
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the local structure of nanoscale actinide oxides: a comparison between PuO2 and ThO2 nanoparticles rules out PuO2+x hypothesis†
Laura
Bonato
 a,
Matthieu
Virot
a,
Thomas
Dumas
b,
Adel
Mesbah
a,
Elodie
Dalodière
a,
Oliver
Dieste Blanco
c,
Thierry
Wiss
c,
Xavier
Le Goff
a,
Michael
Odorico
a,
Damien
Prieur
d,
André
Rossberg
d,
Laurent
Venault
b,
Nicolas
Dacheux
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
a,
Matthieu
Virot
a,
Thomas
Dumas
b,
Adel
Mesbah
a,
Elodie
Dalodière
a,
Oliver
Dieste Blanco
c,
Thierry
Wiss
c,
Xavier
Le Goff
a,
Michael
Odorico
a,
Damien
Prieur
d,
André
Rossberg
d,
Laurent
Venault
b,
Nicolas
Dacheux
a,
Philippe
Moisy
b and
Sergey I.
Nikitenko
a
aICSM, Univ Montpellier, CEA, CNRS, ENSCM, Marcoule, France. E-mail: matthieu.virot@cea.fr
bCEA, DEN, DMRC, Univ Montpellier, Marcoule, France
cEuropean Commission, Joint Research Centre (JRC), Institute for Transuranium Elements (ITU), Postfach 2340, 76125 Karlsruhe, Germany
dHelmholtz-Zentrum Dresden - Rossendorf, Institute of Resource Ecology, Bautzner Landstraße 400, 01328 Dresden, Germany
First published on 27th November 2019
Abstract
Actinide research at the nanoscale is gaining fundamental interest due to environmental and industrial issues. The knowledge of the local structure and speciation of actinide nanoparticles, which possibly exhibit specific physico-chemical properties in comparison to bulk materials, would help in a better and reliable description of their behaviour and reactivity. Herein, the synthesis and relevant characterization of PuO2 and ThO2 nanoparticles displayed as dispersed colloids, nanopowders, or nanostructured oxide powders allow to establish a clear relationship between the size of the nanocrystals constituting these oxides and their corresponding An(IV) local structure investigated by EXAFS spectroscopy. Particularly, the first oxygen shell of the probed An(IV) evidences an analogous behaviour for both Pu and Th oxides. This observation suggests that the often observed and controversial splitting of the Pu–O shell on the Fourier transformed EXAFS signal of the PuO2 samples is attributed to a local structural disorder driven by a nanoparticle surface effect rather than to the presence of PuO2+x species.
1. Introduction
Nanostructured materials can be defined as solid samples with at least one characteristic structural length ranging between 1 and 10 nm. They have attracted considerable interest in recent interdisciplinary research for technological applications related to the nanometric size of their constituent building blocks (e.g., crystalline or atomic and molecular groups).1–3 Indeed, the controlled microstructure of nanoscale materials and nanoparticles at the atomic level offers new physical and chemical properties in comparison to similar bulk materials already applied in, for instance, catalysis, synthesis of luminescent materials, and solar cells.3–6 Such effects are often attributed to the higher surface-to-volume ratio of the nanoparticles that constitute these samples, which increases the number of surface and interface atoms generating stress, strain, and structural perturbations.1,2 In actinide chemistry, the published data dealing with the synthesis or characterization of nanoparticles and nanomaterials are still scarce but are of growing interest due to the reported contribution of actinide nanoparticles in the environment (e.g., migration of actinides) and in industrial processes (e.g., high burn-up structures).7–10 We recently observed nanostructured PuO2 powder prepared by the oxalate route and noticed its outstanding reactivity in pure water under ultrasound irradiation, which stirred up our curiosity concerning the local environment of this oxide at the nanoscale.11PuO2, which was thought to be the most stable Pu oxide phase, was dismissed in 2000 after the observation of an over-stoichiometric oxide when exposed to moist air from 25 to 350 °C. The accompanying hydrogen gas release involved tremendous discussions and concerns about industrial operations and safety for storage.7,12,13 The resulting possible formation of PuO2+x caused controversy and was debated by several authors using a variety of techniques such as thermodynamic calculations, modelling simulations, Raman spectroscopy, X-ray absorption or photoemission spectroscopies.14–21 Particularly, the possible formation of this oxide was supported by the observation in the EXAFS spectrum of a strong distortion and splitting of the first Pu–O shell, which was attributed to trans-dioxo bonds and related to the presence of Pu(V) or Pu(VI) species.17,18,21 Although considered to be “simple” from a crystallographic point of view, the description of PuO2 (and more generally, AnO2 with An = actinides) using EXAFS spectroscopy appears, nevertheless, to be questionable and lacks a standardized procedure for data treatment and comparison. EXAFS data are indeed known to be highly subjective to misinterpretations or artefact generation related to signal post-treatments such as background subtraction, Fourier transform, or curve fitting.22 Furthermore, local geometry and electronic structure effects driven by nanomaterials in comparison to bulk systems have been reported to generate significant spectral features in XANES and EXAFS spectroscopic studies.23–26
The number of sub-shells used to fit, for instance, the first An–O coordination sphere in PuO2 has been found to significantly differ as a function of the studies. Conradson and co-workers reported the use of up to eight Pu–O sub-shell models to explain the experimental EXAFS spectra obtained for ca. 20 PuO2 samples. Such models well reproduced the experimental data but remain difficult to interpret owing to the very high number of varying structural parameters.17,18,21 Moreover, this fit approach is only statistically relevant when assuming constrained or fixed values for several metrical parameters including the energy threshold ΔE0, Debye–Waller factor (DWF), and coordination numbers. Recent single Pu–O shell models allowed acceptable fitting of the experimental EXAFS spectra only for highly fired bulk PuO2 materials. The fits obtained with PuO2 nanoparticles resulted in unrealistically high DWF for the first oxygen shell. As suggested by most of the authors, this high DWF evidences a high degree of local disorder that also gives rise to large errors in the fitted structural parameters.27,28 The use of a reduced number of oxygen sub-shells appears as a good compromise to reproduce the experimental data in both PuO2 bulk materials and nanomaterials with justified and reasonable fitted parameters.11,29–31
The present study focuses on the local structure characterization of PuO2 nanoparticles exhibiting different sizes. In this paper, plutonium is compared to thorium due to the similar properties of both the elements to exist in the (+IV) oxidation state, to crystallize as oxides in the same space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m of the fluorite crystal system and to exhibit close ionic radii (1.05 Å for Th, against 0.96 Å for Pu, with both in 8-fold coordination).32 The reported EXAFS spectra for ThO2 samples already show similar Fourier transforms compared to the An–O and An–An coordination spheres.30,33–35 In addition, Th only exists in the (+IV) oxidation state and therefore, allows the comparison of Pu compounds so as to avoid misinterpretations related to the potential contribution of other oxidation states in the crystalline structure. This paper first reports the preparation and the characterization of AnO2 nanoparticles (An = Pu or Th) in the form of suspended colloids, nanopowders, and nanostructured oxides. Then, relevant characterization of the local structure of a selection of AnO2 nanoparticles by EXAFS spectroscopy is discussed. A clear relationship between the shrinking size of the nanocrystals that constitute the actinide oxides and their related structural defects is established.
m of the fluorite crystal system and to exhibit close ionic radii (1.05 Å for Th, against 0.96 Å for Pu, with both in 8-fold coordination).32 The reported EXAFS spectra for ThO2 samples already show similar Fourier transforms compared to the An–O and An–An coordination spheres.30,33–35 In addition, Th only exists in the (+IV) oxidation state and therefore, allows the comparison of Pu compounds so as to avoid misinterpretations related to the potential contribution of other oxidation states in the crystalline structure. This paper first reports the preparation and the characterization of AnO2 nanoparticles (An = Pu or Th) in the form of suspended colloids, nanopowders, and nanostructured oxides. Then, relevant characterization of the local structure of a selection of AnO2 nanoparticles by EXAFS spectroscopy is discussed. A clear relationship between the shrinking size of the nanocrystals that constitute the actinide oxides and their related structural defects is established.
