
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling the distribution of nanoparticles in hydrogels via interfacial synthesis†
Olivier
Gazil
,
Teodora
Gancheva
,
Michel
Bilodeau-Calame
,
Basil D.
Favis
 and
Nick
Virgilio
*
and
Nick
Virgilio
*
CREPEC, Department of Chemical Engineering, Polytechnique Montréal, C.P. 6079 Succursale Centre-Ville, Montréal, Québec H3C 3A7, Canada. E-mail: nick.virgilio@polymtl.ca
First published on 5th October 2020
Abstract
In this article, a dual-solvent method is presented which allows for precise control over the distribution of nanoparticles (NPs) in hydrogels. The technique is based on the interfacial reaction between a reducing agent (herein THPC) initially solubilized in the hydrogel phase, and an organometallic precursor (herein Au(PPh3)Cl) solubilized in the surrounding organic liquid phase. When the organic phase is completely immiscible with water, the interfacial reaction yields a fragile monolayer film of NPs at the hydrogel surface. Then, the addition of a co-solvent (miscible with both aqueous and organic phases) allows precise tuning over the distribution of NPs, from a fine and well-anchored layer at the interface, to the whole gel volume. As a result, it is possible to independently control the size and concentration of NPs, and their distribution. The impact of such control is demonstrated with the reduction of p-nitrophenol to p-aminophenol catalyzed by gold nanoparticles (AuNPs). When AuNPs are mostly localized at the gel surface, the apparent reaction rate is more than 10× superior compared to AuNPs distributed in the whole gel – at comparable particle content and size. This approach is straightforward, decisive and compatible with broad arrays of NPs and hydrogel chemistries, and solvent combinations.
Introduction
In the last two decades, nanoparticles (NPs) have attracted much interest in many fields of applications such as catalysis, sensors and actuators, biotechnologies, bactericide wound dressing, and imaging, to name a few.1–4 For catalysis applications, this interest stems from the high reaction rates, selectivity and yield for many electron-transfer reactions.2,5 With high surface-to-volume ratio, and surface energy, NPs can catalyze reactions at mild reaction conditions (i.e. at room temperature and pressure) in aqueous solutions.5 Two significant problems encountered when using NPs freely dispersed in solution are their gradual agglomeration and resulting deactivation, and the extensive and costly separation processes involved when the reaction is over.2,6,7 Immobilizing NPs using monoliths can solve these issues, but the synthesis process can be complex and/or costly, and can also lead to decreased catalytic performances.6–8A promising approach to overcome these shortcomings is to immobilize NPs in hydrogels, which can efficiently retain NPs. Hydrogels display liquid-like diffusion/mass transfer properties – interesting for catalysis applications, with solid-like mechanical properties, for ease of manipulation and separation.9 So far, NPs immobilized in hydrogels either show a distribution gradient, or a nearly uniform distribution in the volume of these materials.10 Typically, NPs are either synthesized in situ by using a variety of reduction reactions, or simply by gelling a colloidal suspension.2,11 For catalysis applications, one significant concern when NPs are distributed in the whole gel volume is that diffusion can significantly limit chemical reaction rates, resulting in a large fraction of NPs being either less accessible, or isolated. To increase the reaction rate, selectively locating the NPs near the surface of hydrogels is thus a pertinent strategy. However, there are very few studies addressing the control of NPs spatial distribution within hydrogel materials.12–17 Kim et al. have prepared self-assembled 2D films of gold (Au) NPs by direct deposition onto substrates immersed into AuNPs solutions.18 Planar and curved glass surfaces, inner walls of capillary tubes, silica beads and cotton fabrics were covered with AuNPs.18 While high control over NPs size and film thickness can be achieved, complex 3D structures cannot be covered, and film adhesion can be problematic.
A method that stands out to control the localization and distribution of NPs is periodic precipitation (related to the Liesegang phenomenon).12–17 This method allows formation of concentric rings of NPs in hydrogels, starting from the gel surface/interface. NPs are formed by the reaction between two precursors – one situated in the gel phase, and the second in the surrounding solution, followed by the precipitation of the resulting metallic compound. The resulting periodic ring structure is governed by reactants diffusion, the delicate balance between NPs formation and precipitation, and transport phenomena. The width, intensity, and spacing between the bands or rings can be controlled by the choice of gelling agent and its concentration,12 the application of electric currents,13 and the precursors' concentrations.14 Complex patterns are also possible via wet stamping: a gel stamp containing one of the precursors is applied to a gel film containing the other precursor, resulting in the formation of Liesegang rings.15,16 However, this technique is limited to simple 2D morphologies. Finally, oppositely charged NPs can be used as well to form these periodic patterns.17 In all cases, macroscopic positioning of the bands or uniform distribution of NPs in a certain thickness of the gel is not achievable other than by stopping or limiting the reaction.14,16
In a series of articles,19–21 Rao et al. have developed a method to synthesize NPs at the liquid–liquid interface, i.e. at the interface of two immiscible liquids, typically an organic solvent and an aqueous phase – a much less explored synthesis pathway. The formation of a thin film of nanoparticles (e.g. AuNPs) comes from the reduction reaction at the solvent/water interface between an organometallic precursor and a reducing agent, solubilized respectively in the solvent and aqueous phases.20 The reaction results in the formation of a dense monolayer of NPs, and it is controlled in part by the precursors' concentrations, the reaction time, and temperature.19,21 The NPs monolayers typically display very smooth surfaces with high interfacial coverage (>75%) of fairly uniform nanoparticles.19 An interparticle separation of ≈1 nm is observed and is linked to triphenylphosphine ligands and THPC, acting as capping agents.19 The approach is versatile and simple, as demonstrated with the synthesis of metallic, semi-conductor and chalcogenide films.19,21 Films of multispecies alloys can also be prepared with this technique.22 However, the method has been limited so far to NPs film formation at the liquid–liquid interface or, with the addition of suitable capping agents, to aqueous and organic solutions, yielding respectively hydrosols or organosols.19,20
The objective of this work is to control the localization of NPs in hydrogels by adapting the approach developed by Rao et al. The hypothesis is that the addition of a co-solvent to the organic phase, miscible with both the solvent and aqueous (hydrogel) phases, can result in precise control over the NPs localization, while at the same time ensuring strong NPs anchoring in the gel phase. Ultimately, concentrating the NPs at the gel surface should result, for example, in enhanced catalytic properties compared to NPs dispersed in the whole gel volume.
