported by the National Research Foundation of Korea grant funded by the Korea government (MSIT) (2015M3D3A1A01065440 and 2020R1A2C3004146).Notes and references
- J. B. Goodenough and K. S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS
.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 RSC
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y. K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994–10024 CrossRef CAS
- Y. Lu, L. Yu and X. W. Lou, Chem, 2018, 4, 972–996 CAS
- J. Lee, S. Kim, J.-H. Park, C. Jo, J. Chun, Y.-E. Sung, E. Lim and J. Lee, J. Mater. Chem. A, 2020, 8, 3119–3127 RSC
- K.-N. Jung, J. Kim, Y. Yamauchi, M.-S. Park, J.-W. Lee and J. H. Kim, J. Mater. Chem. A, 2016, 4, 14050–14068 RSC
- J. Li, N. Zhuang, J. Xie, X. Li, W. Zhuo, H. Wang, J. B. Na, X. Li, Y. Yamauchi and W. Mai, Adv. Energy Mater., 2019, 10, 1903455 CrossRef
- M. Wang and Y. Tang, Adv. Energy Mater., 2018, 8, 1703320 CrossRef
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243–257 RSC
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS
- E. C. Evarts, Nature, 2015, 526, S93–S95 CrossRef CAS
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS
- X.-B. Cheng, R. Zhang, C.-Z. Zhao, F. Wei, J.-G. Zhang and Q. Zhang, Adv. Sci., 2016, 3, 1500213 CrossRef
- B. Liu, J.-G. Zhang and W. Xu, Joule, 2018, 2, 833–845 CrossRef CAS
- H. Zhang, G. G. Eshetu, X. Judez, C. Li, L. M. Rodriguez-Martinez and M. Armand, Angew. Chem., Int. Ed., 2018, 57, 15002–15027 CrossRef CAS
- X. Zhang, A. Wang, X. Liu and J. Luo, Acc. Chem. Res., 2019, 52, 3223–3232 CrossRef CAS
- A. Varzi, R. Raccichini, S. Passerini and B. Scrosati, J. Mater. Chem. A, 2016, 4, 17251–17259 RSC
- W. Xu, J. Wang, F. Ding, X. Chen, E. Nasybulin, Y. Zhang and J.-G. Zhang, Energy Environ. Sci., 2014, 7, 513–537 RSC
- W.-G. Lim, C. Jo, J. Lee and D. S. Hwang, Korean J. Chem. Eng., 2018, 35, 579–586 CrossRef CAS
- S. Yang, F. Zhang, H. Ding, P. He and H. Zhou, Joule, 2018, 2, 1648–1651 CrossRef
- S. Kim, M. Ju, J. Lee, J. Hwang and J. Lee, J. Am. Chem. Soc., 2020, 142, 9250–9257 CrossRef CAS
- X. Guo, Y. Ding, H. Gao, J. B. Goodenough and G. Yu, Adv. Mater., 2020, 32, 2000316 CrossRef CAS
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338–2460 RSC
- S. M. Hwang, J.-S. Park, Y. Kim, W. Go, J. Han, Y. Kim and Y. Kim, Adv. Mater., 2019, 31, 1804936 CrossRef
- Y. Kim, K.-H. Ha, S. M. Oh and K. T. Lee, Chem. – Eur. J., 2014, 20, 11980–11992 CrossRef CAS
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef
- H. Moriwake, A. Kuwabara, C. A. J. Fisher and Y. Ikuhara, RSC Adv., 2017, 7, 36550–36554 RSC
- Z.-L. Xu, G. Yoon, K.-Y. Park, H. Park, O. Tamwattana, S. Joo Kim, W. M. Seong and K. Kang, Nat. Commun., 2019, 10, 2598 CrossRef