2. Experimental section
Caution! Th and Pu are α-emitting radionuclides; standard precautions should be followed for handling these radioactive materials.2.1. Reagents and preparation of the oxides
All of the used commercial reagents were of analytical grade and purchased from Sigma-Aldrich. Aqueous solutions were prepared using deionized water having a resistivity higher than 18 MΩ cm at 25 °C. Experiments with 232Th were carried out in the ICSM facility (Marcoule, France) using thorium nitrate pentahydrate (Sigma-Aldrich) as the starting material. Experiments with Pu were performed in the Atalante facility (Marcoule, France) in a dedicated glove box. Pu solution was previously purified through an anion exchange resin in agreement with the previous reports and used as Pu(IV) solution in ca. 1.5 M HNO3.11,36,37Nanostructured ThO2 and PuO2 were prepared by thermal conversion of Th(IV) and Pu(IV) or Pu(III) oxalate precursors, which were precipitated in nitric acid medium by pouring An solutions into continuously stirred oxalic acid solution (in excess), as already reported in the literature.36,38,39 Regarding the Pu(III) oxalate precursors, the reduction of Pu(IV) solution was previously carried out with a slightly over-stoichiometric amount of hydroxylammonium nitrate in the presence of 0.05 M hydrazinium nitrate at ca. 40 °C. One hour after mixing the solutions, An(IV) and An(III) oxalate precipitates were separated from the supernatants by centrifugation, washed several times with water, and dried at room temperature under vacuum (Th) or under air atmosphere of the glove box (Pu). ThO2 and PuO2 nanopowders (n-PWD) were prepared by adding 30 wt% ammonia solution into continuously stirred An(IV) acid solutions in the presence of polyethylene glycol (PEG, M = 3000, 2.5 wt%), in agreement with the previous report.40 The precipitates were then heated under air atmosphere with a constant rate of 10 °C min−1 up to 120 °C (2 °C min−1 for ThO2 at 485 °C) for 1 h. An equivalent heating rate was then used to reach the selected firing temperature, which was maintained for 1 to 17 h depending on the sample. All the samples and respective firing conditions are summarized in Table 1 for the various samples studied in this work. AnO2 samples were also compared to stable Pu(IV) colloidal suspension prepared by controlled hydrolysis in agreement with our previous report, which particularly showed that Pu(IV) intrinsic colloids are composed of stabilized PuO2 nanoparticles of about 2.9 nm diameter.11 Diffuse reflectance spectroscopic measurements confirmed the presence of similar absorption bands for both PuO2 and Pu(IV) intrinsic colloids (Fig. S1, ESI†).
| Sample | Name firing temperature (duration) | Preparation route |
|---|---|---|
| a n-PWD: nanopowder; RT: room temperature. b Preparation conditions fixed on the basis of our previous investigations.11 | ||
| ThO2 | ThO2 485 °C (4 h) | Th(IV) oxalate |
| ThO2 485 °C (12 h) | Th(IV) oxalate | |
| ThO2 600 °C (2 h) | Th(IV) oxalate | |
| ThO2 1000 °C (2 h) | Th(IV) oxalate | |
| ThO2 n-PWD 485 °C (2 h) | PEG/ammonia | |
| PuO2 | PuO2 485 °C (2 h) | Pu(IV) oxalate |
| PuO2 660 °C (2 h) | Pu(IV) oxalate | |
| PuO2 660 °C (17 h) | Pu(IV) oxalate | |
| PuO2 1200 °C (1 h) | Pu(IV) oxalate | |
| PuO2 n-PWD 485 °C (2 h) | PEG/ammonia | |
| PuO2[III] 485 °C (2 h) | Pu(III) oxalate | |
| PuO2[III] 485 °C (12 h) | Pu(III) oxalate | |
| PuO2[III] 660 °C (2 h) | Pu(III) oxalate | |
| Pu intrinsic colloid | Pu(IV) colloid (RT) | Controlled hydrolysisb |
2.2. Raman micro-spectroscopy
μ-Raman spectra were acquired using a Horiba – Jobin Yvon Aramis apparatus calibrated with a Si wafer at 520.7 cm−1 and equipped with an edge filter and Nd:YAG (532 nm) or He–Ne (633 nm) laser. An Olympus BX41 microscope was used to focus the laser beam on the sample previously deposited on a glass slide.2.3. X-ray diffraction
Powder X-ray diffractograms (PXRD) were collected at room temperature on pure silicon (ThO2) or inox (PuO2) sample holders using Bruker D8 Advance X-ray diffractometers, both equipped with a linear Lynx-eye detector (Cu Kα1,2 radiation, λ = 1.54184 Å). PuO2 samples were immobilized in an epoxy resin to prevent any dispersal of harmful radioactive dust. In this latter case, the addition of gold as an internal standard was also carried out (ICSD 03-065-2870). LaB6 measurements were used to extract the instrumental functions. The collected data related to ThO2 and PuO2 crystallization in the fluorite type structure (CaF2) in the space group Fmm of the cubic system were refined by the Rietveld method using the Fullprof_suite package.41 During the treatments, the following profile and structural parameters were refined: zero shift, unit-cell parameters, scale factor, and overall displacement factor. Moreover, anisotropic size and micro-strain models by considering the mm Laue class were applied for ThO2 in order to evaluate the broadening effect. In the case of PuO2, only a size model was applied because of the difficulties to correctly estimate the background because of the used epoxy resin. Fig. S2, ESI† illustrates the method by providing experimental and calculated patterns for a refined ThO2 sample.
2.4. Scanning and transmission electron microscopy
The oxide morphologies were investigated by Scanning Electron Microscopy (SEM) using a FEI Quanta 200 ESEM FEG microscope and a Zeiss Supra 55 device for ThO2 and PuO2, respectively. The samples were deposited on carbon tapes without additional preparation except for PuO2 samples, which required gold coating. SEM characterization images are provided in Fig. S3–S5 (ESI†) for a selection of the synthesized powders. The nanoscale characterization of some oxides was carried out with High-Resolution Transmission Electron Microscopy (HR-TEM) after deposition of a droplet of oxide suspension on carbon coated copper grids. The oxide suspension was previously prepared in pure water and sonicated in an ultrasonic bath for 5 min at 25 kHz. ThO2 samples were either studied with a JEOL JEM-3010 (300 kV, CINAM Marseille, France) or a JEOL JEM-2200FS (200 kV, Montpellier, France). The PuO2 samples were characterized at the Joint Research Centre of the Institute for Transuranium Elements, Karlsruhe, Germany (JRC-ITU) using a TecnaiG2 (FEI™) 200 kV microscope equipped with a field emission gun and modified during its assembly to enable the examination of radioactive samples. The particle size was estimated from the HR-TEM images using Image J software with a high number of measurements to improve the statistics (n > 50).2.5. Atomic force microscopy
The morphology of the ThO2 samples was studied by Atomic Force Microscopy using a MULTIMODE 8 AFM apparatus equipped with a Nanoscope 5 controller from Bruker (Germany). Sample aliquots were dispersed in pure water before a droplet was deposited onto a mica or carbon disk. The samples were imaged in the peak force tapping mode with SNL tips (K = 0.12 N nm−1, f0 = 23 kHz from Bruker) and the applied force was set at about 300 pN.2.6. X-ray absorption spectroscopy
5 to 7 mg of the powdered compounds were finely ground and mechanically mixed with ca. 200 mg boron nitride. EXAFS measurements were performed at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on HZDR-operated Rossendorf Beamline (BM20) using a Si(111) double-crystal monochromator. The experimental spectra were recorded in the fluorescence mode at Th LIII and Pu LIII edges using a 13-element Ge detector at room temperature and pressure. The fit was obtained in the 2 Å−1 < k < 14 Å−1 range (Kaiser–Bessel window, cluster size 1–5 Å).42 The EXAFS data were analysed using Athena and Arthemis software from the IFEFFIT package.43 The theoretical scattering paths for An–O and An–An shells were obtained using the FEFF8.4 program.44In order to avoid artefact and misinterpretation generation, and to rigorously and consistently compare the fitted metrical parameters, the data treatment procedure was standardized for all of the studied samples. The structural parameters were obtained by separately considering the first and second coordination spheres. Two background removal approaches were used as function of the considered shell. For the An–O shells, normalization and background subtraction were performed on the normalized absorption spectra truncated at k = 10 Å−1. After Fourier transformation of the obtained k3-weighted EXAFS signal in the 2.5 Å−1 to 10 Å−1k-range (Kaiser–Bessel window), a back Fourier transform was applied to isolate the first An–O shell selected between R–ϕ = 1.1 Å and R–ϕ = 2.5 Å. The extracted k3-weighted first shell oscillations were then fitted by a single O-shell. The only floating parameter was the DWF while the coordination number (CN = 8) and the An–O distances (2.41 Å or 2.33 Å for Th and Pu, respectively) were fixed in agreement with the ideal fluorite structure type. The An–An shell fits were performed on the Fourier transform of the k3-weighted EXAFS oscillations in the 2 Å−1 < k < 14 Å−1 range where DWF, CN, and RAn–An were considered as floating parameters.
