
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of protein–graphene oxide interactions with varying degrees of oxidation†
Shahid A.
Malik
ab,
Zinia
Mohanta
bc,
Chandan
Srivastava
d and
Hanudatta S.
Atreya
 *ab
*ab
aDepartment of Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore-560012, India. E-mail: hsatreya@iisc.ac.in
bNuclear Magnetic Resonance Research Centre, Indian Institute of Science, Bangalore-560012, India
cCentre for Bio Systems Science and Engineering, Indian Institute of Science, Bangalore-560012, India
dDepartment of Materials Engineering, Indian Institute of Science, Bangalore-560012, India
First published on 27th March 2020
Abstract
The degree of oxidation of graphene oxide (GO) has been shown to be important for its toxicity and drug-loading efficiency. However, the effect of its variations on GO–protein interaction remains unclear. Here, we evaluate the effect of the different oxidation degrees of GO on its interaction with human ubiquitin (8.6 kDa) using solution state nuclear magnetic resonance (NMR) spectroscopy in combination with other biophysical techniques. Our findings show that the interaction between the protein and the different GO samples is weak and electrostatic in nature. It involves fast dynamic exchange of the protein molecules from the surface of the GO. As the oxidation degree of the GO increases, the extent of the interaction with the protein changes. The interaction of the protein with GO can thus be modulated by tuning the degree of oxidation. This study opens up new avenues to design appropriate graphenic materials for use in various biomedical fields such as drug delivery, biomedical devices and imaging.
Introduction
Graphene and graphenic materials have attracted attention owing to their intriguing properties1–5 and applications in various fields.6–9 The chemical modification of graphene yielding amphiphilic sheet-like GO molecules,10 characterized by the presence of a high density of oxygen in the form of hydrophilic functional moieties such carboxylic acids, carbonyls, alcohols and epoxides,11 is believed to modulate its compatibility in biological systems.12 Owing to the rich oxygenic surface,12 large surface area:volume ratio,13 surface enhanced properties14 and presence of hydrophobic sp2 and sp3 carbon on its sheet-like structure,13 GO has been considered for different biomedical applications such as biosensing,15,16 drug delivery,17 bioimaging,18 photothermal therapies,19 tissue engineering,20 cell growth and cell differentiation21 and various biomedical devices.22In general, any biomedical application of GO would involve its interaction with various biomolecules such as nucleic acids,23 lipids24 and proteins.25 The GO surface interacts with various functional groups present on the proteins. The extent of these interactions is expected to depend upon the surface charge, surface energy and hydrophobicity of GO.26 In the case of proteins, a ‘protein corona’27 is formed which may significantly affect not only the protein structure and function but also the biological characteristics of GO.28–30 The formation of the protein corona on the surface affects the fate of the designed nanomaterials.31,32 Thus, a comprehensive study of the effect of surface modifications of GO on its interaction with proteins is required for understanding and improving its biomedical efficacy.12
GO–protein interactions have been studied, using techniques such as Circular Dichroism (CD), fluorescence, UV-Vis spectroscopy, and Fourier transform Infra-Red spectroscopy (FTIR).30,33,34 Using NMR spectroscopy, information about the dynamics of the protein and its individual residues can be obtained across a varying range of time scales.35 This feature renders NMR a powerful spectroscopic technique for probing structural and dynamic perturbations in the protein backbone upon its interaction with GO.25
In the present work, using NMR spectroscopy in combination with other techniques such as isothermal titration calorimetry (ITC), zeta potential (ζ) measurements, FTIR spectroscopy, X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), CD spectroscopy, Atomic Force Microscopy (AFM) and Raman spectroscopy, we have studied the residue specific interaction of a globular protein human ubiquitin with GO prepared with different oxidation degrees.36
We have chosen human ubiquitin as the model globular protein in the present study because it is found in almost all eukaryotes and is an important regulatory protein involved in the ubiquitin proteasome pathway. Its structure comprises both alpha helices and beta strands, thereby serving as an important example for exploring the mechanism of the interaction of GO with structured proteins.25
Experimental
Materials and methods
For non-NMR studies, the unlabelled protein was purified from Luria Broth (LB) in place of a minimal medium following an identical purification procedure as that described above.
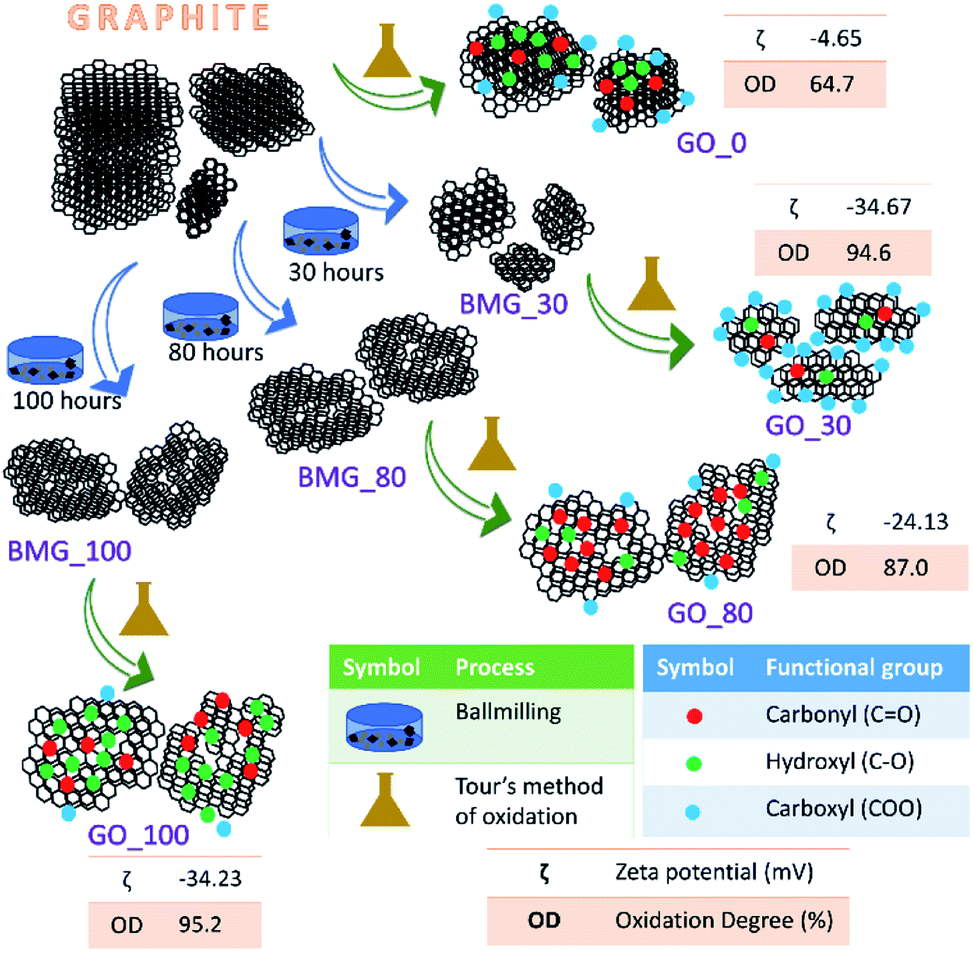 | ||
| Fig. 1 Schematic representation of the synthesis of various GO samples. For each GO sample the number of ball milling hours (X hours), zeta potential (ζ) and percentage of the degree of oxidation (OD%) are also provided. Here BMG_X refers to ball milled graphite for X hours. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, orthophosphoric acid with a total amount of 14 ml was added to it. Both the acids were mixed with the help of a glass rod. Then the beaker containing the mixture of graphite–KMnO4 was placed in the ice bath and the acid solution was poured in the mixture immediately. Adding the solution slowly would result in a highly exothermic reaction. The reaction mixture was constantly stirred with a glass rod to obtain a suspension. A stirring bar was added to the reaction solution which was placed on a hot plate magnetic stirrer to obtain a consistent mixture. The reaction mixture was then continuously stirred for 12 hours at 45 °C. The reaction mixture was slowly poured into another beaker containing 140 ml of DI ice and stirred until the ice melted completely. This was followed by addition of 10 ml of H2O2 to the final mixture. The mixture turned chrome yellow. Once the mixture cooled down, 200 ml of DI water was added and was allowed to decant for 2 days. The sediment was procured and washed using DI water thrice and with ethanol twice. Later it was dried to obtain the GO powder.