- G. Chang, Y. Zhao, L. Dong, D. P. Wilkinson, L. Zhang, Q. Shao, W. Yan, X. Sun and J. Zhang, J. Mater. Chem. A, 2020, 8, 4996–5048 RSC
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS
- S. Fang, D. Bresser and S. Passerini, Adv. Energy Mater., 2019, 10, 1902485 CrossRef
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. L. Notten, Adv. Energy Mater., 2018, 8, 1800079 CrossRef
- H. Wang, E. Matios, J. Luo and W. Li, Chem. Soc. Rev., 2020, 49, 3783–3805 RSC
- B. Lee, E. Paek, D. Mitlin and S. W. Lee, Chem. Rev., 2019, 119, 5416–5460 CrossRef CAS
- Y. Wang, Y. Wang, Y.-X. Wang, X. Feng, W. Chen, X. Ai, H. Yang and Y. Cao, Chem, 2019, 5, 2547–2570 CAS
- L. Fan and X. Li, Nano Energy, 2018, 53, 630–642 CrossRef CAS
- C. Yan, R. Xu, Y. Xiao, J. F. Ding, L. Xu, B.-Q. Li and J.-Q. Huang, Adv. Funct. Mater., 2020, 30, 1909887 CrossRef CAS
- R. Mogensen, D. Brandell and R. Younesi, ACS Energy Lett., 2016, 1, 1173–1178 CrossRef CAS
- W. Liu, P. Liu and D. Mitlin, Adv. Energy Mater., 2020, 2002297 CrossRef CAS
- D. I. Iermakova, R. Dugas, M. R. Palacín and A. Ponrouch, J. Electrochem. Soc., 2015, 162, A7060–A7066 CrossRef CAS
- K. Pfeifer, S. Arnold, J. Becherer, C. Das, J. Maibach, H. Ehrenberg and S. Dsoke, ChemSusChem, 2019, 12, 3312–3319 CrossRef CAS
- Y.-S. Hong, N. Li, H. Chen, P. Wang, W.-L. Song and D. Fang, Energy Storage Mater., 2018, 11, 118–126 CrossRef
- Y. Yang, X. Liu, Z. Dai, F. Yuan, Y. Bando, D. Golberg and X. Wang, Adv. Mater., 2017, 29, 1606922 CrossRef
- B. Michalak, H. Sommer, D. Mannes, A. Kaestner, T. Brezesinski and J. Janek, Sci. Rep., 2015, 5, 15627 CrossRef CAS
- Y. Yui, M. Hayashi and J. Nakamura, Sci. Rep., 2016, 6, 22406 CrossRef CAS
- X. Chen, X. Shen, B. Li, H.-J. Peng, X. B. Cheng, B.-Q. Li, X.-Q. Zhang, J.-Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2018, 57, 734–737 CrossRef CAS
- M. Goktas, C. Bolli, J. Buchheim, E. J. Berg, P. Novak, F. Bonilla, T. Rojo, S. Komaba, K. Kubota and P. Adelhelm, ACS Appl. Mater. Interfaces, 2019, 11, 32844–32855 CrossRef CAS
- Y. Xiang, G. Zheng, Z. Liang, Y. Jin, X. Liu, S. Chen, K. Zhou, J. Zhu, M. Lin, H. He, J. Wan, S. Yu, G. Zhong, R. Fu, Y. Li and Y. Yang, Nat. Nanotechnol., 2020, 15, 883–890 CrossRef CAS
- J. Chazalviel, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 7355–7367 CrossRef CAS
- M. Rosso, C. Brissot, A. Teyssot, M. Dollé, L. Sannier, J.-M. Tarascon, R. Bouchet and S. Lascaud, Electrochim. Acta, 2006, 51, 5334–5340 CrossRef CAS
- C. Brissot, M. Rosso, J. N. Chazalviel and S. Lascaud, J. Power Sources, 1999, 81-82, 925–929 CrossRef CAS