In this work, the aqueous phase previously used by Rao et al. is substituted by a solid and highly permeable hydrogel phase. The liquid organic phase contains the solubilized metal precursor (e.g. Au(PPh3)Cl), while the hydrogel phase contains the reducing agent (e.g. THPC). The hydrogels are then plunged in the dual-solvent phase, which is comprised of one solvent completely immiscible with the gel phase (e.g. toluene), and a co-solvent miscible with both the organic and gel phases (e.g. acetonitrile). AuNPs formation occurs by the reduction reaction between the metal precursor and reducing agent, which is investigated as a function of solvent type, dual-solvent composition, reaction time, and temperature.
Results and discussion
Formation of AuNPs thin film at the toluene/hydrogel interface
In this work, the interfacial synthesis of metallic NPs at liquid/liquid (organic/aqueous) interfaces (Fig. S1a–c†), a method developed by Rao et al., is adapted to control the distribution of NPs in hydrogels.20 Initially the aqueous phase was replaced with sodium alginate (SA) hydrogels loaded with a reducing agent (herein THPC, as described in the Methodology, see ESI†), before plunging the gels in a 1.5 mM solution of Au(PPh3)Cl in toluene (see Fig. S1d†).Fig. 1a displays a ≈1 cm3 cubic SA hydrogel covered with a thin film of AuNPs, prepared at 60 °C during 48 h – the experimental parameters were chosen following a series of initial experiments looking at the effects of reaction time and temperature on NPs film formation. At 60 °C, a complete NPs film is obtained after 24 h, whereas the film remains incomplete after 10 days when the reaction is realized at 30 °C (Fig. S1d†). In this work, all of the following reactions were then conducted at 60 °C. A cross-section of the gel (Fig. 1b) reveals that the AuNPs strictly grow at the toluene/hydrogel interface and form a very thin film. TEM observations (Fig. 1c) show a film entirely composed of a monolayer of closely packed polycrystalline AuNPs with an average size of 14 ± 4 nm – note that the AuNPs show multiple facets resulting from growing in a confined environment. Moreover, the EDX spectra in Fig. 1d confirms the particles composition (pure Au) (the inset confirms the typical face-centered cubic crystal structure of gold).23
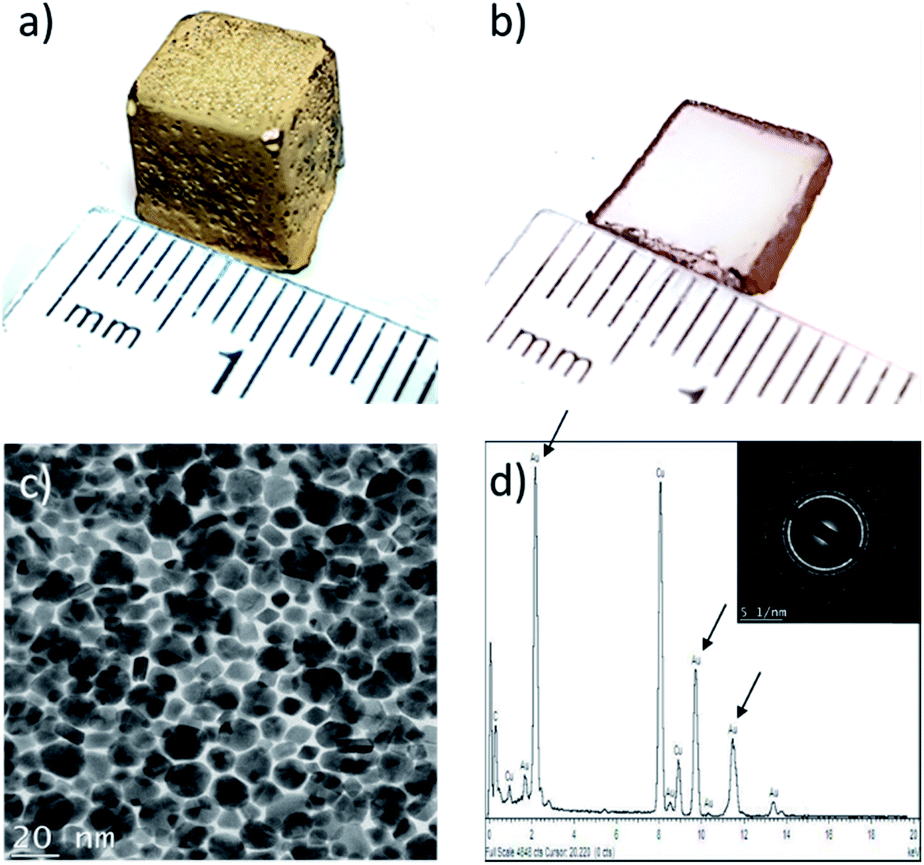 | ||
| Fig. 1 Bulk alginate hydrogel with AuNPs synthesized at the surface using toluene as the organic phase (48 h of growth at 60 °C): (a) alginate hydrogel cube covered with AuNPs; (b) cross-section slice of cube displayed in (a); (c) TEM micrograph of monolayer polycrystalline AuNPs film on the alginate cube. Average particle size = 14 ± 4 nm; (d) energy-dispersive X-ray (EDXS) spectra of AuNPs film with characteristic composition peaks, herein gold (excluding Cu and C which are the constituents of the copper grid), with the corresponding electron diffraction profile (SAED) in inset. | ||
Lastly, a crucial feature is film adhesion – in this case, the film is fragile, easily peelable and fragmented with a spatula, confirming that it is only superficially deposited onto the hydrogel surface. This is a significant concern for applications requiring strong adhesion (e.g. catalysis), but an interesting feature when NP film transfer is required. In order to improve film adhesion and stability onto hydrogel surfaces, the effect of organic solvent miscibility with water was next considered.
Effect of the organic solvent miscibility with water on the selective localization of NPs in hydrogels
To understand how organic solvent miscibility with water impacts the formation of AuNPs, a series of synthesis were undertaken using three solvents miscible with water (acetone, DMF and acetonitrile), and two immiscible solvents (toluene and benzene) – note that Au(PPh3)Cl is soluble in all five solvents. The results are presented in Fig. 2. For acetonitrile and acetone (Fig. 2a and c), NPs synthesis occurs in the whole volume of the hydrogel, which is explained by the diffusion of the solvent and Au(PPh3)Cl in the hydrogel, followed by the reduction of Au(PPh3)Cl with the THPC initially contained in the gel. For DMF, a peculiar NPs distribution is observed (Fig. 2b). This cross-like pattern is caused by gel syneresis (liquid expulsion and gel shrinking) when plunged in DMF, combined with the diffusion of DMF and Au(PPh3)Cl in the gel, and the resulting formation of AuNPs. Once the reaction is completed and the hydrogel is rehydrated (as shown on the picture), the gel expands again and displays this interesting pattern. While difficult to control, this approach could be employed to synthesize hydrogels with certain patterns. For the other two solvents that are immiscible with water (Fig. 2d and e), toluene and benzene display similar results, with NPs strictly growing at the solvent/gel interface, as expected, with low film adhesion. | ||
| Fig. 2 Cross-sections of 1 cm3 cubic AuNPs/SA hydrogels nanocomposites prepared with different organic solvents during the NPs synthesis step (48 h at 60 °C): (a) acetone; (b) N,N-dimethylformamide (DMF); (c) acetonitrile; (d) toluene; (e) benzene. From (a)–(c), NPs are formed in the hydrogels, while in (d) and (e), they are formed at the gel surface. Note that the first three solvents are miscible with water, while the last two are immiscible. | ||
The five cases illustrated above are in fact the two limiting cases of NPs distribution control – (1) solely at the gel surface, or (2) in the whole gel volume. The different colors observed for the first three cases come from the different type of solvents used and their impact on AuNPs synthesis.2 While concentrating the NPs at the surface in Fig. 2d and e is quite interesting for a number of applications, the NPs films are fragile and easy to remove. On the other hand, NPs distributed in the whole volume are strongly bound to the gel due to the binding interactions between the AuNPs and alginate carboxyl groups.1 However, a significant fraction of AuNPs is now isolated or “lost” in the bulk gel phase – as a result, diffusion limits applications such as catalysis.
Following these results, using a co-solvent to control the diffusion of Au(PPh3)Cl in the gel phase, and ultimately the distribution of NPs, was investigated. Also, in order to avoid pattern formation, and corner/edge effects, further NPs synthesis experiments were realized with SA gel cylinders.
Controlling the localization of AuNPs with a co-solvent
To assess the effect of a co-solvent, we started with toluene as the organic phase, which is completely immiscible with the hydrogel phase, to which we gradually added acetonitrile, a co-solvent for both toluene and water. Acetonitrile was also chosen since it has a higher boiling point compared to acetone and does not cause gel syneresis like DMF.Fig. 3 illustrates the distribution of AuNPs in hydrogels when the volume fraction ϕvol of co-solvent in the organic phase gradually increases from 0% acetonitrile (pure toluene) to 100% acetonitrile (no toluene). As seen earlier, these two extreme values yield NPs distributed solely at the interface for the former, and in the whole gel volume for the latter (Fig. 2). At low acetonitrile volume fractions ϕvol (0–30%), no significant effect on the distribution of AuNPs is observed – NPs synthesis occurs solely at the interface. In addition, the film of AuNPs can be easily removed at low acetonitrile contents (0% to 20%), but starts anchoring at the surface at ϕvol = 30%. Then, over 30%, AuNPs are well anchored but still localized near the interface – even after 48 h of synthesis time. Finally, when ϕvol > 80%, AuNPs growth mostly occurs in the whole gel volume.
 | ||
| Fig. 3 Cross sections of AuNPs/SA hydrogel nanocomposites cylinders (diameter dcyl = 11 mm) prepared in toluene/acetonitrile mixtures (48 h of growth at 60 °C), in increasing order of acetonitrile content (vol%). | ||
Adding a co-solvent has a significant effect on the distribution of AuNPs during synthesis. As the composition in acetonitrile and the miscibility of the organic phase with water increase, the diffusion rate of the organic phase containing Au(PPh3)Cl in the hydrogel also increases, gradually leading to the formation of NPs deeper in the gel phase by reaction with THPC. This represents a simple approach to precisely control the distribution of NPs in hydrogels. Note that a similar result is obtained when replacing acetonitrile by acetone, also a co-solvent of toluene and water (Fig. S2†). In that case, tightly closed vials were used to avoid acetone evaporation during synthesis.
Next, the effect of NPs synthesis time (from 4 h to 48 h at 60 °C) for three organic phase compositions – 100% acetonitrile, 70/30 acetonitrile/toluene, and 100% toluene (vol%) (Fig. 4) – was investigated. Note that the time intervals were chosen to clearly illustrate how the gels' appearances evolve during NPs synthesis. Firstly, it was observed that the presence of acetonitrile increases the reaction speed and NPs formation. Indeed, the diffusion of Au(PPh3)Cl in the hydrogel due to acetonitrile facilitates the reaction of the organometallic precursor with THPC. However, the most important feature is the level of control we obtain over AuNPs localization, which becomes nearly independent of the reaction time. When pure acetonitrile is used (Fig. 4a), AuNPs are formed in the whole volume (a concentration of NPs near the surface can be distinguished). For pure toluene, NPs are strictly formed at the gel surface (Fig. 4c). The effect is most striking at intermediate solvent compositions, since at 70/30 acetonitrile/toluene, AuNPs are mostly restricted to a very thin region near the surface. This thin region grows darker with time, suggesting a growing number of AuNPs also increasing in size.
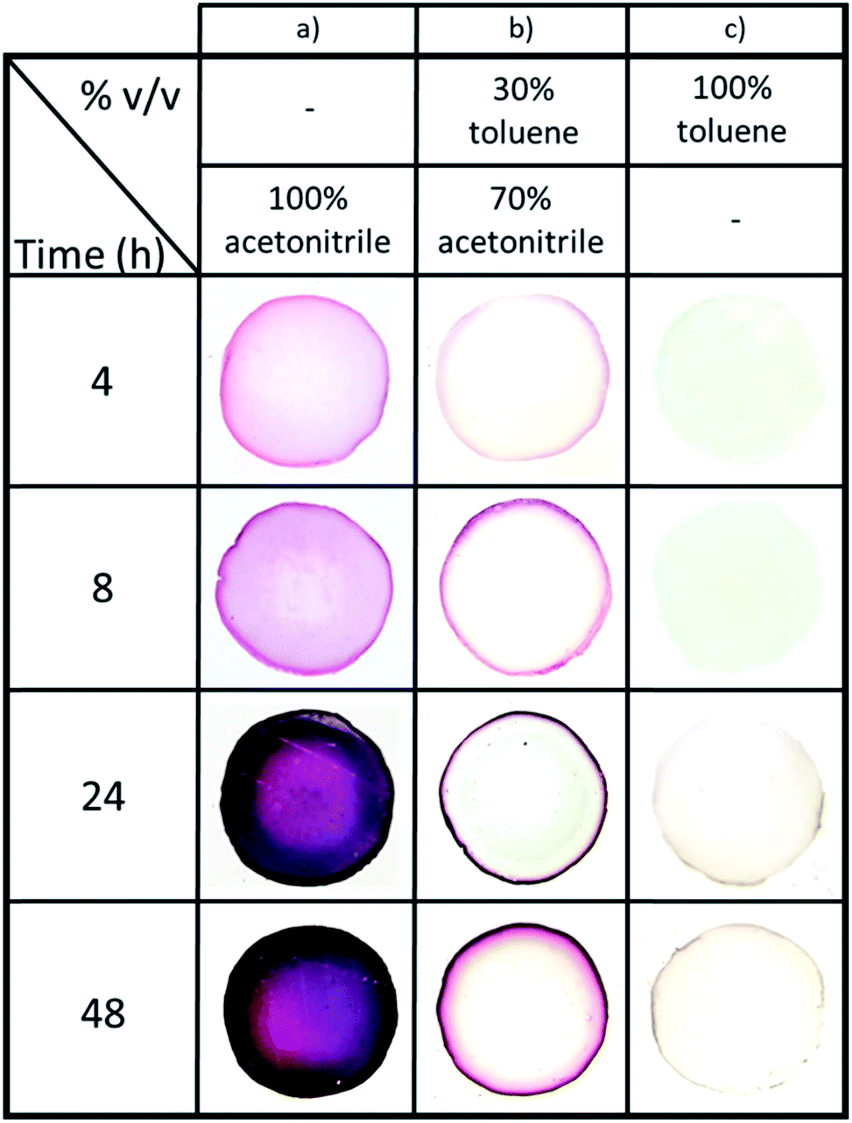 | ||
| Fig. 4 Effect of AuNPs synthesis time (from 4 h to 48 h at 60 °C) on AuNPs distribution (cross sections of alginate hydrogel cylinders with AuNPs), for (a) pure acetonitrile, (b) 70/30 toluene/acetonitrile (vol%) and (c) pure toluene solvents. | ||
This indicates that synthesis time is now decoupled from NPs localization in the gel: this now avoids any limitation due to diffusion. To the best of our knowledge, this is the first study demonstrating such a level of control, without requiring to stop the reaction in order to limit diffusion in the hydrogel. As a result, it is now possible to control the size and concentration of the synthesized particles via reaction time and reactants concentrations, while at the same time controlling precisely their localization in the hydrogel.
Versatility of the dual-solvent interfacial reaction method
We next assessed whether other organometallic precursors could be used. Pd(PPh3)2Cl2 possesses the same phosphine ligands as the gold precursor. Using a 70/30 vol% toluene/acetonitrile organic phase, and a synthesis time of 48 h, we again observe a similar localized concentration of PdNPs near the surface (Fig. S3†). However, the alginate also displays a light yellow-brownish coloration, as opposed to no coloration for Au. We believe this is due to the faster diffusion of the Pd(PPh3)2Cl2 precursor in the hydrogel as compared to Au(PPh3)Cl, for a given solvent mixture composition. Reducing the content in acetonitrile could then better confine PdNPs to the surface of the gel.Enhancing the catalytic properties via control over AuNPs distribution
We next assessed if the synthesis conditions, i.e. solvent composition and, ultimately, AuNPs localization within the hydrogel, had an effect on the catalytic activity. AuNPs were synthesized during 10 h within small hydrogel beads (dbeads ≈ 2.5 mm), in both pure acetonitrile (Fig. 5a) and the acetonitrile/toluene (70/30 vol%) mixture (Fig. 5b) (beads were used because of the glass cuvettes small dimensions). Like gel cylinders (Fig. 3 and 4), synthesis in pure acetonitrile results in the distribution of AuNPs into the whole hydrogel beads volume, whereas synthesis in the solvent mixture yields gel beads with AuNPs localized at the gel surface – as the darker region at the beads periphery confirms.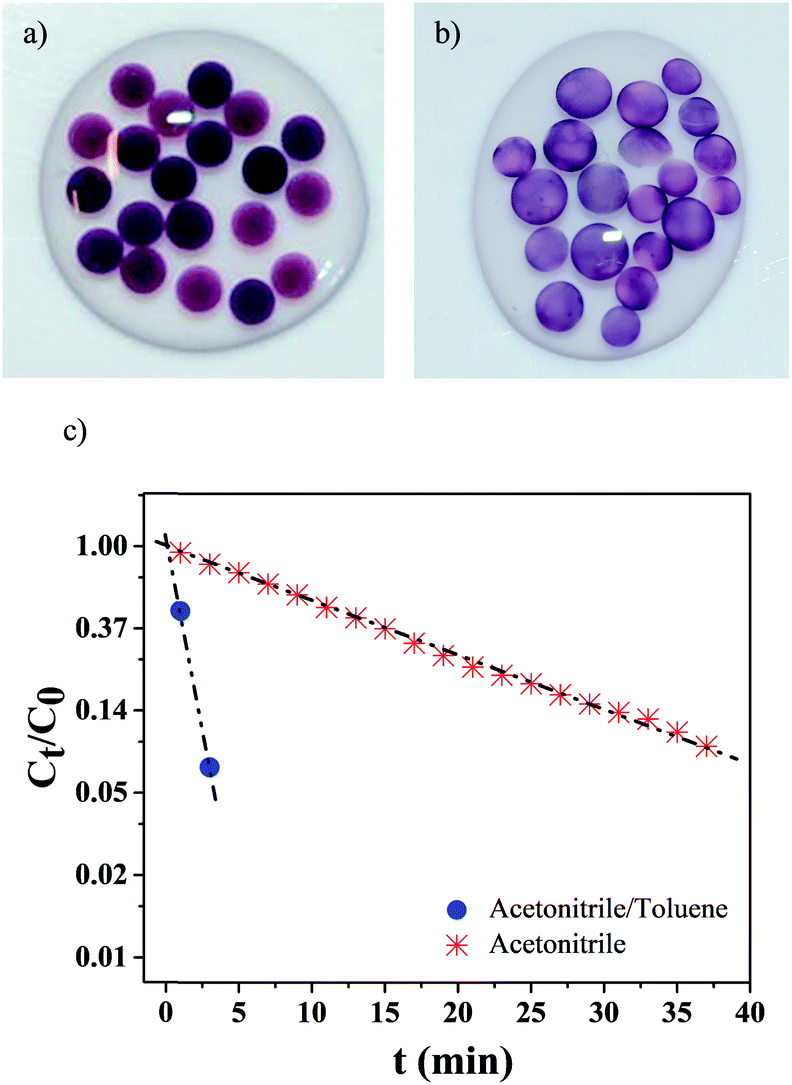 | ||
| Fig. 5 AuNPs/hydrogel beads synthesized during 10 h at 60 °C in: (a) pure acetonitrile (AuNPs dispersed in whole gel volume); (b) in acetonitrile/toluene 70/30 vol%. (c) p-Nitrophenol relative concentration as a function of time t, using gel beads displayed in (a) and (b). Each data point corresponds to the average of 3 samples. The dashed-dot lines are guides for the eye. | ||
The catalytic activity of these gel beads loaded with AuNPs was studied with the model reduction reaction of p-nitrophenol to p-aminophenol.24–27 The conversion was followed by monitoring the absorption spectra at 400 nm of p-nitrophenol. As Fig. 5c illustrates, the catalytic activity is highly dependent on the AuNPs synthesis conditions: for gel beads with AuNPs at the surface, the p-nitrophenol reduction reaction is completed in about 3 min. In comparison, for beads with AuNPs distributed in their whole volume, the full conversion of p-nitrophenol takes more than 40 min. Clearly, selectively locating AuNPs at the surface significantly increases the reaction kinetics.
When AuNPs are embedded into micro-carriers and the reducing agent (herein NaBH4) is in excess compared to p-nitrophenol, the reduction reaction kinetics can be modeled as a pseudo-first-order reaction.24,25,28 In our case, a linear correlation between time t and ln(Ct/C0) is found (Ct corresponds to the p-nitrophenol concentration at time t and C0 at t = 0). The apparent reaction rate constant kapp (calculated from the linear fittings) is strongly dependent on the NPs synthesis conditions: when the solvent mixture is used, kapp = 0.95 min−1, while when only acetonitrile is employed, kapp = 0.065 min−1 – a difference superior to one order of magnitude.
Analysis of AuNPs distributed in hydrogel spheres
Transmission electron microscopy (TEM) was employed to characterize AuNPs size and morphology inside the hydrogel spheres. Fig. 6a–d-(i) displays representative samples of AuNPs grown in hydrogel beads. Many particles show irregular shapes with facets, and they are clearly larger after 48 h of synthesis time, compared to 10 h. High-resolution TEM micrographs (Fig. 6a–d-(ii)) reveal that the particles are polycrystalline, and the [111] planes in the cubic Au crystalline structure are visible, with an interplanar distance of ≈2.36 Å. The average diameters are obtained from the size distribution histograms (Fig. 6a–d-(iii)). For AuNPs synthesized in acetonitrile, the average diameter d clearly depends on the reaction time: d = 15 (±5) nm for 10 h of reaction time, versus d = 31 (±7) nm for 48 h. Reaction time has a less pronounced impact when using the acetonitrile–toluene mixture for AuNPs synthesis. In this case, NPs synthesized during 10 h have average diameter d = 10 (±4) nm, while those synthesized during 48 h have an average value of d = 13 (±9) nm. Moreover, low-magnification TEM micrographs display randomly and uniformly distributed AuNPs, with no significant aggregates (Fig. S4†).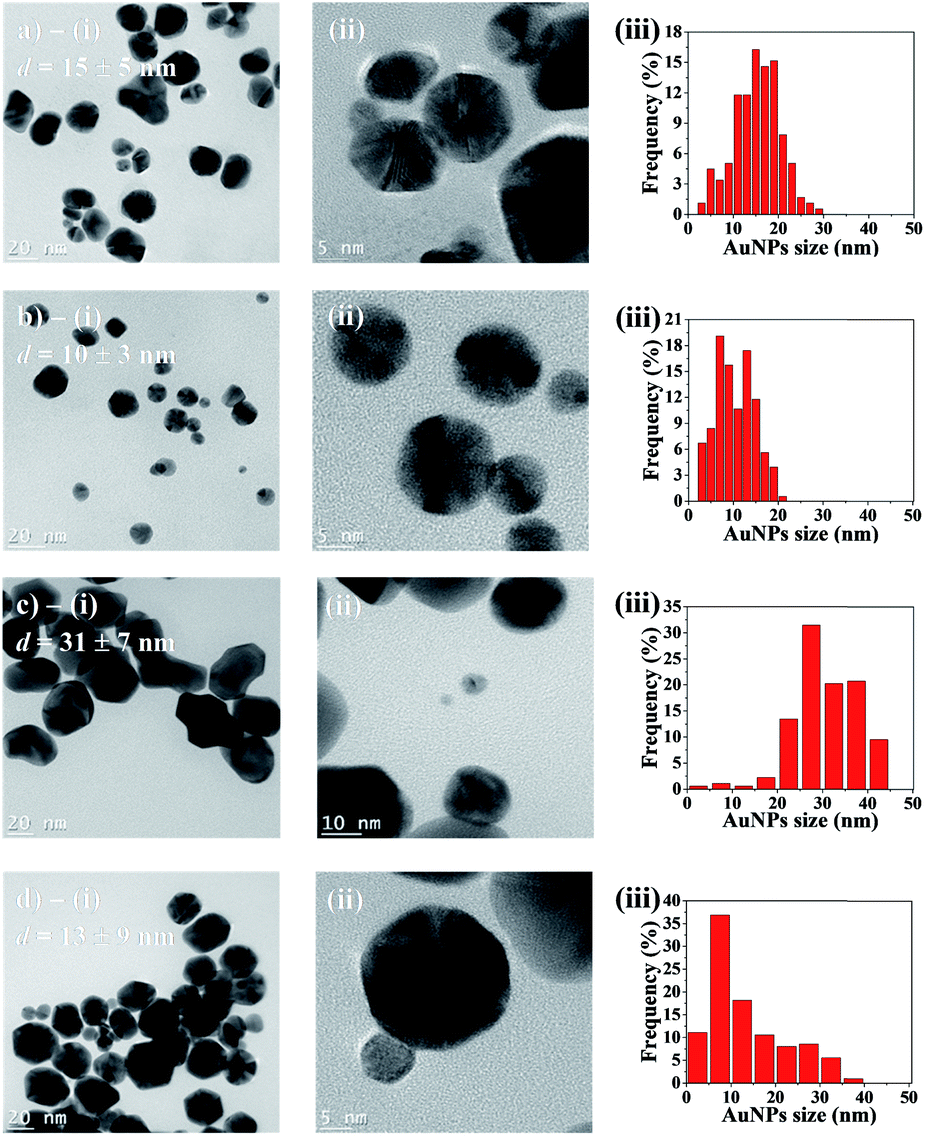 | ||
| Fig. 6 TEM micrographs and analysis of particles synthesized in: (a) acetonitrile during 10 h; (b) acetonitrile/toluene (70/30 vol%) during 10 h; (c) acetonitrile during 48 h; (d) acetonitrile/toluene (70/30 vol%) during 48 h. In (i), micrographs showing representative AuNPs samples, with average diameter d; (ii) high resolution micrographs displaying AuNPs polycrystallinity; (iii) AuNPs size distribution histograms (obtained by measuring between 180 and 200 AuNPs). | ||
Finally, thermogravimetric analysis (TGA) was employed to quantify the AuNPs content in gel beads (Table 1). The results show that both types of beads – synthesized using pure acetonitrile, and with the acetonitrile/toluene mixture – contain comparable amounts of AuNPs (about 5 wt%, based on the dry mass of SA beads).
| Beads type | Remaining massa (%) | AuNPs contentb (%) |
|---|---|---|
| a Based on initial weight of dried beads. b AuNPs content (%) = remaining mass with NPs minus remaining mass without NPs. | ||
| No AuNPs (reference) | 33 ± 1 (N = 3) | — |
| AuNPs – 100 vol% acetonitrile | 38 ± 1 (N = 3) | 5 ± 2 |
| AuNPs – 70/30 vol% acetonitrile/toluene | 38 ± 2 (N = 6) | 5 ± 3 |
Discussion
This work demonstrates that it is possible to precisely control the localization of NPs at hydrogel surfaces by adapting the interfacial synthesis technique developed by Rao et al. for liquid–liquid interfaces.20 The addition of a co-solvent to the organic phase allows for both the control of NPs distribution in the hydrogel as well as a strong anchoring of NPs near the gel surface. In doing so, it is possible to independently control NPs size, distribution, and concentration, as Fig. 3 and 4 illustrate – a unique feature of our approach. In comparison, NPs growth in gels by periodic precipitation (Liesegang rings formation), for example, occurs at a certain depth in the material, and independent control over NPs concentration, size and localization is quite difficult.12,15,16 However, tuning parameters such as reactants concentrations and/or gel composition allows some additional control over NPs localization with this approach.12,14–16 As a result, the Liesegang rings can be further apart or thicker by controlling gel composition, but their formation will not be restricted to a precise localization in the gels.12The results also demonstrate that anchoring the NPs near the surface significantly enhances the catalytic properties of hydrogel monoliths. At comparable NPs contents (Table 1) and NPs average diameters (Fig. 6), the catalytic activity of gel beads with NPs concentrated at the surface (prepared with the toluene/acetonitrile solvent mixture) is more than an order of magnitude superior to beads with NPs distributed in the whole gel volume. This is explained by the much shorter diffusion path to the NPs surface when they are distributed near the gel surface, all other parameters being equal.
The interfacial synthesis method modified with a co-solvent is a straightforward approach compatible with a variety of solvents and gel chemistries. Rao et al. also demonstrated previously that a wide array of precursors and reducing agents can be used to synthesize NPs at liquid–liquid interfaces, including metals, metal oxides and chalcogenides.21 This methodology is simple and relatively inexpensive to prepare high performance catalytic monolithic materials, and more generally hydrogels comprising thin films of NPs at their surface.
From a broader point of view, the possibility of controlling the distribution of NPs in hydrogels could significantly enhance their functional properties. For example, concentrating silver nanoparticles near the surface of hydrogels could improve their bactericidal properties as wound dressing materials.29 Shape-memory hydrogels (for actuators and self-folding materials) containing NPs are typically based on the photothermal effect and require precise control of NPs distribution.4 Finally, synthesis of anisotropic nanoparticles should be explored since particle nucleation and growth occurs at the interface of two immiscible fluids.30
Conclusions
This work presents a methodology to control the localization and distribution of NPs near hydrogel surfaces, with strong NPs anchoring. The interfacial synthesis of NPs is realized by plunging a hydrogel containing a water soluble reducing agent (herein THPC), in an immiscible organic solvent containing an organometallic precursor (herein Au(PPh3)Cl in toluene). In that case, a thin and easily peelable film of AuNPs film is formed. The addition of a co-solvent (herein acetonitrile) to the organic phase allows for both the precise control of NPs distribution near the gel surface and strong NPs anchoring. By varying the solvent phase composition, it is possible to control the NPs distribution, from a thin film located at the gel surface, to NPs distributed in the whole gel volume.The resulting NPs distribution has a strong impact on catalytic properties. At comparable NPs average sizes and amounts, gels with NPs concentrated near the surface display a significantly higher catalytic activity, with a reaction rate constant k more than 10 times superior to the gel with NPs distributed in the whole gel volume – a consequence of the shorter diffusion path to NPs surface. Future work will aim at modelling the diffusion properties inside hydrogels as a function of NPs localization.