:1, orthophosphoric acid with a total amount of 14 ml was added to it. Both the acids were mixed with the help of a glass rod. Then the beaker containing the mixture of graphite–KMnO4 was placed in the ice bath and the acid solution was poured in the mixture immediately. Adding the solution slowly would result in a highly exothermic reaction. The reaction mixture was constantly stirred with a glass rod to obtain a suspension. A stirring bar was added to the reaction solution which was placed on a hot plate magnetic stirrer to obtain a consistent mixture. The reaction mixture was then continuously stirred for 12 hours at 45 °C. The reaction mixture was slowly poured into another beaker containing 140 ml of DI ice and stirred until the ice melted completely. This was followed by addition of 10 ml of H2O2 to the final mixture. The mixture turned chrome yellow. Once the mixture cooled down, 200 ml of DI water was added and was allowed to decant for 2 days. The sediment was procured and washed using DI water thrice and with ethanol twice. Later it was dried to obtain the GO powder.
During the process of synthesis (the ball milling step) of GO samples, the mechanical introduction of defects leads to a decrease in the sp2 carbon content and introduction of oxygenic functional moieties (during the oxidation step). The overall sp2/sp3 ratio of the GO falls as the number of ball milling hours increases and the oxidation state increases (Table S1 in the ESI†).
sinθ) (d is the d-spacing or interlayer spacing, λ is the X-ray wavelength and θ is the diffraction angle), the d-spacings for G_0, G_30, G_80 and G_100 were calculated (Table S1 in the ESI†).
Results
The terms oxidation level and degree of oxidation have been used interchangeably for expressing the extent of oxidation in graphene oxide. However, the two parameters are different and hence have to be used precisely while corroborating a hypothesis to avoid ambiguity. The oxidation level, determined using the O/C ratio gives the oxygen content of the graphite oxide and considers the stoichiometric ratio of the functional groups, that is, it provides information about the amount of oxygen present with respect to the carbon content. The degree of oxidation or oxidation degree (OD) of GO on the other hand is the amount of carbon that is oxidized. The calculation of OD takes two import factors into consideration, the aromatic and non-aromatic carbon ratio and the amount of graphite that undergoes complete oxidation. Pertaining to our study, we have considered degree of oxidation as the chemical property of interest. The degree of oxidation for the four different types of GO prepared is indicated in Fig. 1.The AFM results show that all the GO samples exhibited poly dispersive distribution; distinct differences could be observed. G_0 had a sheet thickness of about 19 nm indicating that the GO prepared was multi-layered. Another interesting observation was that G_30 consisted of much smaller particles and the layer thickness was about 3 nm. This type of distribution has been explained in a previously reported work from our group. G_80 and G_100 show uniform sheet structures with each sheet having a thickness of around 3 nm (Fig. S6 in the ESI†).
First, the quantification of the defect density of each GO sample was carried out by obtaining the ratio of ID and IG bands in the Raman spectra (Fig. S2 in the ESI†) which reveals that the defects introduced during ball milling can help in obtaining GO samples with varying degrees of oxidation and surface charges owing to the presence of different functional moieties (−COOH, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, –COR, etc.) on the surface of the GO as reflected in the FTIR spectra (Fig. S2 in the ESI†) and Solid-state NMR spectra (Fig. S3 in the ESI†) of various GO samples.
O, –COR, etc.) on the surface of the GO as reflected in the FTIR spectra (Fig. S2 in the ESI†) and Solid-state NMR spectra (Fig. S3 in the ESI†) of various GO samples.
On deconvoluting the Raman spectra of GO, we observed distinguishable peaks at 1350, 1532 and 1598 cm−1 (Fig. S2†). The peak at 1350 cm−1 arises from the disorder induced mode, A1g. The Raman bands at 1532 and 1598 cm−1 are a combination of A1g, E1g and E2g vibration modes. The peak at 1598 cm−1 is assigned to the G band. The peak at 1532 cm−1 can be attributed to amorphous carbon.
A broad and wide peak was observed in all the FTIR spectra (Fig. S2†) of GO samples at 3185 cm−1 which is ascribed to O–H stretching vibrations and is due to the presence of –OH functional moieties and intercalated water. Intense peaks at 1730, 1419 and 1041 cm−1 are peaks of ketone, carboxylic and epoxy groups. The FTIR spectra of the synthesized GO samples exhibiting the characteristic peaks of GO for each sample confirm the presence of oxygen containing functional moieties from successful formation of GO in each case.
To understand the nature of carbons and the contribution of sp2 and oxidized states of carbon, the high resolution C 1s scans were obtained through XPS. The notable humps at 292 eV were considered to be because of the π–π* transitions (HOMO–LUMO), characteristic satellite peaks arising from carbon in aromatic rings. The XPS spectra were deconvoluted for sp2 (at ∼285 eV), C–O (at ∼286.7 eV), CO (at ∼287.9 eV), COO (at ∼290.3 eV) and π–π* (at ∼292 eV). The deconvolution of each C 1s spectrum is shown in Fig. S4 in the ESI.†
The oxygen content was quantified using the O/C ratio obtained using the relation:
| O/C = ((2 × I(COO)) + (1 × I(C–O)) + (1 × I(CO)))/ (I(CC) + I(COO) + I(C–O) + I(CO)) |
O, and ICC correspond to the intensities of the peaks corresponding to carboxyl, hydroxyl, carbonyl groups and aromatic carbon, respectively, from deconvolution of high resolution C 1s scans obtained by XPS (Table S1 in the ESI†).