- H. Wu, G. Chan, J. W. Choi, I. Ryu, Y. Yao, M. T. McDowell, S. W. Lee, A. Jackson, Y. Yang, L. Hu and Y. Cui, Nat. Nanotechnol., 2012, 7, 310–315 CrossRef CAS
- Y. Sun, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071 CrossRef CAS
- X. Cao, X. Ren, L. Zou, M. H. Engelhard, W. Huang, H. Wang, B. E. Matthews, H. Lee, C. Niu, B. W. Arey, Y. Cui, C. Wang, J. Xiao, J. Liu, W. Xu and J.-G. Zhang, Nat. Energy, 2019, 4, 796–805 CrossRef CAS
- J. Cui, A. Wang, G. Li, D. Wang, D. Shu, A. Dong, G. Zhu, J. Luo and B. Sun, J. Mater. Chem. A, 2020, 8, 15399–15416 RSC
- J. Sun, M. Zhang, P. Ju, Y. Hu, X. Chen, W. Wang and C. Chen, Energy Technol., 2020, 8, 1901250 CrossRef CAS
- Q. Lu, X. Wang, A. Omar and D. Mikhailova, Mater. Lett., 2020, 275, 128206 CrossRef CAS
- Y. Zhao, X. Yang, L. Y. Kuo, P. Kaghazchi, Q. Sun, J. Liang, B. Wang, A. Lushington, R. Li, H. Zhang and X. Sun, Small, 2018, 14, 1703717 CrossRef
- X. Zheng, P. Li, Z. Cao, W. Luo, F. Sun, Z. Wang, B. Ding, G. Wang and Y. Huang, Small, 2019, 15, 1902688 CrossRef CAS
- Y. Fang, R. Lian, H. Li, Y. Zhang, Z. Gong, K. Zhu, K. Ye, J. Yan, G. Wang, Y. Gao, Y. Wei and D. Cao, ACS Nano, 2020, 14, 8744–8753 CrossRef CAS
- C. D. Fincher, Y. Zhang, G. M. Pharr and M. Pharr, ACS Appl. Energy Mater., 2020, 3, 1759–1767 CrossRef CAS
- C. Bommier and X. Ji, Small, 2018, 14, 1703576 CrossRef
- P. Peljo and H. H. Girault, Energy Environ. Sci., 2018, 11, 2306–2309 RSC
- Y. Li, W. Huang, Y. Li, A. Pei, D. T. Boyle and Y. Cui, Joule, 2018, 2, 2167–2177 CrossRef CAS
- G. G. Eshetu, T. Diemant, M. Hekmatfar, S. Grugeon, R. J. Behm, S. Laruelle, M. Armand and S. Passerini, Nano Energy, 2019, 55, 327–340 CrossRef CAS
- X. Sun, X. Zhang, Q. Ma, X. Guan, W. Wang and J. Luo, Angew. Chem., Int. Ed., 2020, 59, 6665–6674 CrossRef CAS
- R. Dugas, A. Ponrouch, G. Gachot, R. David, M. R. Palacin and J. M. Tarascon, J. Electrochem. Soc., 2016, 163, A2333–A2339 CrossRef CAS
- S. Wang, W. Cai, Z. Sun, F. Huang, Y. Jie, Y. Liu, Y. Chen, B. Peng, R. Cao, G. Zhang and S. Jiao, Chem. Commun., 2019, 55, 14375–14378 RSC
- Q. Shi, Y. Zhong, M. Wu, H. Wang and H. Wang, Angew. Chem., Int. Ed., 2018, 57, 9069–9072 CrossRef CAS
- H. Wang, C. Wang, E. Matios and W. Li, Angew. Chem., Int. Ed., 2018, 57, 7734–7737 CrossRef CAS
- W. Li, H. Yao, K. Yan, G. Zheng, Z. Liang, Y. M. Chiang and Y. Cui, Nat. Commun., 2015, 6, 7436 CrossRef CAS
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J.-G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS
- Y. Zhang, J. Qian, W. Xu, S. M. Russell, X. Chen, E. Nasybulin, P. Bhattacharya, M. H. Engelhard, D. Mei, R. Cao, F. Ding, A. V. Cresce, K. Xu and J.-G. Zhang, Nano Lett., 2014, 14, 6889–6896 CrossRef CAS