Overall, this methodology is simple, compatible with wide libraries of gel and NPs chemistries, allows precise control over NPs distribution, and the possibility of forming NPs films over complex gel shapes.
Experimental
Materials
Food grade sodium alginate (SA) was purchased from La Guilde Culinaire. Tetrakis(hydroxymethyl)phosphonium chloride (THPC; 80% in water), chloro(triphenylphosphine)gold(I) (Au(PPh3)Cl), glucono delta-lactone (GDL), sodium borohydride (NaBH4) and p-nitrophenol (4-NP) were all purchased from Sigma-Aldrich (reference numbers 404![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 861, 254037, G4750, 452874 and 241326 respectively). Calcium carbonate (CaCO3), sodium hydroxide (NaOH) and calcium chloride dihydrate (CaCl2·2H2O) were purchased from Fischer Scientific and are ACS certified (reference numbers C64-500, MSX05901 and C79-500). Toluene, acetone, acetonitrile, benzene and N,N-dimethylformamide (DMF) were also purchased from Fischer Scientific and are ACS graded solvents. All chemicals were used as received without further purification.
861, 254037, G4750, 452874 and 241326 respectively). Calcium carbonate (CaCO3), sodium hydroxide (NaOH) and calcium chloride dihydrate (CaCl2·2H2O) were purchased from Fischer Scientific and are ACS certified (reference numbers C64-500, MSX05901 and C79-500). Toluene, acetone, acetonitrile, benzene and N,N-dimethylformamide (DMF) were also purchased from Fischer Scientific and are ACS graded solvents. All chemicals were used as received without further purification.
Preparation of the SA hydrogels
Sodium alginate hydrogel cylinders (0.55 cm radius × 1.6 cm height) or cubes were prepared with a 2% w/v SA aqueous solution by the internal setting method as described by Draget et al. with 30 mM CaCO3 and 60 mM GDL.31 Briefly, the (SA/CaCO3) suspension was obtained by dissolving SA in deionized water while stirring during 30 min at 75 °C, followed by the addition of the required amount of CaCO3. The GDL solution was freshly prepared and added to the solution, quickly followed by molding of the hydrogel using either a commercial silicon mold or polylactide 3D printed molds (for cubes and cylinders, respectively). The gels were subsequently removed from the molds and were plunged in a 2% w/v CaCl2 solution.For the preparation of SA beads (dbeads ≈ 2.5 mm), gelling was realized with the external setting method by using a 2% w/v CaCl2 solution. Briefly, SA droplets were deposited in a CaCl2 solution, instantly forming hydrogel spheres.
Synthesis of AuNPs in the hydrogels
Synthesis of AuNPs at the surface or in the hydrogels was realized by adapting the technique developed by Rao et al. to synthesize NPs at the interface between two immiscible liquids.20 For the hydrogel cylinders and cubes, each SA hydrogel was first plunged in 8 mL of a THPC basic solution for two days (2 mM, pH = 11.8 adjusted with 1 M NaOH). Afterwards, each gel was plunged in 5 mL of an organic solution of Au(PPh3)Cl (1.5 mM) at 60 °C, using a Thermo Scientific circulating thermostated bath (model 2864), for a period ranging from 4 h to 48 h. The same methodology was employed for the synthesis of AuNPs in the gel beads.Transmission electron microscopy (TEM) characterization
The observations were carried out using a JEOL JEM-2100F field emission electron microscope operated at 200 kV. For the observation of AuNPs films prepared with SA cubes in toluene, TEM grids were slightly pressed onto the NPs film, dried and analyzed. For AuNPs synthesized in acetonitrile/toluene 70/30 vol% solutions, once the catalytic reduction tests were completed (see next section), the beads were kept in the reaction medium solution for ≈72 h to let the beads dissociate, in order to recover the AuNPs with TEM grids. For AuNPs synthesized in pure acetonitrile, between 15 and 20 beads were placed into a vial containing 10 mL of Milli-Q water. The beads were manually squashed to free NPs in solution. Subsequently, one drop of the solution was deposited onto a carbon grid, which was dried at room temperature before analysis.Catalytic properties of AuNPs/hydrogel beads
Prior to the catalysis tests, beads with AuNPs synthesized during 10 h in either pure acetonitrile or in the acetonitrile/toluene 70/30 vol% mixture were cleaned following four steps: (1) approx. 20–25 beads were placed into a vial containing 30 mL of distilled water; (2) then, 3 mL of a 1 M hydrochloric acid (HCl) solution were added to the vial; (3) the vial was next placed into an ultrasonic bath during 15 min; (4) finally, the beads were rinsed with distilled water and were placed in a thermostated bath at 60 °C for 1 h. The procedure was repeated three times for a given set of beads.The catalytic properties of the synthesized AuNPs/hydrogel beads were characterized by monitoring the catalytic reduction of 4-NP to 4-aminophenol (4-AP), as described by Pal et al.32 The reaction was monitored using UV-visible spectroscopy (UV-Vis, SFM-400 spectrometer from BioLogic, spectra recorded over 250–550 nm) by following the relative absorption of 4-NP at 400 nm. The p-nitrophenol reduction reaction was conducted under the following conditions: (1) first, 1.5 mL of a 0.3 mM p-nitrophenol aqueous solution and 1.5 mL of a 50 mM NaBH4 aqueous solution (freshly prepared prior to the experiments) were mixed in a 1 cm path-length quartz cuvette for UV-Vis spectroscopy analyzes, at room temperature. Then, 20 AuNPs/hydrogel beads (cleaned following the procedure described above) were added into the quartz cuvette (the beads sink to the bottom of the cuvette, out of the light path). The catalytic conversion was monitored every 2 min. The reaction medium in the cuvette was manually mixed between each measurement. At least three experiments were realized for each type of tested beads.