The percentage of the oxidation degree (OD%), Table S1 in the ESI,† is calculated using the percentage of aromatic carbon and non-aromatic carbon, calculated from XPS data (Fig. S4 in the ESI†) and the intensities of the GO and graphite peaks in the XRD pattern of GO (as shown in Fig. S3 in the ESI†) using the following relation:
| I(non-aromatic)/(Iaromatic + I(non-aromatic)) × IGO/(IGO + Igraphite) × 100 |
Quantitatively, the extent of the interaction of protein with the various synthesized GO samples was studied by ITC by measuring the dissociation constant (Kd) of the binding as shown in Fig. 2. The values of the dissociation constant (Kd) for ubiquitin with different GO samples having varying oxidation degrees fall in the micro molar range (Table 1); thus a weak electrostatic interaction is expected. This implies that ubiquitin interacts weakly with the different GO samples, indicating dynamic association–dissociation as discussed below. It should be noted that we could not obtain the saturation point in the interaction of ubiquitin with G_30 (Fig. 2) at the defined concentration of the protein (50 μM) and the number of injections (30 injections total) of protein as used for the other GO samples.
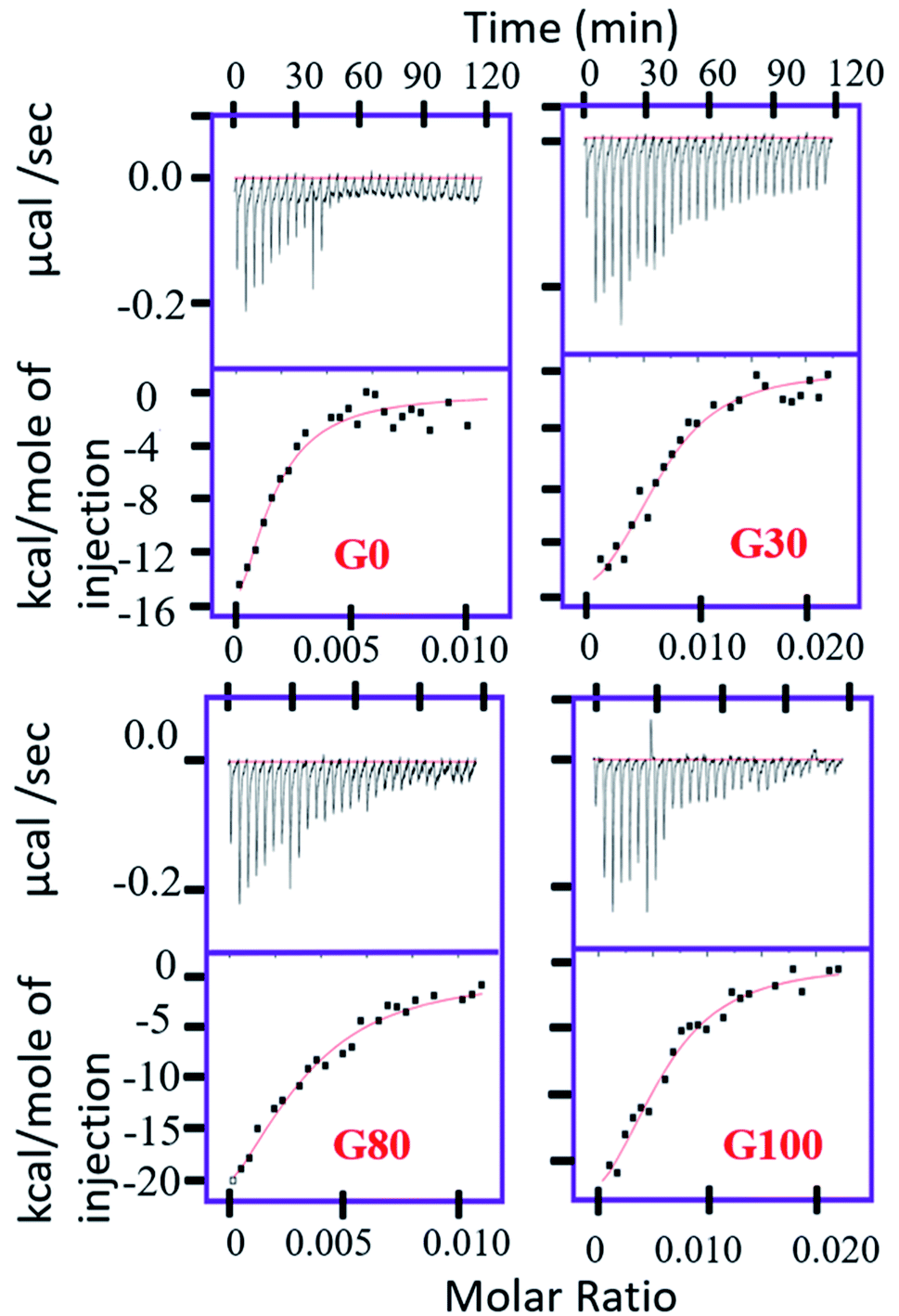 | ||
| Fig. 2 ITC studies of different GO samples with human ubiquitin at 25 °C. | ||
| K d (μM) | ΔH (cal mol−1) | ΔS | |
|---|---|---|---|
| G_0 | 1.4 ± 0.8 | −3.2 × 104 ± 1.2 × 104 | −81.14 |
| G_30 | 1.3 ± 0.35 | −2.05 × 104 ± 2933 | −41.66 |
| G_80 | 2.5 ± 0.75 | −3.55 × 104 ± 9265 | −93.5 |
| G_100 | 1.9 ± 0.4 | −3.04 × 104 ± 1.36 × 104 | −75.64 |
The CD spectra of the ubiquitin conjugates with GO samples having varying degrees of oxidation and surface chemistry show a decreasing ellipticity, which is because of the decrease in the population of free ubiquitin upon interaction with various GO samples. We did not observe any appreciable shift in the CD spectra of the ubiquitin upon conjugation with different GO samples (Fig. S5 in the ESI†), implying that the the secondary structure of the protein is retained upon interaction with various GO samples.
NMR studies of the ubiquitin-GO interaction
To characterize and quantify the dynamic aspects of the interaction of ubiquitin with various types of GO at the residue level, we recorded 2D [15N, 1H] Heteronuclear Single Quantum Coherence (HSQC) NMR spectra and amide proton relaxation times T2 (1HN) of the protein sample in the presence and absence of each GO sample at pH 6.25,37,38 In a 2D [15N, 1H] HSQC NMR spectrum each cross-peak corresponds to a directly bonded 1H–15N spin pair (with the exception of proline residues); thus the individual behaviour of each residue of ubiquitin can be probed using the intensity of its own cross-peak belonging to that amide bond.37Two types of titrations were performed, forward titration and reverse titration.25,37,39,40 In the former (forward) titration, from a stock of 1 mg ml−1, successive amounts of a particular GO sample (20 μl, 30 μl, 40 μl, 50 μl and 60 μl corresponding to a final concentration of each GO sample of 38.4 μg ml−1, 56.6 μg ml−1, 74.1 μg ml−1, 90.9 μg ml−1 and 107.1 μg ml−1 after considering the dilution of the whole volume) were added to 500 μl of the uniformly 15N-labeled ubiquitin (100 μM) dissolved in 50 mM phosphate buffer supplemented with 150 mM NaCl and 5% 2H2O at pH 6. This is referred to as ‘forward titration’.