- F. Ding, W. Xu, X. Chen, J. Zhang, Y. Shao, M. H. Engelhard, Y. Zhang, T. A. Blake, G. L. Graff, X. Liu and J.-G. Zhang, J. Phys. Chem. C, 2014, 118, 4043–4049 CrossRef CAS
- L. Xiao, X. Chen, R. Cao, J. Qian, H. Xiang, J. Zheng, J.-G. Zhang and W. Xu, J. Power Sources, 2015, 293, 1062–1067 CrossRef CAS
- B. Yoon, S. Kim, Y. M. Lee and M.-H. Ryou, ACS Omega, 2019, 4, 11724–11727 CrossRef CAS
- Q. Xu, Y. Yang and H. Shao, Electrochim. Acta, 2016, 212, 758–766 CrossRef CAS
- J.-L. Ma, F.-L. Meng, Y. Yu, D.-P. Liu, J.-M. Yan, Y. Zhang, X.-B. Zhang and Q. Jiang, Nat. Chem., 2019, 11, 64–70 CrossRef CAS
- X. Chen, X. Shen, T.-Z. Hou, R. Zhang, H.-J. Peng and Q. Zhang, Chem, 2020, 6, 2242–2256 CAS
- Q. Zhang, Y. Lu, L. Miao, Q. Zhao, K. Xia, J. Liang, S. L. Chou and J. Chen, Angew. Chem., Int. Ed., 2018, 57, 14796–14800 CrossRef CAS
- F. Hoshyargar, J. Crawford and A. P. O'Mullane, J. Am. Chem. Soc., 2017, 139, 1464–1471 CrossRef CAS
- A. G. M. da Silva, T. S. Rodrigues, S. J. Haigh and P. H. C. Camargo, Chem. Commun., 2017, 53, 7135–7148 RSC
- S. W. Chee, S. F. Tan, Z. Baraissov, M. Bosman and U. Mirsaidov, Nat. Commun., 2017, 8, 1224 CrossRef
- M. H. Oh, T. Yu, S. H. Yu, B. Lim, K. T. Ko, M.-G. Willinger, D.-H. Seo, B. H. Kim, M. G. Cho, J.-H. Park, K. Kang, Y. E. Sung, N. Pinna and T. Hyeon, Science, 2013, 340, 964–968 CrossRef CAS
- X. Wang, S. Chen, G. Reggiano, S. Thota, Y. Wang, P. Kerns, S. L. Suib and J. Zhao, Chem. Commun., 2019, 55, 1249–1252 RSC
- X. Zheng, H. Fu, C. Hu, H. Xu, Y. Huang, J. Wen, H. Sun, W. Luo and Y. Huang, J. Phys. Chem. Lett., 2019, 10, 707–714 CrossRef CAS
- W. Fang, H. Jiang, Y. Zheng, H. Zheng, X. Liang, Y. Sun, C. Chen and H. Xiang, J. Power Sources, 2020, 455, 227956 CrossRef CAS
- J. Luo, C. Wang, H. Wang, X. Hu, E. Matios, X. Lu, W. Zhang, X. Tao and W. Li, Adv. Funct. Mater., 2019, 29, 1805946 CrossRef
- H. Wang, E. Matios, C. Wang, J. Luo, X. Lu, X. Hu, Y. Zhang and W. Li, J. Mater. Chem. A, 2019, 7, 23747–23755 RSC
- C. Wang, H. Xie, L. Zhang, Y. Gong, G. Pastel, J. Dai, B. Liu, E. D. Wachsman and L. Hu, Adv. Energy Mater., 2018, 8, 1701963 CrossRef
- M. Zhu, S. Li, B. Li, Y. Gong, Z. Du and S. Yang, Sci. Adv., 2019, 5, eaau6264 CrossRef CAS
- Z. Wang, X. Zhang, S. Zhou, K. Edström, M. Strømme and L. Nyholm, Adv. Funct. Mater., 2018, 28, 1804038 CrossRef
- S. Tang, Z. Qiu, X.-Y. Wang, Y. Gu, X.-G. Zhang, W.-W. Wang, J.-W. Yan, M.-S. Zheng, Q.-F. Dong and B.-W. Mao, Nano Energy, 2018, 48, 101–106 CrossRef CAS
- S. Tang, Y.-Y. Zhang, X.-G. Zhang, J.-T. Li, X.-Y. Wang, J.-W. Yan, D.-Y. Wu, M.-S. Zheng, Q.-F. Dong and B.-W. Mao, Adv. Mater., 2019, 31, 1807495 CrossRef
- T. Yang, T. Qian, Y. Sun, J. Zhong, F. Rosei and C. Yan, Nano Lett., 2019, 19, 7827–7835 CrossRef CAS
