
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptical writing and single molecule reading of photoactivatable and silver nanoparticle-enhanced fluorescence†
Nicholas P.
Dogantzis‡
 ,
Gregory K.
Hodgson‡
and
Stefania
Impellizzeri
*
,
Gregory K.
Hodgson‡
and
Stefania
Impellizzeri
*
Laboratory for Nanomaterials and Molecular Plasmonics, Department of Chemistry and Biology, Ryerson University, 350 Victoria St., Toronto, ON M5B 2K3, Canada. E-mail: simpellizzeri@ryerson.ca
First published on 28th February 2020
Abstract
We designed a hybrid nanoparticle–molecular system composed of silver nanostructures (AgNP) and a fluorogenic boron dipyrromethene (BODIPY) that can be selectively activated by UVA or UVC light in the presence of an appropriate photoacid generator (PAG). Light irradiation of the PAG encourages the release of p-toluenesulfonic, triflic or hydrobromic acid, any of which facilitate optical ‘writing’ by promoting the formation of a fluorescent species. Metal-enhanced fluorescence (MEF) by AgNP was achieved through rational design of the nano–molecular system in accordance with the principles of radiative decay engineering. In addition to increasing signal to noise, AgNP permitted shorter reaction times and low irradiance – all of which have important implications for applications of fluorescence activation in portable fluorescence patterning, bioimaging and super-resolution microscopy. Single molecule fluorescence microscopy provided unique insights into the MEF mechanism which were hidden by ensemble-averaged measurements, demonstrating that single molecule ‘reading’ is a valuable tool for characterizing particle–molecule interactions such as those responsible for the relative contributions of increased excitation and plasmophoric emission toward overall MEF. This work represents a step forward in the contemporary design of synergistic nano–molecular systems, and showcases the advantage of fusion between classic spectroscopic techniques and single molecule methods in terms of improved quantitative understanding of fluorophore–nanoparticle interactions, and how these interactions can be exploited to the fullest extent possible.
Introduction
Organic molecules can absorb radiation in the ultraviolet and visible regions of the electromagnetic spectrum.1 Some excited species can then radiatively release the absorbed energy in the form of fluorescence.2 Metal nanostructures are known to impact both of these processes via mechanisms such as electron transfer,2,3 thermal effects (i.e. plasmonic heating),4 or near-field electromagnetic interactions (this work).2,3a,c,d,5 The latter are well-described by the theory of metal-enhanced fluorescence (MEF), explaining the effective enhancement or quenching of fluorescence depending upon the optical properties of plasmonic nanostructures and their relationship to those of the dye.2,5a,e The efficiency with which a particular fluorophore absorbs a photon of excitation light is a function of the molecular cross-section, and the likelihood of absorption is known as the extinction coefficient (larger extinction coefficients indicate that absorption at a given wavelength is more probable). The fluorescence quantum yield of the fluorochrome denotes the ratio of the number of quanta emitted compared to the number absorbed.2 Extinction coefficient, quantum yield and fluorescence lifetime (average time a species remains in an excited state prior to returning to the ground state by emitting a photon) all contribute to the utility of chemical systems for fluorescence-based applications, and all are important factors in the rational design of nanoparticle–molecular systems intended to exploit MEF (i.e. radiative decay engineering).5 It follows that the study of nano–molecular interactions governing processes such as MEF is critical to furthering the development of related applications.Organic chromophores can also be designed to switch from a nonemissive state to a fluorescent one under optical control.6 Such photoactivatable systems permit only a small, selective population of molecules to fluoresce at a given time, (those that have been ‘activated’). In turn, external control over the ability of fluorochromes to emit enables a variety of advanced light-based applications in super-resolution microscopy and sensing.7 In this context, the identification and study of mechanisms to photoactivate and enhance fluorescence can greatly facilitate the implementation of cutting-edge optical techniques and devices. These techniques can be designed to ‘read’ information, as in fluorescence microscopy, which exploits the photophysical behaviour of fluorophores and concomitantly acquires information on a specimen or process. Conversely, methods to ‘write’ information, as in lithography, instead rely upon photochemical processes to fabricate a pattern into a material. With respect to the latter, optical writing can be rapid and accurate, with the possibility for even greater performance by incorporating MEF (e.g. increased speed and sensitivity). Fluorescent or pre-fluorescent dyes have been used in microlithography to study the mechanism and efficiency of photoacid8 and free radical initiators,8e,9 and have delivered valuable insights on the generation and spatial distribution of polymerization reactions. Nonetheless, activatable fluorophores could also be used for the development of lithographic-like features through the precise and rationally designed generation of functional fluorescent images. This ingenious strategy ultimately delivers a viable, practical hybridization between imaging and lithography – patterning through fluorescence – provided that the fluorescent features (i) are externally controlled (i.e. by photoactivation) and (ii) have relatively high fluorescence outputs.
Upon the basis of these considerations, we have designed a hybrid system composed of a switchable fluorophore that can be selectively activated by UV light in the presence of a photoacid generator (PAG) and concomitantly enhanced by metallic silver nanoparticles (AgNP). We further investigated the link between nanoparticle-enhanced fluorescence observed at the steady state and single molecule levels, where single molecule reading was expected to be capable of elucidating the mechanism of MEF, laying the groundwork for additional performance optimization in this system and improved design of fluorescence-based nano–molecular systems in general. We foresee that this synergistic combination of photochemical and photophysical properties can become a general strategy for developing exceptional photoresponsive materials, offering advantages that would not be accessible through the separate organic or nanostructured components alone, with the potential for profound implications in lithography, imaging, biotechnology and information storage.
