
3D cultures for modeling nanomaterial-based photothermal therapy
Emilie
Darrigues†
 *a,
Zeid A.
Nima
a,
Robert J.
Griffin
b,
James M.
Anderson
c,
Alexandru S.
Biris†
*a and
Analiz
Rodriguez†
*d
*a,
Zeid A.
Nima
a,
Robert J.
Griffin
b,
James M.
Anderson
c,
Alexandru S.
Biris†
*a and
Analiz
Rodriguez†
*d
aCenter for Integrative Nanotechnology Sciences, University of Arkansas at Little Rock, 2801 S University Avenue, Little Rock, AR 72204, USA. E-mail: exdarrigues1@ualr.edu; asbiris@ualr.edu
bUniversity of Arkansas for Medical Sciences, Winthrop P. Rockefeller Cancer Institute, Arkansas Nanomedicine Center, Department of Radiation Oncology, 4301 W Markham St, Little Rock, AR 72205, USA
cDepartments of Pathology and Biomedical Engineering, Case Western Reserve University, Cleveland, OH 44106, USA
dUniversity of Arkansas for Medical Sciences, Winthrop P. Rockefeller Cancer Institute, Department of Neurosurgery, 4301 W Markham St, Little Rock, AR 72205, USA. E-mail: arodriguez@uams.edu
First published on 9th November 2019
Abstract
Photothermal therapy (PTT) is one of the most promising techniques for cancer tumor ablation. Nanoparticles are increasingly being investigated for use with PTT and can serve as theranostic agents. Based on the ability of near-infrared nano-photo-absorbers to generate heat under laser irradiation, PTT could prove advantageous in certain situations over more classical cancer therapies. To analyze the efficacy of nanoparticle-based PTT, preclinical in vitro studies typically use 2D cultures, but this method cannot completely mimic the complex tumor organization, bioactivity, and physiology that all control the complex penetration depth, biodistribution, and tissue diffusion parameters of nanomaterials in vivo. To fill this knowledge gap, 3D culture systems have been explored for PTT analysis. These models provide more realistic microenvironments that allow spatiotemporal oxygen gradients and cancer cell adaptations to be considered. This review highlights the work that has been done to advance 3D models for cancer microenvironment modeling, specifically in the context of advanced, functionalized nanoparticle-directed PTT.
 Emilie Darrigues | Emilie Darrigues is a PhD student in applied sciences and a graduate assistant at the Center for Integrative Nanotechnology Sciences at the University of Arkansas Little Rock. Her PhD project is based on the development of complex 3D models for nanomedicine application. Her research uses these models to understand cancer interactions with various plasmonic nanoparticles for theragnostic applications. To characterize these complex dynamics between cancer and nanoparticles, her thesis work involves imaging techniques such as Fluorescence, Raman, Photoacoustic, and Photothermal microscopies. She graduated with a BA in chemistry, a BS in material sciences and an MSC in engineering completed by 4-years’ experience for various industrial companies in R&D, process and lean manufacturing. |
 Alexandru S. Biris | Alexandru S. Biris, PhD, is the Director and Chief Scientist of the Center for Integrative Nanotechnology Sciences at the University of Arkansas Little Rock. His main research interest has been focused on nanostructure sciences and engineering of 3D complex organization for biomedical applications. His laboratory has developed nanomaterials for cancer theragnostics and tissue regeneration such as bone, skin, and nerve. His areas of expertise include nanotechnology and nanostructures, nanocomposite materials, nano-biosensors, materials science, and engineering. He is particularly interested in the integration of nano-plasmonics into cancer biology and tissue regeneration. He is also Professor in the Systems Engineering Department at the Donaghey College of Engineering and Information Technology and is the Roy and Christine Sturgis Charitable Trust Nanotechnology Chair. |
 Analiz Rodriguez | Analiz Rodriguez MD, PhD, is the director of Neurosurgical Oncology at University of Arkansas for Medical Sciences. She is currently conducting brain cancer research at the Winthrop Rockefeller Cancer Institute, with a great interest in photothermal ablation techniques. Her research concentrates on the development of 3D organoids made from primary and patient derived brain tumor cell lines to advance personal and individual onco-therapies. She obtained both her MD and PhD by attending the National Institute of Health – Medical Scientist Training Program at Case Western Reserve University. She completed her neurosurgical residency at Wake Forest University Medical Center and her neurosurgical oncology fellowship at City of Hope National Medical Center. |
Introduction
Cancer cell death is the central goal of all cancer therapies. Thermal therapy induces cancer cell death by exposing cells to high temperatures (generally above 40 °C); photothermal therapy (PTT) is a highly useful form of this technique. PTT induces tumor death by converting light-to-heat when its photo-absorbers are exposed to a near-infrared (NIR) laser. PTT can be used as a local therapy for cancer cells in primary tumors or for local metastases.1 In recent decades, multifunctional, tunable nano-based photo-absorbers have been explored to improve PTT's selectivity, efficiency, and heat generation for site-specific cell death. Many types of photo-absorbing NPs exist, including noble metals, transition metals, sulfide/oxide NPs (semi-conductor, rare earth ion-doped nanocrystals), carbonaceous NPs, and other NIR-organic-polymeric nanoagents.2,3 When used for PTT, these NPs convert light-to-heat by absorbing incident photons from laser excitation; the absorbed photons increase NPs’ corresponding energy levels, then this energy is released through heat production in order to maintain equilibrium. Light absorption is dependent on the size, shape/morphology, concentration, and nature of NPs, and researchers are working to optimize these parameters to improve PTT. Laser wavelength excitation, power energy, and time of exposure are also crucial parameters for cancer-oriented PTT. Additionally, the light-to-heat conversion can involve different mechanisms and spectral ranges of excitation.4 PTT-suitable NPs must be biocompatible, highly optically absorbent, potentially able to carry various bio-active agents (such as drugs (chemotherapy) or photosensitizers (photodynamic therapy (PDT))), and have tunable surface chemistry to allow strong conjugations with antibodies, peptides, and proteins for selective tumor targeting.5,6 Additionally, NPs can aid tumor detection and therapy by enabling multimodal imaging,7,13 such as fluorescence,8 magnetic resonance imaging (MRI),9 photoacoustics,10 computed tomography (CT), and Raman spectroscopy.11,12For preclinical testing, PTT has traditionally been investigated in 2D cell culture systems and animal models. 2D or monolayer cell culture models are widely used to predict in vivo behavior despite their many disadvantages, especially their inability to accurately mimic the complex tumor microenvironment. Patient-derived xenograft models can recapitulate tumor characteristics but are expensive and do not easily allow for high scalability.14 Recently, cancer research has utilized 3D in vitro models to provide an environment closer to in vivo cancer conditions and overcome the limitations of 2D models. 3D models can also be used to improve the study of NP-based drug discovery, screening, and development. 3D cell cultures are more complex than 2D and can incorporate the extracellular matrix (ECM)16 and multiple cell types. Thus, 3D models can more closely simulate the physiological complexities present in vivo. Furthermore, these 3D models are more cost effective and have a greater potential for high scalability than animal models.15,17–19
Despite these improvements in modeling, combining NPs and 3D cultures is still in its infancy, as exemplified by a recent PubMed search (Fig. 1A). Few studies have used 3D cultures to study NP-directed PTT (Fig. 1B). While the relevance of 3D cultures to NP-based drug development, PDT, and theranostic applications has been reviewed,20–23 a comprehensive review of the characteristics of NPs and 3D cell cultures for PTT has not been published. Herein, we present a review of the current literature that used 3D cultures to study NP-directed PTT and provide NPs/3D culture improvement considerations to constructively advance this research field.
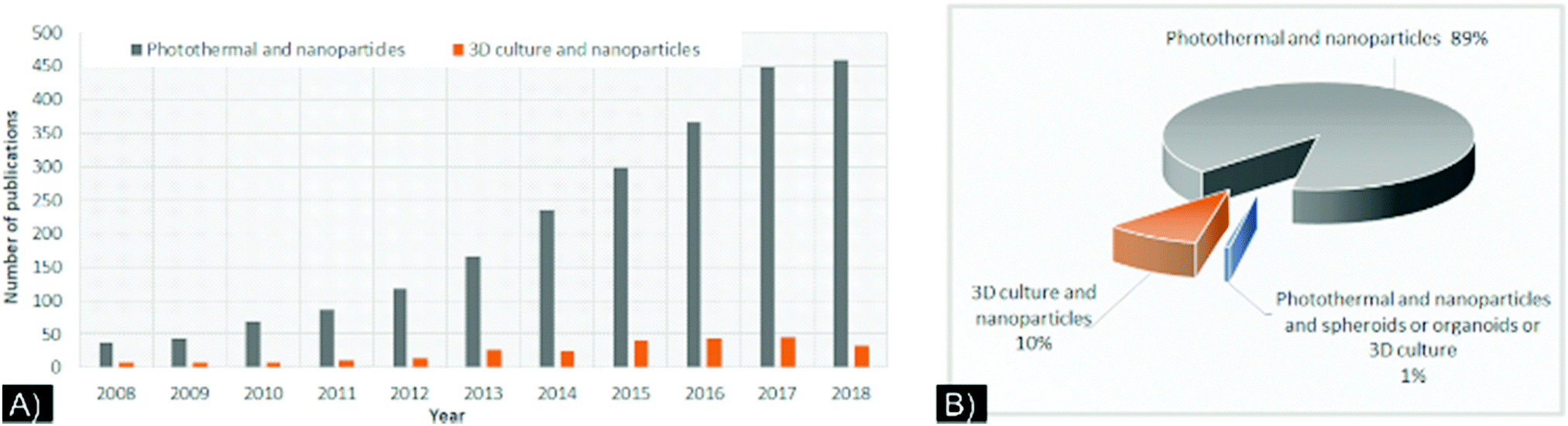 | ||
| Fig. 1 (A) Number of “photothermal,” “nanoparticle,” and “3D culture”-related publications by year, from 2008 to October 2018, based on a PubMed search for the keywords “photothermal and nanoparticles” (grey) and “3D culture and nanoparticles” (orange). (B) Percentage of publications in PubMed database referencing the keywords “photothermal and nanoparticle” (grey), “3D culture and nanoparticles” (orange), and “Photothermal and nanoparticles and spheroids or organoids or 3D culture”, from 2008 through 2018. All data are based on keywords and not on the relevance of the studies. Extracted from PubMed database in.cvs format in October 2018. | ||
Transition from 2D to 3D in vitro cancer models
In a 2D cell culture model, which is not complex enough to mimic the elaborate tumor biology, adherent cells are grown in a monolayer depicting limited cell-to-cell interaction and predominantly cell–surface interactions. Cancerous tissues or tumors are heterogeneous and comprised of a complex microenvironment.24 As a result, crucial interactions that occur in vivo between the cells and the ECM surrounding them cannot be modeled in 2D.25Concern of mono 2D cultures to study nanoparticles
When evaluating cellular processes such as proliferation, migration, invasion, therapeutic resistance, toxicity, clear differential outcomes can be seen between 2D and 3D models.25–27 When NP-based cancer therapies have been tested in 2D models, the results have typically been encouraging.However, these results do not fully take into account biological barriers and NPs’ toxicity (polymeric, metal, or carbon-based) has been considerably overestimated. In 2015, Chia et al.28 analyzed ZnO NPs in 2D and 3D models of colorectal cancer and observed that the NPs had higher toxicity in the 2D model than the 3D model. The 3D model had a more realistic tissue-ECM construct, which likely played a role in determining toxicity: it possessed a mass transfer gradient hypothesized by an “onion like sacrificial model” that protected the inner cells from the ZnO-NPs.
Therapeutics performance of drug-carriers NPs evaluated in a 3D model also showed inaccurate or underestimated effects compared to a conventional 2D model, indicated by a higher IC50 and lower drug delivery efficiency. On a breast cancer 3D model, Privalova et al.29 showed cell viability to be 1.4-fold higher than the monolayer culture, after treatment with dual-drug carried NPs. In contrast, Du et al.30 in 2015, shown that 2D model indicated a relative lack of NP effectiveness as a drug delivery vehicle compared to a free drug, while their 3D model indicated the opposite. The researchers concluded that the drugs’ chemical binding and slow cleavage from the NP in a 2D model caused higher IC50 values, which would have led to the inaccurate assumption that the NP system was not a suitable vehicle. However, the NPs once internalized into the tumors, along with their ability to carry and controllably release drugs could present a major advancement. These studies indicated that NPs interact with cells in dynamic, heterogenic and complex processes. The variability and sometimes opposite results recorded between a 2D vs. 3D model point to the necessity for further investigation of more complex and dynamic in vitro models to mimic in vivo characteristics.
Evolution of ex vivo cancer models: 3D culture as a new addition for patient-derived xenograft (PDX) models
The field of precision medicine is increasingly interested in clinically managing and monitoring patients’ specific therapies ex vivo. In vivo animal models are important tools for cancer drug screening and therapy development, molecular mechanism discovery, cell migration, and evolution of the tumor microenvironment. With the evolution of personalized therapy, patient-derived xenograft models have become a relevant preclinical platform to reflect human tumor biology.14However, PDX growth and inoculation in animals has several disadvantages, including medical facilities to regulate ethical cancer patient tissue access, maintenance, and the long timeframe required for engraftment (2 to 12 months).14 These factors limit the growth and applications of this model. At the same time, the development of microfabrication and biomaterials for tissue engineering and cell biology has promoted the growth of 3D cell culture technologies in many shapes and compositions, such as multicellular (homo or heterocellular) systems grown without or within polymeric scaffolds or hydrogels, which can even be bioprinted with or without the use of magnetic or mechanical accelerated sedimentation31 or used as an ex vivo model, with “organ-on-a-chip” or organoids33 through microfluidic devices.18 3D cultures, although each distinct in their principles, protocols, advantages, and disadvantages, are all meant to mimic human tissue morphology, function, and microenvironment (normoxic, hypoxic, ECM rich and dense, co-culture) (Fig. 2).15,34
 | ||
| Fig. 2 Schematic of 3D culture spheroid engineering and its cancer therapeutic applications. Left: 3D spheroids can be grown to different sizes, generating variation in cell proliferation (senescence and hypoxic core diameter) due to gradients in nutrients, O2, growth factor, hypoxia, CO2, and waste; right: multicellular spheroids can be grown using a monoculture of cancer cells or co-cultures with cancer-related cells (such as cancer-associated fibroblasts, endothelial cells, immune cells). | ||
Complex ex vivo 3D systems are generally called ‘organoids’ but this etymology does not have a clear definition, representing essentially, a self-organizing three-dimensional structure embedded in an artificial or natural ECM to closely mimic the organ of origin. As opposed to spheroids (made mainly from common or commercially available cell lines without the incorporation of ECM), organoids are made from a variety of primary cell sources, including primary tissue, pluripotent stem cells, and adult stem cells.33,35 Compared to PDX, organoids allow stem cells and tissues to be studied in a variety of contexts and can be used and maintained for a long time without genomic alterations. They can also be derived from multiple cell sources, even being generated from a very small amount of starting material, and they can model cancers that are difficult to model in animals or in vitro, such as sarcoma. One of the best assets of the 3D culture model is its ability to reproduce or mimic real tumor functions. The ECM, a major part of the cell microenvironment, can be produced by cells in co-culture or manually introduced with biomimetic functional materials. The ECM has many functions as a structural foundation/scaffold for cells, shaping their biochemical and biophysical traits that alter their behaviors and responses, as observed in vivo. Though artificial ECMs are still being investigated, 3D model ECMs are already showing extremely promising data not only for cancer therapy but also for many other applications, such as tissue regeneration and immune system modulation.36
Despite their advantages, 3D cultures still face limitations,34 including the dependency of their properties on the techniques and parameters used to make them. These concerns will be addressed in the future directions section of this review. We do not think that the 3D system is a “perfect” model, nor that it is a complete substitute for preliminary 2D data or in vivo studies, but we do believe that it can be used as a highly beneficial intermediate step. This would allow pre-clinical and more fundamental research for complex multi-modal imaging or therapies (Fig. 3). We will develop this point in detail in the last part of this review to show how this more complex model can be used for nanoparticle applications.
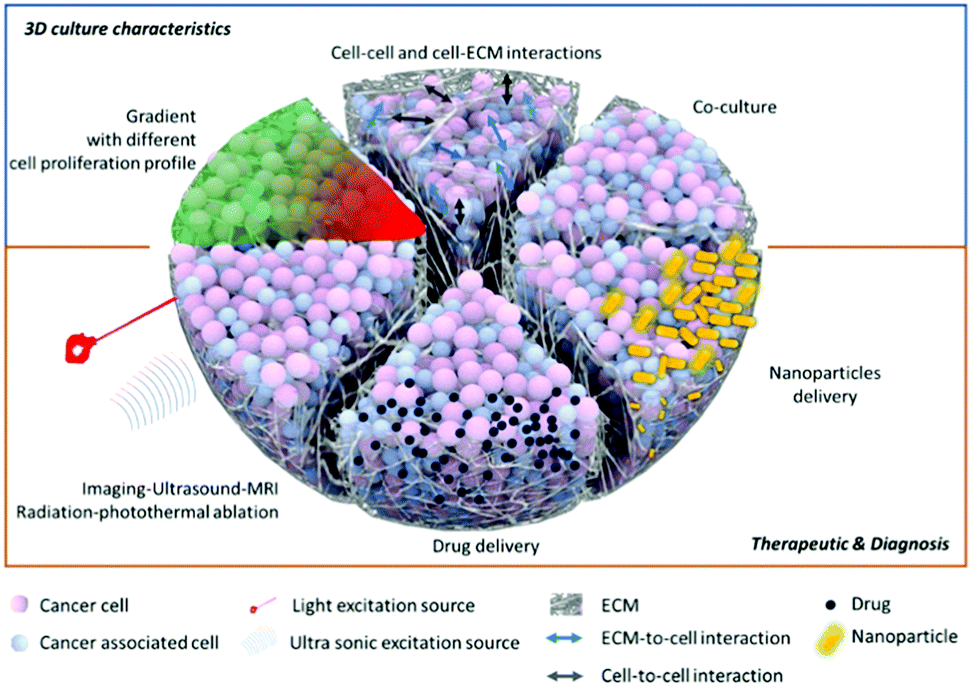 | ||
| Fig. 3 Schematic of slices of a 3D multicellular spheroid, showing the various modeling systems and testing of different therapeutics. | ||
3D cultures for photothermal therapy
Thermal therapy uses a certain level of heat to irreparably disrupt cellular functions or destroy cell components to induce death. Two types of heat techniques can be used. The first—low and moderate hyperthermia—is a slow, lethal heat that gradually increases tissue temperature (>40 °C over a span of hours), resulting in tumor cell death by necrosis and/or apoptosis. The second—thermal ablation or high-temperature hyperthermia—is a more drastic approach that destroys or irreversibly damages tissues or cells by extreme hyperthermia (>50–60 °C) in a short time (minutes), resulting in immediate necrosis of the cells.37,38 Increase in temperature depends on the type of laser power used, the duration of laser irradiation, the type of tissue to be removed, and the surgical context, based on a mathematical approach derived from the Arrhenius equation.39–41Conventional clinical practice is to use lasers, microwaves, radiofrequency, or high intensity-focused ultrasound to cause hyperthermia or ablative thermal therapy. However, in vivo heating is technically challenging because it is not tumor specific. Additionally, the methods used to image tumor edges in order to target only the tumor site and to maintain and monitor temperature are oftentimes inadequate and inaccurate.
Clinical PTT generates heat upon laser excitation. NIR lasers exploit biological wavelength windows (NIR-I or first window: 700–950 nm, NIR-II or second window: 1000–1350 nm, and NIR-III or third window: 1550–1870 nm) favorable for optimal penetration in living tissues.42 In the past few years, to optimize the heat generation and selectivity of PTT, a new kind of photo-absorber has emerged: nanoscale intermediaries, which have an extensive range of classifications, including noble metallic and transition metal sulfide/oxide nanostructures, carbonaceous NPs, and other organic nanoagents.2 The fabrication and surface tunability of NPs makes it possible to modify how they interact with biological systems in the context of hyperthermia. Originally, these photo-nanoabsorbers were mainly designed to work in the first NIR windows. However, technological growth has led to the use of a variety of lasers in routine clinical usage with a range of wavelengths, from 800 to 1060 nm; for example, laser ablation with MRI guidance is possible in neurosurgical applications. In parallel, the last decade of research has also shown that exploiting nanoprobes in the NIR-II and NIR-III windows for biological purposes is promising.42–44 When NPs are used, PTT can be combined with other cancer therapies, such as drugs, and be monitored with multimodal imaging techniques, such as MRI, fluorescence, X-ray, photoacoustic, CT (computed tomography) scan, and, more recently, Raman spectroscopy to (1) diagnose cancer, (2) confirm the delivery and internalization of drugs within the tumor site, and (3) evaluate treatment efficiency.12,45
The ability of 3D models to mimic hypoxia with a necrotic core and contain different kinds of cells by co-culture makes them suitable to investigate in vitro thermo-ablation efficacy and NP optimization. An advantage of thermal therapy is the ability to improve the blood flow preferentially within tumors (vs. normal tissues), resulting in better oxygenation of the low vascularized hypoxic core of the tumor (generally resistant to ionizing radiation).46,47 Cells residing in the hypoxic core tend to be contained in a more acidotic environment, which makes them more sensitive to thermal damage but less sensitive to radiation therapy.48–50
Testing NP enhanced PTT in 3D models that simulate relevant, crucial in vivo biological features were done in a study by Crawford et al.51 The research team developed a 3D model of inflammatory breast cancer (IBC), using cell lines SUM149 and SUM190, that could mimic tumor emboli structures. To enable NP-based imaging and PTT in this system, plasmonic gold nanostars (GNS) were synthesized; these NPs had a large, two-photon luminescence cross-section and enhanced plasmonic behavior. A 2D culture demonstrated endocytosis of the GNS into multiple cancer cell lines. In 3D culture, penetration was also demonstrated by the GNS, as evaluated by two-photon luminescence microscopy (TPL). A 3D emboli model made of SUM149 cell lines indicated homogeneous distribution of the GNS, while a SUM190 model, which had a larger and more compact 3D structure, showed confinement of the GNS to only the outer layer of the emboli. PTT with a continuous 808 nm diode laser was performed at various power densities (9.375, 5, and 2.18 W cm−2). 5 and 9.375 W cm−2 laser power densities caused marked cell death, but the 2.18 W cm−2 power density resulted in decreased cellular death, with tumor ablation confined to the periphery. In contrast, the higher power densities were able to cover the whole spheroid, even in the SUM190 3D system, where the GNS were found mainly in the periphery rather than central area of the spheroid. Thus, the 3D model provided insights into how to optimize laser power parameters for successful treatment.
Metal-based and gold nanomaterials for PTT enhancement
The interaction between metallic nanomaterials and light was initially recognized by Faraday and Mie, who presented the first theories on the electromagnetic properties of light in interaction with a metallic surface. The researchers suggested that during this interaction, light transmits a single or collective and coherent oscillating movement of surface free electrons, inducing polarization inside the metallic NP. This theory became known as surface plasmon resonance (SPR).52–54 Gold (Au)-based NPs are the main metallic PTT enhancers investigated due to their biocompatibility, low cytotoxicity, NIR region or biological light absorption windows and excellent photostability, efficient light-to-heat conversion, and tunability in terms of shapes, sizes and surface functionalities.55,56 Other metals, such as iron, silver, or copper (Cu)-based NPs, despite their possibly higher toxicity, are valuable for their enhancement of surface plasmon or resonance-induced heat conversion.57,58Using a 3D glioma model, Iodice et al.59 studied 6 nm AuNPs agglutinated in larger polymeric nanoconstructs (SPNs) made from a PLGA (poly(lactic-co-glycolic acid)) core and stabilized by a superficial lipid–PEG monolayer, to enhance stability of the system under physiological conditions. The size, optical, and photothermal properties of the nanoconstructs (Au-SPNs) were controlled by modifying the initial Au mass loading which led to variable diameters of the inner core. Specific absorption rate (SAR) was determined ex vivo for each Au-SPN. 3D glioma spheroids were cultured with Au-SPNs and then photothermal ablation was done via an 800 nm wavelength laser. A cell viability test indicated more cell death was induced in the Au-SPN-treated spheroids than in the untreated control. Interestingly, for the same treatment, the toxicity was higher in 3D culture than in 2D, but the authors didn’t provide explanations on why such difference was observed. The interest to study NP–PTT in 3D models are not only based on their ability to convert light-to-heat, but also on the maintenance of their performance in the context of a multidirectional (3D) cancer model with potential different gradients, diffusion and maintain of temperatures than in 2D cell culture.
In order to facilitate the testing of NPs as effective PTT enhancers, 3D cancer models should mimic certain cancer properties, components and features, such as cancer stem cells, which are generally responsible for tumor recurrence and metastasis.60 NPs can be tuned to recognize and interact with such cells. In 2017, Gonçalves et al.61 developed gold nanorods (AuNRs) functionalized with an engineered peptide that can recognize nestin expressed at the surface of glioma stem cells, which allows the peptide to act as a targeting agent. This system was tested in a scaffold-hydrogel 3D culture model. The hydrogel, designed to mimic the ECM and brain stiffness, was made with star-shaped PEG covalently connected to matrix metalloproteinase-susceptible peptide and maleimide-functionalized heparin. The 3D culture was made in a mono and co-culture of nestin-positive (Nes+) and nestin-negative (Nes−) glioma stem cell lines isolated from patient glioblastoma multiform tumor (GMB), X01 GBM (stem cells, mainly all Nes+) and X01 GBM-BMP (differentiated cells), respectively (70% Nes−/30% Nes+, as identified by flow cytometry). It is important to note that the 2D cells were seeded until attachment and proliferation, then incubated with the nanoparticles, while in 3D, the gold nanoparticles (NesPEG-AuNRs or PEG-AuNRs (as a control)) were first mixed at different concentrations inside the hydrogel matrix prior to cell seeding to form 3D colonies. The fluorescent AuNRs were tracked in 2D and 3D models by confocal microscopy. Higher uptake (2D) and penetration (3D) were observed for the peptide-functionalized AuNRs, especially in the stem cell model. The non-functionalized PEG-AuNRs without the targeting peptide showed very low interactive effect in both 2D and 3D. The researchers also found that the active functionalization caused the AuNRs to be taken up into the cells by energy-dependent internalization and caveolae-mediated endocytosis, suggesting primary localization within acidic endosomes. 3D cultures treated with functionalized AuNRs, containing a mixture of X01 GBM and X01 GBM-BM, underwent laser irradiation. Following treatment, 80% of the GBM cells and 30% of the GBM-BMP were dead, corresponding with the 30% Nes+ cell content in the GBM-BMP. Varying the hydrogel stiffness did not cause significant variation in PTT efficiency. Additionally, the overall higher survival rate in the 3D X01 GBM cultures in comparison to the 2D cultures, was likely due to the higher photothermal resistance in the 3D culture microenvironments. To further prove the advantage of using 3D models to mimic in vivo systems, a comparative study utilizing doxorubicin showed higher chemotherapy resistance for the 3D system (∼59% cell viability) than the 2D model (∼13% cell viability), even for X01 GBM. Active functionalization of NPs in this study highlights the ability of PTT to be more selective and, potentially, preserve normal or healthy cells.
Due to the plasmonic properties of metal-based NPs, it is also possible to provide real-time targeted imaging of the tumor site for image-guided photothermal surgery. For this purpose, using a 3D gastric cancer model, Liang et al.62 developed PEGylated gold nanostars (PEG-GNS) to image, detect, and treat cancer cells via PTT. They actively targeted these cells by conjugating CD44v6 on the PEG-GNS. CD44 is a cell surface glycoprotein involved in cell adhesion. Spheroids were developed from CD44+ or CD44− MKN-45Gc cells. Photothermal effect was evaluated on different ranges of laser power. At all treatment timepoints, better cellular uptake and penetration of CD44+ spheroids were observed for GNS-PEG-CD44v6 than GNS-PEG, with the targeted NPs being inside the cytoplasm and a small portion distributed in the cell membrane and nucleus cleft. With laser power superior to 1.5 W cm−2, cell colonies were ablated and completely damaged in contrast with lower power density, 0.3 W cm−2, resulting in dissociation and loss of spheroidal structure. Also targeted NPs led to better penetration in this 3D cancer model. 3D model results were affirmed by in vivo mouse experiments. It was shown that when higher laser power density (1.5 W cm−2) and one single irradiation exposure, induced an effective treatment resulting in full necrosis. However, this study led to the speculation that the higher the laser intensity the more the surrounding tumor normal tissue may be affected by the resulting thermal zone. Lower irradiation performed every two days for two weeks, also showed significant tumor volume diminution for a high survival rate. Consequently, usage of GNS-PEG-CD44v6 indicate the ability of PTT to be use as an ablative technique even for lower power density settings. Photoacoustic imaging was conducted on two different in vivo mice models: orthotopic and subcutaneous xenografts, confirming the higher accumulation of GNS-PEG-CD44v6 compared to unfunctionalized GNS.
As an additional comment, in the last two studies reviewed,61,62 the stem cell behavior of cancer was a concern. It is important to note that 3D culture allows the study of PTT for local therapy but can also support the evaluation of this therapy to kill cancer stem cells and avoid treatment resistance, tumor progression, and metastasis development.
PTT can also be improved by combining it with other therapeutic solutions, such as drugs, gene silencing delivery or PDT. These dual therapies can help overcome drug resistance, promote higher treatment success rates, and avoid cancer recurrence and metastasis by combining the strengths of multiple therapies. With this goal, in 2014, Mohamed et al.63 complemented their study64 by synthesizing conjugated AuNPs to combine the curcin toxin effect (a type 1 ribosome inactivating protein) with PTT. They functionalized AuNPs (∼10–15 nm) DSPE-PEGylated (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-PEG or DSPE-PEG) to favor conjugation with multiple targeting agents: folate and transferrin by EDC/NHS reaction (N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride/N-hydroxysuccinimide).65 The release of the curcin from the NPs was controlled by pH variation. A 3D culture was made by seeding 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 glioma cells onto 100 μl Geltrex-coated wells then culturing them for 4 days until 3D colonies developed, to be used to evaluate the drug effect, independently of the PTT effect. The study demonstrated that the conjugated nanoparticles were able to shrink 1/3 of the 3D colony and prevent the formation of cellular protrusions responsible for migration and proliferation. In this case, the drug was able to flow in all the spheroids, which showed ∼100% cellular death. Next, PTT was tested on the glioma 3D cultures and temperature elevation was only seen in the presence of NP treated cultures and not in the control group indicating the potential for specificity. Laser stimulation for drug released was also evaluated in a 3D structure of osteosarcoma by Li Volsi et al.,66 who functionalized two sets of AuNRs, each with different coatings: (1) an amphiphilic polysaccharide-based graft-copolymer (INU-LA-PEG-FA) and (2) an amino derivative of the α,βpoly(N-2-hydroxyethyl)-D,L-aspartamide (PHEA-EDA-FA), both functionalized with folic acid. The researchers also encapsulated the drug nutlin-3 on the NPs. The ability to release the drug from the NPs under pH stimulation was also evaluated by varying the pH from 7.4 to 5.5 to mimic the variation of pH inside the tumor microenvironment. The results indicated higher drug release by the AuNRs@PHEA-EDA-FA than by the AuNRs@INU-LA-FA. Drug release was also evaluated with the synergistic influence of pH and laser stimulation with different exposure times. After laser irradiation, the amount of nutlin-3 released was two times higher for the AuNRs@INU-LA-FA (from 3.8 to 10%), and four times higher (from 6 to 24%) for AuNRs@PHEA-EDA-FA compared to untreated NPs. Toxicity and efficiency of the NPs in a 3D model vs. a 2D model were studied, revealing higher IC50. The profile of drug release analysis also indicated a difference between the 2D and the 3D model: around 0.35 to 1.68 μM of drugs were released over 24 to 48 hours of incubation in 2D vs. around 50 μM over 72 hours in 3D. AlexaFluor647-labeled AuNRs allowed NP penetration to be tracked inside the organoids. Laser therapy on cells containing targeted-drug NPs, achieved 100% therapy efficiency, as confirmed by toxicity assay carried out 48 hours after treatment (Fig. 4). Additionally, in comparing 2D and 3D resistance to therapy, the influence of the polymer used to functionalize the NPs for drug encapsulation/bonding on the drug release was highlighted. Indeed, for 10% variation in terms of drug encapsulation efficiency and 1% (w/w) in polymer mass, AuNRs@PHEA-EDA-FA showed 3 to 4 times more drug release and, as a result, higher organoid destruction ability than AuNRs@INU-LA-FA. Loading ability and release efficiency are two key parameters for NP-drug delivery: the last one, release efficiency, whether stimulated or not by PPT, should be investigated closely, especially in the case of 3D cultures, where the delivery of the drug can be hampered.
000 glioma cells onto 100 μl Geltrex-coated wells then culturing them for 4 days until 3D colonies developed, to be used to evaluate the drug effect, independently of the PTT effect. The study demonstrated that the conjugated nanoparticles were able to shrink 1/3 of the 3D colony and prevent the formation of cellular protrusions responsible for migration and proliferation. In this case, the drug was able to flow in all the spheroids, which showed ∼100% cellular death. Next, PTT was tested on the glioma 3D cultures and temperature elevation was only seen in the presence of NP treated cultures and not in the control group indicating the potential for specificity. Laser stimulation for drug released was also evaluated in a 3D structure of osteosarcoma by Li Volsi et al.,66 who functionalized two sets of AuNRs, each with different coatings: (1) an amphiphilic polysaccharide-based graft-copolymer (INU-LA-PEG-FA) and (2) an amino derivative of the α,βpoly(N-2-hydroxyethyl)-D,L-aspartamide (PHEA-EDA-FA), both functionalized with folic acid. The researchers also encapsulated the drug nutlin-3 on the NPs. The ability to release the drug from the NPs under pH stimulation was also evaluated by varying the pH from 7.4 to 5.5 to mimic the variation of pH inside the tumor microenvironment. The results indicated higher drug release by the AuNRs@PHEA-EDA-FA than by the AuNRs@INU-LA-FA. Drug release was also evaluated with the synergistic influence of pH and laser stimulation with different exposure times. After laser irradiation, the amount of nutlin-3 released was two times higher for the AuNRs@INU-LA-FA (from 3.8 to 10%), and four times higher (from 6 to 24%) for AuNRs@PHEA-EDA-FA compared to untreated NPs. Toxicity and efficiency of the NPs in a 3D model vs. a 2D model were studied, revealing higher IC50. The profile of drug release analysis also indicated a difference between the 2D and the 3D model: around 0.35 to 1.68 μM of drugs were released over 24 to 48 hours of incubation in 2D vs. around 50 μM over 72 hours in 3D. AlexaFluor647-labeled AuNRs allowed NP penetration to be tracked inside the organoids. Laser therapy on cells containing targeted-drug NPs, achieved 100% therapy efficiency, as confirmed by toxicity assay carried out 48 hours after treatment (Fig. 4). Additionally, in comparing 2D and 3D resistance to therapy, the influence of the polymer used to functionalize the NPs for drug encapsulation/bonding on the drug release was highlighted. Indeed, for 10% variation in terms of drug encapsulation efficiency and 1% (w/w) in polymer mass, AuNRs@PHEA-EDA-FA showed 3 to 4 times more drug release and, as a result, higher organoid destruction ability than AuNRs@INU-LA-FA. Loading ability and release efficiency are two key parameters for NP-drug delivery: the last one, release efficiency, whether stimulated or not by PPT, should be investigated closely, especially in the case of 3D cultures, where the delivery of the drug can be hampered.
 | ||
| Fig. 4 (A) Schematic of functionalized AuNR-folate in a 3D sarcoma model before and after NIR laser exposure. (B) Brightfield images of U-2 OS organoids incubated with AuNRs@INU-LA-PEG-FA/Nut, AuNRs@PHEA-EDA-FA/Nut, and free nutlin-3 at equivalent concentrations of 10 μM of drug and of 52 μg mL−1 of Au0, taken before and after 30, 60, and 200 seconds of NIR laser exposure. Figure and caption reproduced with permission.66 Copyright 2017, American Chemical Society. | ||
Chen et al.67 also investigated a triple negative breast cancer (TNBC) 3D culture to test PTT combined with drug delivery; this time, the metal core was made from copper instead of Au. They developed a CuS (copper(II) sulfide) core embedded as a micelle in a bi-thermosensitive amphiphilic poly(acrylamide-acrylonitrile)–PEG block copolymer, encapsulating the experimental drug “AF”. The CuS-micelles were characterized at a final size of 63 nm (hydrodynamic diameter) and 37 nm (dry micelle in TEM analysis), with a negative zeta potential and a strong absorption band in the NIR region at 989 nm. Drug release was again triggered by pH variation and heat from NIR laser exposure. However, in this case, the amphiphilic block polymer was designed to have an upper critical solution temperature (UCST), meaning the release is also managed by a transition from hydrophobic to hydrophilic when the temperature rises above the UCST, to avoid premature drug release by the human body temperature of 37.4 °C. To increase uptake and penetration in 2D and 3D models, CuS-micelles were functionalized with the GE11 peptide, which targets an epidermal growth factor receptor overexpressed in TNBC. Untargeted NPs showed less penetration inside the 3D model than targeted ones after 2, 6, and 24 hours of incubation. Combination of chemotherapy and PTT for non-targeted and targeted micelles induced 54 and 75% cell death, respectively. Interestingly in this study, as for the study of Liang et al.62 with gold nanostructures, it is important to notice the lower power selected: 0.75 W cm−2 showing the ability to combine the therapeutics effect to reduce potential negative thermal effects on the healthy surrounding cells. The nanoparticles showed interesting photoacoustic properties, as they were investigated in the 3D system to support imaging of penetration or diffusion.
In 3D models, NPs are primarily delivered into cancer cells via direct incubation inside the cell culture media or matrix for various lengths of time. Another method is to pre-incubate the NPs or develop NPs able to target tumor-associated macrophages (TAMs), which accumulate inside the tumor site to allow efficient PTT. The other advantage of using TAMs in addition to their fast ingestion of NPs, is their ability to infiltrate brain tumor.68 The thermal therapy of both brain tumor and TAMs might be beneficial for TAMs depletion and/or reprogramming to overcome tumor growth.69 Thus far, studies indicate that the efficiency of macrophage use for PTT depends on two parameters: laser power intensity and the number of macrophages loaded (directly correlated with NP concentration) in the 3D model structures. In 2012, Madsen et al.70 investigated this type of targeting and delivery with macrophages and a 3D glioma culture. Two-photon fluorescence microscopy confirmed the migration potential of macrophages and macrophage-loaded NPs in 3D culture. The Au nanoshell's light-to-heat conversion occurred with an 800 nm wavelength laser; increasing laser power density decreased spheroid growth, with complete suppression of growth at 14 W cm−2 and complete explosive dissociation at 28 W cm−2. Concerning the macrophage number, significant spheroid growth suppression was observed with 10% macrophages, and complete growth cessation was seen at a concentration of 20%. In 2017, Christie et al.71 used a glioma 3D model to compare how the penetration and maturation of macrophages change when the morphology and composition of the NPs are modified. They compared gold–silica nanoshells (AuNS) with AuNRs. PTT of spheroids was achieved by light exposure from an 810 nm diode laser at irradiances ranging from 0 to 28 W cm−2 for 10 minutes. PTT efficacy was analyzed by tracking spheroid growth for 14 days after therapy. Macrophage uptake of PEGylated AuNR (3.9 ± 0.9%) was two times higher than its uptake of PEGylated AuNS (7.9 ± 0.7%). However, the growth inhibition was consequential for the AuNS, while no inhibition was seen with the AuNR. It is important to note that the authors chose laser irradiation of 810 nm for NPs possessing maximal absorptions of around 819 nm (AuNS) and 765 nm (AuNR). Thus, the laser emission and absorption for light conversion or cross-section are closer for the AuNS than the AuNR, which could explain the low therapeutic efficiency of the AuNRs despite their high uptake.
In the last two studies, a new delivery method of NPs for PTT was investigated using macrophages as the carrier rather than the conventional method of tumor targeting, which may be beneficial to reduce the number of macrophages in brain tumors and improve its therapy. However, the laser power used was higher compared to the other studies, and therefore performance of macrophages as NPs carrier for PTT still need to be evaluated under lower laser power.
Carbon-based nanomaterials for PTT enhancement
Though their cytotoxicity is complex and not fully understood, carbon-based nanomaterials should also be considered for PTT, given their high surface area and resonance properties. Investigating their use for PTT in 3D models can further our knowledge of their PTT behavior in more complex systems, including penetration, accumulation, activity, and more especially toxicity, considering the underestimation of IC50 defined in 2D model, to potentially enable clinical applications. Carbon-based nanostructures, including single-walled carbon nanotubes (SWCNT), multi-walled carbon nanotubes (MWCNT), and, more recently, graphene, are investigated for PTT to support cancer treatment. Carbon nanostructures are semi-conductors or metallic in function of the chirality of their carbon ring. Consequently, heat is either generated by the light-induced collective mobility of free carriers—the π plasmon excitation and the relaxation of the surface—or by the de-excitation processes between Van Hove states, which involves luminescence and/or non-radiative relaxation caused by the defective carbon-ring structures.4 Several projects have combined the properties of metal and carbon NPs for PTT, such as Au and SWCNTs72 or Au and graphene to improve theranostic abilities.73,74In 3D glioma spheroids made from U-87 glioma cancer cells, Eldridge et al.75 evaluated the diffusion, penetration, and light-to-heat conversion of MWCNTs for PTT. They first analyzed and modified the NPs’ surface to improve their biocompatibility, comparing the diffusion and penetration of MWCNTs with a dense or low coating of phospholipid-PEG (DSPE-PEG) or with Pluronic® coating, through brain phantoms that mimicked the ECM in the brain. The researchers showed that for a 100 μg mL−1 concentration of MWCNTs, those with 2% DSPE-PEG penetrated 4–5 mm deep in all directions, while all the other MWCNT preparations diffused no more than 2 mm. All maintained ablative temperatures (above 40 °C) after laser exposure (970 nm diode laser, 3 W cm−2, 30 s). Then, the team evaluated the potential of carbon nanotube-mediated thermal therapy (CNMTT) to treat GBM multicellular spheroids compared to conventional heat delivery. They performed two experimentations: one with 24 hour incubation of the NP (5 to 10 μg mL−1), to allow enough time for NPs to interact with the spheroids, followed by washing and exposure to NIR-irradiation (Fig. 5A), and another with immediate irradiation after incubation with a higher concentration of NPs (20 μg mL−1) (Fig. 5B). In the case of Fig. 5A, penetration inside the spheroids was estimated at 100 μm (by TEM imaging). Surprisingly, in the 3D study, with or without nanotubes, after 90–120 seconds of laser exposure, PTT was unable to stop the growth of GBM spheroids (followed for 12 days after treatment), even though considerable cell death was quantified in the 2D study. To provide data on the efficiency of the NPs, the researchers performed the experimentation described in Fig. 5B, with an increase in NP concentration, which inhibited spheroid growth. In this study, the 2D culture's under-evaluation of IC50 is highlighted, with cell survival noted in the 3D systems, for the same drug concentration. However, they added a pertinent, complemental study (done in 2D culture) on the phenomena of “heat shock response” or HSR. Indeed, thermal therapies, in general, can be compromised due to reactive stress-induced chaperone molecules known as heat shock proteins (HSPs), appearing in areas of the tumor where the thermal dose did not exceed the thermo-ablation threshold (temperature elevation or heating duration was insufficient). Acting on protein reshaping and apoptotic signaling, activation of HSPs may induce tumor recurrence and resistance, especially HSP27, HSP70, and HSP90.76,77 Quantifying these three HSPs by Western-blot analysis, the researchers discovered that DSPE-PEG-MWCNTs did not induce HSR. Due to the nature and properties of the 3D culture, studying the activity of HSPs for targeting and/or delivering inhibitors might be an advanced study to consider in order to understand HSP functions better before in vivo work.
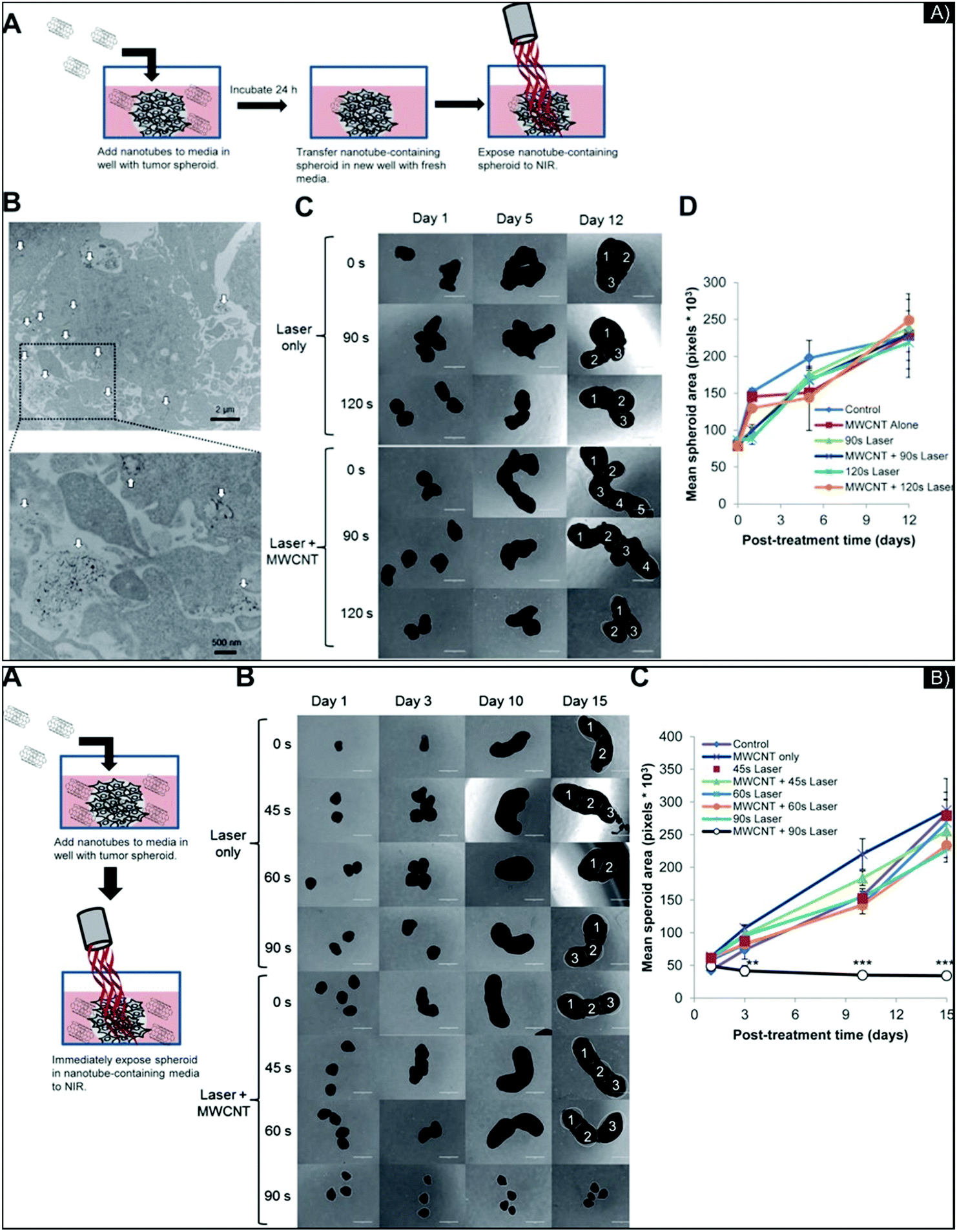 | ||
| Fig. 5 (A) Images of 3D GBM spheroids incubated overnight with 2% DSPE-PEG MWCNTs and treated by laser. (A) Schematic of the experiment; (B) MWCNTs (indicated by white arrows) penetrating the spheroids visualized by TEM image at low and high magnification; (C) representative photomicrographs at different days post-laser treatment with different exposure times and with or without MWCNT; (D) mean surface area per spheroid, quantified by ImageJ software. No major differences in spheroid growth were seen among the treatment groups. (B) Treatment of GBM spheroids by CNMTT with extracellular 2% DSPE-PEG MWCNTs. (A) Schematic illustrating the experimental design; (B) representative photomicrographs of spheroid growth over time; (C) mean surface area per spheroid, quantified by ImageJ software. Images of spheroids in addition to those shown in (B) were used to calculate area measurements. Significant spheroid growth differences were seen between spheroids treated with MWCNTs and laser for 90 s and all other treatment groups. Figure and caption reproduced with permission.75 Copyright 2016, American Chemical Society. | ||
As with metal-based NPs, carbon nanostructures make possible the combination of PTT with other therapies. Zhang et al.78 created bimodal therapeutic NPs to support PTT-TPPDT:PTT and two-photon PDT (TPPDT). In this therapy, reactive oxygen species (ROS) are generated once by a two-metal d-complex: Ru(II), Ru1, and Ru2, co-assembled with SWCNTs with non-covalent π–π stacking, are released by PTT irradiation and start to produce ROS (808 nm diode laser with a power density of 0.25 W cm−2). Doping the SWCNT with Ru also increases its photothermal properties: 50 μg mL−1 of Ru@SWCNT in water exposed for 300 seconds presented a ∼ΔT = 20 °C and 40 °C compared to SWNCT and water only, respectively. Additionally, exposure to the laser induced a release of Ru: 52.5% for Ru1 and 55.4% for Ru2; switching off the laser stops the release immediately, making it tunable in function of exposure. PDT and TPPDT were evaluated in vitro in a 2D culture, and the ability of Ru@SWCNTs to induced death in HeLa cancer cells was analyzed. Dual PTT-TPPDT was verified in 3D culture and in vivo on tumor-bearing mice. The 3D model was generated with a liquid overlay technique on agarose gel, plated in a 96-well plate, 6000 cells per well, and grown for 3 days to reach ∼400 μm. A dose of 50 μg mL−1 of SWCNTs or Ru@SWCNTs was incubated with the model for 48 hours, then treated with different laser exposure. Cell viability and diameter were extremely reduced: >70% in size compared to the control, and only 5% viability remained (at 5 min of laser exposure time).
In 2017, Liu et al.79 reported the first evaluation of the combined use of nano-based chemotherapy, PDT, and PTT in a 3D glioma model, with the NP supported by a reduced graphene oxide (rGO)–PEG coating to enhance biodistribution and loaded with Dox for chemotherapy and chlorin e6 (Ce6) for PDT. Evaluating each therapy—PTT (laser at 808 nm), chemotherapy (no laser), and PDT (laser at 661 nm)—in a 2D cell culture presented similar results. However, in the 3D model, only the PTT showed significant cell death, and even 10 μg ml−1 of rGO–PEG–Dox and rGO–PEG/Ce6(L+) were unable to induce less than 40% cell viability. An additional TUNEL assay for apoptosis detection confirmed the results. To understand these variations in therapeutic efficiency, the researchers hypothesized that NP penetration was the root of the issue, theorizing that PTT does not necessarily need efficient deep penetration inside a tumor system to be useful. A computer modeling simulation based on a 500 μm multicellular spheroid with heat transmission, with and without rGO, confirmed the theory, showing a temperature increase of 58 °C with rGO and just 3 °C without rGO, even though the rGO penetrated the outer layer of the spheroids. This experiment revealed that tumor hypoxia and the tumor microenvironment can highly limit the efficacy of chemotherapy, PDT, and radiation therapy but not PTT. Comparable results were seen in another cancer model on an A549 cell line (lung cancer). An additional in vivo study was performed on female nude mice (6–8 weeks) with U-87 tumor xenografts. Microscopic fluorescent pictures of 4 μm tumor slices revealed that PTT had higher efficiency than Dox or PDT, even if the cancer cells were not in a closed blood vessel area. This last point emphasizes the extreme relevance of using 3D cultures to mimic avascular and vascular systems in nanomedicine. Indeed, functionalization, size, charge, and properties of the NPs, which improve their cellular interaction, penetration, and diffusion, will be directly correlated to cancer angiogenesis modulation, and as a result, predict their therapeutic abilities in a dynamic biological context. The most pressing concern is to define if these nanoparticles properties are relevant or not for PTT, and these results may be obtained only through in vivo studies or with the usage of complex 3D cancer model.
New polymeric-based nanomaterials for PTT enhancement
Inorganic materials, such as Au and carbon-based NPs, are commonly used for their ability to convert light-to-heat and enhance PTT. However, a variety of NIR-absorbing organic materials have also been synthesized in recent years, which may be more suitable due to their biodegradability. These organic materials can be loaded and delivered by polymeric NPs such as micelles, protein-based agents, conjugated polymers, and organic/inorganic nanocomposites.8,80In 2018, Li et al.81 designed micelles loaded with a loading efficiency of 87% (w/w) equivalent to 25% (w/w) of loading content of the photothermal agent, IR780,82 and evaluated their photothermal performance on MCF-7 breast cancer cell lines. The team also analyzed the tunability of sphere, rod, and worm-shaped micelles to determine the relationship between their shape and bio-behaviors in 2D, 3D, and in vivo models. In a 3D culture, confocal laser scanning microscopy revealed that the overall fluorescence intensity (quantified by IR-780 intensity) from the rod-like treated cells was highest. In mice, after irradiation with an 808 nm wavelength laser, the tumor temperature was recorded to be about 55.6 °C for those injected with rod-like micelles vs. 49 °C for free IR-780. The same year, Rajendrakumar et al.,83 also using micelles and IR-780, evaluated the double stimuli response of controlled gene release by hyperthermia activation on a 4T1 breast cancer spheroid model. They combined disulfide-crosslinked polyethylenimine (ssPEI) conjugated with a tumor-specific cell-penetrating peptide (DS4-3) (SPD) polyplex and bovine serum albumin (BSA)-loaded IR780 (BI) NPs, resulting in a dual-stimulus-triggered, tumor-penetrating, and gene-carrying nano-assembly (BI-SPD) via electrostatic complexing (∼460 nm NPs). BI-SPD-loaded IR-780 NPs were placed in solution and irradiated with an 808 nm laser at 1 W cm−2; the nano-assembly induced a temperature of 46 °C at 11.25 μg ml−1 concentration. In 3D spheroids, the BI-SPD nano-assembly containing DS4-3 peptide penetrated and spread uniformly throughout the tissue to a depth of 37.03 μm.
Critical analysis, challenges and future expectations
PTT-mediated NPs have been tested in 3D cancer models made from many different cancer cell lines. Various techniques and nanoparticles have been used, including metal-based, carbon-based, and organic polymeric NPs. Different laser parameters have also been tested: wavelength, power density, and time of exposure. Overall, the literature has confirmed the ability of nanoparticles to facilitate light-to-heat conversion in 3D systems; investigated the influence of active targeting on the performance of PTT; showed the theragnostic use of NPs for imaging and therapy; indicated the possibility of combining PTT with other therapies, such as drugs or PDT; and even explored and diversified PTT-NP delivery to cancer cells via macrophages. These preliminary studies have demonstrated the potential of PTT-mediated NP techniques in complex 3D systems and highlighted the multifactorial parameters that should be considered to prove their suitability for clinical applications. They are extremely important for a thorough understanding of this important area of cancer research; however, as in all new areas of science, more research is still needed in order to fully take advantage of the promise of this approach. To constructively advance this field, the following considerations should be investigated:(1) The advantages of 3D systems over 2D cultures make it possible to evaluate a new parameter—NP penetration and diffusion over physical/chemical barriers. Some studies indicate that PTT does not necessarily need deep NP penetration inside a tumor system in order to be efficient, while others assert the opposite. It remains unclear if the efficacy of NP-enhanced PTT is due to the increased NP diffusion, penetration, and homogenic/heterogenic accumulation or to the cumulative effect of recognition, cellular uptake, and “heat wave” release, propagation, and accumulation within the spheroids. Additional testing of NPs in 3D cultures can clarify these points, especially by comparing NPs’ nature, size, charge, surface functionalization (active or passive targeting), and protein corona.
(2) Most studies incorporate NPs after the 3D system is formed. Comparative studies of this technique vs. NPs embedded in the matrix prior to cell seeding would be beneficial because they may reveal different mechanisms of NP flow, diffusion, and interaction with cells.
(3) The laser wavelengths used thus far have mainly been in the NIR-I window. 3D systems can also support research for NIR-II and NIR-III nanophotosensitizers to overcome technical, safety, and regulatory barriers and accelerate their clinical translation, without using expensive animal models.
(4) The advantage of 3D cultures over 2D is their ability to more closely mimic cancer cell-to-cell interactions, as well as NPs’ interactivity with other cells, such as endothelial, stromal, adipose, blood brain barrier, and immune cells. In vivo, PTT may act on these other cellular systems, and new mechanisms should be explored to quantify its impact on them, with the ultimate goal of taking advantage of its overall benefits. Also, studies have shown variations in the efficiency of PTT by itself or with drug therapies. This is primarily because the techniques used to generate 3D systems vary drastically (size, density, stiffness, organization, nature of the cells studied). Too often, these parameters are neglected or unspecified. The performance of NPs should be evaluated and compared on 3D spheroids, colony cultures, and 3D systems with or without addition of ECM or with or without co-cultures.
(5) Over 2D cell cultures, 3D models could be maintained for longer periods of time (weeks) under observation and, thus, are suitable for exploring long-term factors, such as how heat shock proteins can impact cancer generation, recurrence, and recovery.
(6) Finally, using consistent 3D cultures could enable more precise evaluation of PTT temperature diffusion and the “thermal zone” through a multidirectional system. It could also enable NP design to be adjusted to make PTT a combination of ablative and moderate hyperthermia techniques, allowing the evaluation of the immediate impact of the treatment (ablative technique with high power laser density and low exposure time vs. low to moderate hyperthermia with longer time of exposure for lower laser power density, or even the cumulation of both techniques through generation of temperature gradient ability of NPs) and the resultant action on the cancer cells (and potential “normal” cells in a co-culture model) with long term experimentation (weeks of potential observation after PTT). The longer evaluation periods made possible by 3D cultures may also be useful to characterize the mechanisms of cancer cell death, resistance, and recovery and, potentially, to evaluate the role played in cancer cell behavior by other components, such as stromal or immune cells, in the context of PTT. It might also highlight why some studies found PTT more beneficial than other therapies when combined with drugs or PDT.
Optimization guidelines for NP performances and characteristics for PTT in 3D cancer models
Evaluation of the performance of the size, charge and surface chemistry for NPs investigated in PTT
Although the use of NPs of different compositions in 3D model systems is relatively new, an impressive number of studies on the subject have already been published on polymeric and/or protein-based NPs composed of lipids84 polyethylene glycol (PEG) alone or mixed;85–94 polyamine;95 chitosan;96,97 poly(lactic-co-glycolic acid) (PLGA) alone or conjugated;98–101 and poly-L-lysine (PLL) and derivates102 Other NPs include metal-based NPs composed of Au61,66,71,103–105 titanium,106 zinc,107,108 iron,109–111 or even metalloids with silica;112,113 quantum dots;114,115 and carbon-based NPs75,78,116,117 were also investigated. The properties of NPs can be tuned to meet specific needs, especially when they can interfere with PPT abilities. The effect of charge, size, surface chemistry, and protein corona on 2D models have been heavily investigated to understand how cells interact with and internalize NPs.118 However, these parameters should also be analyzed in 3D cancer models in order to more fully understand the constraints created by the tumor micro-environment, including the influence of the ECM, proteins, matrix, and adhesion on NP penetration, diffusion, and penetrability throughout the system (Fig. 6). | ||
| Fig. 6 Schematic of the interaction between 3D culture and NPs. The nature, composition, properties, and function of NPs can be diverse and intentionally varied. Each variation of the parameters can be studied in 3D culture to simulate theirs in vivo abilities. | ||
• Size influence
To study the effect of NP size, Yu et al.112 developed an ultra-small (∼10 nm) hybrid silica/polymeric nanosystem and evaluated its dispersion and diffusion into 3D glioma spheroids and then compared with the results observed for ∼100 nm mesoporous (non-hybrid) silica NPs. The researchers tracked the NPs’ using fluorescence labeling and found that both kinds of NPs reached the core of the spheroids, but the heterogeneity of the distribution inside the spheroids was higher for the smaller NPs, especially on the outer edge of the spheroids, correlating that the size of the nanoparticle influence its penetration. The role of size in penetration/diffusion efficiency was also investigated by Mikhail et al.119 in 2014. They showed that distribution variation can be caused by size/incubation time synergy and influenced by the cell lines studied (HeLa vs. HT29 cancer cell lines). The researchers designed different lengths of block copolymer micelles composed of PEG-b-PCL, varying the size by increasing the chain of PEG or PCL (from ∼15 to ∼55 nm, with similar negative charges). They then studied these chains’ penetration on 500 μm spheroids for 1 and 24 hours, comparatively. The NPs were tracked by fluorescence imaging and evaluated according to fluorescence intensity in three specific areas of the spheroids: periphery, intermediate, and core. Results showed higher distribution and accumulation in the spheroids after 24 hours than after 1 hour, especially for the biggest NPs. The researchers also observed higher fluorescence intensity in HeLa than in HT29 spheroids, correlating with the fact that penetration and diffusion can be influenced by spheroid density—HeLa spheroids are less dense and possess greater extracellular volume than HT29 spheroids and the incubation time. The team then demonstrated the predicted penetration trend for nanoparticle interstitial diffusion in tumor xenografts.
In 2016, Arranja et al.120 also sought to evaluate the effect of NP size on spheroid penetration in function of different cell lines (HeLa vs. U-87). They showed that Pluronic® (triblock copolymers of poly(ethylene oxide)-b-poly(propylene oxide)-b-poly(ethylene oxide) (PEO–PPO–PEO)) nano-micelles with varying PEO block mass (F127 constituted of PEO96–PPO56–PEO96 for a MW = 11700 g mol−1vs. P94 made of PEO26–PPO48–PEO26 for a MW = 5000 g mol−1) and aggregation states (unimers vs. cross-linked micelles called SPM-F127 and SPM-P94) can penetrate and accumulate differently, most likely because they may activate different internalization pathways. The researchers also presented a correlation between the size and penetrability of different NPs in function with the nature of the cells used to make the spheroids, related to their cellular interstitial space and intracellular mechanism. Results showed that SPM-F127 micelles (with hydrodynamic radius RH ∼ 16.9 nm) were more efficiently delivered across the tumor spheroids than their unimer version F127 (RH ∼ 2.9 nm); deeper penetration was observed for SPM-127 (around 2 times deeper for 4 time points) in HeLa and U87 spheroids. The researchers also mentioned that compared to the 2D culture, the 3D model is dependent on the aggregation states (meaning micelle-sized NPs). However, it was already theorized that the penetration of NPs through the tumor interstitial space is related to the diffusion coefficient, which decreases as the molecular weight increases. Additionally, other cellular mechanisms, such as internalization and subsequent exocytosis from one layer of cells to another, cellular binding affinity, cell density, and spheroid compactness, may also govern NP penetration depth in tumor systems, as might the model being an avascular system, in our opinion.
• Charge and surface chemistry
The effect of surface chemistry, and more specifically, cationic and anionic charge was investigated in 2010 by Kim et al.121 with human colon carcinoma cell cylindroids. Using mathematical models and 3D culture, they revealed that cationic AuNPs improved drug delivery to most cells (higher uptake and dissociation by viable cells), while the anionic AuNPs showed better drug deliver deep into tissues (faster diffusion). In 2013, England et al.122 showed that varying the size, zeta potential, and morphology of PEG-coated AuNPs via functionalization or layering affects their penetration and diffusion differently in different 3D cancer models. They studied liver, lung, and pancreatic cancer cell lines with similar incubation times, using dark field microscopy to determine the intensity of NPs on the periphery and center region of the 3D models. In 2015, Agarwal et al.123 tested an additional parameter to the size and charge considerations by quantifying the impact of the geometry (shape and aspect ratio) of polymeric NPs (PEG anionic nanohydrogels) in terms of penetration and distribution inside 3D spheroids of kidney cancer cell line, showing that “disc-like” cylindrical NPs with a low aspect ratio had maximal, uniform intratissue penetration (>50% increase) compared to nanorods or smaller NPs (220 nm diameter) of the same shape. This additional parameter resulted in data that is in total contrast with previous findings, in which smaller spherical NPs (polymeric or metal based) resulted in deeper and more uniform penetration.
In 2016, Bugno et al.124 hypothesized that the size and surface chemistry of poly(amidoamine) dendrimers (PAMAM) with controlled size and charge were key parameters for their distribution, accumulation, and penetration inside KB and MCF-7 spheroids and on an external ECM (collagen I gel) model, which was used to mimic an additional barrier within the tumor microenvironment. The researchers found that smaller dendrimers diffused faster in the model with ECM and inside the spheroid core than the larger dendrimers. Additionally, higher accumulation in MCF-7 spheroids was seen for the positively charged NPs with –NH3+ functionalization or cationic termination than for either the anionic or neutral NPs.
The influence of NP size, charge, and surface chemistry on penetration was also investigated by Priwitaningrum et al.125 in 2016, who used silica and PLGA NPs with a 3D co-culture spheroidal array that mimicked breast tumors and pancreatic cancer with stromal cells. To characterize the impact of the ECM on the penetration and diffusion of different NPs, the researchers modified the ratio of cancer cells to fibroblasts (1:1 and 5:1). Fabrication and analysis of the spheroids were done using a microwell array platform. Silica NPs were synthesized to have various sizes and charges (30 nm, 70 nm, or 100 nm; zeta potential: −40 mV and −20 mV). The spheroids were incubated with the silica NPs, then fluorescent microscopy of spheroid cryosections was performed, showing high penetration by the 30 nm NPs—about 75–80%—into the 4T1 homospheroids within 24 hours but almost no penetration into the fibroblast models. A similar experiment conducted with another composition of NPs, PEGylated PLGA (200 nm, −7 mV), had analogous results. Additionally, as the negative charge of the silica increased, so did its penetration inside the spheroids. Another study on human homospheroids (made of only cancer cells) and heterospheroids (cancer cells co-cultured with fibroblasts or stromal cells) confirmed the same trend. In addition to the differences between low/high charge and size-varied NPs, this study highlighted the ability to modulate the extracellular stroma in 3D multicellular spheroids, which can act as a strong barrier to NP penetration.
Some studies could show better penetration of negatively charged NPs, and others for positively charged NPs. Both trends might be relevant, as penetration is multifactorial (function of the composition, size and shape of the NPs) and can be dependent on the cell line chose and cell incubation time. Given the differential tumor cell lines used and culturing conditions in these model systems, it is difficult to ascertain which functionalized surface portends better penetration or tumor interaction clinically. More investigation needs to be done in this area of research.
• Protein corona consideration
NPs’ incubation with cell media or biological fluids can affect their surface chemistry, charge, and size due to the proteins present in these environments. This well-established surface chemistry modification process, in which the NPs get covered by a protein corona, can impact their cytotoxicity, treatment efficiency, targeting recognition, and immune response.126 Therefore, in addition to the parameters already described in the previous sub-sections, the impact of protein corona formation around NPs on tumor penetration should be considered, as this can modify their size, shape, bio/chemical activity, and biodistribution. To prove this point, in 2017, Huang et al.127 designed a chip to study the effect of surface charge and protein corona on fixed-size NPs (100 nm polystyrene NPs), positively or negatively charged, without and with serum proteins to induce protein corona, in a constantly moving liquid flow. Accumulation and penetration were quantitatively tracked by confocal fluorescence laser microscopy. The results indicated that the negatively charged NPs had easier surface accumulation and penetration into the spheroids; the best results under flow were seen from negatively charged NPs without a protein corona. Indeed, the induced protein corona modified NP-cell affinity, affecting their interactions with cells and resulting in lower concentration of NPs on the outer shell/surface of the spheroid. However, the corona might be responsible for deeper penetration. (Fig. 7).
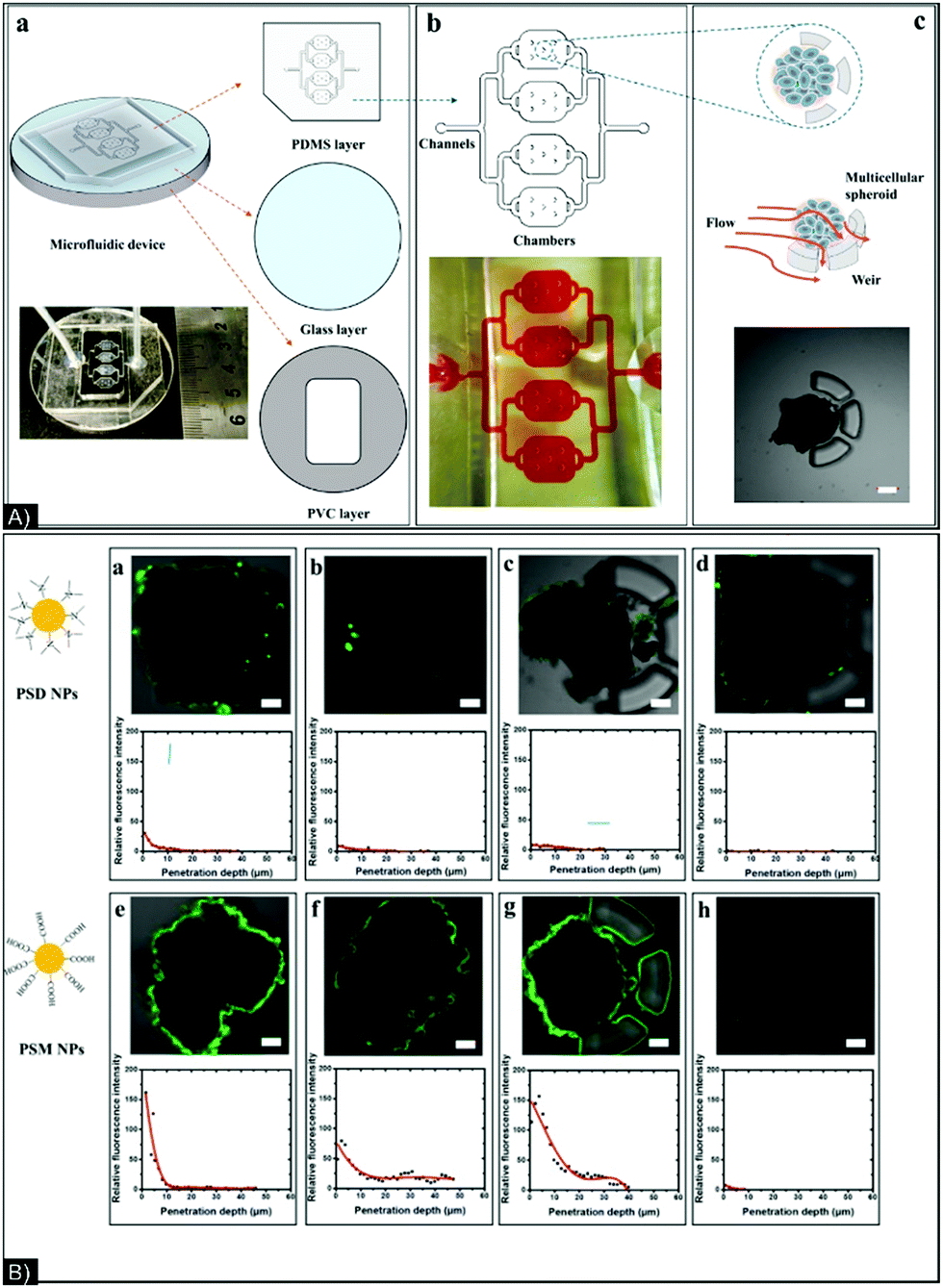 | ||
| Fig. 7 (A) (a) Structure of chip composed of 3 layers: PDMS (upper), glass (middle), and PVC (bottom); (b) picture and schematic of a four-chamber chip; (c) schematic and microscope photo of spheroid trapping. (B) Confocal images of spheroids after 2 hours of incubation with (a) PSD NPs with no corona and in static conditions, (b) PSD NPs with a corona and in static conditions, (c) PSM NPs with no corona and in static conditions, (d) PSM NPs with a corona and in static conditions, (e) PSD NPs with no corona and in flow conditions, (f) PSD NPs with a corona in flow conditions, (g) PSM NPs with no corona in flow conditions, and (h) PSM NPs with a corona in flow conditions. PDMS: polydimethylsiloxane, PVC: polyvinyl chloride, PSD: p(styrene-co-dimethylaminoethyl methacrylate) and PSM: p(styrene-co-methacrylic acid). Figure and caption reproduced with permission.127 Copyright 2017, American Chemical Society. | ||
• Active functionalization of the nanoparticle surface
Size, composition, surface chemistry and/or charge play a crucial role in NPs interaction and accumulation in tumors via passive targeting with enhanced permeability and retention (EPR) effect.128 Though much of the EPR effect is caused by the characteristics of the tumor vasculature, this mechanism has been heavily investigated in 2D, 3D, and in vivo models to predict the advantages of using NPs in therapeutic applications. However, the EPR effect has already started to show its limitations, even in vivo models with intact blood circulation. As mentioned earlier, unlike organs, cancer can make solid tumors with low vasculature and complex microenvironments—i.e., stroma, extracellular matrix, and immune environment—which can affect the transport and distribution of drugs.129 Another path investigated for the delivery of nanoparticles to a tumor site is active targeting, in which NPs are functionalized with biological objects that recognize and bind to specific markers on the cell surface.130–132 In active targeting, targeting ligands are added to the surface of NPs to guide their delivery into only cancer cells (or subcellular cancerous sites), which reduces undesirable systemic exposure and cytotoxicity to healthy tissues. Active targeting increases the specificity of the interactions between the receptors expressed within or on the surface of cancer cells and the ligands on the surface of nanocarriers by triggering receptor-mediated endocytosis. Receptor-based active targeting has led to the development of novel targeted NPs for therapeutic, diagnostic, and imaging applications. Various kinds of targeting ligands have been employed to functionalize NPs, including peptides and whole proteins (e.g., transferrin, integrin, matrix metalloproteinase), aptamers, antibodies or antibody fragments, and different receptor ligands (such as folic acid).131,133
Beyond cellular recognition, 3D culture integrates the concept of penetration and diffusion; qualification of these two last parameters should be determined in the context where the NPs are modified to enhance cellular recognition and affinity. Ran et al.134 developed a microfluidic system to make multifunctional plain, PEGylated (DSPE-PEG2000), and folic acid-functionalized (DSPE-PEG2000-folate) liposomes, all conjugated with a fluorescent dye (DiI or (2Z)-2-[(E)-3-(3,3-dimethyl-1-octadecylindol-1-ium-2-yl)prop-2-enylidene]-3,3-dimethyl-1-octadecylindol) encapsulated for imaging. The recorded zeta potentials were −9.5 mV ± 0.9, −0.8 mV ± 0.4, and −0.9 mV ± 0.1 mV for, respectively, plain, DSPE-PEG2000, and DSPE-PEG2000-folate. The DSPE-PEG2000 was 50 nm, and the plain and DSPE-PEG2000-folate were 100 nm. 3D cultures of SKOV3 (FA+) and MCF7 (FA−), formed over 7 days, were exposed to the plain, DSPE-PEG2000, and DSPE-PEG2000-folate for 4 hours for a quantitative cell uptake analysis and for 24 hours for a 3D culture penetration analysis. The quantitative analyses and fluorescence penetration in 3D showed logical and similar results: DSPE-PEG2000-folate had higher uptake in and effect on SKOV3 (FA+) than on MCF7 (FA−). This study highlights the concept of differentiation and characterization between interactions at the cellular level (cellular uptake) and on the scale of a 3D tumor with penetration and flow of the nanoparticles through interstitial space. We believe that these are two important concepts to distinguish, and 3D culture is a key model to allow the synergistic optimization of both.
This notion of interstitial flow was mentioned in a study by Wang et al.,135 who investigated the transport of carbon nanotubes (CNT) and their diffusion in Hep-G2 hepatocellular carcinoma (made with polyacrylamide hydrogel inverted colloidal scaffold (ICC)). CNTs were functionalized with transforming growth factor β1 (TGFβ1) and with the fluorescent dye FITC (fluorescein isothiocyanate), CNT-TGFβ1-FITC, added for the dual function of imaging and drug-model delivery system. Diffusion and transport through the spheroids were evaluated with CNT-FITC. Both nanoparticles possessed a negative charge but different molecular weights—CNT-FITC was 5.83 ± 0.74 × 104 Dalton lighter than CNT-TGFβ1-FITC. Mathematical prediction via the Stokes–Einstein equation evaluated a similar diffusion coefficient between both NPs with a rate of 5.7 × 10−16 m2 s−1. However, based on a 3D culture assay, it was discovered that apparent diffusion coefficients of CNTs in 3D were higher than similarly charged molecules with molecular weights, which were 10000 times lower. CNT diffusion in tissues was enhanced when they were functionalized, which the researchers attributed to planar diffusion (gliding) of CNTs along cellular membranes. This gliding reduced the effective dimensionality of the diffusional space. In this study, TGFβ1 improved the transport process and diffusion inside the spheroids, increasing the penetration but not the cellular interaction. Additionally, the implicit conclusion of this study was that the nature of cancer or the technique used to generate 3D culture will influence the interstitial space, which can involve NPs diffusion. This supports the idea that NPs can be functionalized to control transport processes at the cellular interface by affecting parameters such as adsorption/desorption to the cellular membrane, surface diffusion, and cellular endocytosis/exocytosis. With the same strategic concept of balancing NP cellular internalization vs. tissue penetration, Sims et al.136 designed NPs made of poly(lactic-co-glycolic) acid with PEG, MPG peptide, a mix MPG/PEG, and vimentin (VIM). This time, 2D and 3D cultures were used to differentiate NP interaction from NP penetration and diffusion. In a HeLa 2D culture after 24 hours of incubation, the MPG, MPG/PEG, PEG, and VIM NPs showed 66×, 24×, 30×, and 15× more interaction, respectively, than the unmodified NPs. However, in HeLa 3D spheroids, for the same incubation time, all nanoparticles were severely hindered: MPG and MPG/PEG NPs were internalized only 2× and 3× times more than PEG and VIM NPs. Additionally, MPG NPs were mainly localized at the outer layer of the sphere, while PEG NPs penetrated 2× farther. This study emphasizes the balance between interaction and penetration, where active functionalization with MPG enhances cellular interaction and internalization and when PEG enhances 3D penetration. We can easily understand how this balance is important when studying systems with high or low vascularization. To mimic this hyper/hypo-vascularization and evaluate the transport and longevity of NPs during local or systemic administration, with the same nanoparticle, they developed, in another study, 3D spheroids made by two different methods: hanging drop (HD), producing smaller and regularly shaped avascular tissue, and liquid overlay (LO). Additionally, they chose three cell lines—HeLa, CaSki, and SiHa—to compare NP distribution within the same type of tissue as a function of different cancer types. In HD-made spheroids, enhanced distribution of unmodified NPs in HeLa was recorded, whereas SiHa spheroids showed positive penetration results for all modified NPs. Interestingly, in spheroids made by LO, opposite results were seen, with a higher distribution of MPG and MPG/PEG NPs in HeLa and of PEG and MPG/PEG NPs in SiHa spheroids.99 Here, as we concluded previously, the researchers demonstrated the influence of penetration in function of the cancer cells selected, and also that the technique used to make a 3D culture can induce different NP penetration results, even when the NPs are functionalized for active targeting or enhanced peptide recognition.
The goal of the review is not to provide a state of art of the active targeting in 3D culture but to illustrate potential insight for optimization of NPs with active targeting for PTT application. Consequently, we have focused the next papers on active targeting recognition that can be able to target cancer but also its ECM components or cancer resultant as stem cells, and present the different outputs.
Integrins have been long investigated for active targeting. Typically, they are transmembrane proteins that work as mechanosensors, supporting multiple biological processes through adhesion molecules and signal transduction platforms. Several inhibitive integrin-targeted drugs have shown clinical efficiency to treat and prevent diseases, especially cancers.137 Short, artificial peptides that have the RGD sequence can replicate the integrin-binding activity of adhesion proteins. These peptides can bind at the integrin attachment site, either on cell surface proteins or the ECM. Because integrin-mediated cellular attachment impacts and controls cell proliferation, differentiation, dispersion, and death, RGD peptides can be employed to trigger integrin functions and support new cancer drugs discovery based on RGD recognition.138 In 2015, Ni et al.139 developed a peptide iRGD (specific integrating RGD: 9-amino acid cyclic peptide)-functionalized nanodots (iND ∼10 nm) and nanospheres (iNP ∼70 nm), loaded with PTX at a high drug loading capacity (>80 wt%) by encapsulation in different polymers combinations (PPG, PEG and Pluronic F127). These systems were tested with a breast cancer cell line. In a monolayer model, the tumor-penetrating peptide facilitated better uptake of both kinds of nanosystems into the tumor cells than the non-targeted nanosystems or free PTX. Confocal laser scanning revealed no significant difference in relative internalization of iND and iNP or in cell viability after 48 hours of incubation in 2D cell culture. However, when tested in a 3D multicellular tumor spheroid model, the iND's performance was markedly superior to the iNP's in terms of inward penetration and spheroid elimination. Penetration tracking was done after 24 hours of incubation and during the following 3, 5, and 7 days, and toxicity was evaluated in comparison with the relative volume of a control treated with PBS. These results suggest that, even when active targeting is involved, NP size is an influential, determinant parameter for cellular penetration and resulting cytotoxicity. In vivo experiments were performed in mouse models of orthotopic mammary adenocarcinoma, confirming that only the PTX-iND efficiently reached the tumor site, while the PTX-iNP, non-targeting NPs, and PTX alone did not (Fig. 8).
 | ||
| Fig. 8 (A) Schematic and TEM images of the synthetized NPs showing the composition of iRGD-conjugated PTX-iNPs/iNDs, their distribution, and effects on different cells, and cancer stem cell elimination post-IV. (B) In vitro spheroid images of tumor tissue penetration and antitumor activity. (a) CLSM images indicate drug distribution in spheroids after 24 hours of incubation with 5 μg mL−1 of Cy5-doped PTX/NDs/NPs/iNDs/iNPs; corresponding fluorescence signal intensities across the spheroids are shown along the dotted lines. (b) Representative CLSM images of TUNEL analysis of spheroid cryosections post-treatment with different PTX formulations (5 μg mL−1) for 24 hours; red: dead cells; green: live cells. (c) Spheroid growth inhibition caused by various PTX formulations (5 μg mL−1). Abbreviations: iNPs: nanoparticles, iNDs: nanodots, PTX: paclitaxel, IV: intravenous injection, TEM: transmission electron microscopy, CLSM: confocal laser scanning microscopy. Figure and caption reproduced with permission.139 Copyright 2015, John Wiley and sons. | ||
Waite et al.140,141 delivered micro-RNA in a 3D brain cancer model via RGD targeting. They designed a new generation of 5 PAMAM dendrimers functionalized with cyclic RGD-targeting peptides (∼200 nm) and analyzed their ability to associate with siRNA and be delivered to cancer cells. The researchers demonstrated that siRNA delivery was a function of the number of RGD ligands per dendrimer. PAMAM-RGD NPs delivered inside 3D spheroids had higher penetration performance than non-targeted PAMAM dendrimers, most likely by interfering with the integrin-ECM contacts in the 3D tumor model. The number of ligands attached or present on the nanoparticles is, in general, not evaluated and, thus, unknown. Methods of analysis should be created to improve active targeting efficiency, especially in the context of 3D culture experimentation, to balance cellular interaction and penetration abilities of the NPs.