- L. Suo, Y. S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef
- Y. Yamada and A. Yamada, J. Electrochem. Soc., 2015, 162, A2406–A2423 CrossRef CAS
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS
- J. Wang, Y. Yamada, K. Sodeyama, C. H. Chiang, Y. Tateyama and A. Yamada, Nat. Commun., 2016, 7, 12032 CrossRef CAS
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Chem, 2018, 4, 174–185 CAS
- C. Geng, D. Buchholz, G. T. Kim, D. V. Carvalho, H. Zhang, L. G. Chagas and S. Passerini, Small Methods, 2018, 3, 1800208 CrossRef
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, J. Am. Chem. Soc., 2014, 136, 5039–5046 CrossRef CAS
- K. Sodeyama, Y. Yamada, K. Aikawa, A. Yamada and Y. Tateyama, J. Phys. Chem. C, 2014, 118, 14091–14097 CrossRef CAS
- J. Lee, Y. Lee, J. Lee, S. M. Lee, J.-H. Choi, H. Kim, M. S. Kwon, K. Kang, K. T. Lee and N. S. Choi, ACS Appl. Mater. Interfaces, 2017, 9, 3723–3732 CrossRef CAS
- L. Schafzahl, I. Hanzu, M. Wilkening and S. A. Freunberger, ChemSusChem, 2017, 10, 401–408 CrossRef CAS
- M. He, K. C. Lau, X. Ren, N. Xiao, W. D. McCulloch, L. A. Curtiss and Y. Wu, Angew. Chem., Int. Ed., 2016, 55, 15310–15314 CrossRef CAS
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS
- S. Chen, J. Zheng, D. Mei, K. S. Han, M. H. Engelhard, W. Zhao, W. Xu, J. Liu and J.-G. Zhang, Adv. Mater., 2018, 30, 1706102 CrossRef
- X. Ren, S. Chen, H. Lee, D. Mei, M. H. Engelhard, S. D. Burton, W. Zhao, J. Zheng, Q. Li, M. S. Ding, M. Schroeder, J. Alvarado, K. Xu, Y. S. Meng, J. Liu, J.-G. Zhang and W. Xu, Chem, 2018, 4, 1877–1892 CAS
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 CrossRef CAS
- X.-R. Chen, Y.-X. Yao, C. Yan, R. Zhang, X.-B. Cheng and Q. Zhang, Angew. Chem., Int. Ed., 2020, 59, 7743–7747 CrossRef CAS
- C. Monroe and J. Newman, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS
- Y. Zhao, L. V. Goncharova, A. Lushington, Q. Sun, H. Yadegari, B. Wang, W. Xiao, R. Li and X. Sun, Adv. Mater., 2017, 29, 1606663 CrossRef
- W. Luo, C.-F. Lin, O. Zhao, M. Noked, Y. Zhang, G. W. Rubloff and L. Hu, Adv. Energy Mater., 2017, 7, 1601526 CrossRef
- Y. Zhao, L. V. Goncharova, Q. Zhang, P. Kaghazchi, Q. Sun, A. Lushington, B. Wang, R. Li and X. Sun, Nano Lett., 2017, 17, 5653–5659 CrossRef CAS
- S. Choudhury, S. Wei, Y. Ozhabes, D. Gunceler, M. J. Zachman, Z. Tu, J. H. Shin, P. Nath, A. Agrawal, L. F. Kourkoutis, T. A. Arias and L. A. Archer, Nat. Commun., 2017, 8, 898 CrossRef
- M. Zhu, G. Wang, X. Liu, B. Guo, G. Xu, Z. Huang, M. Wu, H.-K. Liu, S.-X. Dou and C. Wu, Angew. Chem., Int. Ed., 2020, 59, 6596–6600 CrossRef CAS
- S. Wei, S. Choudhury, J. Xu, P. Nath, Z. Tu and L. A. Archer, Adv. Mater., 2017, 29, 1605512 CrossRef