3. Results and discussion
3.1. Characterization of oxide nanoparticles
PXRD patterns presented in Fig. 1 confirm that the studied samples crystallize in the fluorite-type structure (space group Fmm) typical of ThO2 (ICSD 01-071-6407) and PuO2 (ICSD 01-073-7903). More intense and sharpened diffraction peaks could be noticed on increasing the firing temperatures and heating durations. Such observations are generally attributed to crystallite growth, elimination of amorphous domains and crystal defects, and to porosity closure.39,45–47 The broad XRD peaks observed for ThO2 and PuO2 powdered samples prepared at the lowest temperatures agree well with the presence of nanometric coherent domains (referred to as crystallites).45,46,48 The selected electron diffraction patterns obtained with HR-TEM (Fig. 2) confirm that the expected fluorite structure for all of the studied samples are in agreement with the PXRD observations.
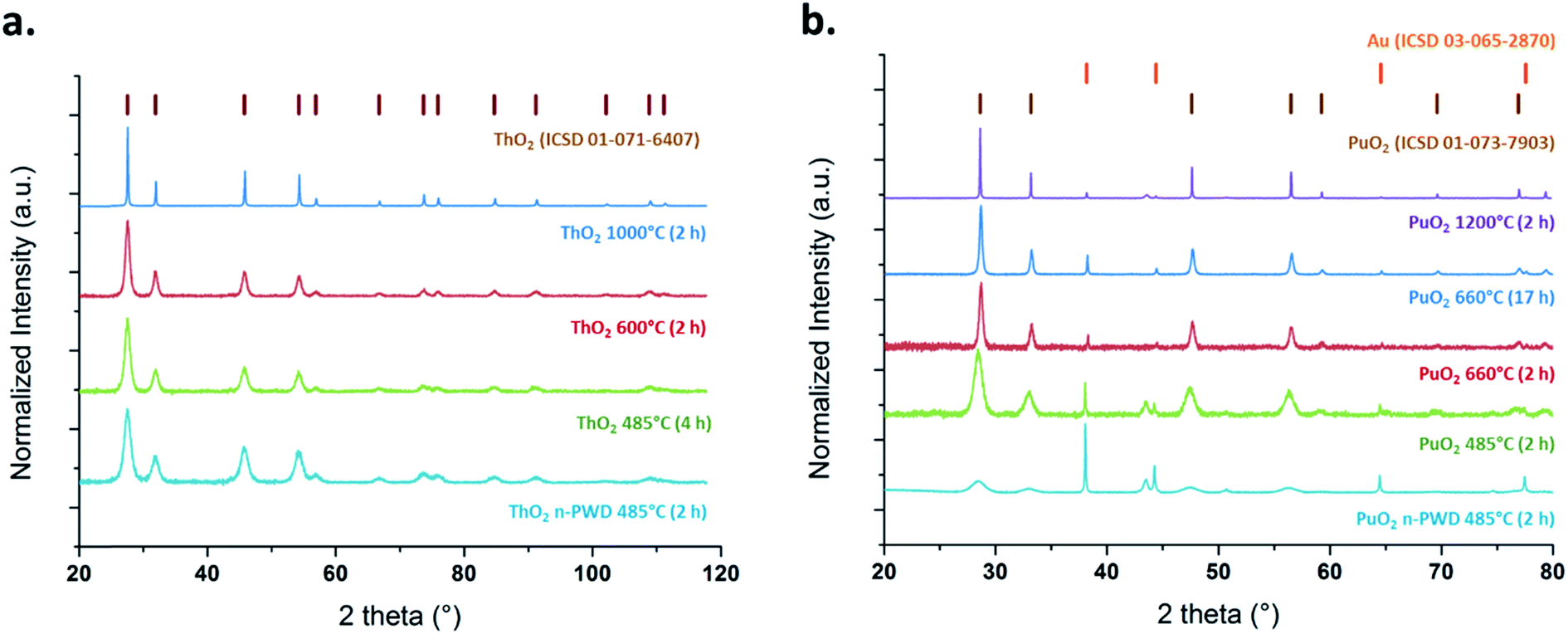 | ||
| Fig. 1 Background-corrected PXRD patterns of ThO2 (a) and PuO2 (b) samples. | ||
 | ||
| Fig. 2 HR-TEM images and corresponding electron diffraction patterns (insets) for ThO2 (left) and PuO2 (right) samples. | ||
Rietveld refinements allowed the determination of corresponding crystallite sizes and lattice parameters, which were found to be in good agreement with the nanoparticle size obtained by direct measurements from the HR-TEM images (Table 2). The HR-TEM images show that the actinide oxide powders synthesized in basic conditions (n-PWD) are composed of agglomerates of well-defined nanoparticles that are found to be spherical, monodispersed, and crystalline. Statistical measurements made on the images gave an average nanoparticle size of ca. 7.7 ± 1.4 nm for ThO2 and 4.6 ± 1.0 nm for PuO2 (Table 2). The oxides prepared by the thermal conversion of oxalate precursors at relatively low temperature (485–600 °C) show a nanostructured architecture, which disappears due to large grains that appear on increasing the heating temperature. Particularly, the oxides obtained at 485 °C exhibit micrometric squared tablets that are also formed with monodispersed, spherical, and crystalline nanoparticles. The average nanoparticle size is about 7.6 ± 2.1 nm and 9.8 ± 2.5 nm for the powdered ThO2 samples prepared at 485 °C (4 h) and 600 °C (2 h), respectively. The powdered PuO2 samples follow almost the same trend sizes of 5.2 ± 1.1 nm and 12.2 ± 3.3 nm when heated at 485 °C (2 h) and 660 °C (2 h), respectively (Table 2). It is worth noting that the PuO2 samples obtained from Pu(III) oxalates exhibit a similar nanostructure. The nanoscale organization observed at 485 °C and its loss (coalescence of nanodomains) at 600 °C is clearly illustrated in Fig. 2 for this sample (particle sizes of 7.3 ± 1.9 nm and 16.0 ± 4.5 nm, respectively).
| Sample | Particle size from HR-TEM (nm) | Particle size from XRD (nm) | Lattice parameter (Å) |
|---|---|---|---|
| a Arbitrary value taken from the literature and obtained for conversion of cerium and plutonium oxalate precursors at >1000 °C;47,49,50 the reported lattice parameters are 5.396–5.3975 Å for bulk PuO2 (ref. 12, 17, 20 and 51) and 5.592–5.597 Å for bulk ThO2 (ref. 33, 39, 46, 52 and 53). | |||
| ThO2 n-PWD 485 °C (2 h) | 7.7 ± 1.4 | 5.7 ± 0.1 | 5.6009(3) |
| ThO2 485 °C (4 h) | 7.6 ± 2.1 | 6.4 ± 0.7 | 5.6016(3) |
| ThO2 485 °C (12 h) | 3.8 ± 0.9 | 8.0 ± 0.4 | 5.6025(2) |
| ThO2 600 °C (2 h) | 9.8 ± 2.5 | 8.7 ± 0.9 | 5.5993(2) |
| ThO2 1000 °C (2 h) | 66.6 ± 27.1 | 63.6 ± 1.5 | 5.5963(4) |
| Pu(IV) colloid | 2.9 ± 0.4 | — | — |
| PuO2 n-PWD 485 °C (2 h) | 4.6 ± 1.0 | 5.1 ± 0.1 | 5.4014(3) |
| PuO2 485 °C (2 h) | 5.2 ± 1.1 | 7.6 ± 0.3 | 5.3980(2) |
| PuO2 660 °C (2 h) | 12.2 ± 3.3 | 17.0 ± 1.2 | 5.4021(1) |
| PuO2 660 °C (17 h) | — | 25.4 ± 1.7 | 5.3970(1) |
| PuO2 1200 °C (1 h) | 200.0a | 193.0 ± 12.4 | 5.3938(1) |
| PuO2[III] 485 °C (2 h) | 6.2 ± 1.3 | 7.4 ± 0.4 | 5.3979(2) |
| PuO2[III] 485 °C (12 h) | 7.3 ± 1.9 | 7.9 ± 0.2 | 5.3969(1) |
| PuO2[III] 660 °C (2 h) | 16.0 ± 4.5 | 14.3 ± 1.1 | 5.4005(1) |
The data displayed in Fig. S6 (ESI†) confirm the good correlation between the small and larger values of particle size determined by both HR-TEM and XRD. The distinction between the terms “nanoparticle” or “nanocrystal” is not discussed in this study and we assume that both terms can be equally used based on the analytical characterization developed in this study (Table 2). The Rietveld refinement, however, offers a more standardized procedure by avoiding the contribution of “human eye” errors on the measurements and improves the statistics by taking the whole diffracted signal as a source of data (measurements of HR-TEM images can only be made on well-defined and contrasted particles). The discrepancy noted for the particle size of ThO2 at 485 °C (12 h) is attributed to such kind of mistakes.
A slightly increasing trend of the lattice parameter is generally observed when decreasing the size of the PuO2 and ThO2 nanocrystals that compose the samples (in comparison to the reported bulk PuO2 and ThO2, Table 2). Such a phenomenon has been already attributed to negative surface stress due to coordination and bond differences at the surface of the materials.54 The resulting unit cell volume may compress or expand as a function of the material studied.25,35,54 For instance, an increase in the lattice parameter has been observed for CeO2 nanoparticles smaller than 20 nm.55,56 The lattice parameter of a0 = 5.600(6) Å has been reported for 14 nm ThO2 particles;52 Bouexiere et al. reported the lattice parameter a0 of 5.4042(2) Å for 3.7 ± 1.0 nm PuO2 nanoparticles prepared by hydrothermal decomposition of oxalates.45 Popa et al. reported the unit cell parameter values ranging from 5.611(1) to 5.613(1) Å for 6.1 ± 0.7 to 7.1 ± 0.9 nm sized ThO2 nanoparticles, respectively, and 5.397(1) for 3.7 ± 1.0 nm PuO2 particles.48 Recently, the expansion of the lattice parameter has been observed for ThO2 nanoparticles that exhibit different sizes. Carbonate and hydroxyl surface groups have been suggested to exhibit a tensile effect on the crystalline lattice.35
Generally, the lower nanoparticle sizes observed for PuO2 samples compared to ThO2 when using similar firing conditions can be attributed to the different mechanisms and temperatures related to the decomposition of oxalate precursors for both the oxides.52,57 The nanostructure of the oxides obtained by the oxalate route appears to be controlled by the firing temperature and its duration, which involves a loss of nanoscale organization when they both increase. The duration of the heating treatment at the given temperature appears, nevertheless, to be less significant, as evidenced by the nanoparticle sizes measured, for instance, for PuO2 samples obtained from the thermal conversion of Pu(III) oxalate at 485 °C for 2 h (6.2 ± 1.3 nm) and 12 h (7.3 ± 1.9 nm). Note that a slow nanograin growth has been reported for UO2 and ThO2 prepared by the oxalate route below 700 °C.52 The differences in the crystallite growth rates have also been reported for AnO2 powders (An = Th, Np, and Pu).45,48 Finally, the nanoscale organization and morphology observed at low firing conditions for oxalate compounds has been already reported to occur with the oxalate route for PuO2, UO2, and ThO2 samples.11,52
The selected AFM images shown in Fig. 3 confirm the nanoscale architecture obtained for the ThO2 samples prepared at low-temperature. Interestingly, these images clearly demonstrate the absence of nanostructure for the oxalate precursors, thus confirming that such an organization is mainly related to the firing conditions (50–100 nm grains are observed on the oxalate precursor). Note that attempts related to the HR-TEM characterization of oxalates failed due to its degradation under the electron beam. The Raman spectra of the ThO2 samples allowed to observe a red shift and enlargement in the T2g band located at 465 cm−1, which is correlated with the decrease in the annealing temperature and duration (Fig. S7, ESI†). Such an observation is attributed to phonon confinement in agreement with the literature, which can be considered as a fingerprint for nanocrystals and related nanostructured materials.1,48,52,58 These Raman spectral features have already been reported by several authors who have studied ThO2 nanocrystals.48,52,59,60 The enlargement of the FWHM parameter was directly related to the decreasing size of the nanocrystals that constitute the oxide, whereas the red shift was attributed to volume expansion, grain size distribution, presence of defects, Grüneisen parameter, etc.52
 | ||
| Fig. 3 AFM images emphasizing the nanoscale architecture of ThO2 samples, which is lost on increasing the firing temperature. The absence of nanostructure is evident for Th oxalate precursors, confirming their temperature-formation dependency. | ||
3.2. Probing the local structure
X-ray Absorption Near-Edge Structure (XANES) spectra acquired for a selection of ThO2 and PuO2 samples exhibit a white line at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 307.5 ± 0.5 eV and 18067.6 ± 0.5 eV, respectively (Fig. S8, ESI†). XANES has been reported to be sensitive to the chemical state and the local structure of a photoabsorbing atom, which can be different from the core to the bulk of a surface.23 Similarly, the shape of a particle (and the related bond distances, angles, and symmetry) may cause modifications in the XANES spectra.23,25 Nevertheless, the absence of significant variation of the white line position unequivocally indicates the predominance of the (+IV) oxidation state for all of our studied samples (Fig. S8, ESI†).28 A small decrease in the white line intensity (and shape of the post edge region) can, however, be noticed for PuO2 and ThO2 nanopowders, suggesting a possible variation of the local symmetry around some absorbing Th and Pu atoms for the smallest particles. A similar nanoparticle effect on the white line intensity has already been observed for PuO2 nanocrystals by Hudry et al.28
307.5 ± 0.5 eV and 18067.6 ± 0.5 eV, respectively (Fig. S8, ESI†). XANES has been reported to be sensitive to the chemical state and the local structure of a photoabsorbing atom, which can be different from the core to the bulk of a surface.23 Similarly, the shape of a particle (and the related bond distances, angles, and symmetry) may cause modifications in the XANES spectra.23,25 Nevertheless, the absence of significant variation of the white line position unequivocally indicates the predominance of the (+IV) oxidation state for all of our studied samples (Fig. S8, ESI†).28 A small decrease in the white line intensity (and shape of the post edge region) can, however, be noticed for PuO2 and ThO2 nanopowders, suggesting a possible variation of the local symmetry around some absorbing Th and Pu atoms for the smallest particles. A similar nanoparticle effect on the white line intensity has already been observed for PuO2 nanocrystals by Hudry et al.28
The experimental k3-weighted Extended X-ray Absorption Fine Structure (EXAFS) spectra are given in Fig. S9 (ESI†) in the interval 2 Å−1 < k < 14 Å−1 for the selected ThO2 and PuO2 samples. Whatever the nature of the oxide, these functions can be visually decomposed into lower frequency oscillations that dominate the signal between 2 and 10 Å−1 characteristic from the An–O shell and higher frequency oscillations becoming predominant after k = 10 Å−1 that are assigned to An–An interactions. In agreement with these observations, two peaks (uncorrected for the phase shift) centred at R–ϕ = 1.93 Å and 3.80 Å for ThO2 and R–ϕ = 1.84 Å and 3.71 Å for PuO2 were observed after Fourier transformation of the k3-weighted signal (Fig. 4a and b for ThO2 and PuO2, respectively). These two peaks are respectively assigned to the An–O and An–An coordination spheres in the AnO2 fluorite crystal system.11,16–18,27,33,35
 | ||
| Fig. 4 Fourier transform of the k3-weighted EXAFS spectra for a selection of ThO2 (a) and PuO2 (b) samples. | ||
A strong decrease in the intensity of both An–O and An–An shell peaks is noticed on decreasing the firing temperature for both ThO2 and PuO2 samples. This modification in the local actinide structure can be ascribed to the decrease in crystallinity of the sample, which affects the local order in bulk materials (increase of DWF), or to the particle size variation, which significantly affects the coordination number in the nanometer range.25,61 Note that particle size effect is clearly noticed in the long range order from R–ϕ = 6 to 10 Å, where distant coordination shells are highly pronounced for high fired ThO2 and PuO2, whereas they become less defined on decreasing the particle size of the sample. XANES and EXAFS spectroscopic studies are known to be sensitive to size effects and the reduced number of neighbouring atoms at the surface of a particle in comparison to the bulk, which becomes significant for the smallest particles.23,26,35,62–64 As an instance, previous calculations showed that ca. 27% of Pu atoms are present at the surface of a PuO2 particle of 2.9 nm in diameter.11 Amidani et al. reported that a model ThO2 particle of 2.1 nm exhibits 68% of the total cations at its surface.23 Whatever the considered actinide oxide, these observations are accompanied with a striking distortion and splitting of the An–O shell correlated with the decreasing annealing temperature (Fig. S10 in ESI† provides a magnification of the An–O shells). This feature is emphasized for the samples composed with the smallest particles, particularly for both Th and Pu nanopowders and for Pu(IV) intrinsic colloids.
Such a phenomenon has been already reported for PuO2 samples and previously attributed to the contribution of several Pu–O distances in the oxide.7,11,17,18,21 More precisely, Pu–O shell splitting has been tentatively explained by the contribution of short oxo bonds found, for instance, in molecular Pu(V) or Pu(VI) ions, suggesting the formation of PuO2+x. The similar features observed for the ThO2 samples in this current study cannot be attributed to such an effect since Th only exists at the (+IV) oxidation state. Thus, the analogous distortion and splitting of both Pu–O and Th–O shells support another explanation. In our previous study focused on the synthesis and characterization of Pu(IV) intrinsic colloids, we correlated the Pu–O shell distortion to the size of the PuO2 nanoparticles constituting the colloidal suspensions. A stronger alteration of the EXAFS spectra was indeed observed with the shrinking size of the particles, which are known to exhibit higher atomic surface-to-volume ratio. The rising concentration of the uncoordinated atoms, hydrolysed moieties, and μ3-oxo bonds (measuring ca. 2.2 Å) on the surface of a size-decreasing oxide may indeed be more sensitive to EXAFS spectroscopy. At first glance, this hypothesis agrees with the observations reported in this current work for the various Th and Pu nanoparticles in the form of colloids, nanopowders, or “more conventional” nanostructured oxides.
In order to avoid misinterpretations related to EXAFS signal reading and improve the fit quality, the An–O coordination shells were fitted separately and the data treatments were standardized to treat all the data in a similar way (see experimental section). Such a procedure allowed the investigation of the Debye–Waller factor (DWF) variation for the first oxygen shell, whereas the corresponding coordination number and distance parameters were fixed, in agreement with the ideal fluorite-type Fmm structure (CN = 8 and An–O = 2.41 Å (Th) or 2.33 Å (Pu)).27 The resulting DWF variations represent the relative local structural disorder from a material merging in a single parameter, the variations in crystalline domains and shapes, lattice defects, and environment of nanomaterials (all known to affect the DWF parameter).61 By fixing the coordination number and An–O distance in our fitting procedure, the DWF parameter somehow indicates how high the fitted local structural disorder has to be for the An–O shell to fit with the ideal AnO2 structure. In contrast, the extracted structural parameters for the An–An coordination shell offer complementary information, determined by fitting the long range EXAFS spectra. Table 3 gathers the structural parameters calculated according to this procedure for the two coordination shells of both the actinide oxides.
| Sample | O-shell DWF (10−3 Å2) | An–An shell CN | An–An shell DWF (10−3 Å2) | An–An distance (Å) |
|---|---|---|---|---|
| ThO2 n-PWD 485 °C (2 h) | 9.5 ± 1.1 | 6.3 ± 1.9 | 6.2 ± 0.4 | 3.97 ± 0.02 |
| ThO2 485 °C (4 h) | 6.9 ± 1.1 | 7.8 ± 1.3 | 5.3 ± 0.3 | 3.96 ± 0.01 |
| ThO2 485 °C (12 h) | 6.4 ± 1.1 | 8.8 ± 1.4 | 4.9 ± 0.3 | 3.97 ± 0.01 |
| ThO2 600 °C (2 h) | 6.1 ± 1.1 | 8.9 ± 1.6 | 4.9 ± 0.3 | 3.97 ± 0.01 |
| ThO2 1000 °C (2 h) | 4.9 ± 1.1 | 12.4 ± 1.2 | 3.9 ± 0.4 | 3.97 ± 0.01 |
| Pu(IV) colloid | 12.1 ± 0.9 | 5.6 ± 2.1 | 5.6 ± 1.6 | 3.80 ± 0.01 |
| PuO2 n-PWD 485 °C (2 h) | 8.9 ± 1.0 | 9.5 ± 1.3 | 4.9 ± 0.7 | 3.80 ± 0.02 |
| PuO2 485 °C (2 h) | 7.0 ± 1.1 | 11.8 ± 1.8 | 5.6 ± 0.6 | 3.81 ± 0.01 |
| PuO2 660 °C (2 h) | 6.8 ± 1.2 | 10.4 ± 2.0 | 4.3 ± 0.7 | 3.81 ± 0.03 |
| PuO2 660 °C (17 h) | 6.1 ± 1.2 | 12.1 ± 1.7 | 4.9 ± 0.6 | 3.82 ± 0.01 |
| PuO2 1200 °C (2 h) | 5.9 ± 1.2 | 12.0 ± 2.9 | 4.2 ± 0.9 | 3.84 ± 0.02 |
The corresponding O-shell DWF parameters show a striking increase with the decrease in the annealing temperature for various considered oxides (with fixed coordination number and An–O distance of 2.41 Å and 2.33 Å for Th–O and Pu–O, respectively). For the ThO2 sample fired at 1000 °C, the first O-shell DWF parameter equals 4.9 ± 1.1 10−3 Å2, whereas it increases to 6.9 ± 1.1 10−3 Å2 when heated at 485 °C (oxalate route) and reaches 9.5 ± 1.1 10−3 Å2 for the nanopowder. A similar trend is noted for PuO2 samples, for which the first O-shell DWF value reaches 5.9 ± 1.2 10−3 Å2 when fired at 1200 °C and 7.0 ± 1.1 10−3 Å2 when heated at 485 °C. This parameter drastically increases for the sample composed of the smallest nanoparticles, i.e., for PuO2 nanopowder (8.9 ± 1.0 10−3 Å2) and for Pu(IV) intrinsic colloid (12.1 ± 0.9 10−3 Å2).
Fig. 5 plots the first O-shell DWF parameter as a function of the An oxide particle size determined by XRD through Rietveld refinement (the similar curves are separately displayed in Fig. S11, ESI†). Both PuO2 and ThO2 samples exhibit the same trend and are featured with an increase in the O-shell DWF, which is correlated with the shrinking particle size of the oxides. This phenomenon is strongly accentuated for the smallest particles, i.e., when the particle size reaches a value below 10 to 20 nm. These observations can be also correlated with the structural parameters calculated for the An–An coordination spheres (Table 3 and Fig. 5b). In contrast, the An–An DWF and the distance parameters are found to be quite stable, whereas the related coordination numbers dramatically decrease with the decrease in the nanocrystal size of the oxides. The corresponding trends illustrated in Fig. 5 clearly demonstrate a significant particle size dependency for powders composed of particles smaller than ca. 10 to 20 nm in diameter, which agrees well with the first O-shell investigations.
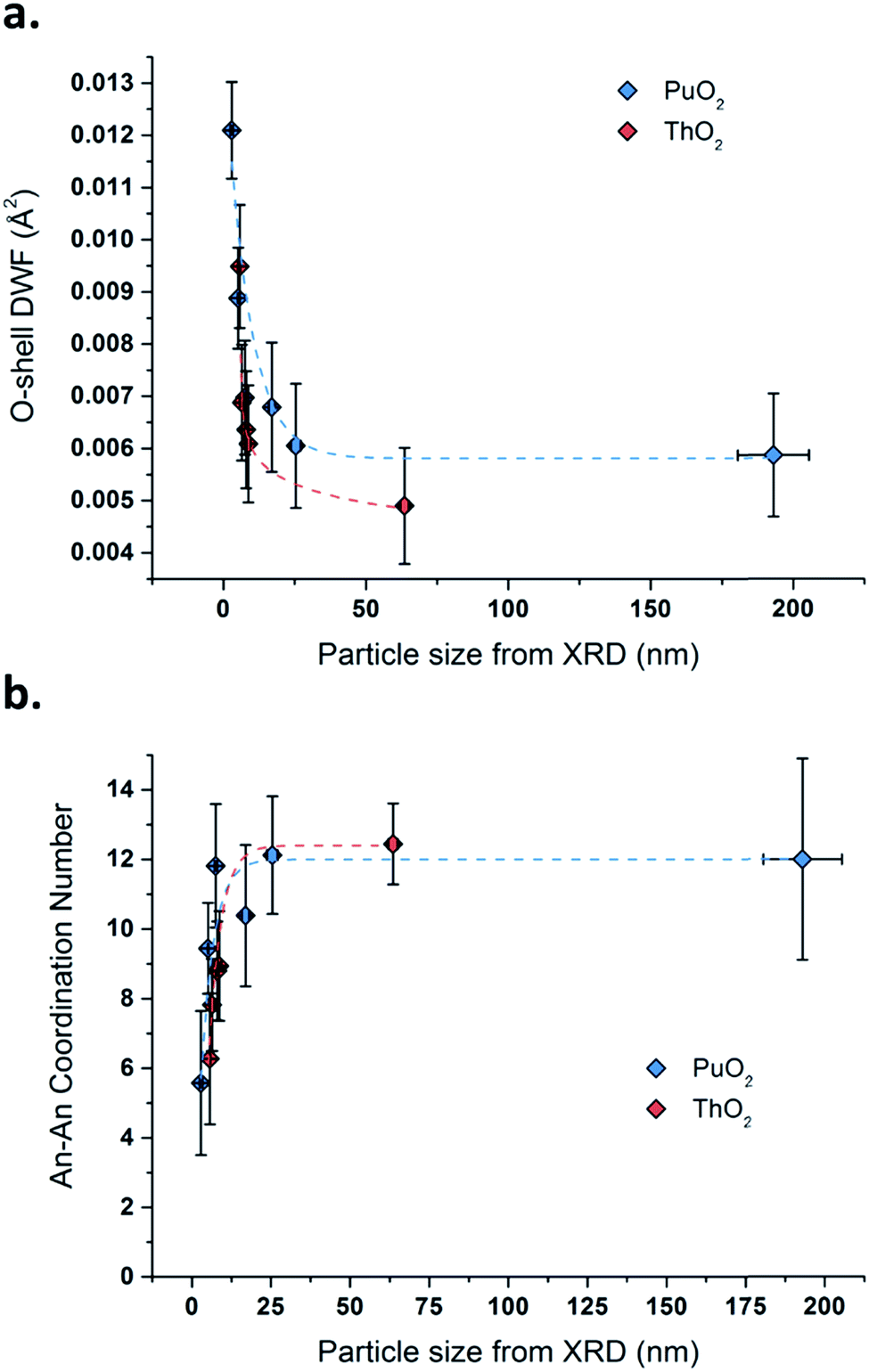 | ||
| Fig. 5 Variation of the O-shell DWF (a) and the An–An coordination number (b) as a function of the particle size for PuO2 (blue data) and ThO2 (red data) determined by XRD Rietveld refinement. | ||
3.3. Discussion
EXAFS spectroscopy is known to be more sensitive to the local structure of the absorbing atoms in comparison to XRD, the latter being more sensitive to periodic features and leading to an average positions of atoms in the structure.17 The crystalline nature of AnO2 nanoparticles is confirmed in this work through XRD and HR-TEM techniques but also with EXAFS spectroscopy, which points out the absence of significant variation in the An–An lattice. By contrast, the above-described observations reveal a striking correlation between the shrinking nanocrystal sizes of the samples and their respective local structure. The extracted DWF parameter, which provides a measurement of the crystallographic disorder, increases in the presence of nanoparticles and is dramatically amplified for the samples composed of the smallest ones (e.g., colloid or nanopowders). In comparison to bulk, the surfaces and interfaces of nanoparticles are highly disordered and their reducing size involves an increasing number of An(IV) with lower coordination numbers (Fig. 5). A good description of this phenomenon has been described by Kuzmin et al.25 or Frenkel et al.26In addition to the reduced number of neighbouring atoms standing at the surface of a nanoparticle, surface effects such as bond stress, boundaries, local strains, and lattice distortions involve crystallographic defects that increase the DWF parameter. Such an assertion is emphasized for ThO2 samples when plotting the Rietveld-extracted strain value as a function of the nanoparticle size (Fig. S12, ESI.† Note that the PuO2 strain parameter was not extracted due to artefact generation attributed to the epoxy resin used to fix the powdered samples). The strain value, which provides a measurement of the lattice stress, dramatically increases with the shrinking size of the nanoparticles in agreement with the DWF parameter determined by EXAFS. The increase in the lattice parameter with decreasing particle size also supports this explanation.35 The DWF parameter was found to be strongly correlated with the increase in the surface-to-volume ratio when decreasing the size of nanoparticles in this work.
The surface Debye temperature, which is inversely proportional to the Debye–Waller coefficient, has been reported to be about 50% lower than that of the bulk value in metals.61 In addition, molecular dynamics has shown that the atomic square displacement continuously increased from the bulk to the surface of a nanoparticle. The presence of surface low-coordinated atoms plays a significant role in the atomic displacement and vibrational properties. These variables have also been reported to increase according to the environment of a nanoparticle. Indeed, the presence of hydrolysed moieties, μ3-oxo bonds, and adsorbed water molecules in the Pu(IV) intrinsic colloidal species may be viewed as structural defects and therefore, enhance DWF. The very high proportion of surfaces and interfaces driven by free nanoparticles and their boundaries in nanostructured oxides also enter in this category. Therefore, the distortion and splitting of the O-shell on Fourier transformation of the EXAFS weighted spectra can be convincingly assigned to structural defects driven by the nanoparticle surfaces, which appears to be correlated to their decreasing sizes. Finally, the analogous behaviour of ThO2 and PuO2 nanoparticles supports this hypothesis and discards the hypothesis of PuO2+x formation suggested in the literature. Indeed, such an assertion is clearly invalid for the redox free thorium actinide, which is only stabilized in the (+IV) oxidation state.
4. Conclusion
A strong correlation between the size of PuO2 and ThO2 nanocrystals that constitute the corresponding oxides and their local structure probed by EXAFS spectroscopy has been established. The ThO2 samples exhibit a very similar behaviour as that of the PuO2 samples in term of nanoparticle size vs. local structure relationship. The data described in this paper strongly support that this correlation is related to structural defects in the nanoparticles, which drastically increase with the shrinking size of the particles that constitute the oxides. This suggests that the first shell distortion and splitting of Pu–O observed with EXAFS spectroscopy cannot be attributed to the existence of PuO2+x. Beyond the still-open ongoing discussions related to the realness of PuO2+x, these results also raise the question of the local structure and related physico-chemical properties of oxide nanomaterials crystallizing in the fluorite structure, which are of paramount importance for engineering applications such as nuclear energy, solid oxide fuel cells, catalysis, and sensors. Particularly, this study demonstrates that the nanostructure of actinide oxides can be controlled not only by the synthetic procedure but also by the firing duration and temperature. The oxalate route, which constitutes a reference method for the nuclear industry, may therefore lead to nanostructured PuO2 in a relatively large range of firing temperature.Author contributions
Two groups situated in ICSM and CEA Marcoule conceived the study. Sergey Nikitenko (head of ICSM/LSFC group) and Matthieu Virot from ICSM collaborated with Philippe Moisy and Laurent Venault (head of Pu lab) from CEA Marcoule for the development of the study. Moisy and Venault also managed the access to appropriate infrastructures and provided purified Pu solutions. Laura Bonato and Elodie Dalodière prepared and characterized most of the samples during their PhD theses. Matthieu Virot supervised the two related PhD theses. Matthieu Virot, Sergey Nikitenko and Laura Bonato wrote the manuscript. Damien Prieur and André Rossberg from ESRF performed the synchrotron characterizations. Thomas Dumas from Marcoule performed the EXAFS fits. Adel Mesbah and Nicolas Dacheux from ICSM gave their expertise in actinide materials and crystallography. Adel Mesbah performed the Rietveld refinements. Xavier Le Goff from ICSM performed the HR-TEM characterizations on Th samples. Oliver Dieste-Blanco and Thierry Wiss from JRC Karlsruhe performed the HR-TEM characterizations on Pu samples. Michael Odorico from ICSM performed the AFM characterizations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by CEA/DEN and european TALISMAN research program (Grant No. TALI-C05-17). The authors gratefully acknowledge Florent Bernard, Virginie Brethenoux, Julie Hennuyer, Charles Hours, Mireille Guigue, Julia Hidalgo, Joseph Lautru, Renaud Podor, Cyrielle Rey, Emilie Russello, Christelle Tamain, Victor Trillaud, Jackie Vermeulen, and Eleonore Welcomme for help in experiments, characterization, and useful discussions.References
- M. Fernandez-Garcia, A. Martinez-Arias, J. C. Hanson and J. A. Rodriguez, Chem. Rev., 2004, 104, 4063–4104 CrossRef CAS PubMed.
- H. Gleiter, Acta Mater., 2000, 48, 1–29 CrossRef CAS.
- H. Goesmann and C. Feldmann, Angew. Chem., Int. Ed., 2010, 49, 1362–1395 CrossRef CAS PubMed.
- J. M. Teulon, C. Godon, L. Chantalat, C. Moriscot, J. Cambedouzou, M. Odorico, J. Ravaux, R. Podor, A. Gerdil, A. Habert, N. Herlin-Boime, S. W. W. Chen and J. L. Pellequer, Nanomaterials, 2019, 9, 18 CrossRef PubMed.
- W. J. Stark, P. R. Stoessel, W. Wohlleben and A. Hafner, Chem. Soc. Rev., 2015, 44, 5793–5805 RSC.
- G. R. Patzke, Y. Zhou, R. Kontic and F. Conrad, Angew. Chem., Int. Ed., 2011, 50, 826–859 CrossRef CAS PubMed.
- D. Clark, S. Hecker, G. Jarvinen and M. Neu, in The Chemistry of the Actinide and Transactinide Elements, ed. L. Morss, N. Edelstein and J. Fuger, Springer Netherlands, 2011, ch. 7, pp. 813–1264, DOI:10.1007/978-94-007-0211-0_7.
- D. A. Costanzo, R. E. Biggers and J. T. Bell, J. Inorg. Nucl. Chem., 1973, 35, 609–622 CrossRef CAS.
- C. Walther and M. A. Denecke, Chem. Rev., 2013, 113, 995–1015 CrossRef CAS PubMed.
- V. V. Rondinella and T. Wiss, Mater. Today, 2010, 13, 24–32 CrossRef CAS.
- E. Dalodière, M. Virot, V. Morosini, T. Chave, T. Dumas, C. Hennig, T. Wiss, O. Dieste Blanco, D. K. Shuh, T. Tyliszcak, L. Venault, P. Moisy and S. I. Nikitenko, Sci. Rep., 2017, 7, 43514 CrossRef PubMed.
- J. M. Haschke, T. H. Allen and L. A. Morales, Science, 2000, 287, 285–287 CrossRef CAS PubMed.
- C. Madic, Science, 2000, 287, 243–244 CrossRef CAS.
- P. A. Korzhavyi, L. Vitos, D. A. Andersson and B. Johansson, Nat. Mater., 2004, 3, 225–228 CrossRef CAS PubMed.
- B. Y. Ao, R. Z. Qiu, H. Y. Lu, X. Q. Ye, P. Shi, P. H. Chen and X. L. Wang, J. Phys. Chem. C, 2015, 119, 101–108 CrossRef CAS.
- P. Martin, S. Grandjean, M. Ripert, M. Freyss, P. Blanc and T. Petit, J. Nucl. Mater., 2003, 320, 138–141 CrossRef CAS.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, L. A. Morales, M. P. Neu, P. Paviet-Hartmann, W. Runde, C. D. Tait, D. K. Veirs and P. M. Villella, J. Am. Chem. Soc., 2004, 126, 13443–13458 CrossRef CAS PubMed.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. den Auwer, M. Ding, P. K. Dorhout, F. J. Espinosa-Faller, P. L. Gordon, R. G. Haire, N. J. Hess, R. F. Hess, D. W. Keogh, G. H. Lander, D. Manara, L. A. Morales, M. P. Neu, P. Paviet-Hartmann, J. Rebizant, V. V. Rondinella, W. Runde, C. D. Tait, D. K. Veirs, P. M. Villella and F. Wastin, J. Solid State Chem., 2005, 178, 521–535 CrossRef CAS.
- J. D. Farr, R. K. Schulze and M. P. Neu, J. Nucl. Mater., 2004, 328, 124–136 CrossRef CAS.
- M. J. Sarsfield, R. J. Taylor, C. Puxley and H. M. Steele, J. Nucl. Mater., 2012, 427, 333–342 CrossRef CAS.
- S. D. Conradson, B. D. Begg, D. L. Clark, C. Den Auwer, F. J. Espinosa-Faller, P. L. Gordon, N. J. Hess, R. Hess, D. W. Keogh, L. A. Morales, M. P. Neu, W. Runde, C. D. Tait, D. K. Veirs and P. M. Villella, Inorg. Chem., 2003, 42, 3715–3717 CrossRef CAS PubMed.
- S. Calvin, XAFS for Everyone, CRC Press, 2013 Search PubMed.
- L. Amidani, T. V. Plakhova, A. Y. Romanchuk, E. Gerber, S. Weiss, A. Efimenko, C. J. Sahle, S. M. Butorin, S. N. Kalmykov and K. O. Kvashnina, Phys. Chem. Chem. Phys., 2019, 21, 10635–10643 RSC.