Thermogravimetric analysis (TGA) of AuNPs/hydrogel beads
The AuNPs content in hydrogel beads was measured by thermogravimetric analysis (TGA) using a Q-500 instrument from TA Instruments. Beads without AuNPs (reference material), and with AuNPs synthesized in either pure acetonitrile or in the acetonitrile/toluene 70/30 vol% mixture during 10 h, were dried in an oven for 3 days at 100 °C. Then, ≈15 mg of each type of beads were heated up to 800 °C under a nitrogen flow (60.0 mL min−1) at a heating rate of 10 °C min−1. At least three samples were analyzed for each type of beads.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Alexander Graham Bell Canada graduate scholarships program from the Natural Sciences and Engineering Research Council of Canada (NSERC), and with a B1X scholarship from the Fonds de Recherche du Québec – Nature et Technologies (FRQNT). This work was also supported by the TransMedTech Institute (NanoBio Technology Platform) and its main funding partner, the Canada First Research Excellence Fund. The authors would like to acknowledge support from the Centre de Caractérisation Microscopique des Matériaux (CM2) for access to their facilities. We finally thank Dr Benoît Liberelle, from the NanoBio Technology Platform, for fruitful discussions.References
- S. Sarkar, E. Guibal, F. Quignard and A. K. SenGupta, J. Nanopart. Res., 2012, 14, 715 CrossRef
.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS
- L. K. Bogart, G. Pourroy, C. J. Murphy, V. Puntes, T. Pellegrino, D. Rosenblum, D. Peer and R. Lévy, ACS Nano, 2014, 8, 3107–3122 CrossRef CAS
- H. Guo, J. Cheng, K. Yang, K. Demella, T. Li, S. R. Raghavan and Z. Nie, ACS Appl. Mater. Interfaces, 2019, 11, 42654–42660 CrossRef CAS
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS
- Z. J. Jiang, C. Y. Liu and L. W. Sun, J. Phys. Chem. B, 2005, 109, 1730–1735 CrossRef CAS
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2010, 12, 144–149 RSC
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS
- W. Zhang, C. Ma and M. Ciszkowska, J. Phys. Chem. B, 2001, 105, 3435–3440 CrossRef CAS
- P. Thoniyot, M. J. Tan, A. A. Karim, D. J. Young and X. J. Loh, Adv. Sci., 2015, 2, 1400010 CrossRef
- S. R. Sershen, S. L. Westcott, N. J. Halas and J. L. West, Appl. Phys. Lett., 2002, 80, 4609–4611 CrossRef CAS
- I. Lagzi, Langmuir, 2012, 28, 3350–3354 CrossRef CAS
- I. Bena, M. Droz, I. Lagzi, K. Martens, Z. Racz and A. Volford, Phys. Rev. Lett., 2008, 101, 075701 CrossRef CAS
- I. Yoon, A. M. Zimmerman, C. C. Tester, A. M. DiCiccio, Y. Jiang and W. Chen, Chem. Mater., 2009, 21, 3924–3932 CrossRef CAS
- I. T. Bensemann, M. Fialkowski and B. A. Grzybowski, J. Phys. Chem. B, 2005, 109, 2774–2778 CrossRef CAS
- H. Nabika, M. Sato and K. Unoura, Langmuir, 2014, 30, 5047–5051 CrossRef CAS
- I. Lagzi, B. Kowalczyk and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 58–60 CrossRef CAS
- K. Kim, J. W. Lee, H. B. Lee and K. S. Shin, Langmuir, 2009, 25, 9697–9702 CrossRef CAS
- C. N. Rao, G. U. Kulkarni, V. V. Agrawal, U. K. Gautam, M. Ghosh and U. Tumkurkar, J. Colloid Interface Sci., 2005, 289, 305–318 CrossRef CAS
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas, V. V. Agrawal and P. Saravanan, J. Phys. Chem. B, 2003, 107, 7391–7395 CrossRef CAS
- C. N. Rao and K. P. Kalyanikutty, Acc. Chem. Res., 2008, 41, 489–499 CrossRef CAS
- V. V. Agrawal, P. Mahalakshmi, G. U. Kulkarni and C. N. Rao, Langmuir, 2006, 22, 1846–1851 CrossRef CAS
- S. A. Aromal, K. V. Babu and D. Philip, Spectrochim. Acta, Part A, 2012, 96, 1025–1030 CrossRef
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS
- P. Herves, M. Perez-Lorenzo, L. M. Liz-Marzan, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 41, 5577–5587 RSC
- F.-h. Lin and R.-a. Doong, J. Phys. Chem. C, 2011, 115, 6591–6598 CrossRef CAS
- T. Gancheva and N. Virgilio, ACS Appl. Mater. Interfaces, 2018, 10, 21073–21078 CrossRef CAS
- G. Marcelo, M. López-González, F. Mendicuti, M. P. Tarazona and M. Valiente, Macromolecules, 2014, 47, 6028–6036 CrossRef CAS
- B. A. Aderibigbe and B. Buyana, Pharmaceutics, 2018, 10, 42 CrossRef
- N. D. Burrows, A. M. Vartanian, N. S. Abadeer, E. M. Grzincic, L. M. Jacob, W. Lin, J. Li, J. M. Dennison, J. G. Hinman and C. J. Murphy, J. Phys. Chem. Lett., 2016, 7, 632–641 CrossRef CAS
- K. Ingar Draget, K. Østgaard and O. Smidsrød, Carbohydr. Polym., 1990, 14, 159–178 CrossRef
- N. Pradhan, A. Pal and T. Pal, Colloids Surf., A, 2002, 196, 247–257 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of AuNPs at water/toluene interface, AuNPs distribution in alginate gels when synthesized in acetone/acetonitrile mixtures, synthesis of palladium NPs in alginate gel, TEM micrograph of NPs distribution in gel. See DOI: 10.1039/d0na00488j |
| This journal is © The Royal Society of Chemistry 2020 |