- D. Zhang, B. Li, S. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 40265–40272 CrossRef CAS
- W. Zhou, Y. Li, S. Xin and J. B. Goodenough, ACS Cent. Sci., 2017, 3, 52–57 CrossRef CAS
- Y. Zhao, J. Liang, Q. Sun, L. V. Goncharova, J. Wang, C. Wang, K. R. Adair, X. Li, F. Zhao, Y. Sun, R. Li and X. Sun, J. Mater. Chem. A, 2019, 7, 4119–4125 RSC
- X. Liang, Q. Pang, I. R. Kochetkov, M. S. Sempere, H. Huang, X. Sun and L. F. Nazar, Nat. Energy, 2017, 2, 17119 CrossRef CAS
- Z. Tu, S. Choudhury, M. J. Zachman, S. Wei, K. Zhang, L. F. Kourkoutis and L. A. Archer, Nat. Energy, 2018, 3, 310–316 CrossRef CAS
- Q. Chen, H. He, Z. Hou, W. Zhuang, T. Zhang, Z. Sun and L. Huang, J. Mater. Chem. A, 2020, 8, 16232–16237 RSC
- X. Jin, Y. Zhao, Z. Shen, J. Pu, X. Xu, C. Zhong, S. Zhang, J. Li and H. Zhang, Energy Storage Mater., 2020, 31, 221–229 CrossRef
- H. Wang, C. Wang, E. Matios and W. Li, Nano Lett., 2017, 17, 6808–6815 CrossRef CAS
- H. Tian, Z. W. Seh, K. Yan, Z. Fu, P. Tang, Y. Lu, R. Zhang, D. Legut, Y. Cui and Q. Zhang, Adv. Energy Mater., 2017, 7, 1602528 CrossRef
- Y.-J. Kim, H. Lee, H. Noh, J. Lee, S. Kim, M.-H. Ryou, Y. M. Lee and H.-T. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 6000–6006 CrossRef CAS
- X. Xiang, K. Zhang and J. Chen, Adv. Mater., 2015, 27, 5343–5364 CrossRef CAS
- Y. Jiang, S. Yu, B. Wang, Y. Li, W. Sun, Y. Lu, M. Yan, B. Song and S. Dou, Adv. Funct. Mater., 2016, 26, 5315–5321 CrossRef CAS
- P.-F. Wang, H. Xin, T.-T. Zuo, Q. Li, X. Yang, Y.-X. Yin, X. Gao, X. Yu and Y. G. Guo, Angew. Chem., Int. Ed., 2018, 57, 8178–8183 CrossRef CAS
- Y. Dong, S. Li, K. Zhao, C. Han, W. Chen, B. Wang, L. Wang, B. Xu, Q. Wei, L. Zhang, X. Xu and L. Mai, Energy Environ. Sci., 2015, 8, 1267–1275 RSC
- H. Wang, Y. Xiao, C. Sun, C. Lai and X. Ai, RSC Adv., 2015, 5, 106519–106522 RSC
- S. Xin, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, Adv. Mater., 2014, 26, 1261–1265 CrossRef CAS
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS
- J. Fally, C. Lasne, Y. von Lazennec, Y. Le Cars and P. Margotin, J. Electrochem. Soc., 1973, 120, 1296–1298 CrossRef CAS
- Y.-X. Wang, B. Zhang, W. Lai, Y. Xu, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Energy Mater., 2017, 7, 1602829 CrossRef
- M. S. Syali, D. Kumar, K. Mishra and D. K. Kanchan, Energy Storage Mater., 2020, 31, 352–372 CrossRef
- Y. X. Ren, H. R. Jiang, T. S. Zhao, L. Zeng and C. Xiong, J. Power Sources, 2018, 396, 304–313 CrossRef CAS
- Y.-X. Wang, W.-H. Lai, S.-L. Chou, H.-K. Liu and S.-X. Dou, Adv. Mater., 2020, 32, 1903952 CrossRef CAS
- I. Kim, J.-Y. Park, C. Kim, J.-W. Park, J.-P. Ahn, J.-H. Ahn, K.-W. Kim and H.-J. Ahn, J. Electrochem. Soc., 2016, 163, A611–A616 CrossRef CAS