Results and discussion
Steady state spectroscopy
Compound 1 is based on a boron-dipyrromethene (BODIPY) fluorophore where a catecholate chelator was used to replace the two fluoro groups on the boron centre (Scheme 1). Spectroscopic data from our and other10 laboratories demonstrated that this process suppresses fluorescence of the BODIPY almost completely, due to the transfer of one electron from an occupied molecular orbital centred on the catecholate chelator to an unoccupied molecular orbital localized on the adjacent BODIPY platform. The absorption spectrum of species 1 (Fig. 1, a) shows the typical band of the BODIPY chromophore at λAbs = 505 nm, while its emission spectrum (Fig. 1, b) reveals that the fluorescence is strongly quenched. Nevertheless, the catecholate chelator of the nonemissive species 1 can be replaced by two methoxide groups to form the fluorescent species 2 in methanol under acidic conditions (Scheme 1), as demonstrated by the appearance of an intense emission band at λEm = 518 nm upon the addition of ten equivalents of p-toluenesulfonic acid (PTSA, ‘c’ in Fig. 1). 1H nuclear magnetic resonance (NMR) spectra recorded before and after the addition of PTSA in deuterated methanol are also consistent with the transformation of 1 into 2. After addition of PTSA to 1, it was observed that the resonance of the two pairs of protons H1 and H2, originally centred at δH = 6.77 ppm (Fig. S3, a†) in the spectrum of 1, splits into two new sets of resonances (δH = 6.76 and δH = 6.65 ppm respectively, Fig. S3, b†). These observations are fully consistent with the formation of free catechol (Fig. S3, c†), as demonstrated by the presence of the identical resonance pattern in the 1H NMR spectrum of catechol recorded under the same conditions. 11B NMR spectra of a CD3OD solution of 1 recorded before (Fig. S4, a†) and after (Fig. S4, b†) treatment with PTSA are in accordance with the proposed transformation.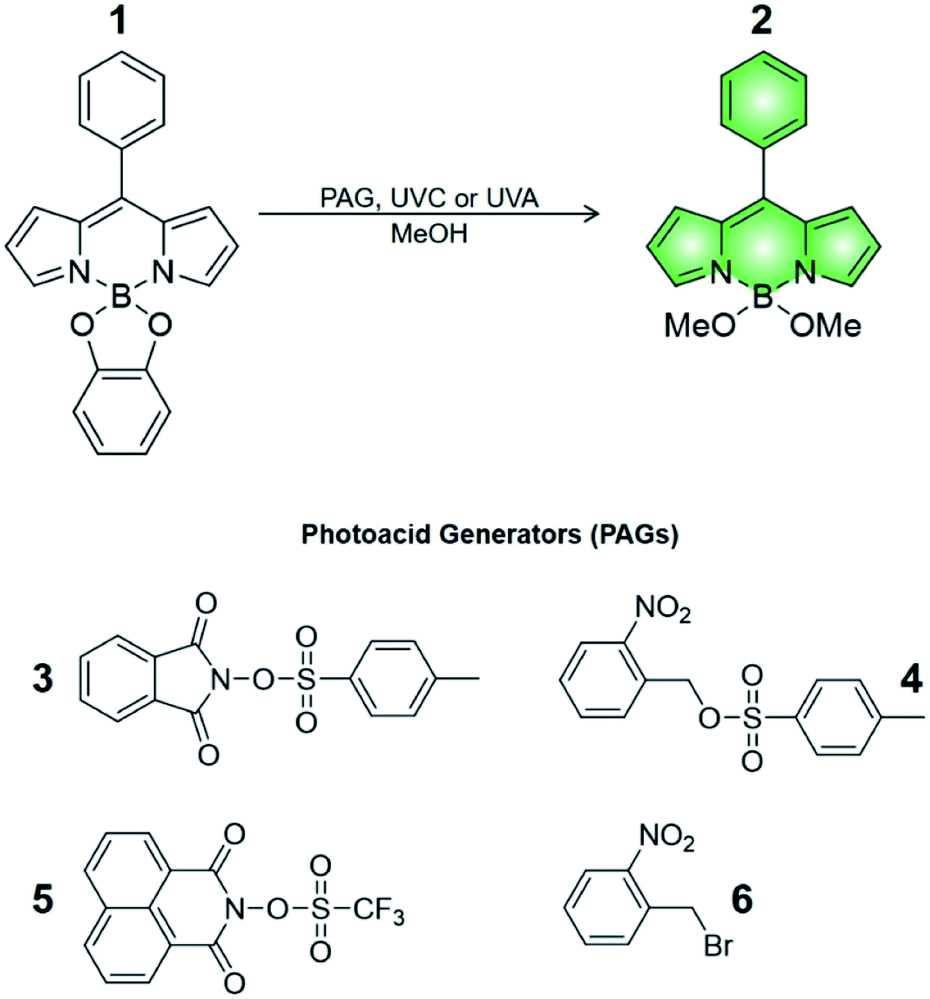 | ||
| Scheme 1 Structures of compounds 1–6. | ||
 | ||
| Fig. 1 Absorption spectrum (a) of a solution of 1 (10 μM, 20 °C) in MeOH. Emission spectra of a solution of 1 (10 μM, 20 °C, λEx = 490 nm) before (b) and after (c) the addition of 10 equivalents of PTSA in the dark. | ||
The transformation of 1 into 2 can also be encouraged with the assistance of photoacid generators 3 and 4, both reported herein (Scheme 1). These compounds are able to release PTSA under illumination at an appropriate wavelength (254 nm for 3 and 365 nm for 4). Thus, compounds 3 and 4 can mediate the photochemical formation of 2 and hence activate its fluorescence. The photochemically-induced generation of PTSA from aromatic N-oxyimidosulfonates such as 3 has been previously investigated by absorption spectroscopy11 and laser flash photolysis;12 these studies revealed that the excited state involved in the process is the singlet state of the PAG and that the photoreaction proceeds through homolytic cleavage of the N–O bond. In parallel, we adapted the established UVA photochemistry of the 2-nitrobenzyl group13 to photogenerate acid via compound 4. Specifically, ultraviolet illumination of 4 results in the formation of 2-nitrosobenzaldehyde and PTSA. In the presence of 10 equivalents of either 3 (Fig. 2) or 4 (Fig. 3) and after illumination at 254 or 365 nm, respectively, the emission spectra of solutions of 1 show acid-induced conversion into 2 with concomitant fluorescence ‘turn-on’ comparable to that observed after direct addition of PTSA. While small changes are observed in the ultraviolet region of the absorption spectrum of a mixture of 1 and 3 (‘a’ and ‘b’ in Fig. 2) upon illumination at 254 nm, the absorption spectrum of a solution of 1 and 4 recorded before (Fig. 3, a) and after (Fig. 3, b) irradiation at 365 nm shows a dramatic decrease of the band associated with 4 (260 nm) and the concomitant appearance of a new band for 2-nitrosobenzaldehyde (ca. 310 nm), in agreement with literature precedents14 and with our photolysis experiments performed upon 4 alone (Fig. S5†). Therefore, the PTSA released during the photolysis of the acid generators encourages the conversion of 1 into 2 and, as a result, activates the fluorescence of the probe. In comparison, UVC or UVA irradiation of 1 alone only partially encourages the self-activation of 1 (Fig. S6 and S7†), confirming that the PAGs are required to significantly turn on the fluorescence. Importantly, the absorption band in the visible region remains essentially unaffected under ultraviolet illumination, demonstrating that the BODIPY moiety is stable under these irradiation conditions.
 | ||
| Fig. 2 Absorption (a and b) and emission (c and d) spectra of a solution of 1 (10 μM, 20 °C, λEx = 490 nm) and 3 (100 μM) before (a and c) and after (b and d) irradiation at 254 nm (0–20 min). | ||
 | ||
| Fig. 3 Absorption (a and b) and emission (c and d) spectra of a solution of 1 (10 μM, 20 °C, λEx = 490 nm) and 4 (100 μM) before (a and c) and after (b and d) irradiation at 365 nm (0–20 min). | ||
The behaviour of our switchable probe 1 in the presence of PTSA-generators prompted us to investigate the fluorescence activation dynamics with hydrobromic (HBr) and trifluoromethanesulfonic acids (TFSA, or triflic acid), respectively. Indeed, a previous literature report10b has shown that 1H NMR and mass spectra of 1 obtained before and after treatment with HBr are consistent with the replacement of the catecholate receptor of 1 with the two methoxide groups of 2, which we observed upon acidification with PTSA. The fluorogenic probe 1 can therefore be activated with the assistance of different acids. Upon the basis of these considerations, we investigated whether such activation could also be achieved under optical control with the aid of photoacid generators 5 and 6 (Scheme 1). Compound 5 is a commercially available naphthalimide-based PAG that can be irradiated at 254 nm to release TFSA in solution. Similar to compound 3, laser flash photolysis studies showed that the excited state involved in the process is the singlet state of the PAG; however, unlike 3, the photoreaction proceeds through heterolytic cleavage of the N–O bond, likely due to the stronger leaving group character of the triflate anion.12 On the other hand, PAG 6, also available commercially, incorporates the 2-nitrobenzyl moiety which prompts the release of HBr to occur concomitantly with the photolysis of the 2-nitrobenzyl group and formation of 2-nitrosobenzaldehyde after irradiation at 365 nm. In agreement with these observations, the emission spectrum of 1 in the presence of 5 or 6 under photoirradiation (Fig. S8†) show the growth of a band for the photogenerated chromophore 2. All results are summarized in Fig. 4. It is important to notice that the PAG-induced fluorescence enhancements in Fig. 4 (entries D, E, H and I) have been compared against control experiments consisting of 1 in the dark (entry A, Fig. 4) as well as 1 irradiated with either 254 or 365 nm in the presence and absence of PTSA, rather than simply the addition of acid in the dark. Although acid will convert 1 into 2 without light, performing these two control experiments (entries C and G, Fig. 4) under reaction conditions conveniently accounted for additional non-PAG-induced conversion resulting from: (i) self-sensitization of 1 (Fig. S6†); and (ii) thermal effects (due to the difference in heating by the UVA LEDs vs. the UVC photoreactor, vide infra), both of which affect the kinetics of the ligand exchange process.