In the second type of titration (referred to as ‘reverse titration’), 15N-labeled ubiquitin (stock concentration 500 μM) was gradually added to 500 μl of each GO (70 μg ml−1) taken in an NMR tube. The protein was added in increments of 10 μl, 20 μl, 30 μl, 40 μl, 50 μl, 60 μl and 70 μl corresponding to a final concentration of the protein of ∼9.4 μM, 18.5 μM, 27.3 μM, 35.7 μM, 44.0 μM, 51.7 μM and 59.3 μM. This titration helps to obtain the approximate amounts of protein loaded onto each GO surface and hence the effect of varying the surface chemistry of the GOs on the protein loading tendency can be probed as described below.
Forward titration: addition of GO to ubiquitin
Fig. 3a and b, respectively, show an overlay of the spectra of the free ubiquitin (blue) with that in the presence of the G_0 sample (red) and the plot of normalized 2D [15N, 1H] HSQC peak intensities (with respect to the free protein) in the presence of indicated amounts of G_0, at a pH of 6.0 and 298 K. Fig. 3c–h depict similar interactions for G_30, G_80 and G_100.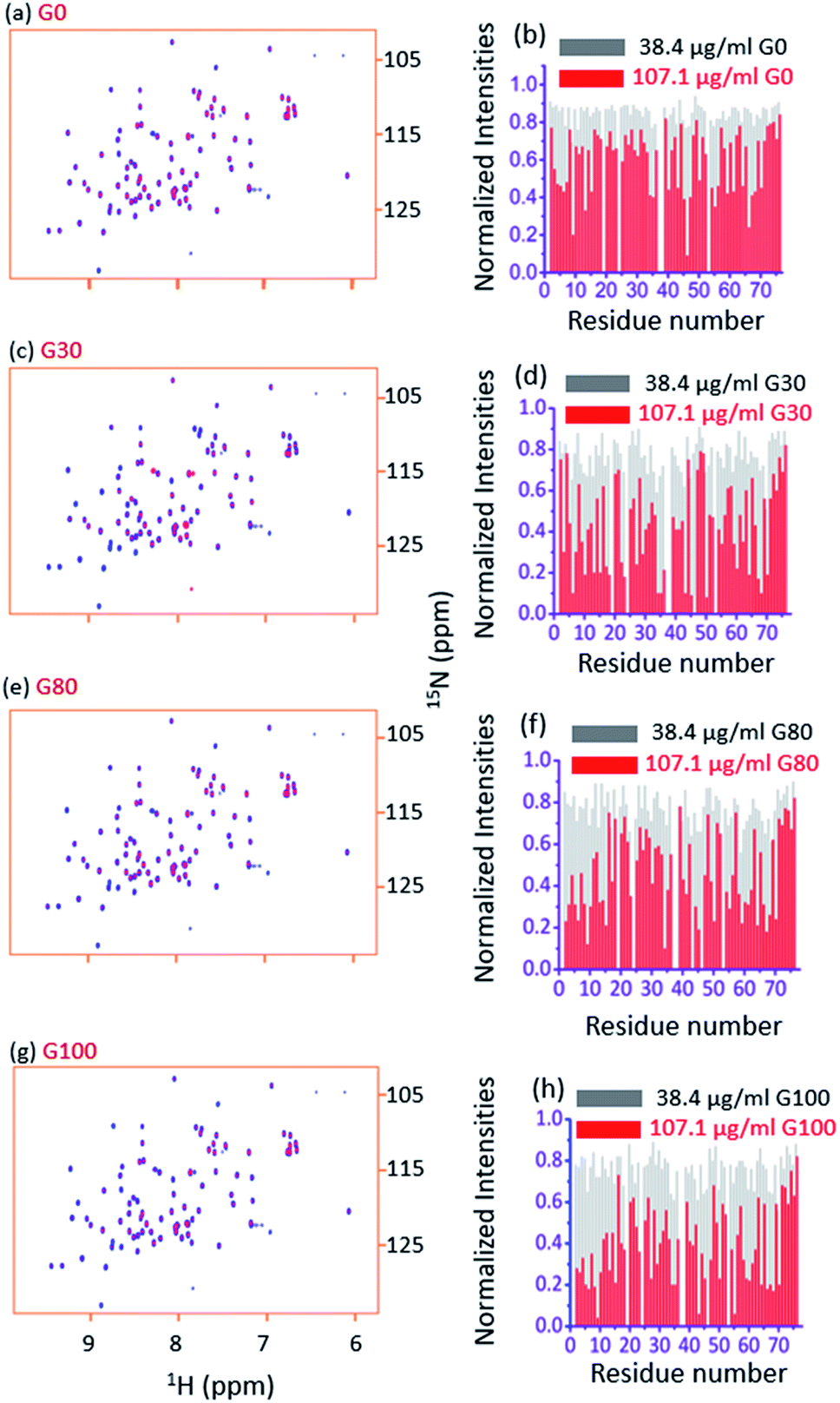 | ||
| Fig. 3 (a and b) The overlay of the 2D [15N, 1H] HSQC spectra of free ubiquitin (blue) with that in the presence of 107.1 μg ml−1 of G_0 (red) and the plot of normalized 2D [15N, 1H] HSQC peak intensities (with respect to the free protein) in the presence of the indicated amounts of G_0, at a pH of 6.0 and 298 K, respectively. Figures (c), (d), (e), (f), (g), and (h) represent the same for G_30, G-80 and G_100, respectively. | ||
Two observations can be made by looking at the spectra. First, no appreciable chemical shift perturbations of the peaks were observed upon addition of different GOs, suggesting that the protein retains its structural integrity upon interaction with different GOs.
Second, for a number of peaks the intensities are appreciably reduced with complete broadening of some of the cross peaks upon successive addition of GO, suggesting the potential residues of the protein that are involved in the interaction with the GO surface.
If the protein interacts strongly with the oppositely charged functional groups on the surface of the GO, there will be a significant fall in the T2 values of the protein because of the large molecular weight of the protein-GO conjugate formed (slow exchange). The larger the molecular weight of the conjugate, the smaller the T2 values. The residues which interact directly with the GO surface will be completely broadened out upon addition of the GO. Thus, the observed signals, if any, in the spectrum will have arisen from the free (unbound) protein molecules in the solution. In such a case, further addition of the GO should not vary the T2 (1HN) values of the unbound protein significantly. In the other scenario, if the increasing amounts of the GO lead to a decrease in the T2 (1HN) values of the protein, it implies that the free (unbound) protein molecules undergo a “fast exchange” with the protein molecules bound to the surface of the GO (ΔR2 ≪ kex). This observed decrease in the T2 (1HN) values of the protein is a consequence of the increase in the relative population of the bound protein molecules as the titration proceeds, hence resulting in a population weighted average of the observed R2 (1HN) as:
| R2(obs) = 1/T2(obs) = pfree × 1/T2free + pbound × 1/T2bound |
where pfree, 1/T2free and pbound, 1/T2bound are the relative populations and 1/T2 (1HN) values of free and bound ubiquitin, respectively. In order to measure the amide proton transverse relaxation times T2 (1HN) of the free and the bound form of ubiquitin, we used a two-point HSQC method, a modified version of the pulse sequence described previously,41 which was used for the measurement of the paramagnetic relaxation enhancement (Γ) in proteins. The modification involves the introduction of an additional delay during the first INEPT and acquisition of two spectra (two-point HSQC), each with an additional delay time (16 ms in the first (referred to as “with delay spectrum” and 20 μs in the second (referred to as “without delay spectrum”))), while keeping all the parameters constant.25,37
The residue specific exponential decay of the signals of amide protons (1HN) as observed in a 2D [15N, 1H] HSQC NMR experiment due to the transverse relaxation of spins can be written as:
| I = exp (−τ/T2 (1HN)) |
Here ‘τ’ represents the period during which the amide protons (1HN) are in the transverse plane. Next, we acquired two 2D [15N, 1H] HSQC spectra such that all other parameters are kept constant, except for the two additional delay times introduced, as a result of which the NMR signal shows an extra decay.
The ratio of the NMR signal intensities of the two HSQC spectra acquired with (τ2) and without delay (τ1) will be:
| Iwith delay/Iwithout delay = exp (−(τ2 − τ1)/T2 (1HN)) |
| T2 (1HN) = (τ2 − τ1)/ln (Iwithout delay/Iwith delay) |
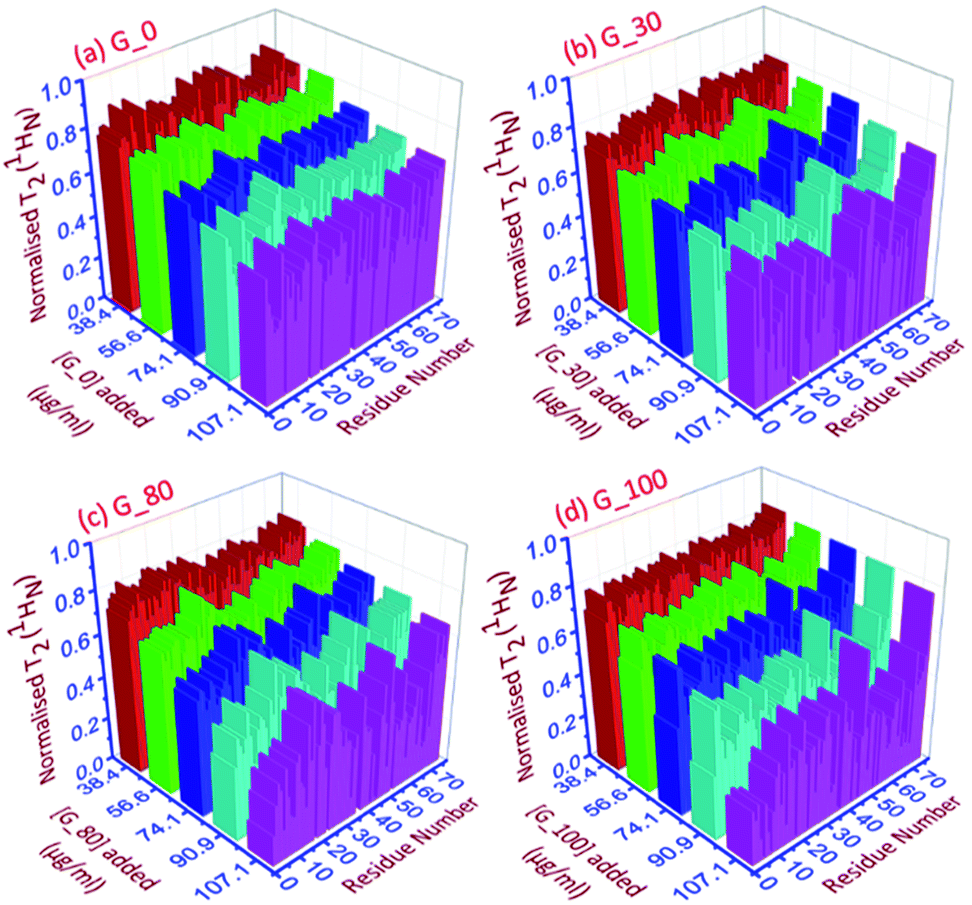 | ||
| Fig. 4 The 3D representation of the relative normalized T2 (1HN) values of each residue for each ubiquitin-GO conjugate at different indicated additions of (a) G_0, (b) G_30, (c) G_80 and (d) G_100. | ||
As can be seen in Fig. 4a–d, for all the residues the observed T2 (1HN) of ubiquitin falls as we gradually titrate with a particular GO sample. This trend is observed for all GOs. This further shows that the fraction of the bound form of ubiquitin ( fbound) gradually increases upon gradual addition of the GO to the protein solution, and hence a fast exchange of protein on the surface of the GO is implied. As the oxidation degree of the GO increases, the number of residues broadening out also increases, confirming that the GO with a higher oxidation degree binds more protein molecules. The residues that undergo the highest reduction (i.e., ≥75%) in T2 (1HN) values are:
For G_0: T9, A46, T66.
For G_30: K6, T7, G10, I13, L15, V17, E18, T22, I23, K27, K29, E34, G35, I36, L43, F45, A46, L50, N60, E64, L67, H68.
For G_80: Q2, K6, T9, G10, L15, E34, L43, I44, F45, A46, L50, Y59, E64, L67, H68, V70.
For G_100: Q2, I3, V5, K6, T7, L8, T9, G10, I13, L15, K27, E34, G35, L43, F45, A46, L50, T55, Y59, N60, E64, T66, L67, H68, V70.
Thus, as the oxidation degree on the GO surface increases going from G_0 to G_100, the extent of the interaction with the protein also increases. Since an increase in the degree of oxidation of the graphene oxide increases the negatively charged oxygen bearing functional moieties on its surface, which interact with the residues located at the positively charged surface of human ubiquitin through electrostatic interactions, more residues interact with the G_100 than with other GO samples.