- T. Yim, M.-S. Park, J.-S. Yu, K. J. Kim, K. Y. Im, J.-H. Kim, G. Jeong, Y. N. Jo, S.-G. Woo, K. S. Kang, I. Lee and Y.-J. Kim, Electrochim. Acta, 2013, 107, 454–460 CrossRef CAS
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 11722 CrossRef CAS
- D. Kumar, Solid State Ionics, 2018, 318, 65–70 CrossRef CAS
- D. Zhou, Y. Chen, B. Li, H. Fan, F. Cheng, D. Shanmukaraj, T. Rojo, M. Armand and G. Wang, Angew. Chem., Int. Ed., 2018, 57, 10168–10172 CrossRef CAS
- J. Wu, J. Liu, Z. Lu, K. Lin, Y.-Q. Lyu, B. Li, F. Ciucci and J.-K. Kim, Energy Storage Mater., 2019, 23, 8–16 CrossRef
- V. Kumar, A. Y. S. Eng, Y. Wang, D.-T. Nguyen, M.-F. Ng and Z. W. Seh, Energy Storage Mater., 2020, 29, 1–8 CrossRef
- V. Kumar, Y. Wang, A. Y. S. Eng, M.-F. Ng and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100044 CrossRef
- P. Hartmann, C. L. Bender, M. Vracar, A. K. Durr, A. Garsuch, J. Janek and P. Adelhelm, Nat. Mater., 2013, 12, 228–232 CrossRef CAS
- C. L. Bender, P. Hartmann, M. Vračar, P. Adelhelm and J. Janek, Adv. Energy Mater., 2014, 4, 1301863 CrossRef
- Y. Hu, X. Han, Q. Zhao, J. Du, F. Cheng and J. Chen, J. Mater. Chem. A, 2015, 3, 3320–3324 RSC
- J.-H. Kang, W.-J. Kwak, D. Aurbach and Y.-K. Sun, J. Mater. Chem. A, 2017, 5, 20678–20686 RSC
- S. Zhang, Z. Wen, K. Rui, C. Shen, Y. Lu and J. Yang, J. Mater. Chem. A, 2015, 3, 2568–2571 RSC
- B. Lee, D.-H. Seo, H.-D. Lim, I. Park, K.-Y. Park, J. Kim and K. Kang, Chem. Mater., 2014, 26, 1048–1055 CrossRef CAS
- C. Xia, R. Black, R. Fernandes, B. Adams and L. F. Nazar, Nat. Chem., 2015, 7, 496–501 CrossRef CAS
- N. Ortiz Vitoriano, I. Ruiz de Larramendi, R. L. Sacci, I. Lozano, C. A. Bridges, O. Arcelus, M. Enterría, J. Carrasco, T. Rojo and G. M. Veith, Energy Storage Mater., 2020, 29, 235–245 CrossRef
- Y. Mao, H. Cheng, J. Xie, W. Zheng, G. Zhang and X. Zhao, Sustainable Energy Fuels, 2020, 4, 4108–4116 RSC
- S. Wu, Y. Qiao, K. Jiang, Y. He, S. Guo and H. Zhou, Adv. Funct. Mater., 2018, 28, 1706374 CrossRef
- X. Hu, J. Sun, Z. Li, Q. Zhao, C. Chen and J. Chen, Angew. Chem., Int. Ed., 2016, 55, 6482–6486 CrossRef CAS
- C. Fang, J. Luo, C. Jin, H. Yuan, O. Sheng, H. Huang, Y. Gan, Y. Xia, C. Liang, J. Zhang, W. Zhang and X. Tao, ACS Appl. Mater. Interfaces, 2018, 10, 17240–17248 CrossRef CAS
- L. Guo, B. Li, V. Thirumal and J. Song, Chem. Commun., 2019, 55, 7946–7949 RSC
- X. Hu, Z. Li, Y. Zhao, J. Sun, Q. Zhao, J. Wang, Z. Tao and J. Chen, Sci. Adv., 2017, 3, 1602396 CrossRef
- X. Wang, X. Zhang, Y. Lu, Z. Yan, Z. Tao, D. Jia and J. Chen, ChemElectroChem, 2018, 5, 3628–3632 CrossRef CAS
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |