
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSub-monolayer Au9 cluster formation via pulsed nozzle cluster deposition†
Jesse
Daughtry
ab,
Gunther G.
Andersson
 *ab,
Gregory F.
Metha
d,
Siriluck
Tesana
e and
Tomonobu
Nakayama
*c
*ab,
Gregory F.
Metha
d,
Siriluck
Tesana
e and
Tomonobu
Nakayama
*c
aFlinders Institute for NanoScale Science and Technology, Flinders University, Adelaide, SA 5001, Australia. E-mail: gunther.andersson@flinders.edu.au
bFlinders Microscopy and Microanalysis, College of Science and Engineering, Flinders University, Adelaide, SA 5042, Australia
cNational Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: nakayama.tomonobu@nims.go.jp
dDepartment of Chemistry, The University of Adelaide, Adelaide, SA 5005, Australia
eThe MacDiarmid Institute for Advanced Materials and Nanotechnology, School of Physical and Chemical Sciences, University of Canterbury, Christchurch 8041, New Zealand
First published on 20th July 2020
Abstract
Submonolayer coverages of chemically synthesised triphenylphosphine-protected Au9 clusters on mica and TiO2 substrates were achieved through the development of a Pulsed Nozzle Cluster Deposition (PNCD) technique under high vacuum conditions. This method offers the deposition of pre-prepared, solvated clusters directly onto substrates in a vacuum without the potential for contamination from the atmosphere. AFM and TEM were used to investigate the rate of gold cluster deposition as a function of cluster solution concentration and the number of pulses, with pulse number showing the most effective control of the final deposition conditions. TEM and XPS were used to determine that the clusters retained their unique properties through the deposition process. Methanol solvent deposited in the PNCD process has been shown to be removable through post-deposition treatments. A physical model describing the vapour behaviour and solvent evaporation in a vacuum is also developed and presented.
Introduction
The properties of atomically precise gold nanoclusters have been found to have novel properties distinct from those found in bulk Au and gold nanoclusters. Gold nanoclusters can be defined by their small size (<2 nm) and consist of fewer than 246–279 Au atoms1 with clusters below this threshold exhibiting properties that are highly dependent on the number of atoms constituting the nanocluster.1–4 These properties have proven to be associated with their unique electronic density of states.5,6 Utilising the properties of atomically precise clusters for use in catalysis, for example, in water-splitting7,8 and CO oxidation,9,10 has become a promising field of research. The interactions between atomically precise nanoclusters and substrates have been shown to be crucial to stabilising and retaining nanocluster properties and can also enhance catalysis.11–13Metal nanoclusters can also be generated in gas phase cluster sources and deposited on substrates; however, they must be used immediately and cannot be stored for any period of time. While vacuum generated nanoclusters can be deposited directly onto treated substrates for further analysis,14,15 chemically synthesised nanoclusters have required any deposition to take place outside of vacuum, thus introducing contamination through the exposure to of the surface to atmosphere and solvents from solution based deposition methods and potentially affecting any surface treatments made in vacuum.16,17 Thus, a method of depositing chemically synthesised nanoclusters directly onto a substrate under vacuum conditions would have the advantage to strongly reduce contaminations through exposure to air and solvents.
The usefulness of pulsed vapour depositions to achieve controlled thin film growth have been demonstrated in surface coating systems, particularly metal oxides utilising ultrasonically vibrated vacuum pulse nozzles to inject packets of the solvated precursor from high pressure to vacuum.18–20
These reports raised the question of whether a similar technique utilising pulsed valves could achieve the desired characteristics for deposition of metal nanoclusters through the injection of solvated nanoclusters into high vacuum. Injection over a large pressure differential in such a manner means liquid would undergo a supersonic expansion, evaporating the solvent and allowing deposition of nanoclusters. Such a deposition would ideally have the following characteristics:
• Being able to deposit only fractions of a monolayer of clusters onto the substrate surface
• Distribution of clusters should be as homogeneous as possible
• Deposition of individual clusters and avoiding agglomeration of clusters or droplets of solvent
• Retaining the cluster properties and characteristics
• Verifiable decrease in solvent contamination compared to existing methods of deposition from solution
High vacuum pulse valves offer control of deposition conditions through the valve opening time (pulse width), the time between pulses (pulse frequency), solution concentration and the pressure differential between vacuum and backing pressure of the valve. In this paper, we present the operation and design of a pulsed cluster vacuum deposition system for ligand-protected, atomically-precise gold nanoclusters, specifically [Au9(PPh3)8]3+, from a methanol solution. Optimisation of the system was investigated for several deposition parameters towards the desired cluster deposition conditions stated previously. Atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS) and transmission electron microscopy (TEM) have been used to characterise the surfaces after pulsed nozzle deposition of the gold clusters.
Experimental methodology
To investigate the usefulness of Pulsed Nozzle Cluster Deposition (PNCD) for chemically synthesised nanoclusters, a representative nanocluster was selected for the study, [Au9(PPh3)8]3+ (hereafter referred to as Au9). Chemically-synthesised, atomically-precise gold clusters were synthesised and purified as reported previously,21,22 before recrystallisation and the use of nuclear magnetic resonance spectroscopy, mass spectrometry and X-ray crystallography to confirm the size specificity of the clusters.23 Au9 was stored at −17 °C in the dark before use in the deposition process. For PNCD, the Au9 was weighed before dissolution in ultra-pure methanol (HPLC grade, Scharlau) to the desired concentration. The concentration range used was 1 mM, 0.5 mM, 0.25 mM, 0.125 mM, 0.063 mM.This solution was placed into the liquid reservoir of the pulse nozzle in 0.5 mL aliquots before being flushed with and then back pressured by argon to 2 bar. The Au9 was deposited onto (i) mica (grade V-4 Muscovite, SPI supplies) for atomic force microscopy (AFM) measurements, (ii) lacey carbon film TEM microgrid (#10-1003 RO-C15, Okenshoji) for transmission electron microscopy (TEM), and (iii) RF sputtered, nanoparticulate TiO2 for X-ray photoelectron spectroscopy (XPS). While the substrates differ in chemical makeup and surface roughness, they are not expected to alter the deposited cluster's properties without further treatment such as heating (vide infra).
While the chosen substrates have different surface properties, the interaction of ligated gold clusters with mica and TiO2 is known to be similarly weak. As a consequence, it is assumed that examining particle size and pulsed deposition topography with mica through AFM and elemental composition and chemical properties with TiO2 trough XPS is a reasonable approach. The mica was freshly cleaved along the [001] plane before deposition, with both mica and clean nanoparticulate TiO2 cut into 10 × 10 mm samples and mounted to sample holders. TEM microgrids were held by a custom sample holder. After mounting, all substrates were placed into a high vacuum sample load lock and were pumped to 10−6 mbar over 6–8 h. Each sample was deposited with a set number of pulses of equal pulse length (250 μs, unless otherwise stated). After waiting for the ‘settling time’, the system can be pulsed again to inject further material until the desired amount has been injected. For gold clusters, the ideal case is that a certain amount of pulses delivers specific coverage in a controllable manner. The desired depositions for this process would be capable of producing a controllable, sub-monolayer coverage. Up to 2 samples could be prepared in the load lock chamber at the same time, allowing for identical deposition conditions across different substrates. The PNCD system design is schematically represented in Fig. 1.
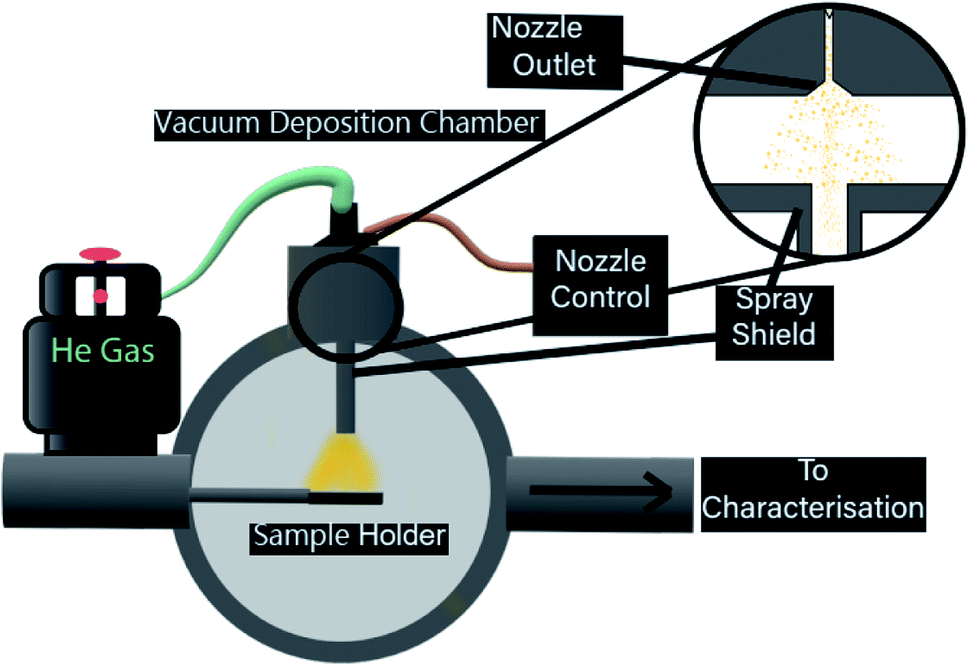 | ||
| Fig. 1 Representation of the PNCD system, mounted to a high vacuum load lock (cutaway). Au9 is loaded into the liquid reservoir and then backed with argon to 2 bar of pressure. The pulse nozzle control unit was operated with a programmed frequency to drive the opening/closing of the solenoid valve. Inside the vacuum chamber, a fabricated metal channel allows the pulsed vapour to reach the sample via line of sight while collecting the more dispersed vapour. The sample is held in a sample manipulator in the path of the vapour pulse and once deposited, can be inserted into the ultra-high vacuum for XPS measurement. AFM measurement requires removal to atmosphere. | ||
The pulse vapour deposition system is comprised of three main components:
• High vacuum pulsed valve
• Pulsed valve control unit
• Cold-wall vacuum chamber with sample manipulator
The high vacuum pulse valve used is a miniature high-speed, high vacuum dispense solenoid valve (Parker Hannifin, USA) with a 0.51 mm orifice, cone-shaped outlet flange and PTFE poppet. The valve has an internal 3 mL reservoir and is fitted for a gas line for providing backing pressure. This valve is mounted such that it is in a vertical orientation, pulsing vertically downwards. The nozzle is not a ultrasonic vibrational nozzle. The latter is probably not suitable for the deposition of clusters because the ultrasound could lead to disintegration of the clusters.
The pulse valve control unit (Iota One valve driver, Parker Hannifin, USA) used at NIMS was specifically designed for operating high-speed solenoid valves. Depositions performed at Flinders utilised a custom built pulse driver to control identical high-speed solenoid valves. The control unit allows the selection of pulse width (nozzle open time) to the microsecond range as well as the ability to make single pulses or automatically triggered pulse sequences at determined intervals. A successful pulse would register an audible click from the high-speed vacuum valve followed by a rapid increase in observed pressure, peaking in the high 10−5 to low 10−4 mbar. The pressure would return to the starting value within approximately 30 seconds, and after a further 30 seconds, the next pulse was injected, giving a 60 second pulse interval.
The load lock used for deposition is attached to a separate UHV chamber, allowing deposited samples to be easily introduced, or removed to atmosphere for further analysis. A custom-designed vapour shielding channel (see Fig. 1) was used in the load lock in order to restrict the path of injected vapour, allowing only those nanoclusters with a line of sight to the substrate to reach it, it was then removed for cleaning in between depositions.
AFM
All AFM measurements presented were performed at NIMS. Mica substrates were selected for AFM measurements due to their low surface roughness to easily distinguish the sub-nanometer deposited species.24,25 These samples were removed from vacuum and transferred directly to a vibrationally isolated AFM (SPA400, Seiko Instruments, Japan) and scanned with tapping mode (SI-DF20 cantilevers, Hitachi, Japan) rated at k = 42 (N m−1), 250–390 kHz with 125 μm cantilever length. Scans were run in atmospheric conditions at multiple sites across the surface of a sample. Scans were made from 10 × 10 μm down to 200 × 200 nm.AFM imaging allows plotting of the distribution of deposited particles as well as their height for analysis. AFM tip curvature means that surface species smaller than the AFM resolution cannot be accurately resolved laterally but their height measurements are unaffected by tip curvature and remain accurate. Raw topography scan data was processed with the Gwyddion software package. Once the scan was levelled, deposited particles were masked using the mask by watershed feature, with each watershed region containing approximately one particle. The masked particles were then filtered to remove any particles larger than known cluster size, as well as to have a minimum height cut-off to remove substrate regions with a roughness that had been incorrectly marked as particles. The maximum and mean particle heights could then be extracted through the in-built grain analysis package.
XPS
All XPS measurements were performed at Flinders University (FU) under UHV conditions. While mica substrate was found to be suitable for AFM measurements, test samples used in XPS showed charging effects that interfered with elemental identification. TiO2 has been shown to be appropriate support material for electron spectroscopy.26 Therefore, all samples deposited onto mica for AFM were simultaneously deposited onto TiO2 for XPS measurements to identify the chemical composition of the sample surface. Samples prepared at NIMS (series A & B) were transported to Flinders for measurement using XPS. These samples were transported in individual light-blocking sample holders and stored at −17 °C when not in transit. XPS was performed as soon as possible to avoid unnecessary sample degradation. Samples for the investigation of solvent removal were deposited and investigated at FU under UHV conditions without exposure to atmosphere.X-ray spectra were recorded at 1253.6 eV (Mg Kα radiation) using a SPECS PHOIBOS-HSA300 hemispherical analyser as previously described.27 Due to the limited mean free path of the electrons, only the top ∼5–10 nm of the sample is probed. These measurement conditions have been shown not to damage samples of this type.16
High-resolution XP spectra of the C 1s, O 1s, P 2p, Ti 2p and Au 4f regions were collected at 10 eV pass energy. All XP spectra were analysed with Casa XPS using a Shirley background and a hybrid Gaussian (30%) and Lorentzian (70%) function to fit all peaks in a method described previously.23 Elemental sensitivity factors for an X-ray source at 54.7° were used is the analysis process. The adventitious carbon peak found at 285 eV binding energy was used as the calibration peak for all samples.28
TEM
TEM measurements were undertaken at NIMS on samples prepared using lacey, amorphous carbon film TEM grids that were subsequently dried under vacuum for several days prior to measurement. Measurements were performed on a JEOL JEM-2100F instrument with a 200 kV accelerating voltage in both dark and light field modes. Energy dispersive X-ray (EDX) measurements were also taken on specific samples for elemental analysis. Recorded images were processed using the DigitalMicrograph (Gatan) software package to determine particle sizes. It is worthwhile noting that initial testing on TiO2 nanosheet microgrids showed that pulse deposition was capable of destroying the nanosheet integrity, making measurement difficult or impossible. Subsequently 6 nm amorphous carbon microgrids were used for deposition and were far more resistant to damage by the deposition process. Vacuum drying was used as a pre-treatment of the TEM sample and holder. This reduces the outgassing of hydrocarbon sources such as CH4 or carbon oxides, which would normally induce contamination of the samples with C in the presence of an electron beam.Results
Controlling and optimising deposition
Initial attempts to deposit gold clusters using a differing number of pulses at a range of pulse widths resulted in either very heavy coverages or no discernible change in sample surface as viewed in AFM. This was attributed initially to the pulse length as pulses around 150 μs resulted in inconsistent pressure increases, and longer pulses (>300 μs) showed clear circular features assigned to the impact of a droplet of particles on the substrate, similar investigations using ultrasonically vibrated nozzles had reported imperfect pulse conditions lead to large droplet formation.18 From AFM, it was clear that the depositions were higher than a monolayer coverage and therefore undesirable (Fig. S1–S3†). The pulse width was optimised to produce consistent peak pressures (10−5 mbar) and reduction of circular features at an open time of 250 μs was used for all final measurements.Control depositions were performed using only methanol in a freshly cleaned pulse nozzle set-up. These depositions were performed onto freshly cleaved mica using 250 μs pulse width, 50× pulses at 60 seconds intervals allowing deposition chamber pressure to recover to low 10−7 mbar pressure between pulses. The mica used for deposition was measured in AFM directly after cleaving and then again straight after removal from the deposition chamber. The AFM scans of the before and after deposition conditions can be seen in Fig. 2a and b, respectively (also, Fig. S4†). The mica before a pulsed deposition can be seen to have a reasonably homogenous coverage of 2–3 nm high particles. Similar features have been found in previous studies on mica cleavage and assigned to crystallite growth from surface contaminants.25 AFM scans of post-deposition methanol exhibited the continued observation of the 2–3 nm features, with a small number of larger features in the 3–5 nm range being introduced. As both prior and post-deposition scans show large regions of picometer roughness (attributed to flat mica) with observable features being of a size range easily distinguishable from deposited gold clusters (0.75–1.25 nm),29,30 the use of PNCD on mica was determined to be appropriate for the study of Au9 cluster.
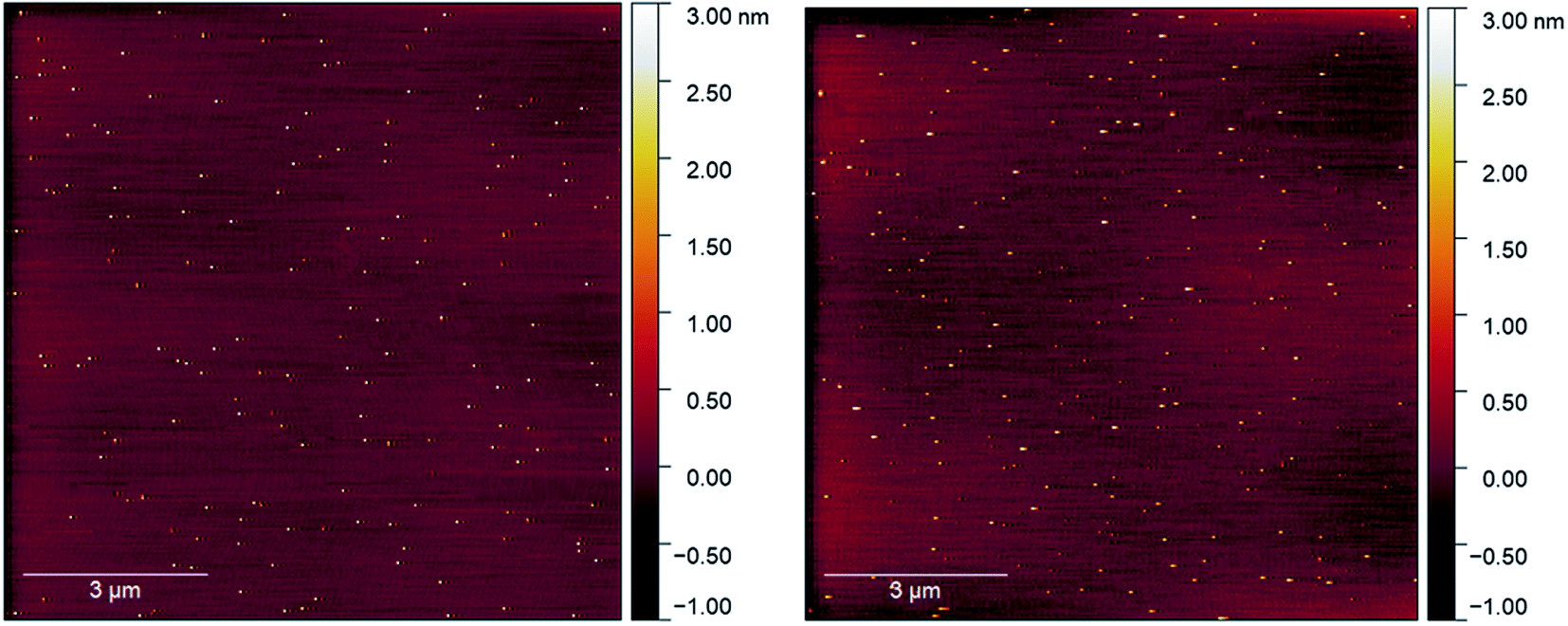 | ||
| Fig. 2 (Left) AFM scan on freshly cleaved mica, (right) AFM scan on mica after 50 pulses of methanol, some larger features are clipped out of scale range. | ||
As such, a series of measurements were undertaken to investigate the effect of PNCD solution concentration and pulse number (see Table 1) onto mica and TiO2 placed side by side in the load-lock.
| Deposition series A | Deposition series B | ||
|---|---|---|---|
| a Sample A4 & B4 come from the same deposition. | |||
| A1 | 1 mM | B1 | 100× |
| A2 | 0.5 mM | B2 | 50× |
| A3 | 0.25 mM | B3 | 40× |
| A4 | 0.125 mM | B4 | 30× |
| A5 | 0.061 mM | B5 | 20× |
| B6 | 10× | ||
| B7 | 1× | ||
The amount of gold cluster deposited for each sample was determined through quantification with XPS. It has been determined through previous studies that ligated Au9 clusters have a 4f7/2 peak with characteristic binding energy (BE) at 84.9 ± 0.3 eV contrasting with the same peak for agglomerated clusters found at the binding energy for bulk gold at 84 ± 0.2 eV.16,23 For samples A4, A5, B2–5, agglomeration is fairly consistent at a ratio around 70/30 cluster/bulk, as shown in Fig. S5† for sample A4. All other samples showed no signs of agglomeration, as shown in Fig. 3 for sample B6, suggesting that agglomeration in the other samples may have been caused by transport or sample handling. The P 2p region was also investigated, but no discernible peaks were found, probably due to partial ligand loss during the multiple vacuum pumping events, as found by us previously.30 The results of the analysis can be seen for all species in Table 2, with the trends of gold cluster deposition across series A and B shown in Fig. 3.
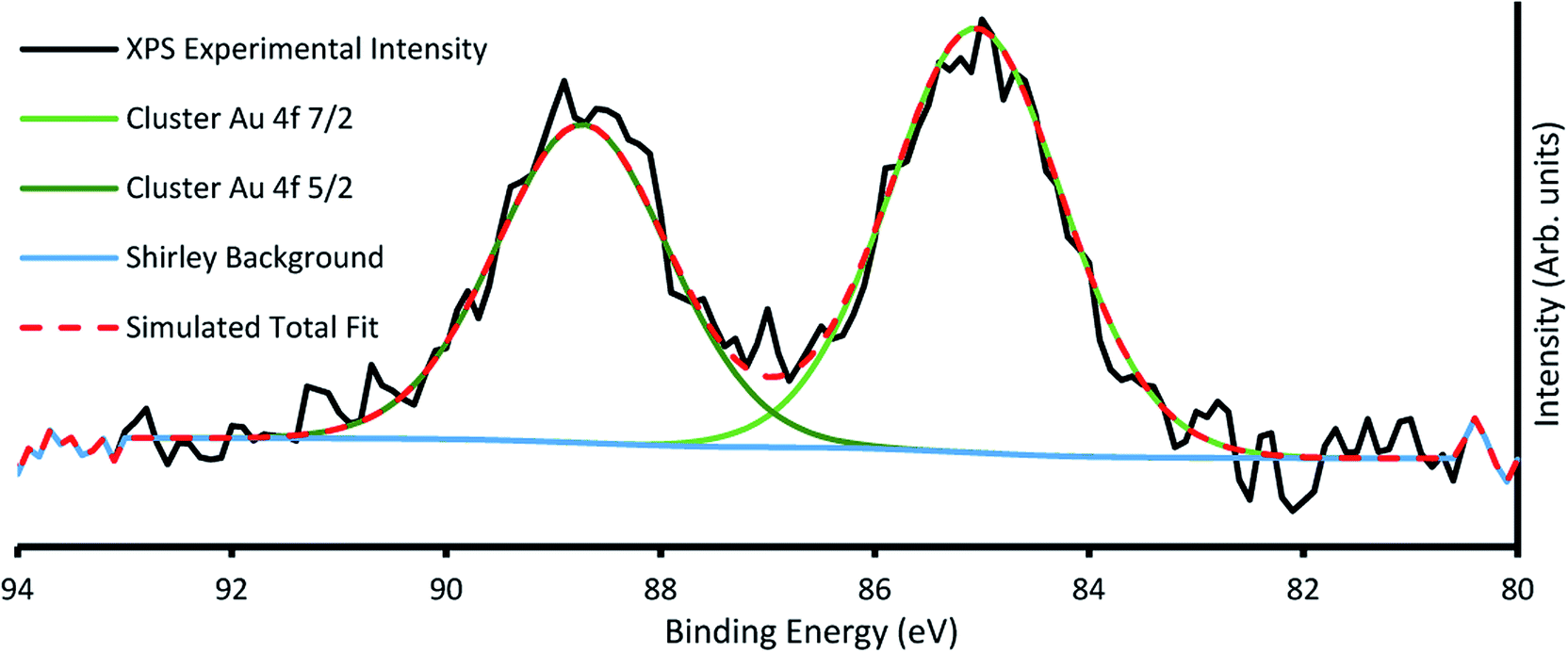 | ||
| Fig. 3 Example XP spectra of sample B6 showing Au 4f region, with the characteristic cluster Au 4f7/2 peak at 85.2 eV. | ||
| Carbon (285 eV) | Oxygen (530.5 ± 0.2 eV) | Titanium (459 ± 0.2 eV) | Gold (cluster, 85 ± 0.2 eV) | Gold (bulk, 84 ± 0.2 eV) | |
|---|---|---|---|---|---|
| A1 | 22.4 ± 0.1 | 57.6 ± 0.1 | 19.9 ± 0.1 | 0.1 ± 0.01 | — |
| A2 | 23.8 ± 0.1 | 57.6 ± 0.1 | 19.9 ± 0.1 | 0.3 ± 0.01 | — |
| A3 | 33.8 ± 0.1 | 49.0 ± 0.1 | 16.3 ± 0.1 | 0.8 ± 0.01 | — |
| A4 | 17.6 ± 0.1 | 61.0 ± 0.1 | 21.2 ± 0.1 | 0.07 ± 0.01 | 0.03 ± 0.01 |
| A5 | 19.2 ± 0.1 | 60.1 ± 0.1 | 20.5 ± 0.1 | 0.07 ± 0.01 | 0.03 ± 0.01 |
| B1 | 24.0 ± 0.1 | 56.6 ± 0.1 | 19.1 ± 0.1 | 0.2 ± 0.01 | — |
| B2 | 25.2 ± 0.1 | 56.3 ± 0.1 | 18.4 ± 0.1 | 0.11 ± 0.01 | 0.03 ± 0.01 |
| B3 | 23.9 ± 0.1 | 57.1 ± 0.1 | 18.9 ± 0.1 | 0.8 ± 0.01 | 0.06 ± 0.01 |
| B4 | 17.6 ± 0.1 | 61.1 ± 0.1 | 21.2 ± 0.1 | 0.07 ± 0.01 | 0.03 ± 0.01 |
| B5 | 25.7 ± 0.1 | 56.0 ± 0.1 | 18.2 ± 0.1 | 0.034 ± 0.01 | 0.016 ± 0.01 |
| B6 | 21.1 ± 0.1 | 58.7 ± 0.1 | 20.2 ± 0.1 | 0.02 ± 0.01 | — |
| B7 | 17.2 ± 0.1 | 58.5 ± 0.1 | 24.3 ± 0.1 | 0.014 ± 0.01 | — |
It can be seen that the carbon contribution across all samples is very high, as would be expected for samples having been exposed to the atmosphere. Cleaning of the samples with heating/sputtering was not undertaken as it has been shown to affect Au9 stability, causing agglomeration. Sample A3 had unexplained, particularly high carbon contribution (10% higher) and concomitantly lower Ti and O contribution. Otherwise, the samples were quite similar with the exception of gold cluster contribution, which showed marked differences depending on deposition conditions. Across series A, the amount of gold cluster deposited seemed to follow no discernible trend for different solution concentrations, with the highest concentration of gold cluster in methanol, A1 giving the lowest gold coverage overall. The 0.25 mM sample A3 meanwhile had the highest gold contribution of any measured sample.
AFM scans on series A samples showed highly variable coverages with many circular, droplet-like features (see Fig. S3–S5†). These results suggest that solution concentration, at least over multiple pulses is not an effective method of controlling sub-monolayer depositions of gold clusters.
In series B, while the elemental concentration of Au does not scale in a perfectly linear fashion with the number of pulses applied, the response in Au relative XPS contribution results in a near-linear decrease with decrease in pulse number. However, when the relative Au XPS contribution per pulse is examined (orange line in Fig. 4), the amount of Au deposited per pulse is almost constant, except for the sample B7. This single pulsed sample shows nearly 5 times the amount of Au deposited per pulse. The reasons for this are not fully understood but as the first pulse is injected into a vacuum achieved through hours of pumping it undergoes the greatest pressure differential, which may also explain why the most dispersed, homogenous depositions were achieved from such depositions. The first pulse could change the local pressure at the nozzle, or the seal of the nozzle poppet, affecting how much material is drawn through in subsequent pulses.
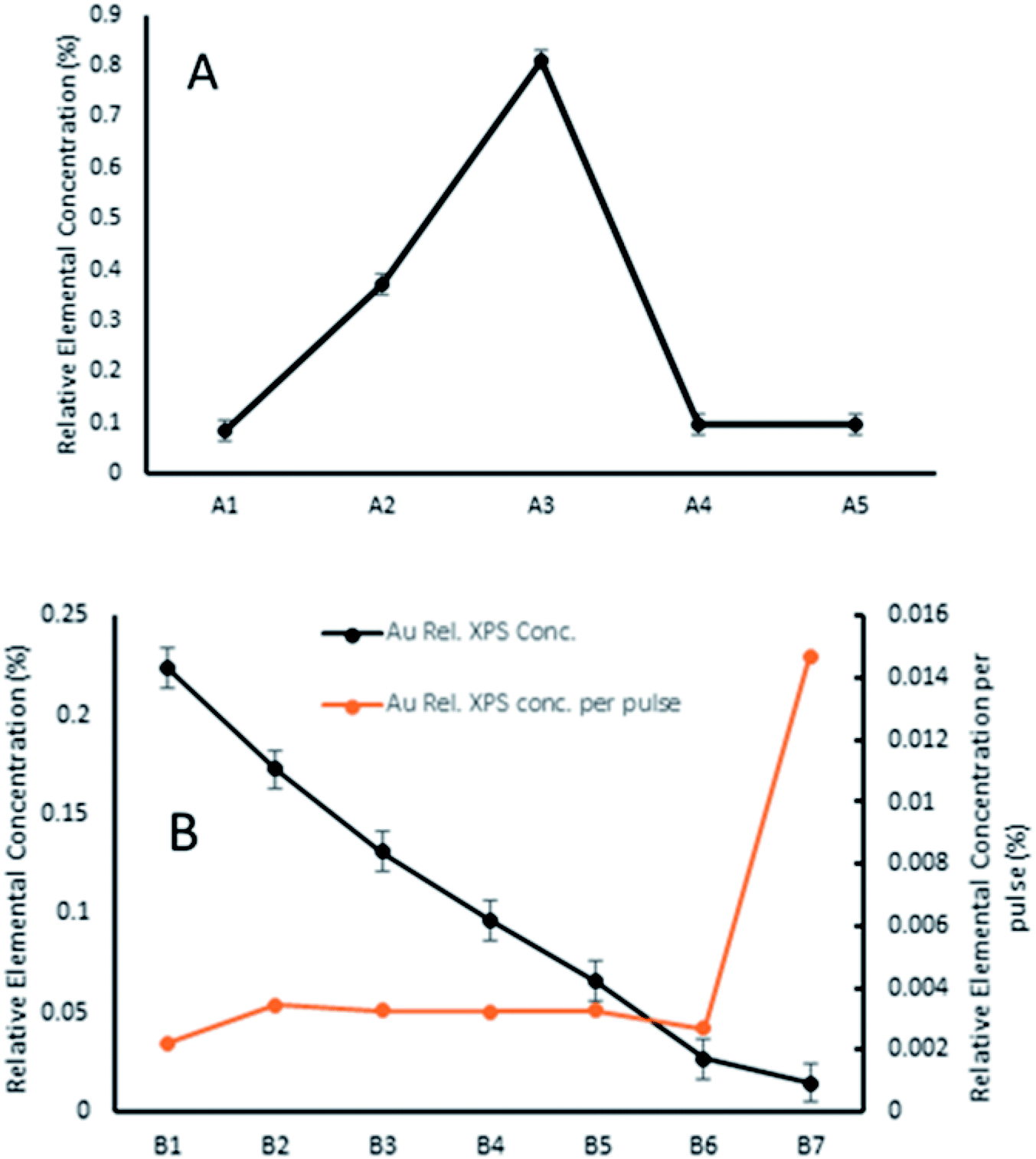 | ||
| Fig. 4 Relative elemental concentration of Au9 clusters from XPS analysis across series A (concentration change (A)) and series B (pulse number change (B)). No trend is evident in series A, whereas series B shows a clear reduction in gold cluster conc. with lower pulse no. and, when normalised per pulse, reveals that the initial pulse deposits far more Au than subsequent pulses. | ||
While the amount of gold cluster deposited is a key criterion for assessing PNCD, another is the dispersion and coverage of a sample with Au9 clusters. Especially given that size-specific clusters are observed to agglomerate more readily in concentrated depositions from solution. The AFM images of samples B1–5 show patchy areas of very high coverage resembling features of closely packed small particles along with some much larger particles (2–4 nm) dispersed throughout (see Fig. S1–S3†). Scans of the samples B6 & B7 showed many more isolated, cluster-like features and very few regions of closely packed clusters. An AFM scan from series B7 can be seen in the top half of Fig. 5 displaying the types of coverages seen in a 1× pulsed deposition. No highly concentrated regions were detected in the scanned areas; instead, disperse particulate coverage exist across the scan, with much less variation in particle concentration. The particle height distribution for the scans on sample B7 can also be seen in Fig. 5. The particle size analysis revealed a distribution of maximum particle heights peaking between 0.85–0.98 nm while taking the mean particle height gave the largest number of particles at 0.90 nm with a broader overall distribution.
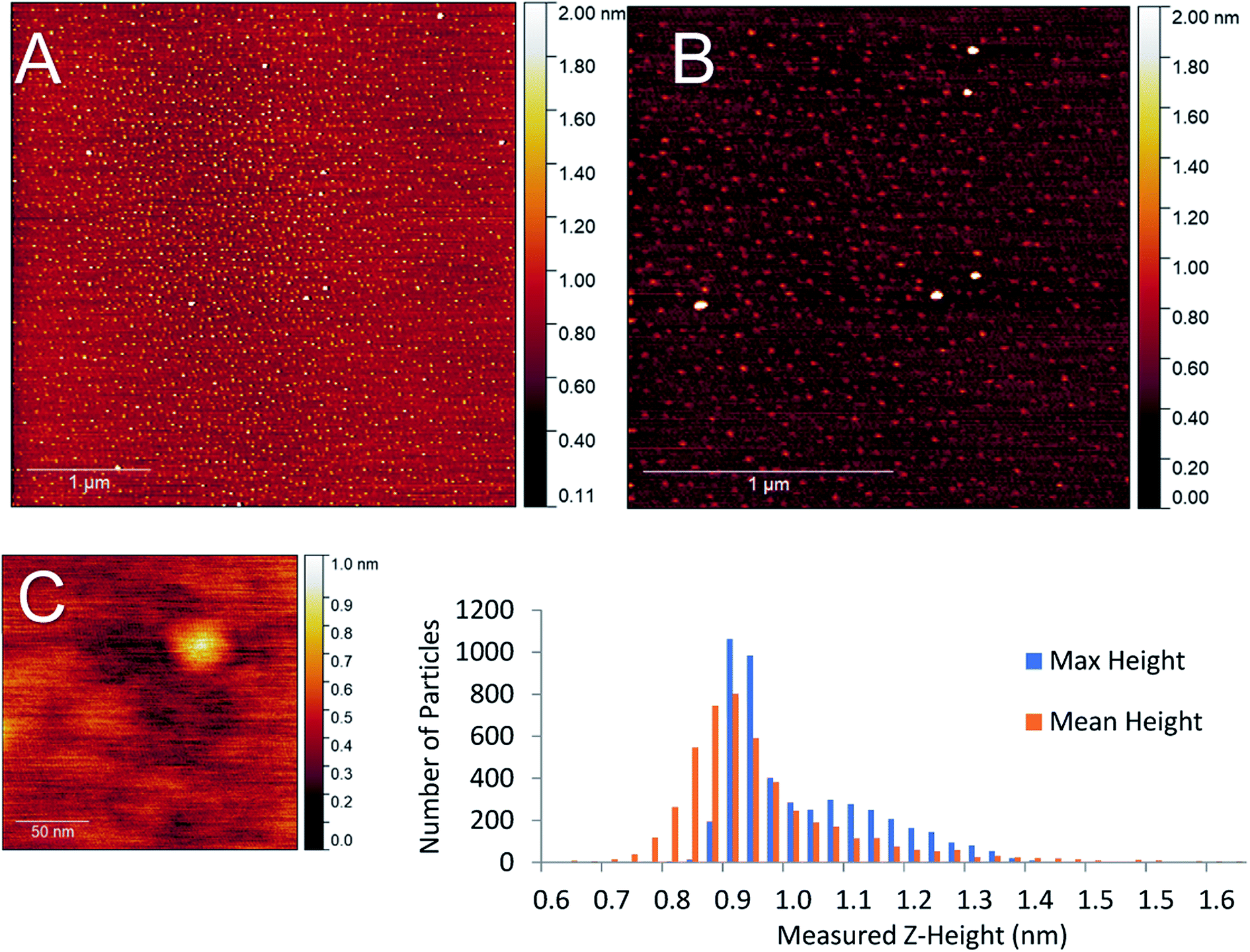 | ||
| Fig. 5 Top – (A) 0.125 mM Au9/methanol single pulse (sample B7) on mica. (B) higher magnification scan of (A). Bottom – (C) High magnification scan on a single cluster-sized object found on the same sample. Particle height distribution for 5 μm2 scan of 0.125 mM from one pulse. Some larger particles are clipped out of scale range. | ||
AFM analysis was undertaken on sample B6 pulse as this sample also produced dispersed coverages with a large fraction of single cluster sized objects. The AFM results for this deposition can be seen in the supplementary information (Fig. S6),† with the particle size analysis of the 5 μm2 scan (Fig. 5, scan A). The maximum and mean height distributions show maxima, both between 0.65–0.91 nm. Both samples B6 and B7 measurements showed that particle max height analysis contained a small distribution of larger particles.
Particle sizes of 0.65–0.98 nm are in good agreement with results previously reported by Al Qahtani et al. from AFM of the same gold clusters on TiO2 nanosheets deposited through immersion, where they showed the average z-height of deposited clusters was 0.75 nm (ref. 29) albeit on a rougher substrate. Previous DFT modelling of ligated gold clusters proposed larger sizes between 1.47–1.66 nm when ligated but did not take into account the change in cluster orientation and arrangement, as seen in high-resolution STEM, gold clusters are capable of ‘laying flat’ when interacting with surfaces,30 or after the loss of some ligands in a vacuum environment, as suggested by XPS P results. Thus, it can be assumed that particle sizes of 0.65–0.98 nm correspond to the height of single Au9 clusters on mica.
Further AFM measurements on the single pulsed sample showed that under certain scan conditions, the gold clusters could be moved by AFM tip contact (see Fig. S7†) as has been previously observed for gold cluster depositions on TiO2 nanosheets.24
TEM results
Samples for TEM measurements were prepared only under conditions closely resembling samples B3–B6 (i.e. conditions similar to series A were not examined with TEM). This involved depositing the samples with 0.125 mM Au9/methanol, at 250 μs pulse time for 40–10 pulses at 60 second intervals.The samples were imaged using darkfield TEM. Several sites were chosen for imaging across each sample. The TEM scans from a 10-pulse deposited microgrid can be seen in Fig. 6. The well-dispersed bright spots indicate individual gold clusters on the carbon substrate. The coverage observed is homogenous, and while there is a variety of particles sizes evident, the TEM intensity line profile (Fig. 6) clearly shows that lateral particle size is in good agreement with complementary AFM height measurements for gold clusters at 0.8–0.95 nm. Several larger particles appear to be irregular in shape and may indicate a grouping of multiple deposited gold clusters. EDX was used to attempt identification of the metallic species but due to low sensitivity was only effective on heavily deposited samples, where it identified Au as present (Fig. S8†).
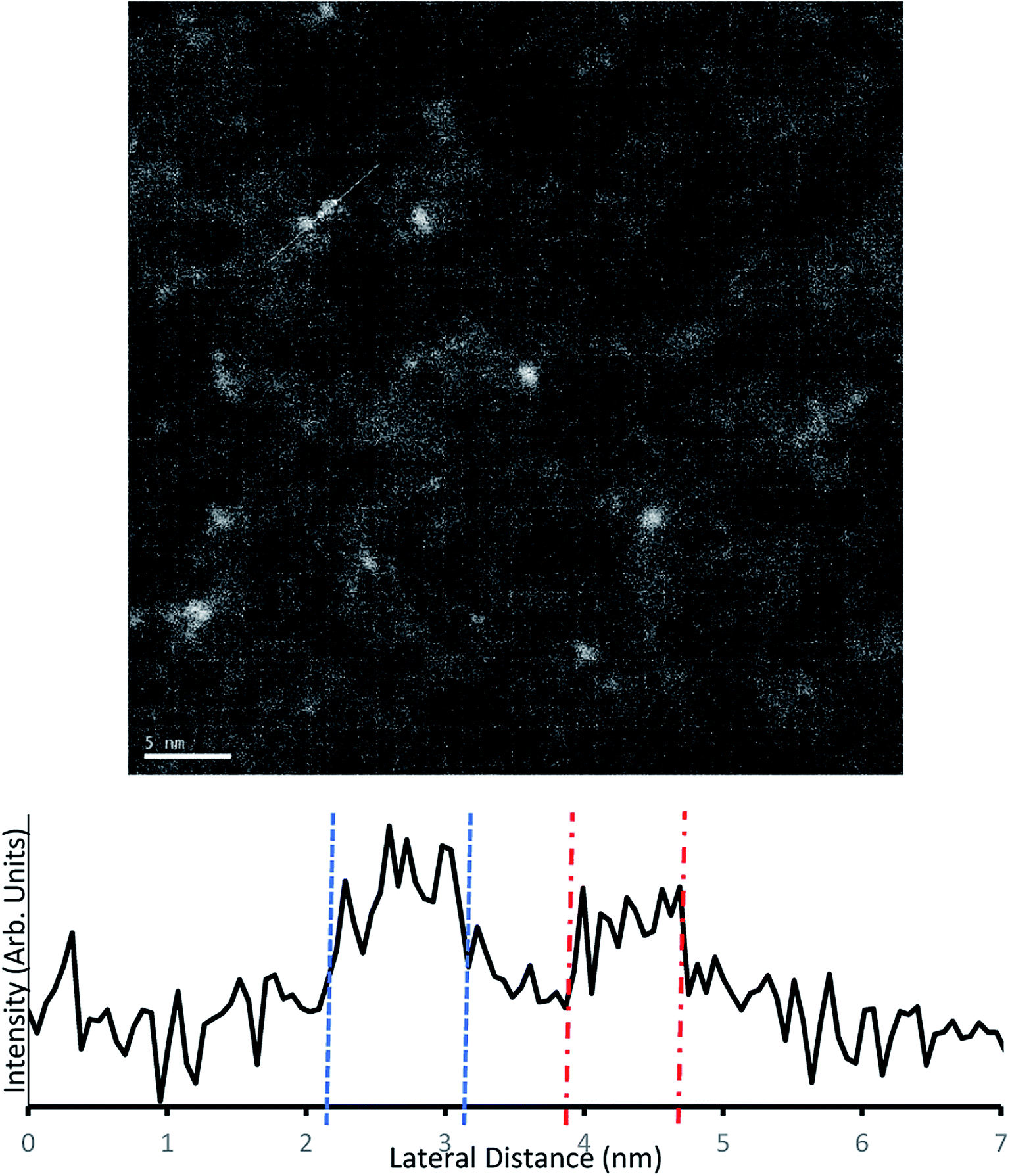 | ||
| Fig. 6 Above – Darkfield TEM of microgrids deposited with 10 pulses of 0.125 mM Au9/methanol solution. Below – Intensity profile along the white trace of the image. | ||
Solvent removal – effect of heating
In order to determine the presence of solvent from the PNCD process on the samples, the system detailed in the experimental section was set up on a load lock with direct access to XPS measurements. The TiO2 substrate was inserted into the vacuum and cleaned of adventitious carbon and other contaminants through heating to 300 °C and sputtering with Ar. It was then deposited with 3 pulses of gold cluster/methanol solution (0.125 mM) before being heated to 200 °C all while remaining in at least 10−7 mbar vacuum.It was observed that by measuring the high-resolution C 1s region in XPS between the vacuum deposition the growth of the carbon peak at 286.7 ± 0.1 eV binding energy appears after the pulses are deposited onto the sample and correspond to C–O bonds from methanol (see Fig. S9†). The procedure described above mirrors that undertaken for previously reported attachment of gold cluster to TiO2 through liquid immersion.16 The critical distinction being that deposition through liquid immersion requires the vacuum treated sample to be removed into atmosphere, introducing sample contamination and altering it's treated state. A comparative sample prepared using liquid immersion of an identically prepared TiO2 substrate in the same gold cluster/methanol solution for 30 minutes was also measured using high-resolution XPS of the C 1s region.
It can be seen from this comparison, presented in Table 3, that the total C 1s intensity after the pulsed nozzle deposition is only around 6% of the total XPS intensity, only 2% higher than C 1s intensity of vacuum heated and sputtered TiO2 with no deposition. The immersed sample deposition by comparison showed a total C 1s intensity of more than 30%, representing a five-fold increase in intensity over the PNCD prepared sample. For reference, the typical C 1s intensity of in situ heated and sputtered titania exposed to air is typically larger than 25% of the total XPS intensity31 and thus at least four times larger than the total C 1s content of the samples after pulsed nozzle deposition. These measurements confirm that pulse deposition is a method of depositing gold clusters from solution with significantly lower solvent contamination than existing methods.
| TiO2 sample process | Relative XPS % (C 1s) |
|---|---|
| Heated 300 °C, Ar sputtered (in situ) | 3.9 ± 0.2 |
| Pulsed w/Au9/methanol (0.125 mM) | 6.4 ± 0.2 |
| Immersed, 30 min in Au9/methanol (0.125 mM) | 31.0 ± 0.2 |
Pulsed nozzle cluster deposition modelling
The PNCD process involves the injection of a pulsed ‘packet’ of gold clusters/solvent into the chamber. This packet undergoes a supersonic expansion in a pressure differential which causes prompt solvent evaporation. Removal of vapour through surface deposition and constant pumping returns the vacuum chamber to its initial pressure after ∼30 seconds.The evaporation rate of the solvent upon supersonic expansion is important to determine the distance a solvent droplet can survive before vaporisation in the vacuum. Versteeg et al. proposed a model for a droplet of given radius to evaporate utilising partial pressure from a droplet of solvent combined with a curvature of the solvent surface (assuming a round droplet)18 as well as the resulting monolayers deposited per pulse and velocity of the injected pulse. Their modelling was for an ultrasonically active nozzle but was adapted for the nozzle used in these investigations.
Eqn (1) integrates the time to evaporate a methanol droplet from an initial radius (riniital taken as radius of the nozzle bore) to a final radius of a single cluster (i.e..an isolated cluster).
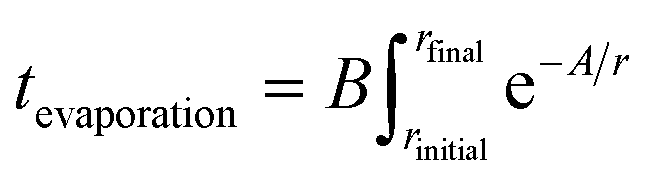 | (1) |
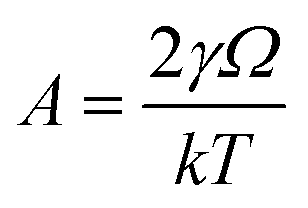 ,
, 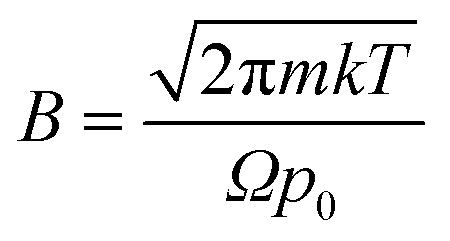
Evaluating (1) using the values from Table 4 gives tevaporation = 0.000196 seconds and is used below to estimate the distance over which a droplet evaporates.
| γ | 2.25 × 10−2 | Surface tension of the solvent (N m−1) |
| Ω | 1.76 × 10−28 | Molecular volume (cm3) |
| k | 1.38 × 10−23 | Boltzmann's constant |
| T | 293.15 | Temperature (K) |
| m | 1.53 × 10−25 | Mass of evaporated particle (kg) |
| p 0 | 1.89 × 104 | Vapour pressure (Pa)32 |
| r final | 4.00 × 10−9 | Final droplet radius (m) |
| r initial | 5.10 × 10−4 | Initial droplet radius (nozzle radius) (m) |
Monolayers deposited per pulse are with the parameters defined in Table 4.
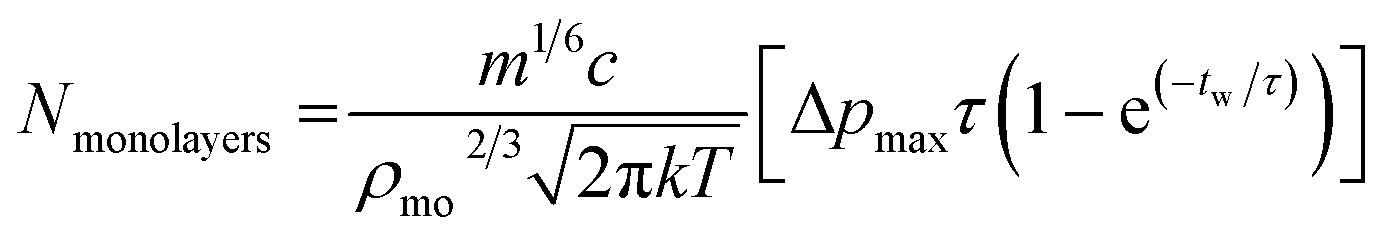 | (2) |
This model is drawn from kinetic gas theory coupled with the assumption that a pulse causes an infinitely fast rise time in chamber pressure at pulse injection followed by exponential decay until the chamber returns to its base pressure. Infinitely fast means here in comparison with the time characteristics of the pulse. This modelling gives a pressure curve that can be integrated over the time from 1st to 2nd pulsing to give the fraction of monolayers per pulse deposited by the valve. Coupling this with the assumption that the partial pressure of the gold cluster is proportional to its concentration in solution and that the growth of monolayer is proportional to adsorbed molecular area, approximated as a cube. Thus eqn (1) can be used to estimate coverage for a single pulse at a given waiting time.
Using eqn (2) and the values from Table 5 gives Nmonolayers = 0.003 per pulse. This value, while relying on some approximations, is approximately in agreement with gold XPS measured for sample B6 (10 pulses) giving 0.02 monolayers and could provide a framework for achieving specific sub-monolayer coverages.
| t w | 60 | Inter-pulse waiting time (s) |
| T | 293.15 | Temperature (K) |
| k | 1.38 × 10−23 | Boltzmann's constant |
| ρ mo | 19.3 | The density of gold (g m−3)33 |
| τ | 1.58 × 10−4 | Decay constant (s−1, experimentally determined) |
| Δp | 4.32 × 10−4 | Pressure maximum (mbar) |
| m | 3870.9828 | Mass of cluster (g mol−1) |
| c | 0.125 | Cluster concentration (mM) |
Estimating the velocity of the solution on injection assumes the pressure drop and resulting acceleration occur entirely within the nozzle bore. Modelling for this can be done using the formula for head loss in a circular pipe, assuming a turbulent flow. This injection velocity, hf, can be determined from eqn (3):
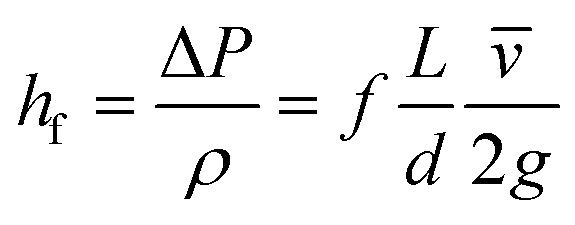 | (3) |
Eqn (3) evaluated using values from Table 6 gives 30 m s−1 as the final velocity on entering the vacuum chamber, combining this value with the lifetime of a 10 μm droplet evaluated from (1) gives droplet evaporation within 5.7 × 10−3 m from the nozzle bore. Given the evaporation chamber has dimension 8 × 10−2 m from the nozzle to sample, only (methanol) vapour will reach the sample surface. Thus, deposition of solvent onto the substrate only occurs from the gas phase. The total amount of solvent attaching to the surface through the deposition method thus is significantly less than when depositing clusters by dipping the substrate into solution.13,16 The circular feature identified on some heavily deposited surfaces in AFM (Fig. S4†) may be accounted for by vacuum degradation over lengthy depositions affecting the solvent evaporation rate. Alternatively, in some pulsed CVD modelling20,34 it has been proposed that particles can aggregate in a vacuum as the solvent droplets containing the particles evaporate and contract, clumping solvated contents into more concentrated groups. [Note: the Mathematica code used for this modelling can be found in Fig. S10.†]
| ρ | 791 | Density of methanol kg m−3 |
| ΔP | 2.0 × 105 | Pressure drop from nozzle head to chamber, Pa |
| d | 0.51 | Nozzle diameter, mm |
| L | 1.25 | Nozzle orifice length, mm |
| f | 0.035 | Friction factor, drawn steel, 0.0015 mm roughness, Reynolds number > 104 |
| g | 9.8 | Acceleration due to gravity, m s−2 |
It is worth noting that while the conditions for this model are relevant for a single pulse, and that the predicted monolayer coverage per pulse aligns well with samples B1–6; the observed coverage on sample B7, a single pulsed sample is in worse agreement. For B1–6, this model's predictions are within a factor of 2 of observed monolayer coverages. For B7 it is more like a factor of 5. While this is still a useful result, it does reinforce the idea that the conditions of the first pulse may interact in a manner not wholly captured in this model.
Conclusions
Au9 clusters were deposited onto mica and titania substrates via pulsed nozzle cluster deposition (PNCD) using a variety of cluster concentrations and pulse conditions and the surfaces analysed with AFM, XPS and TEM. AFM allowed for analysing the height distribution of the clusters on the surfaces and thus a measure for the degree of agglomeration. Through XPS, it was possible to determine the degree of agglomeration via the Au binding energy. It was found that the conditions for the deposition affect the dispersion of the clusters over the substrate surface. Change in Au9 concentration was found not to be a suitable method for controlling the number of deposited clusters. Varying the number of pulses at an Au9 concentration investigated (0.125 mM) resulted in effective control over the concentration of clusters deposited.Under conditions of homogeneous dispersion of the Au9 clusters over the surface, it was found that the vast majority of features found with AFM had a height corresponding to the size of a single Au9 cluster. Additionally, TEM images show Au particles whose lateral size is consistent with single Au9 clusters. XPS showed that most of the Au is at a BE of 85.0 ± 0.3 eV, giving evidence that cluster agglomeration is largely suppressed through the deposition process. XPS also showed that the minimal addition of hydrocarbons from the solvent used in the process (i.e. methanol) to the sample compared very favourably to existing methods of cluster deposition, resulting in a more than five-fold decrease in carbon contamination.
In summary, PNCD with single pulse deposition at low concentrations is a suitable method for depositing ligand protected clusters onto substrates at fractions of a monolayer be deposited and produces deposited clusters that are distributed as individual and isolated clusters over the surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank A/Prof Vladimir Golovko (Canterbury University) for providing access to the Au9(PPh3)8(NO3)3 clusters. We also wish to acknowledge the efforts of Dr Toshiaki Takei (NIMS-MANA) in assisting with TEM measurements. The work is supported by the US army project FA5209-16-R-0017. The work is also supported by the Australian Solar Thermal Research Initiative (ASTRI) program, which is supported by the Australian Government, through the Australian Renewable Energy Agency (ARENA). The authors acknowledge the expertise, equipment, and support provided by the Australian Microscopy and Microanalysis Research Facility (AMMRF) and the Australian National Fabrication Facility (ANFF) at Flinders University.References
- T. Higaki, Q. Li, M. Zhou, S. Zhao, Y. Li, S. Li and R. Jin, Toward the Tailoring Chemistry of Metal Nanoclusters for Enhancing Functionalities, Acc. Chem. Res., 2018, 51(11), 2764–2773 CrossRef CAS PubMed.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, Controlled Loading of Small Au n Clusters (n = 10–39) onto BaLa4Ti4O15 Photocatalysts: Toward an Understanding of Size Effect of Cocatalyst on Water-Splitting Photocatalytic Activity, J. Phys. Chem. C, 2015, 119(20), 11224–11232 CrossRef CAS.
- Y. Negishi, W. Kurashige, Y. Niihori and K. Nobusada, Toward the creation of stable, functionalized metal clusters, Phys. Chem. Chem. Phys., 2013, 15(43), 18736–18751 RSC.
- T. Bernhardt, U. Heiz and U. Landman, Chemical and Catalytic Properties of Size-Selected Free and Supported Clusters, in Nanocatalysis, Springer, 2007, pp. 1–191 Search PubMed.
- M. Chen, Y. Cai, Z. Yan and D. W. Goodman, On the origin of the unique properties of supported Au nanoparticles, J. Am. Chem. Soc., 2006, 128(19), 6341–6346 CrossRef CAS PubMed.
- M. Chen and D. W. Goodman, Catalytically active gold on ordered titania supports, Chem. Soc. Rev., 2008, 37(9), 1860–1870 RSC.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, Controlled loading of small Aun clusters (n = 10–39) onto BaLa4Ti4O15 photocatalysts: Toward an understanding of size effect of cocatalyst on water-splitting photocatalytic activity, J. Phys. Chem. C, 2015, 119(20), 11224–11432 CrossRef CAS.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase and S. Yamazoe, Au25-Loaded BaLa4Ti4O15 Water-Splitting Photocatalyst with Enhanced Activity and Durability Produced Using New Chromium Oxide Shell Formation Method, J. Phys. Chem. C, 2018, 122(25), 13669–13681 CrossRef CAS.
- W. Li, Q. Ge, X. Ma, Y. Chen, M. Zhu, H. Xu and R. Jin, Mild Activation of CeO2-Supported Gold Nanoclusters and Insight into the Catalytic Behavior in CO Oxidation, Nanoscale, 2016, 8(4), 2378–2385 RSC.
- B. C. Gates, Supported gold catalysts: new properties offered by nanometer and sub-nanometer structures, Chem. Commun., 2013, 49(72), 7876–7877 RSC.
- R. H. Adnan, G. G. Andersson, M. I. J. Polson, G. F. Metha and V. B. Golovko, Factors influencing the catalytic oxidation of benzyl alcohol using supported phosphine-capped gold nanoparticles, Catal. Sci. Technol., 2015, 5(2), 1323–1333 RSC.
- G. Portale, L. Sciortino, C. Albonetti, F. Giannici, A. Martorana, W. Bras, F. Biscarini and A. Longo, Influence of metal–support interaction on the surface structure of gold nanoclusters deposited on native SiOx/Si substrates, Phys. Chem. Chem. Phys., 2014, 16(14), 6649–6656 RSC.
- G. G. Andersson, V. B. Golovko, J. F. Alvino, T. Bennett, O. Wrede, S. M. Mejia, H. S. Al Qahtani, R. Adnan, N. Gunby, D. P. Anderson and G. F. Metha, Phosphine-stabilised Au-9 clusters interacting with titania and silica surfaces: the first evidence for the density of states signature of the support-immobilised cluster, J. Chem. Phys., 2014, 141(1), 12 CrossRef PubMed.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Electronic structure controls reactivity of size-selected Pd clusters adsorbed on TiO2 surfaces, Science, 2009, 326(5954), 826–829 CrossRef CAS PubMed.
- D. M. Foster, R. Ferrando and R. E. Palmer, Experimental Determination of the Energy Difference Between Competing Isomers of Deposited, Size-Selected Gold Nanoclusters, Nat. Commun., 2018, 9(1), 1323 CrossRef CAS PubMed.
- G. Krishnan, N. Eom, R. M. Kirk, V. B. Golovko, G. F. Metha and G. G. Andersson, Investigation of Phosphine Ligand Protected Au13 Clusters on Defect Rich Titania, J. Phys. Chem. C, 2019, 123(11), 6642–6649 CrossRef CAS.
- T. Bennett, R. H. Adnan, J. F. Alvino, R. Kler, V. B. Golovko, G. F. Metha and G. G. Andersson, Effect of Gold Nanoclusters on the Production of Ti3+ Defect Sites in Titanium Dioxide Nanoparticles under Ultraviolet and Soft X-ray Radiation, J. Phys. Chem. C, 2015, 119(20), 11171–11177 CrossRef CAS.
- V. A. Versteeg, C. T. Avedisian and R. Raj, Metalorganic chemical vapor deposition by pulsed liquid injection using an ultrasonic nozzle: titanium dioxide on sapphire from titanium(IV) isopropoxide, J. Am. Ceram. Soc., 1995, 78(10), 2763–2768 CrossRef CAS.
- S. Krumdieck, Pulsed-Pressure MOCVD Science, Materials and Technology, ECS Trans., 2009, 25(8), 1209–1219 CrossRef CAS.
- H. M. Cave, S. P. Krumdieck and M. C. Jermy, Development of a Model for High Precursor Conversion Efficiency Pulsed-Pressure Chemical Vapor Deposition (PP-CVD) Processing, Chem. Eng. J., 2008, 135(1), 120–128 CrossRef CAS.
- V. D. Borman, M. A. Pushkin, V. N. Tronin and V. I. Troyan, Evolution of the Electronic Properties of Transition Metal Nanoclusters on Graphite Surface, J. Exp. Theor. Phys., 2010, 110(6), 1005–1025 CrossRef CAS.
- J.-Y. Ruzicka, F. Abu Bakar, C. Hoeck, R. Adnan, C. McNicoll, T. Kemmitt, B. C. Cowie, G. F. Metha, G. G. Andersson and V. B. Golovko, Toward Control of Gold Cluster Aggregation on TiO2 via Surface Treatments, J. Phys. Chem. C, 2015, 119(43), 24465–24474 CrossRef CAS.
- D. P. Anderson, J. F. Alvino, A. Gentleman, H. Al Qahtani, L. Thomsen, M. I. J. Polson, G. F. Metha, V. B. Golovko and G. G. Andersson, Chemically-synthesised, atomically-precise gold clusters deposited and activated on titania, Phys. Chem. Chem. Phys., 2013, 15(11), 3917–3929 RSC.
- H. S. Al Qahtani, G. F. Metha, R. B. Walsh, V. B. Golovko, G. G. Andersson and T. Nakayama, Aggregation Behavior of Ligand-Protected Au9 Clusters on Sputtered Atomic Layer Deposition TiO2, J. Phys. Chem. C, 2017, 121(20), 10781–10789 CrossRef CAS.
- F. Ostendorf, C. Schmitz, S. Hirth, A. Kühnle, J. J. Kolodziej and M. Reichling, How Flat is an Air-Cleaved Mica Surface?, Nanotechnology, 2008, 19(30), 305705 CrossRef CAS PubMed.
- J. Mayer, U. Diebold, T. Madey and E. Garfunkel, Titanium and Reduced Titania Overlayers on Titanium Dioxide (110), J. Electron Spectrosc. Relat. Phenom., 1995, 73(1), 1–11 CrossRef CAS.
- R. G. Acres, A. V. Ellis, J. Alvino, C. E. Lenahan, D. A. Khodakov, G. F. Metha and G. G. Andersson, Molecular Structure of 3-Aminopropyltriethoxysilane Layers Formed on Silanol-Terminated Silicon Surfaces, J. Phys. Chem. C, 2012, 116(10), 6289–6297 CrossRef CAS.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-ray Photoelectron Spectroscopy, Physical Electronics, Inc., Eden Prairie, 1995 Search PubMed.
- H. S. Al Qahtani, R. Higuchi, T. Sasaki, J. F. Alvino, G. F. Metha, V. B. Golovko, R. Adnan, G. G. Andersson and T. Nakayama, Grouping and aggregation of ligand protected Au9 clusters on TiO2 nanosheets, RSC Adv., 2016, 6(112), 110765–110774 RSC.
- H. S. Al Qahtani, K. Kimoto, T. Bennett, J. F. Alvino, G. G. Andersson, G. F. Metha, V. B. Golovko, T. Sasaki and T. Nakayama, Atomically resolved structure of ligand-protected Au9 clusters on TiO2 nanosheets using aberration-corrected STEM, J. Chem. Phys., 2016, 144(11), 114703 CrossRef PubMed.
- G. Krishnan, H. S. Al Qahtani, J. Li, Y. Yin, N. Eom, V. B. Golovko, G. F. Metha and G. G. Andersson, Investigation of Ligand-Stabilized Gold Clusters on Defect-Rich Titania, J. Phys. Chem. C, 2017, 121(50), 28007–28016 CrossRef CAS.
- D. F. Dever, A. Finch and E. Grunwald, The Vapor Pressure of Methanol, J. Phys. Chem., 1955, 59(7), 668–669 CrossRef CAS.
- D. R. Lide, CRC Handbook of Chemistry and Physics, Taylor & Francis, 86th edn, 2005 Search PubMed.
- R. Boichot and S. Krumdieck, Numerical Modeling of the Droplet Vaporization for Design and Operation of Liquid-pulsed CVD, Chem. Vap. Deposition, 2015, 21(10–12), 375–384 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further AFM scans showing sub-optimal depositions; experimental high resolution XPS showing Au 4f region with partial agglomeration and C 2s region; Particle/deposition analysis of further PNCD sample; evidence and discussion of manipulating deposited clusters by AFM tip; EDX data for TEM investigations; Mathematica modelling code; investigations of larger size cluster disperse depositions. See DOI: 10.1039/d0na00566e |
| This journal is © The Royal Society of Chemistry 2020 |