- A. Y. Romanchuk, T. V. Plakhova, A. V. Egorov, T. B. Egorova, P. V. Dorovatovskii, Y. V. Zubavichus, A. A. Shiryaev and S. N. Kalmykov, Dalton Trans., 2018, 47, 11239–11244 RSC.
- A. Kuzmin and J. Chaboy, IUCrJ, 2014, 1, 571–589 CrossRef CAS PubMed.
- A. I. Frenkel, C. W. Hills and R. G. Nuzzo, J. Phys. Chem. B, 2001, 105, 12689–12703 CrossRef CAS.
- C. Ekberg, K. Larsson, G. Skarnemark, A. Odegaard-Jensen and I. Persson, Dalton Trans., 2013, 42, 2035–2040 RSC.
- D. Hudry, C. Apostolidis, O. Walter, A. Janssen, D. Manara, J. C. Griveau, E. Colineau, T. Vitova, T. Prussmann, D. Wang, C. Kubel and D. Meyer, Chem.–Eur. J., 2014, 20, 10431–10438 CrossRef CAS PubMed.
- L. Soderholm, P. M. Almond, S. Skanthakumar, R. E. Wilson and P. C. Burns, Angew. Chem., Int. Ed., 2008, 47, 298–302 CrossRef CAS PubMed.
- J. Rothe, M. A. Denecke, V. Neck, R. Muller and J. I. Kim, Inorg. Chem., 2002, 41, 249–258 CrossRef CAS PubMed.
- V. Neck, R. Muller, M. Bouby, M. Altmaier, J. Rothe, M. A. Denecke and J. I. Kim, Radiochim. Acta, 2002, 90, 485–494 CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Found. Crystallogr., 1976, 32, 751–767 CrossRef.
- S. Hubert, J. Purans, G. Heisbourg, P. Moisy and N. Dacheux, Inorg. Chem., 2006, 45, 3887–3894 CrossRef CAS PubMed.
- J. Purans, G. Heisbourg, N. Dacheux, P. Moisy and S. Hubert, Phys. Scr., 2005, T115, 925–927 CrossRef CAS.
- T. V. Plakhova, A. Y. Romanchuk, D. V. Likhosherstova, A. E. Baranchikov, P. V. Dorovatovskii, R. D. Svetogorov, T. B. Shatalova, T. B. Egorova, A. L. Trigub, K. O. Kvashnina, V. K. Ivanov and S. N. Kalmykov, J. Phys. Chem. C, 2019, 123(37), 23167–23176 CrossRef CAS.
- X. Beaudoux, M. Virot, T. Chave, G. Leturcq, G. Jouan, L. Venault, P. Moisy and S. I. Nikitenko, Dalton Trans., 2016, 45, 8802–8815 RSC.
- E. Dalodière, M. Virot, T. Dumas, D. Guillaumont, M. C. Illy, C. Berthon, L. Guerin, A. Rossberg, L. Venault, P. Moisy and S. I. Nikitenko, Inorg. Chem. Front., 2018, 5, 100–111 RSC.
- X. Beaudoux, M. Virot, T. Chave, G. Durand, G. Leturcq and S. I. Nikitenko, Green Chem., 2016, 18, 3656–3668 RSC.
- L. Claparede, N. Clavier, N. Dacheux, A. Mesbah, J. Martinez, S. Szenknect and P. Moisy, Inorg. Chem., 2011, 50, 11702–11714 CrossRef CAS PubMed.
- V. Morosini, T. Chave, M. Virot, P. Moisy and S. I. Nikitenko, Ultrason. Sonochem., 2016, 29, 512–516 CrossRef CAS PubMed.
- C. Frontera and J. Rodriguez-Carvajal, Phys. B, 2003, 335, 219–222 CrossRef CAS.
- W. Matz, N. Schell, G. Bernhard, F. Prokert, T. Reich, J. Claussner, W. Oehme, R. Schlenk, S. Dienel, H. Funke, F. Eichhorn, M. Betzl, D. Prohl, U. Strauch, G. Huttig, H. Krug, W. Neumann, V. Brendler, P. Reichel, M. A. Denecke and H. Nitsche, J. Synchrotron Radiat., 1999, 6, 1076–1085 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- J. J. Rehr, J. J. Kas, M. P. Prange, A. P. Sorini, Y. Takimoto and F. Vila, C. R. Phys., 2009, 10, 548–559 CrossRef CAS.
- D. Bouexiere, K. Popa, O. Walter and M. Cologna, RSC Adv., 2019, 9, 6542–6547 RSC.
- L. Claparede, F. Tocino, S. Szenknect, A. Mesbah, N. Clavier, P. Moisy and N. Dacheux, J. Nucl. Mater., 2015, 457, 304–316 CrossRef CAS.
- L. Claparede, N. Clavier, N. Dacheux, P. Moisy, R. Podor and J. Ravaux, Inorg. Chem., 2011, 50, 9059–9072 CrossRef CAS PubMed.
- K. Popa, O. Walter, O. Dieste Blanco, A. Guiot, D. Bouexiere, J. Y. Colle, L. Martel, M. Naji and D. Manara, CrystEngComm, 2018, 20, 4614–4622 RSC.
- X. MachuronMandard and C. Madic, J. Alloys Compd., 1996, 235, 216–224 CrossRef CAS.
- G. I. N. Bouala, N. Clavier, J. Lechelle, A. Mesbah, N. Dacheux and R. Podor, Ceram. Int., 2015, 41, 14703–14711 CrossRef.
- B. L. Scott, A. L. Pugmire, J. T. Stritzinger, D. K. Veirs, L. E. Wolfsberg and M. P. Wilkerson, J. Nucl. Mater., 2019, 521, 155–160 CrossRef CAS.
- V. Tyrpekl, J. F. Vigier, D. Manara, T. Wiss, O. Dieste Blanco and J. Somers, J. Nucl. Mater., 2015, 460, 200–208 CrossRef CAS.
- D. Horlait, N. Clavier, N. Dacheux, R. Cavalier and R. Podor, Mater. Res. Bull., 2012, 47, 4017–4025 CrossRef CAS.
- P. M. Diehm, P. Agoston and K. Albe, ChemPhysChem, 2012, 13, 2443–2454 CrossRef CAS PubMed.
- F. Zhang, S. W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127–129 CrossRef CAS.
- S. Tsunekawa, R. Sahara, Y. Kawazoe and K. Ishikawa, Appl. Surf. Sci., 1999, 152, 53–56 CrossRef CAS.
- R. M. Orr, H. E. Sims and R. J. Taylor, J. Nucl. Mater., 2015, 465, 756–773 CrossRef CAS.
- A. K. Arora, M. Rajalakshmi, T. R. Ravindran and V. Sivasubramanian, J. Raman Spectrosc., 2007, 38, 604–617 CrossRef CAS.
- S. Dash, A. Singh, P. K. Ajikumar, H. Subramanian, M. Rajalakshmi, A. K. Tyagi, A. K. Arora, S. V. Narasimhan and B. Raj, J. Nucl. Mater., 2002, 303, 156–168 CrossRef CAS.
- F. Cappia, D. Hudry, E. Courtois, A. Janssen, L. Luzzi, R. J. M. Konings and D. Manara, Mater. Res. Express, 2014, 1, 025034 CrossRef.
- P. Scardi, L. Rebuffi, M. Abdellatief, A. Flor and A. Leonardi, J. Appl. Crystallogr., 2017, 50, 508–518 CrossRef CAS PubMed.
- J. Timoshenko, A. Anspoks, A. Kalinko and A. Kuzmin, Phys. Status Solidi A, 2015, 212, 265–273 CrossRef CAS.
- A. I. Frenkel, Y. Feldman, V. Lyahovitskaya, E. Wachtel and I. Lubomirsky, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 024116 CrossRef.
- A. Anspoks, A. Kalinko, J. Timoshenko and A. Kuzmin, Solid State Commun., 2014, 183, 22–26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00662a |
| This journal is © The Royal Society of Chemistry 2020 |