Furthermore, the different GO samples did not show any preferential binding to the C or N-terminal part of the protein but the whole amino acid sequence of the protein was seen to be affected. Upon mapping these residues (involved in the interaction with the different GO samples) on the structure of ubiquitin, we found that the beta strands are more affected than the alpha helices of the protein (Fig. 5). We have mapped these residues undergoing a significant reduction (≥75%) in T2 (1HN) values on the electrostatic surface of ubiquitin. We found that these residues are located towards the positively charged surface of ubiquitin (Fig. 6).
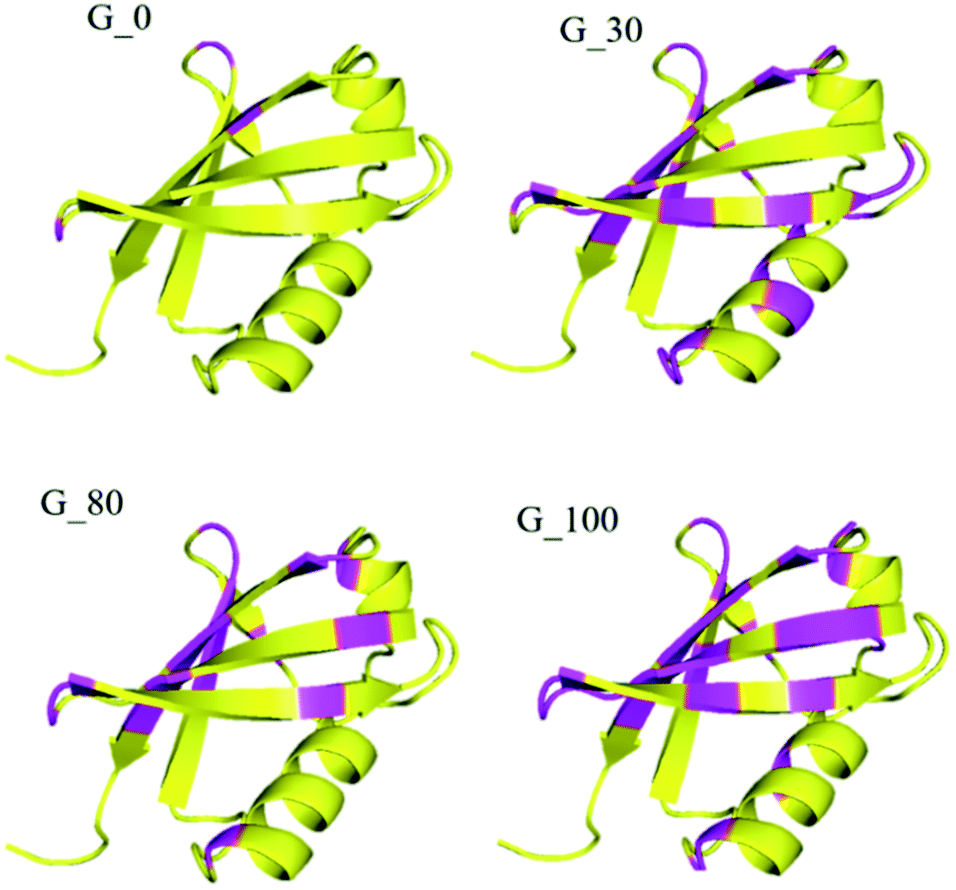 | ||
| Fig. 5 Mapping the residues on the surface of the ubiquitin involved in the interaction with various GO samples. The preferential binding sites of the different GO samples with the beta strands compared to the helices are highlighted in pink on the structure of ubiquitin. | ||
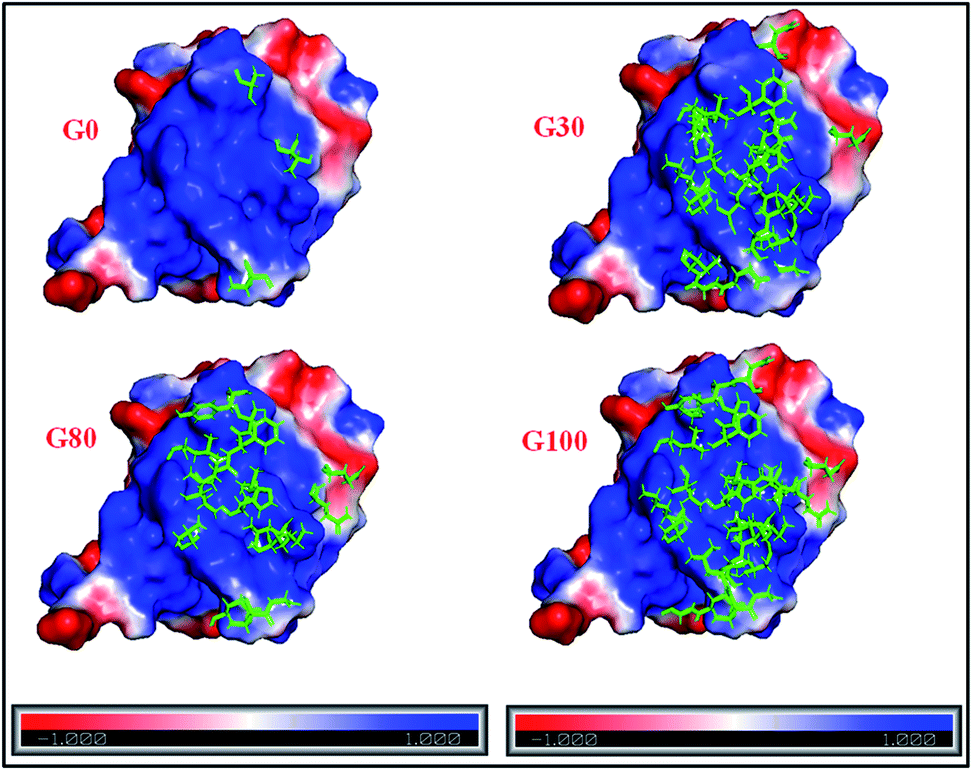 | ||
| Fig. 6 Electrostatic surface potential of human ubiquitin. The red and blue coloured areas correspond to negatively and positively charged areas, respectively; residues marked in green represent the protein backbone residues (shown in Fig. 5) involved in the interaction with different GO samples. | ||
The interaction of negatively charged functional moieties on the GO surface with the positively charged surface of ubiquitin is electrostatically favoured. For obtaining the surface plots as shown in Fig. 6, we have used implicit solvent methods to calculate the electrostatic potential on the protein structure considering the solvent to be a dielectric continuum and using the APBS (Adaptive Poisson–Boltzmann Solver) in PYMOL together with a web-based service provided39,42 at http://www.poissonboltzmann.org/. This further supports the hypothesis that the protein–GO interaction is largely electrostatic in nature and the interacting residues lie on the positively charged surface of the protein.