 | ||
| Fig. 4 Fluorescence emission intensities (λEm = 518 nm) in the absence or presence of PAGs in MeOH solutions. | ||
Within UVC-operating PAGs, our own compound 3 (entry D, Fig. 4) exhibited a level of photoactivation of fluorescence that is on par with chemical activation by direct addition of PTSA (entry C, Fig. 4) – over 30-fold enhancement. In contrast, the commercially available TFSA-generator 5 (entry E, Fig. 4) was less successful in activating the fluorescence of 1. This is consistent with the higher fluorescence quantum yield of 5 relative to 3 (Fig. S9†), which has been shown to be inversely proportional to the quantum yield of photoacid generation by N-oxyimidosulfonates as a result of competition between the two processes.11 The instability of triflic acid in MeOH may also have contributed to the lower fluorescence enhancement obtained using 5. For UVA-operating PAGs, our compound 4 (entry H, Fig. 4) efficiently enhanced fluorescence (65-fold) upon PTSA photorelease, and the commercial halogenated compound 6 (entry I, Fig. 4) induced a 75-fold increase. While 4 is expected to exhibit a slightly higher rate of photoacid generation than 6 due to the superior leaving group ability of the resonance-stabilized tosylate anion, kinetic studies have found that the rate of photodecomposition of 2-nitrobenzyls is largely independent of the acid moiety.15 Consequently, it appears that the moderately better performance of 6 is due to a lower pKa and less steric bulkiness of HBr vs. PTSA, allowing it to more readily convert 1 into 2 within a practical experimental duration of 20 minutes.
Overall, the results summarized in Fig. 4 indicate that UVA illumination is preferred over UVC when PTSA is photogenerated in solution. While self-sensitization of 1 is consistent at 12-fold fluorescence enhancement using either wavelength (entries B and F in Fig. 4), the maximum theoretical fluorescence enhancement (represented by the direct addition of 10 equivalents of PTSA) is 40-fold under UVC light vs. 95-fold under UVA light (entries C and G, Fig. 4). We investigated the possibility that PTSA itself might be affected differently by irradiation at the two wavelengths. Photostability tests on PTSA under 254 and 365 nm working irradiation conditions, performed by monitoring its absorption profile with increasing irradiation time, seem in part to corroborate this hypothesis: data shows that the maximum of absorption of PTSA at 220 nm decreases by ∼14% under UVC light while maintaining stability under UVA illumination (Fig. S10†). In addition, we found that in solution, the higher operating temperature of the UVA LED illumination system encourages the exchange of the catecholate receptor in 1 with the methoxide ligands. Inserting a thermocouple into a cuvette containing MeOH revealed the operating temperature of the UVA LEDs to be a significant 8.8 °C higher than that of the UVC photoreactor under experimental conditions (35.3 °C vs. 26.5 °C, respectively). Indeed, the fluorescence intensities of mixtures of 1 and PTSA (10 equiv.) progressively heated in MeOH confirmed our hypothesis (Fig. S11†). Interestingly, the relative efficiencies of UVA and UVC illumination are effectively reversed in thin polymer films, where the rigid polymeric matrix imposes stringent limitations upon the diffusion of the species (vide infra).
Photoactivated fluorescence in thin films
The photoinduced transformation of 1 into 2 assisted by PAGs can be reproduced within a polymer matrix. Emission spectra of thin polyvinylpyrrolidone (PVP) films (‘a’ and ‘c’ in Fig. 5) doped with 1 and either PAG 3 or 4 (spin coated on glass microscope slides from a MeOH solution as described in the ESI†) show a band (λEm = 520 nm) for the quenched BODIPY chromophore 1. Following irradiation at 254 nm (for films containing 1 and 3) or 365 nm (for films containing 1 and 4) for 40 minutes, we observed an increase in fluorescence intensity consistent with the transformation of the non-emissive moiety 1 into the fluorescent species 2. Residual MeOH in the spin coated formulation is sufficient to allow the reaction to proceed in good yield. Thus, the acid generated during the photochemical uncaging of 3 and 4 can encourage the transformation of 1 into 2 within the polymer matrix with concomitant fluorescence activation. Notably, in contrast to experiments performed in MeOH solution, PAG-induced fluorescence enhancement in thin films was more effective using 3 and UVC light relative to 4 and UVA light over an equivalent duration of illumination. | ||
| Fig. 5 Top: Emission spectra of a PVP (55 kDa, 20% w/v in MeOH) film containing 1 (500 μM, λEx = 490 nm) and 3 (10 mM) before (a) and after (b) exposure at 254 nm for 40 min. Bottom: Emission spectra of a PVP (55 kDa, 20% w/v in MeOH) film containing 1 (500 μM, λEx = 490 nm) and 4 (10 mM) before (c) and after (d) exposure at 365 nm for 40 min. | ||
Photographs of doped thin PVP films before and after illumination visually confirm the photoinduced release of PTSA from PAGs 3 and 4 and the formation of the fluorescent product 2. Specifically, illumination of PVP films of 1 and 3 at 254 nm through a 3D printed mask for 20 minutes converts 1 into 2 exclusively in the irradiated area, producing a defined fluorescent image (‘b’ in Fig. 6). In contrast, no pattern is formed under the same illumination conditions in the absence of PAG 3 (Fig. S12†). Remarkably, the acquisition of an additional photograph (‘c’ in Fig. 6) 10 minutes after exposure reveals no noticeable change in the shape or geometry of the imprinted pattern. This outcome demonstrates that, from a macroscale perspective, diffusion of the photogenerated fluorophores within the rigid polymeric matrix is significantly restricted, preventing distortion of the pattern on this time scale. The written pattern can thus be available for optical reading over longer periods of time. Similar results were obtained upon irradiating PVP films of 1 and 4 at 365 nm, again through the RU-shaped 3D printed mask (Fig. S13 and S14†).
 | ||
| Fig. 6 Photographs of a thin PVP (55 kDa, 20% w/v in MeOH) film containing 1 (500 μM) and 3 (10 mM) before (a), immediately (b) and 10 minutes (c) after exposure at 254 nm for 20 min through a RU-shaped 3D printed mask. | ||
Nanoparticle-enhanced fluorescence
The utilization of metallic nanostructures is a very promising means of improving the efficiency of photochemical and photophysical processes of organic fluorophores. Near-field interactions (i.e. occurring at distances less than the wavelength) between chromophores and the electron clouds present in metal colloids can augment the photophysical processes responsible for far-field emission through MEF.2,5 Indeed, metal nanoparticle surfaces can respond to the oscillating dipole of radiating fluorophores by effectively modifying the rates of excitation and emission, ultimately improving brightness. The latter is the product of the molecular extinction coefficient and fluorescence quantum yield; MEF can improve brightness by acting on either component (excitation or emission rates) alone or in concert. Fluorochromes up to about 200 nm away from AgNP may experience MEF,5a,e and are more readily excited due to the concentrated electric field around the metal. A greater number of excited fluorophores naturally leads to more fluorescence. Fluorochrome–nanoparticle interactions are further known to improve fluorescence quantum yields, through the formation of a NP–fluorophore complex (a ‘plasmophore’). Relative to the fluorophore alone, NPs endow the corresponding plasmophore with a much lower excited state lifetime, increasing the radiative decay rate and thereby improving the fluorescence quantum yield.2,5 The two mechanistic components of MEF can thus be exploited to increase steady state brightness. Though indistinguishable at the ensemble level, each component should manifest differently at the single molecule level (vide infra). From an application perspective, multiple avenues to achieving MEF offer a versatile means of judiciously engineering increased fluorescence intensity for patterning, energy harvesting, fluorescence imaging and single molecule bioassays.Upon the basis of these considerations, we sought to determine whether fluorescence could be enhanced through near-field, non-covalent interactions with metallic silver nanostructures. Triangular AgNP were synthesized through a photochemical seed-growth method by irradiating a solution of silver seeds with 590 nm LEDs (details in the ESI†). The resulting nanostructures have an average diameter of 84 ± 4 nm measured by scanning electron microscopy (Fig. S15†). This morphology and size regime were selected upon the basis of the reasonable expectation that it would lead to MEF for fluorophores emitting in the 400–600 nm range (complementary to 1 and 2). That is, the scattering component of the extinction spectrum is generally higher for larger, non-spherical nanostructures, and the scattering efficiency of AgNP is known to become high for diameters exceeding 60 nm.5a,e According to the radiative plasmon model of MEF, dominance of NP scattering over NP absorption at the fluorophore emission wavelength is critical to the observation of plasmophore emission (recall that the latter is the component of MEF responsible for increasing fluorescence quantum yield).5 In this case, the AgNP surface plasmon (SP) band centred at 665 nm is located away from the emission of 1 and 2 (compare Fig. 2d and 3d with Fig. S16,† right). Depending upon the relative contributions of scattering and absorption in the region of spectral overlap between fluorophore emission and AgNP extinction, this design may assist or diminish the overall efficiency of MEF: assist when the fluorophore emission overlaps with scattering, and diminish with absorption. Nonetheless, our results provide experimental evidence that radiating surface plasmons can indeed be indirectly generated by excited state fluorophores when the far-field excitation wavelength (in this case, λEx = 490 nm) does not interact with the metallic nanostructures directly. Remarkably, the successful observation of MEF in this system (vide infra) experimentally verifies published finite-difference time-domain (FDTD) calculations which illustrate the unique electric field distribution induced by fluorophores located in the near-field of AgNP,5e implying that MEF can indeed occur via indirect nanoparticle excitation and highlighting one of the complexities of nano–molecular interactions.
Thin films for MEF experiments were prepared as previously described, with the exception that glass slides were first coated with concentrated AgNP prior to depositing 1 in PVP. Specular reflectance was used to verify that the PVP layer is approximately 57–82 nm thick (Fig. S17† and associated discussion), consistent with previous research conducted using similarly prepared samples.16c This thickness represents a maximum in terms of nanoparticle–fluorophore distance; fluorophores are distributed throughout the polymer film and thus are located at various distances from AgNP, up to just over 80 nm. No further attempt was made to optimize the AgNP–fluorophore distance for MEF. Given the importance of donor–acceptor distance in other Coulombic energy transfer mechanisms (e.g. FRET), future work may investigate the link between MEF efficiency and nanoparticle–fluorophore distance in greater detail, by synthetically functionalizing fluorogenic species to facilitate non-covalent nano–molecular interactions or even covalent linkage of fluorophores to the AgNP surface. However, we also note that in the trade-off between optimization and over-complication, simpler systems can sometimes offer greater practical accessibility while still providing significant performance gains. In this case, ease of sample preparation combined with experimental evidence of successful MEF represented a convenient and effective route to studying AgNP-enhanced photoactivation of 1 at the bench-scale and single molecule levels.
The UV-visible extinction spectrum of the sample shows the absorption band of the BODIPY chromophore 1 (λAbs = 505 nm) and the surface plasmon band of AgNP at 665 nm (Fig. S16†). A solution of 1 and 4 in PVP was then spin coated atop AgNP-functionalized glass slides and irradiated at 365 nm. Emission spectra of the films reveal that the PAG-assisted photoconversion of 1 into 2 can also be performed in the presence of AgNP. Moreover, fluorescence intensity atop AgNP is visibly enhanced with respect to the same experiment conducted in the absence of the nanostructures (Fig. 7). In agreement with MEF theory, the effect is most dramatic for lower quantum yield fluorophores because AgNP can only increase the quantum yield of fluorescence to a maximum of unity.2,5 In our case, this translates into 1 being more enhanced than 2, and thus we observed higher enhancement factors for lower irradiation times as a result of a smaller concentration of the photogenerated species 2 (e.g. 265% enhancement at time zero versus 125% after 60 minutes, Fig. 7). Nevertheless, conversion of 1 into 2 in the presence of AgNP still results in a net enhancement to the steady state fluorescence intensity while allowing for the utilization of fluorescence activation. Notably, the fluorescence output obtained after only 20 min of irradiation in the presence of AgNP is comparable to that achieved after irradiating for 40 min in the absence of nanoparticles, effectively reducing the necessary irradiation time by half. Nanomaterial-based strategies can thus meaningfully improve the performance of fluorescence-based optical techniques by allowing for faster and superior results. Intrigued by the potential for further performance enhancements by maximizing the MEF effect, we turned to single molecule fluorescence microscopy to obtain a more detailed understanding of the multifaceted MEF mechanism at work in this system.
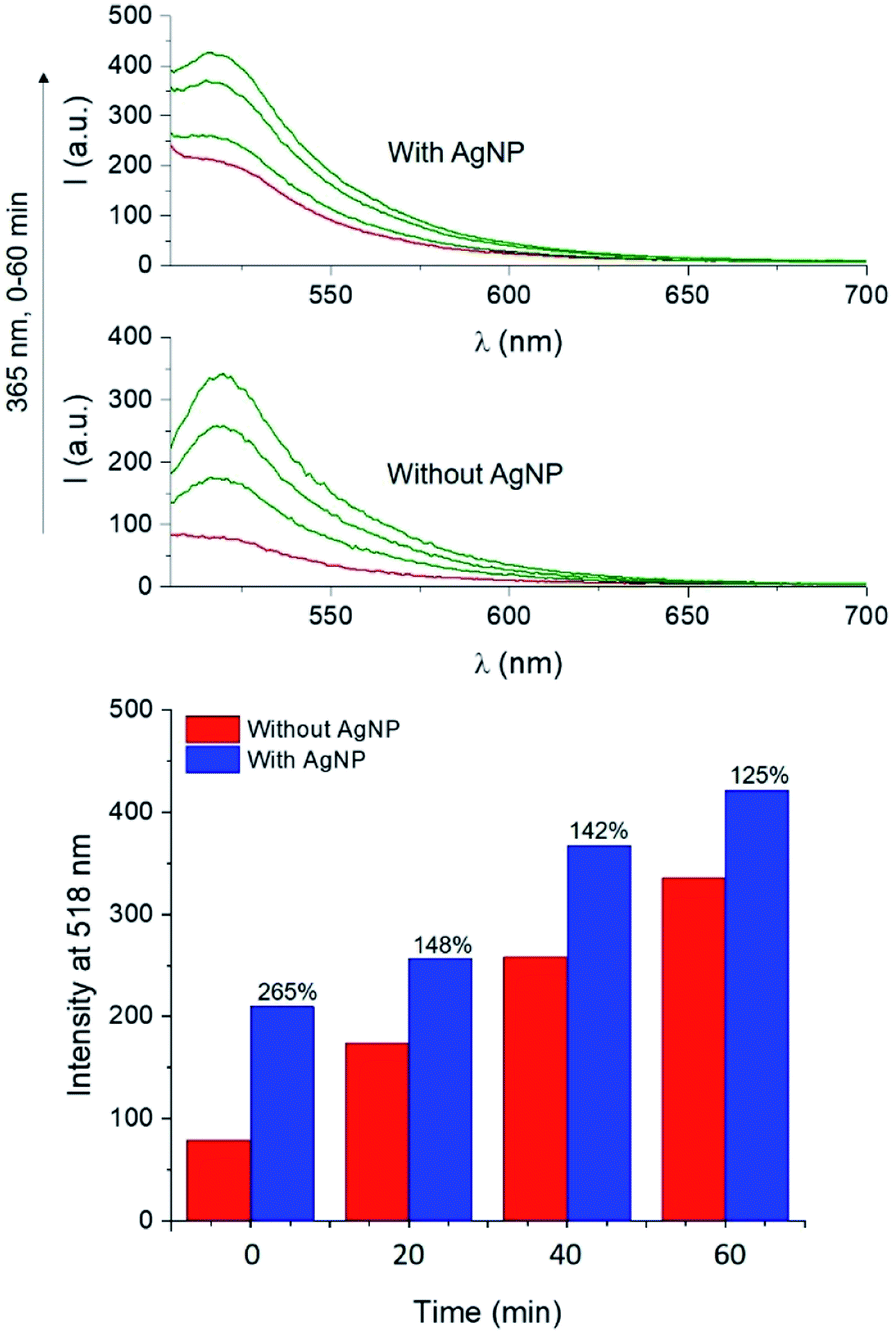 | ||
| Fig. 7 Top: Fluorescence spectra of PVP (55 kDa, 20% w/v in MeOH) films containing 1 (500 μM, λEx = 490 nm) and 4 (10 mM) in the presence or absence of AgNP, irradiated at 365 nm (0–60 min). Bottom: Fluorescence intensity recorded at 518 nm for the same films. | ||
Single molecule fluorescence imaging
Powerful imaging techniques traditionally employed in the biological sector are becoming increasingly available to chemists,16 providing continuity between ensemble-averaged bench-scale spectroscopic methods and single molecule level investigations.Our objective in studying this system at the single molecule level is two-fold: (1) to unequivocally confirm that MEF is responsible for the steady state fluorescence enhancement observed at the macroscale; (2) to set the stage for performance optimization by first ascertaining whether one or both components of the MEF phenomenon (enhanced excitation or quantum yield) contributed to the overall fluorescence enhancement. This single molecule investigation was conducted by performing total internal reflection fluorescence microscopy (TIRFM) upon thin PVP films containing 1 and 4 on glass coverslips, where approximately half of the substrate area was functionalized with AgNP prior to film deposition. Here, the total sample thickness (PVP atop AgNP) of approximately 100 nm conveniently situated molecules of 1 within the MEF region while at the same time fitting well within the region of enhanced signal to noise provided by TIRFM (ESI†).
Regarding the first objective, Fig. 8 shows that fluorescence intensity is significantly higher and less uniform in the sample region containing AgNP (as much as five fold higher in certain ‘hot-spots’ where MEF is most efficient). This semi-quantitative measure provides visual confirmation of nanoparticle-mediated fluorescence enhancement at the single molecule level. In fact, the presence of ‘hot-spots’ highlights the heterogeneity of the fluorescence enhancement which becomes evident at the single molecule level, and rules out the possibility that steady state fluorescence enhancement could be purely the result of increased dye loading stemming from a higher sample surface area in the presence of AgNP.5fFig. 9 illustrates single molecule intensity–time trajectories for discrete locations within the AgNP area of the sample shown in Fig. 8. Sharp signal increases in fluorescence intensity–time trajectories correspond to emission from single molecules of 1 or 2, or AgNP-1 or AgNP-2 plasmophores. For simplicity, we refer to these events as ‘fluorescence bursting’ or ‘bursts’. As shown in Fig. 10, the relative number of occurrences of these bursting events is indeed higher in the presence of AgNP (even considering 60% NP coverage by area), consistent with an increased rate of fluorophore excitation caused by MEF.
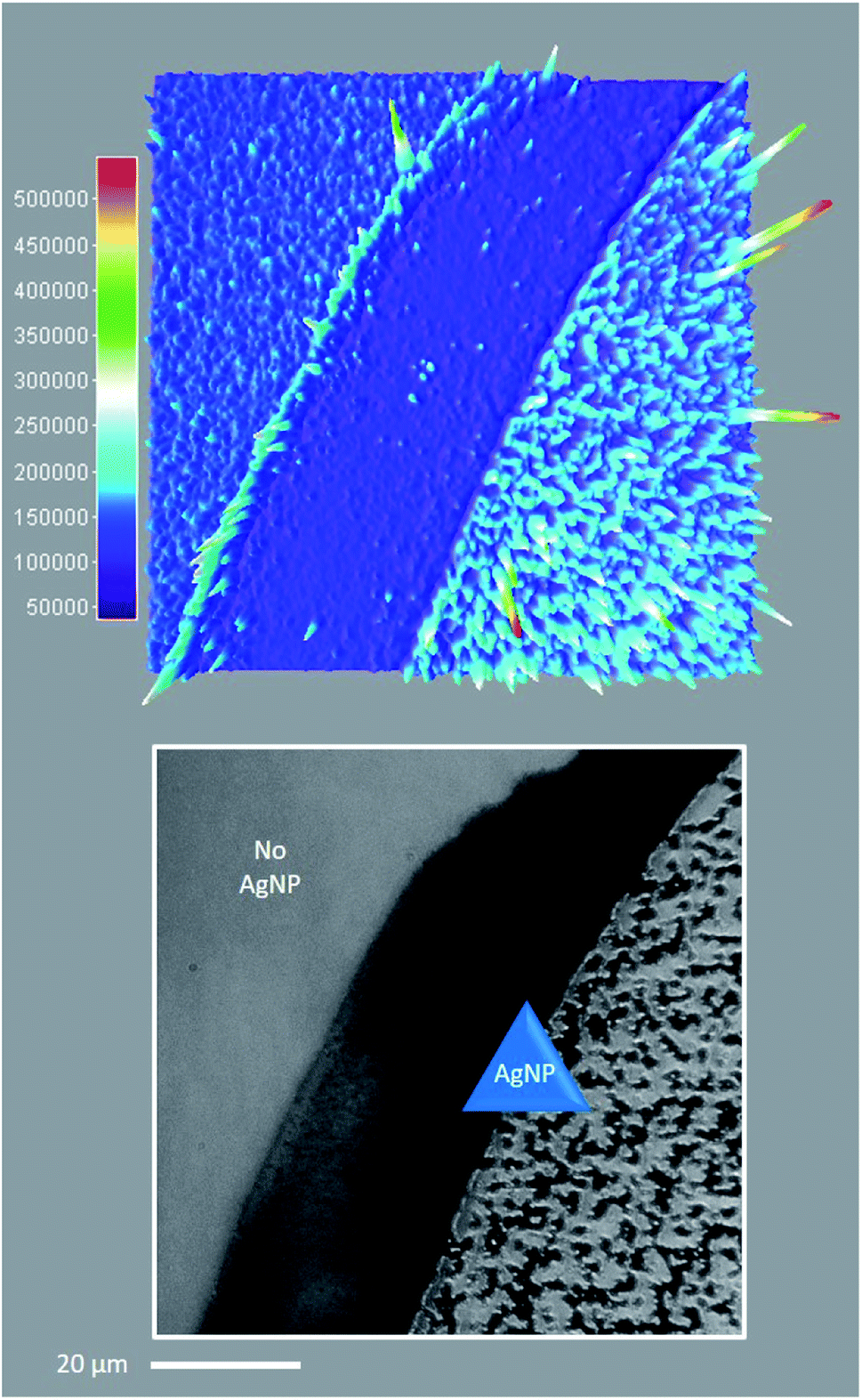 | ||
| Fig. 8 Single molecule TIRFM of thin PVP (55 kDa, 20% w/v in MeOH) films containing 1 (100 nM) and 4 (2 μM) in the presence or absence of AgNP, irradiated at low intensity 365 nm for 1 min. See ESI Video.† Top: 3D surface projection (top-down view) of the TIRFM region of interest, illustrating the spatial distribution of cumulative fluorescence intensity per pixel over 1000 frames. Bottom: Corresponding brightfield image showing the distribution of AgNP across the sample area. Note that the scale bar applies to both top and bottom, and 60% of the sample area contains AgNP. | ||
 | ||
| Fig. 9 Single molecule intensity–time trajectories in the presence of AgNP. | ||
 | ||
| Fig. 10 Numbers of single molecule fluorescence bursting events in the presence and absence of AgNP. | ||
Moving to the second objective, mechanistic insights into reaction dynamics and nano–molecular interactions uniquely obtained through single molecule techniques are no longer uncommon, but most relate to model reactions and/or nanoparticle catalysis.2,5e,f,16 Here instead, additional understanding of a complex system of non-catalytic, photoactivated florescence combined with MEF was gleaned by statistical analysis of the distribution of single molecule fluorescence bursting intensities, which can be related back to the relative contributions of increased excitation and fluorescence quantum yield toward overall MEF. As shown in Fig. 11, burst intensities (average 703 ± 453) after 1 minute of irradiation at 365 nm in the presence of AgNP roughly follow a log-normal distribution and exhibit a high relative standard deviation of 64.4%. The latter is characteristic of both MEF5f and log-normal distributions,17 which are actually quite prevalent across the sciences and have even been shown to describe ensemble-averaged fluorophore emission.18 Whereas Gaussian distributions result from the additive uncertainties of several independent variables, log-normal distributions describe variability when such effects are multiplicative.17 Among other factors (e.g. variable fluorophore–NP distances and orientations, AgNP polydispersity), the burst intensities summarized in Fig. 11 are proportional to the brightness of single molecules, the variability of which is the product of the uncertainties in molecular extinction coefficient and fluorescence quantum yield. It follows that the variability in burst intensity should follow a log-normal distribution.
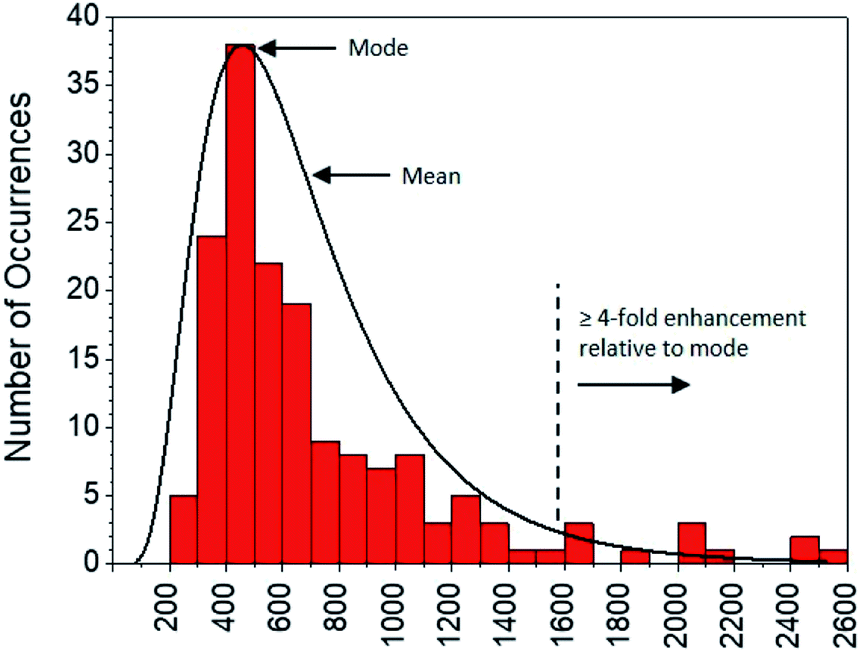 | ||
| Fig. 11 Distribution of burst intensities in the presence of AgNP after exposure to 365 nm for 1 min. | ||
The majority of burst intensities in Fig. 11 are within the 400–500 range (mode = 400). Importantly, the most intense bursts in Fig. 11 were not simply caused by the higher intrinsic fluorescence intensity of 2 relative to 1: high burst intensities (e.g. ≥4-fold enhancement relative to the mode) only occurred in the presence of AgNP (compare Fig. 9 and S18†), and AgNP also increased steady state fluorescence prior to UVA irradiation (Fig. 7, 0 min). In fact, less than 7% of the bursts in Fig. 11 fall outside the log-normal distribution, coinciding with at least a 4-fold intensity enhancement factor. This is somewhat low with respect to the limited number of comparable studies in the literature,5e,f and suggests that ‘hot-spots’ corresponding to plasmophoric emission (the tallest signals in Fig. 9) may represent a proportionately low fraction of the MEF observed after 1 minute of irradiation at 365 nm under TIRFM conditions. In accordance with MEF theory, the balance of fluorescence enhancement is therefore the result of an increased rate of excitation caused by the presence of a concentrated electric field near the AgNP,2,5e which is consistent with the dramatically higher number of bursting events detected in the presence of AgNP (Fig. 10). Remarkably, this distribution between the two components of MEF could be observed at the single molecule level after just 1 minute of low intensity irradiation from a TLC lamp (0.4 mW cm−2), despite being obscured by ensemble-averaged measurements. This investigation showcases the utility of single molecule techniques and demonstrates the practical value of MEF for applications based on fluorescence activation (e.g. rapid fluorescence activation-based patterning using portable, low-power light sources).
This single molecule study suggests that performance might be improved by optimizing the physical and optical properties of AgNP to increase the plasmophoric component of MEF which, as stated earlier, is directly related to the balance between scattering and absorption by NP at the fluorophore emission wavelength.5a At this time, we have focused on introducing the use of single molecule techniques to characterize a snapshot of near-field nano–molecular interactions in complex chemical systems, and also have yet to examine the possibility that AgNP may affect reaction kinetics (e.g. enhancing the conversion of 1 to 2 by facilitating PAG cleavage). In order to maximize the utility of MEF, future work will therefore concentrate on elucidating the increasingly complex relationship between scattering and absorption for non-spherical NPs, and will complete the picture by also examining MEF as a function of reaction progress as well as the possible additional impact of AgNP upon the efficiency of photochemical fluorescence activation.
Conclusions
We have designed a molecular system composed of a fluorogenic BODIPY that can be selectively activated by UVA or UVC light in the presence of a photoacid generator. Two PAGs designed to release PTSA were synthesized and their spectroscopic behaviour investigated in the presence of the switchable BODIPY chromophore. Selective ultraviolet irradiation of PAGs urges the release of PTSA, which promotes the replacement of a catecholate chelator of the nonemissive species by two methoxide ligands in a methanolic environment, thus forming a fluorescent species. The behaviour of our PAGs in solution was compared to that of two commercially available UV-activated PAGs. Our analysis revealed that PAG-assisted, photoactivated fluorescence has enhanced tolerance to UVA illumination conditions in solution, due in part to a built-in thermal effect that encourages the ligand exchange. In polymer films however, fluorescence is more efficiently turned on by the UVC-activated PAGs used in this work, due to the limited diffusion of the reacting species imposed by the rigid polymer matrix.This investigation demonstrated that MEF by AgNP can be employed to improve performance in applications of fluorescence activation, both by increasing steady state fluorescence intensity and by reducing experimental duration and the intensity of the light source required. In addition to facilitating fluorescent patterning using low intensity, portable light sources, this work may have implications in bioimaging and super-resolution microscopy (e.g. Photoactivated Localization Microscopy), where increased sensitivity broadens the range of useable synthetic organic dyes to include many with lower intrinsic fluorescence quantum yields and/or alternative spectroscopic properties (e.g. excitation and emission wavelengths), and where working at lower irradiation intensities can limit sample damage during in vitro or in vivo imaging. In addition, this work expands the utility of MEF by illustrating the possibility of pairing metallic nanoparticles with fluorophores where the latter are excited at wavelengths which do not readily generate surface plasmons by direct far-field irradiation.
Fluorescence microscopy not only demonstrated that PAG-assisted photoactivation of fluorescence can be monitored at the single molecule level, it firmly established the role of MEF and even provided unique insights into the relative contributions of plasmophoric ‘hot-spot’ emission and increased fluorophore excitation toward overall MEF. Single molecule reading suggested that further efforts to optimize MEF in this system should focus on increasing plasmophoric emission, which could be accomplished by improved radiative decay engineering.2,5a Additional optimization of MEF might be realized by designing systems that promote non-covalent nanoparticle–fluorophore interactions. This development marks a significant advancement for applied single molecule fluorescence microscopy in the context of applications of fluorescence activation.
Overall, this study represents a step forward in the rational design of nano–molecular systems, and highlights the benefit of linking single molecule behaviour with bench-scale observations in order to better understand, and ultimately improve, the exploitation of photophysical and photochemical phenomena associated with nano–molecular interactions. In addition to extending our bench-scale and single molecule level investigations to include the possible impact of AgNP upon the photochemical efficiency of PAG-assisted fluorescence activation, as well as the optimal nanoparticle–fluorophore distance for maximum MEF efficiency, next steps will also include using single molecule techniques to characterize the MEF mechanism as a function of reaction progress. More generally, future work will be directed toward applying the principles discussed in this report to the thoughtful development of strategies for optical reading and writing of information based upon synergy between metallic nanoparticles and switchable fluorophores. We foresee that new, complementary strategies to spatiotemporally control emission will deliver practical improvements to the fields of fluorescence spectroscopy, microscopy and lithography.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada (Discovery Grant) and Ryerson University (start-up funds) for supporting this research program. This work has also been supported by the Ryerson University Faculty of Science Dean's Research Fund. N. P. Dogantzis acknowledges the receipt of a NSERC Undergraduate Student Research Award and a Ryerson Graduate Scholarship.Notes and references
- (a) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, Herndon, 2009 Search PubMed; (b) V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, Weinheim, 2014 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed.
- (a) S. A. El-Daly, I. A. Salem, M. A. Hussein and A. M. Asiri, J. Fluoresc., 2015, 25, 379 CrossRef CAS PubMed; (b) A. M. Queiroz, A. V. Mezacasa, D. E. Graciano, W. F. Falco, J.-C. M'Peko, F. E. G. Guimaraes, T. Lawson, I. Colbeck, S. L. Oliveira and A. R. L. Caires, Spectrochim. Acta, Part A, 2016, 168, 73 CrossRef CAS PubMed; (c) C. A. Borges, R. E. Samad, K. O. Goncalves, D. P. Vieira and L. C. Courrol, J. Nanopart. Res., 2018, 20, 165 CrossRef; (d) S. Sharma, R. Uttam, A. S. Bharti and K. N. Uttam, Anal. Lett., 2019, 52, 1539 CrossRef CAS.
- (a) A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30 CrossRef; (b) M. Fedoruk, M. Meixner, S. Carretero-Palacios, T. Lohmuller and J. Feldmann, ACS Nano, 2013, 7, 7648 CrossRef CAS PubMed; (c) G. E. Jonsson, V. Miljkovic and A. Dmitriev, Sci. Rep., 2014, 4, 5111 CrossRef CAS PubMed; (d) L. Jauffred, A. Samadi, H. Klingberg, P. M. Bendix and L. B. Oddershede, Chem. Rev., 2019, 119, 8087 CrossRef CAS PubMed.
- (a) J. R. Lakowicz, Anal. Biochem., 2005, 337, 171 CrossRef CAS PubMed; (b) K. Aslan, Z. Leonenko, J. R. Lakowicz and C. D. Geddes, J. Fluoresc., 2005, 15, 643 CrossRef CAS PubMed; (c) P. Anger, P. Bharadwaj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef PubMed; (d) Y. Fu, J. Zhang and J. R. Lakowicz, J. Fluoresc., 2007, 17, 811 CrossRef CAS PubMed; (e) J. R. Lakowicz, K. Ray, M. Chowdhury, H. Szmacinski, Y. Fu, J. Zhang and K. Nowaczyk, Analyst, 2008, 133, 1308 RSC; (f) S. D. Choudhury, R. Badugu, K. Ray and J. R. Lakowicz, J. Phys. Chem. C, 2012, 116, 5042 CrossRef PubMed; (g) J. W. Liaw, H. Y. Wu, C. C. Huang and M. K. Kuo, Nanoscale Res. Lett., 2016, 11, 1 CrossRef CAS PubMed.