Reverse titration of ubiquitin with various GO samples
In this titration, 15N-labeled ubiquitin (stock concentration 500 μM) was gradually added to 500 μl of each GO sample (70 μg ml−1) present in the NMR tube. Carrying out reverse titration has two benefits: First, reverse titration helps to verify that the protein bound to the GO surface forms a high molecular weight conjugate having a very short T2 and hence is invisible to NMR as such. We have taken 70 μg ml−1 of each GO sample (500 μl) in a 5 mm NMR tube and the protein was gradually added to the GO. The first few additions of protein to the GO did not show peaks in the HSQC spectra, and hence a control experiment was carried out where the same concentration of ubiquitin was added to the same volume of buffer in the absence of GO. The protein at this concentration yielded a spectrum with the all the cross peaks present. This rules out the possibility that the absence of the peaks in the GO containing sample is due to the lower sensitivity of the protein at this concentration (as shown in Fig. S7 in the ESI†). This implies that in case of GO containing samples the absence of the signal can be attributed to a major population of ubiquitin bound to the GO surface, which is present in excess compared to the free protein in the initial stages of the titration. Even at a relatively higher protein concentration (∼35.7 μM), weak signal intensities were observed in the protein-GO conjugates (Fig. S7†). The subsequent additions of the protein led to an increase in the intensities of the cross peaks and more and more peaks showed up in the spectrum. Secondly, for different GO samples bearing different surface chemistries, the amount of bound protein also varies, as calculated theoretically from the reverse titration (as described in the ESI†).25,371 μg of G_0 binds ∼3 μg of Ubq
1 μg of G_30 binds ∼5.7 μg of Ubq
1 μg of G_80 binds ∼4.3 μg of Ubq
1 μg of G_100 binds ∼5.7 μg of Ubq
The difference in the loading capacity of the different GO samples is confirmed by the BCA assay.43 This variable loading capacity of different GO samples is dependent upon their surface charge which in turn depends on the degree of oxidation.
Discussion
In recent decades, many studies have been carried out to understand the underlying mechanism of the protein–nanomaterial interactions which remain the crucial step in therapeutic applications of GO-based nanomaterials.44,45 The binding of GO with biomolecules may involve electrostatic, hydrophobic, covalent and non-covalent, hydrogen bonding, and π–π stacking interactions depending upon the surface charge, energy and hydrophobicity of the interacting partners. It is a well-known fact that the degree of oxidation of GO has a significant role to play in its drug-loading efficiency, the stability of the loaded drug, and also the release of the drug from the GO surface.46 The higher degrees of oxidation of GO are related to its highly toxic nature under in vivo conditions.47 The toxicity of graphene oxide depends upon the type of functional groups at the surface and the extent of the degree of the oxidation.12 The effects of graphene-based materials on different globular proteins like lysozyme, trypsin, bovine haemoglobin and ubiquitin have been studied. It has been shown that proteins may undergo a conformational change upon interaction with these graphene-based materials. Yitong B. et al., using Th-T, UV-Vis and CD spectroscopy, showed that exposure to GO induces severe secondary conformational changes in lysozyme and hence the loss of enzymatic activity in lysozyme was observed.48 G. Fang et al. utilized NMR spectroscopy and observed that Graphene Quantum Dots (GQDs) have the potential to interact with the hydrophobic residues of ubiquitin and reduce the beta sheet content of ubiquitin.49 In another study Yanqing Wang et al. studied the effect of GO on the secondary structure of bovine haemoglobin (BHb). The interactions were found to be non-covalent including hydrophobic, van der Waals, hydrogen bonding, and electrostatic interactions. These studies suggested that there is a decrease in the thermal stability and loss of the protein secondary structure upon interaction with GO. In another study, interactions of GO with human serum albumin (HSA) were found to increase at lower pH, and above the isoelectric point of the protein, the interactions were found to decrease because of the electrostatic nature.50 Thus, it seems that electrostatic interactions are majorly involved in protein and GO binding. These interactions are weak in nature, and so a dynamic exchange of the proteins on the surface of GO is expected.25In our studies, we have used solution sate NMR spectroscopy as a principal technique to probe the underlying mechanism of the interaction of human ubiquitin (at the individual residue level) with GO having different degrees of oxidation. Our results suggest that the extent of interaction of human ubiquitin with the negatively charged surface of GO depends on the degree of oxidation. The interacting residues are found to be located on the beta strands of the protein structure at the positively charged surface of ubiquitin. Thus, the interaction between human ubiquitin and different GO samples having different defect densities and oxidation degrees at a pH of 6 is electrostatic in nature (the pI of ubiquitin is ∼6.8). Upon binding, human ubiquitin does not show any change in the chemical shift of the peaks in the 2D [15N–1H] HSQC spectrum which shows that there is no secondary structure change in the protein upon binding which is supported by CD spectroscopy (Fig. S5 in the ESI†). Moreover, we found that binding to the different GO samples leads to a decrease in the T2 (1HN) values of the protein, and also the Kd falls in the micromolar range, and hence a weak interaction between interacting partners is seen. Thus, a fast-dynamic exchange of the protein molecules on the surface of each GO sample with the free protein molecules in the solution is observed. Also the reverse titration shows that the amount of the protein loaded on the GO surface is a function of the degree of oxidation and the surface charge of both the interacting partners.
Conclusions
In summary, ubiquitin (a globular protein) interacts electrostatically with graphene oxide having varying degrees of oxidation and undergoes a dynamic and reversible exchange (fast exchange regime) from the surface of the GO as revealed by NMR relaxation studies and ITC. The interaction does not involve any change in the secondary structure of the protein. This study helps in understanding the mechanism of the interaction of a globular protein with graphene-based nanomaterials with different defect densities and surface chemistry and thus will help in designing appropriate graphenic materials for use in various biomedical fields like drug delivery, biomedical devices and imaging.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the NMR Research Centre at IISc Bangalore, Department of Science and Technology (DST) (IR/S0/LU-007/2010) and the University Grants Commission (UGC), India for the financial support. The Departments of the Centre for Nano Science and Engineering and MBU are highly acknowledged for their help in recording the Raman spectra and ITC data, respectively.Notes and references
- C. Chen, S. Rosenblatt, K. I. Bolotin, W. Kalb, P. Kim, I. Kymissis, H. L. Stormer, T. F. Heinz and J. Hone, Nat. Nanotechnol., 2009, 4, 861–867 CrossRef CAS PubMed.
- T. Shimizu, J. Haruyama, D. C. Marcano, D. V. Kosinkin, J. M. Tour, K. Hirose and K. Suenaga, Nat. Nanotechnol., 2011, 6, 45–50 CrossRef CAS PubMed.
- F. Léonard and A. A. Talin, Nat. Nanotechnol., 2011, 6, 773–783 CrossRef PubMed.
- C. N. R. Rao, H. S. S. R. Matte and K. S. Subrahmanyam, Acc. Chem. Res., 2013, 46, 149–159 CrossRef CAS PubMed.
- L. Vicarelli, S. J. Heerema, C. Dekker and H. W. Zandbergen, ACS Nano, 2015, 9, 3428–3435 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027–6053 CrossRef CAS PubMed.