- (a) L. M. Wysocki and L. D. Lavis, Curr. Opin. Chem. Biol., 2011, 15, 752 CrossRef CAS PubMed; (b) D. Puliti, D. Warther, C. Orange, A. Specht and M. Goeldner, Bioorg. Med. Chem., 2011, 19, 1023 CrossRef CAS PubMed; (c) F. M. Raymo, J. Phys. Chem. Lett., 2012, 3, 2379 CrossRef CAS PubMed; (d) W. H. Li and G. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460 RSC; (e) F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840 RSC.
- (a) P. J. Wu, J. L. Chen, C. P. Chen and Y. H. Chan, Chem. Commun., 2013, 49, 898 RSC; (b) K. Hwang, P. Wu, T. Kim, L. Lei, S. Tian, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2014, 53, 13798 CrossRef CAS PubMed; (c) V. Grenier, A. S. Walker and E. W. Miller, J. Am. Chem. Soc., 2015, 137, 10894 CrossRef CAS PubMed; (d) J. M. Goldberg, F. Wang, C. D. Sessler, N. W. Vogler, D. Y. Zhang, W. H. Loucks, T. Tzounopoulos and S. J. Lippard, J. Am. Chem. Soc., 2018, 140, 2020 CrossRef CAS PubMed; (e) H. Li and J. C. Vaughan, Chem. Rev., 2018, 118, 9412 CrossRef CAS PubMed; (f) E. Kozma and P. Kele, Org. Biomol. Chem., 2019, 17, 215 RSC; (g) L. Wang, M. S. Frei, A. Salim and K. Johnsson, J. Am. Chem. Soc., 2019, 141, 2770 CrossRef CAS PubMed.
- (a) G. Pohlers, S. Virdee, J. C. Scaiano and R. Sinta, Chem. Mater., 1996, 8, 2654 CrossRef CAS; (b) G. D. Feke, D. Hessman, R. D. Grober, B. Lu and J. W. Taylor, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2000, 18, 136 CrossRef CAS; (c) G. D. Feke, R. D. Grober, G. Pohlers, K. Moore and J. F. Cameron, Anal. Chem., 2001, 73, 3472 CrossRef CAS PubMed; (d) G. Pistolis, S. Boyatzis, M. Chatzichristidi and P. Argitis, Chem. Mater., 2002, 14, 790 CrossRef CAS; (e) C. Coenjarts, O. García, L. Llauger, J. Palfreyman, A. L. Vinette and J. C. Scaiano, J. Am. Chem. Soc., 2003, 125, 620 CrossRef CAS PubMed; (f) K. Ray, M. D. Mason, R. D. Grober, G. Pohlers, C. Staford and J. F. Cameron, Chem. Mater., 2004, 16, 5726 CrossRef CAS; (g) M. Frenette, C. Coenjarts and J. C. Scaiano, Macromol. Rapid Commun., 2004, 25, 1628 CrossRef CAS; (h) A. J. Berro, A. J. Berglund, P. T. Carmichael, J. S. Kim and J. A. Liddle, ACS Nano, 2012, 6, 9496 CrossRef CAS PubMed.
- (a) R. Popielarz, A. M. Sarker and D. C. Neckers, Macromolecules, 1998, 31, 951 CrossRef CAS; (b) A. Aspée, O. García, L. Maretti, R. Sastre and J. C. Scaiano, Macromolecules, 2003, 36, 3550 CrossRef; (c) J. Kabatc, B. Jȩdrzejewska and J. Pączkowski, J. Appl. Polym. Sci., 2006, 99, 207 CrossRef CAS; (d) J. Ortyl and R. Popielarz, J. Appl. Polym. Sci., 2013, 128, 1974 CAS; (e) I. Kamińska, J. Ortyl and R. Popielarz, Polym. Test., 2015, 42, 99 CrossRef.
- (a) C. Tahtaoui, C. Thomas, F. Rohmer, P. Klotz, G. Duportail, Y. Mély, D. Bonnet and M. Hibert, J. Org. Chem., 2007, 72, 269 CrossRef CAS PubMed; (b) S. Shaban Ragab, S. Swaminathan, E. Deniz, B. Captain and F. M. Raymo, Org. Lett., 2013, 15, 3154 CrossRef CAS PubMed.
- T. Aoai, K. Kodama, T. Yamanaka and M. Yagihara, J. Photopolym. Sci. Technol., 1998, 11, 409 CrossRef CAS.
- F. Ortica, J. C. Scaiano, G. Pohlers, J. F. Cameron and A. Zampini, Chem. Mater., 2000, 12, 414 CrossRef CAS.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef PubMed.
- M. Gaplovsky, Y. V. Il'ichev, Y. Kamdzhilov, S. V. Kombarova, M. Mac, M. A. Schwörer and J. Wirz, Photochem. Photobiol. Sci., 2005, 4, 33 RSC.
- M. S. Kim and S. L. Diamond, Bioorg. Med. Chem. Lett., 2006, 4007 CrossRef CAS PubMed.
- (a) G. K. Hodgson, S. Impellizzeri and J. C. Scaiano, Chem. Sci., 2016, 7, 1314 RSC; (b) G. K. Hodgson, S. Impellizzeri and J. C. Scaiano, Catal. Sci. Technol., 2016, 6, 7113 RSC; (c) S. Impellizzeri, S. Simoncelli, G. K. Hodgson, A. E. Lanterna, C. D. McTiernan, F. M. Raymo, P. F. Aramendia and J. C. Scaiano, Chem.–Eur. J., 2016, 22, 7281 CrossRef CAS PubMed; (d) F. C. Hendriks, F. Meirer, A. V. Kubarev, Z. Ristanovic, M. B. J. Roeffaers, E. T. C. Vogt, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Am. Chem. Soc., 2017, 139, 13632 CrossRef CAS PubMed; (e) G. Chen, N. Zou, B. Chen, J. B. Sambur, E. Choudhary and P. Chen, ACS Cent. Sci., 2017, 3, 1189 CrossRef CAS PubMed; (f) R. Ye, X. Mao, X. Sun and P. Chen, ACS Catal., 2019, 9, 1985 CrossRef CAS; (g) A. Layek, J. Van Loon, M. B. J. Roeffaers and A. V. Kubarev, Catal. Sci. Technol., 2019, 9, 4645 RSC; (h) Q. T. Easter and S. A. Blum, Acc. Chem. Res., 2019, 52, 2244 CrossRef CAS PubMed.
- E. Limpert, W. A. Stahel and M. Abbt, Bioscience, 2001, 51, 341 CrossRef.
- E. A. Burstein and V. I. Emelyanenko, Photochem. Photobiol., 1996, 64, 316 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic protocols, spectra, additional photos of patterned films, electron microscopy images, nanoparticle size distribution, single molecule experiments and image analysis methodology, film thickness, supplementary TIRFM video. See DOI: 10.1039/d0na00049c |
| ‡ N. P. Dogantzis and G. K. Hodgson made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2020 |