- C. Chung, Y. Kim, D. Shin, S. Ryoo, B. H. E. E. Hong and D. Min, Acc. Chem. Res., 2013, 2211–2224 CrossRef CAS PubMed.
- L. Feng, L. Wu and X. Qu, Adv. Mater., 2013, 25, 168–186 CrossRef CAS PubMed.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- D. W. Lee, L. V. De Los Santos, J. W. Seo, L. L. Felix, A. D. Bustamante, J. M. Cole and C. H. W. Barnes, J. Phys. Chem. B, 2010, 114, 5723–5728 CrossRef CAS PubMed.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef CAS PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- H. Lai, F. Xu, Y. Zhang and L. Wang, J. Mater. Chem. B, 2018, 6, 4008–4028 RSC.
- C. H. Lu, H. H. Yang, C. L. Zhu, X. Chen and G. N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785–4787 CrossRef CAS PubMed.
- J. Balapanuru, J. X. Yang, S. Xiao, Q. Bao, M. Jahan, L. Polavarapu, J. Wei, Q. H. Xu and K. P. Loh, Angew. Chem., Int. Ed., 2010, 49, 6549–6553 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. Sun and H. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- Q. Liu, L. Wei, J. Wang, F. Peng, D. Luo, R. Cui, Y. Niu, X. Qin, Y. Liu, H. Sun, J. Yang and Y. Li, Nanoscale, 2012, 4, 7084–7089 RSC.
- D. Jaque, L. Martínez Maestro, B. Del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- T. R. Nayak, H. Andersen, V. S. Makam, C. Khaw, S. Bae, X. Xu, P. L. R. Ee, J. H. Ahn, B. H. Hong, G. Pastorin and B. Özyilmaz, ACS Nano, 2011, 5, 4670–4678 CrossRef CAS PubMed.
- W. C. Lee, C. H. Y. X. Lim, H. Shi, L. A. L. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS PubMed.
- J. Wang, Chem. Rev., 2008, 108, 814–825 CrossRef CAS PubMed.
- Y. He, B. Jiao and H. Tang, RSC Adv., 2014, 4, 18294–18300 RSC.
- L. Wu, L. Zeng and X. Jiang, J. Am. Chem. Soc., 2015, 137, 10052–10055 CrossRef CAS PubMed.
- S. Mondal, R. Thirupathi, P. Rao and H. S. Atreya, RSC Adv., 2016, 52539–52548 RSC.
- Y. Zhang, C. Wu and J. Zhang, Nanotechnol. Rev., 2013, 2, 27–45 CAS.
- X. Liu, C. Yan and K. L. Chen, Environ. Sci. Technol., 2019, 53, 8631–8639 CrossRef CAS PubMed.
- K. Yao, P. Tan, Y. Luo, L. Feng, L. Xu, Z. Liu, Y. Li and R. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 12270–12277 CrossRef CAS PubMed.
- Kenry, K. P. Loh and C. T. Lim, Nanoscale, 2016, 8, 9425–9441 RSC.
- B. Sun, Y. Zhang, W. Chen, K. Wang and L. Zhu, Environ. Sci. Technol., 2018, 52, 7212–7219 CrossRef CAS PubMed.
- F. B. Bombelli, D. Walczyk, M. Monopoli, I. Lynch and K. A. Dawson, Eur. Cells Mater., 2010, 20, 10 Search PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- X. Q. Wei, L. Y. Hao, X. R. Shao, Q. Zhang, X. Q. Jia, Z. R. Zhang, Y. F. Lin and Q. Peng, ACS Appl. Mater. Interfaces, 2015, 7, 13367–13374 CrossRef CAS PubMed.
- F. Emadi, A. Amini, A. Gholami and Y. Ghasemi, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- A. Ceccon, V. Tugarinov, A. Bax and G. M. Clore, J. Am. Chem. Soc., 2016, 138, 5789–5792 CrossRef CAS PubMed.
- Z. Mohanta, H. S. Atreya and C. Srivastava, Sci. Rep., 2018, 1–6 CAS.
- S. A. Malik, S. Mondal and H. S. Atreya, RSC Adv., 2019, 9, 28746–28753 RSC.
- P. K. Mandal and A. Majumdar, Concepts Magn. Reson., Part A, 2004, 20, 1–23 CrossRef.
- V. P. Brahmkhatri, K. Chandra, A. Dubey and H. S. Atreya, Nanoscale, 2015, 7, 12921–12931 RSC.
- I. Pal, V. P. Brahmkhatri, S. Bera, D. Bhattacharyya, Y. Quirishi, A. Bhunia and H. S. Atreya, J. Colloid Interface Sci., 2016, 483, 385–393 CrossRef CAS PubMed.
- J. Iwahara, C. Tang and G. Marius Clore, J. Magn. Reson., 2007, 184, 185–195 CrossRef CAS PubMed.
- L. Calzolai, F. Franchini, D. Gilliland and F. Rossi, Nano Lett., 2010, 10, 3101–3105 CrossRef CAS PubMed.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS.
- S. S. Chou, M. De, J. Luo, V. M. Rotello, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2012, 134, 16725–16733 CrossRef CAS PubMed.
- D. Kim, J. M. Yoo, H. Hwang, J. Lee, S. H. Lee, S. P. Yun, M. J. Park, M. Lee, S. Choi, S. H. Kwon, S. Lee, S. Kwon, S. Kim, Y. J. Park, M. Kinoshita, Y. Lee, S. Shin, S. R. Paik, S. J. Lee, S. Lee, B. H. Hong and H. S. Ko, Nat. Nanotechnol., 2018, 13, 812–818 CrossRef CAS PubMed.
- C. W. Liu, F. Xiong, H. Z. Jia, X. L. Wang, H. Cheng, Y. H. Sun, X. Z. Zhang, R. X. Zhuo and J. Feng, Biomacromolecules, 2013, 14, 358–366 CrossRef CAS PubMed.
- M. C. Duch, G. R. S. Budinger, Y. T. Liang, S. Soberanes, D. Urich, S. E. Chiarella, L. A. Campochiaro, A. Gonzalez, N. S. Chandel, M. C. Hersam and G. M. Mutlu, Nano Lett., 2011, 11, 5201–5207 CrossRef PubMed.
- Y. Bai, Z. Ming, Y. Cao, S. Feng, H. Yang, L. Chen and S. T. Yang, Colloids Surf., B, 2017, 154, 96–103 CrossRef CAS PubMed.
- G. Fang, B. Luan, C. Ge, Y. Chong, X. Dong, J. Guo, C. Tang and R. Zhou, Carbon, 2017, 121, 285–291 CrossRef CAS.
- Y. Wang, Z. Zhu, H. Zhang, J. Chen and B. Tang, Mater. Chem. Phys., 2016, 182, 272–279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00807a |
| This journal is © The Royal Society of Chemistry 2020 |