Targeting with ligands such as RGD, folate or transferrin is considered active targeting because malignant cells or tumor tissue possessed overexpressed recognition for these molecules, while their expression is more limited in healthy organs.142–144 Antibodies and aptamer recognition generally promote better recognition/binding affinity (as showed by Kd value (around 10−9 for RGD145vs. 10−12 for antibody146)), and can be used in combination with different peptide, folate, and transferrin targeting agents for both intra and extracellular targeting.147 It is also interesting to compare these active markers, able of higher cellular recognition, in terms of penetration and diffusion through 3D cancer model. Based on the specificity of hyaluronic acid to recognize and fix on cancer cells, particularly cancer stem cells, that overexpress CD44 markers, Rao et al.148 developed a complex nanoparticle made of PEG–PPG–PEG functionalized 4-nitrophenyl chloroformate (4-NPC), chitosan, and pluronic F-127, arranged in micelle shape by emulsion to allow the encapsulation of Dox. CD44, known to support transport between endosomes and the cell surface, is a single chain transmembrane glycoprotein that possesses a “link module” or “proteoglycan tandem repeat” made of ∼100 amino acids, i.e., a hyaluronan-binding site for proteins such as hyaluronic acid or an analog such as chitosan.149 It is important to note that HA–CD44 may not be the only recognition site, and sequences outside the link as well as cell-specific factors and multiple CD44/HA engagement by weak receptor–ligand interaction might achieve functional avidity. The advantages of such recognition are that the polymer coating on the nanoparticle hyaluronic derivates, preliminarily used to increase biocompatibility and stability, can target CD44+ cancer cells. CD44 binding to HA takes place at the cell surface, where many close CD44 receptor molecules can interact with HA's repeating disaccharide chain.150,151 The investigated nanosystem was designed to release encapsulated Dox when in acidic surroundings, such as those found within cellular endosomes/lysosomes, thus controlling the drug's release until the system has been internalized. The researchers also developed MCF-7 3D breast cancer model or mammospheres that could modulate CD44 expression and the ratio of CD24/CD44 expression when cultured in 2D or 3D. Using this model, the researchers found that, due to the CD44 expression, the 3D mammary tumor was 6 times more sensitive to the Dox-loaded chitosan-NPs than to free Dox, as indicated by cytotoxicity and growth inhibition analyses. Additionally, NPs efficiently inhibited the formation and growth of spheroids made from a single cell suspension, showing the relevance of HA or CD44+ to target stem cells and evaluate their impact on 3D culture formation. Investigations in an orthotopic xenograft tumor model confirmed the results (Fig. 9).
 | ||
| Fig. 9 (A) MCF-7 stem cell 3D mammospheres characterization after cellular uptake of 1 μg mL−1 nDox vs. fDox at 37 °C for microscopic images with fluorescence staining of cell Nuclei (blue) and endo/lysosomes (green), fluorescence Dox tracking (red) and merging picture of nuclei (blue), endo/lysosomes (green), Dox (red) after 3 h of incubation/exposure (B) cell viability and % of mammosphere cells remaining after treatment. (a) Micrographs indicate the variation in morphology of the cells/mammospheres after treatments for two types of 3D culture protocols/techniques (single cell 3D, mammosphere 3D culture); (b) survival (IC50) of mammospheres (single cell 3D, mammosphere 3D culture, and 2D cultured parent MCF-7 cancer cells) after treatments (10 days exposure presented only). For the mammosphere 3D model, the percentage of (c) CD44+CD133+, (d) CD44+, and (e) CD133+ cell subpopulations after the treatments for three different times (3 h, 3 days, and 10 days) Abbreviation: nDox = nanoparticle loaded with Dox, fDox = free Dox. Figure and caption reproduced with permission.148 Copyright 2015, American Chemical Society. | ||
HA–CD44 affinity was also investigated by El-Dakdouki et al.152,153 in an ovarian cancer cell line in a 3D culture. The researchers developed silica NPs covered by hyaluronan (HA) and with a highly fluorescent core. The authors showed that this nanosystem had an enhanced ability to be taken up into cells, exit the cells, and, therefore, transition from one cell to another, enabling penetration deep inside the spheroid. The therapeutic potential of this nanosystem was evaluated by loading it with Dox. The 3D ovarian cancer model was cultured until it reached a spheroid shape and a size of 250–350 μm, then it was exposed to the NPs. The penetration of SNP vs. HA-SNP was evaluated by fluorescence confocal microscopy and the number of cells labeled by flow cytometry (FACS) after 6 hours of incubation. The HA-SNPs led to a 4.5-fold increase in the number of labeled cells and increased deep penetration into the spheroids; additionally, drug loading showed a significant therapeutic improvement. Interestingly, they also found that the CD44 underwent recycling between the cell surface and the interior of the cells, allowing efficient drug delivery through receptor-mediated transcytosis.
These studies revealed the importance of active targeting to increase recognition and cellular accumulation. They also show that designing optimal NPs for targeting cells and for penetration/delivery inside a spheroid or 3D model might be two different concepts and require different approaches. The best NPs for cellular targeting and distribution/penetration in a 3D culture might produce diametrically opposed results in 2D. Consequently, NPs with certain surface chemistry, charges, size, and composition that have already been heavily investigated in 2D cultures and judged inefficient compared to active targeting might provide different data in 3D cultures or humans. Developing PTT nanoparticles can benefit this dual approach by allowing comparative studies of size, charge, and surface chemistry with different targeting agents. In conclusion, targeting recognition does not necessarily mean nanoparticle penetration in 3D: an efficient cellular recognition and internalization do not necessarily mean an optimal tumor penetration. It is difficult to fully understand if the penetration of NPs inside a 3D system indicates interaction with the cells. The boundary between uptake and penetration inside the spheroids and interaction with individual cells is not clearly identifiable. More research needs to be done to differentiate between these two states. Studying 3D cancer models and NP design together in relation to the desired application may fill the gap between fundamental research and clinical application development, leading to more efficient NP-enhanced cancer therapies.
As mentioned in the introduction of this section, NPs suitable for PTT depend on their wavelength and absorption ability to produce optimized heat conversion. These parameters are extremely influenced by the size, shape/morphology, concentration, nature, and aggregation state of the NPs. The preceding studies highlight the fact that these criteria can be modified in the context of a biological system. More interestingly, compared to previous studies done in 2D, 3D systems revealed that penetration, diffusion, cellular affinity (with or without active targeting) and, consequently, final aggregation state, concentration, and overall shape of the NPs might be influenced not only by their coefficient of diffusion but also by the nature, cell density and compactness, cellular binding affinity, and cellular mechanism of the system studied. As this area of study progresses, PTT and NPs must be directly optimized in function of the particular 3D spheroid at hand, and performance and therapeutic efficacy must be evaluated in the context of a particular 3D model, both short and long term.
Therapeutic applications of NPs in 3D cultures for potential PTT combinations
The goal of the following section is to highlight the achievements made in each therapy and develop their potential combination with PTT in 3D applications. Combining cancer therapies can provide three key benefits: (1) the dual effects of a short therapy (PTT) and a longer one (drug or gene delivery), (2) attacking remaining cancer cells that were not removed by PTT, (3) limiting the adverse effects of treatment while still administering and delivering effective doses of anticancer agents for total cancer therapy (local and metastasis) and (4) evaluate if PTT need to be combined with other therapies to be beneficial, in function of a specificity of the cancer studied. The tunability of NPs’ physicochemical traits and functionalities make them suitable to carry chemotherapies or small molecule inhibitors (proteins, cytokines) or conduct gene delivery; this tunability also makes NPs uniquely suited for PDT and PTT.• Chemotherapeutic combination approach
Chemotherapy generically refers to cancer treatments that utilize a synthetic or natural drug; they can be used alone and/or in a combination of drugs or other therapies. The action of a chemotherapeutic drug against cancer cells depends on its nature, mechanisms, and/or “functional groups”. These functionalities allow classification as following: alkylating agents, antimetabolites, mitotic inhibitors, topoisomerase inhibitors, anti-angiogenesis or plant alkaloids.154 However, chemotherapeutic drugs can harm healthy cells, affecting the patient's whole body; possess low pharmacokinetics and low plasma concentration retention; and, sometimes, cannot be delivered to the cancer site due to low solubility, generally caused by their high hydrophobicity. Lee et al.155 discussed the importance of viability and efficiency to conventional chemotherapies and how these parameters can be increased with delivery method optimization, including by nano “packaging” for drugs. The drugs can be carried by the NPs by encapsulation or attached or trapped with non-covalent or covalent bonds directly onto the NP surface; the functionalization decision is made based on the chemical groups or the delivery process.
Drug nanocarriers can be taken up at the tumor site by a variety of processes, including endocytosis.156 Additionally, the drug(s) can be released from the NPs inside cancer cells by different mechanisms, including heat (from an internal or external source), redox reaction, pH changes, and enzymatic or protein actions.157
Compared to 2D models, 3D models have shown that they may be able to successfully model one of the major hurdles of chemotherapy: cancer drug-resistance. In 2014, Kang et al.158 developed concave microwells of different diameters (300, 500, and 700 μm) to generate uniformly shaped spheroids of breast cancer. Analysis of the 3D culture generated with the same initial cell seeding showed a lower cell number in the 300 μm-made spheroids, while the 700 μm-made spheroids possessed a higher cell number but also a necrotic core, modeling a nutrient an oxygen diffusion to their center. They developed a thermosensitive polymeric nanoparticle made of poly(N-isopropylacrylamide) (or PNIPAM)-co-acrylic (co-AA) loaded with doxorubicin, modifying their size and wettability as the temperature was increased (>34 °C). After 2, 24, and 96 hours of incubation, the 300 μm-made spheres were losing cohesive structure and had a significant reduction in diameter. Interestingly, the 300 μm-made spheres treated with free doxorubicin showed the same percentage of toxicity as those treated with the drug-loaded NPs. In contrast, the size of the largest tumor spheroids did not change, even when treated with the drug-loaded NPs. Cell viability assays revealed that the largest spheroids cultured with doxorubicin-loaded PNIPAM-co-AA NPs were very drug-resistant. Here, the researchers showed that variation in spheroid size causes drug diffusion to vary, inducing drug resistance due to the features of the 3D system studied. It was also proven that in vivo, dose-limiting systemic toxicity might be responsible for the development of multidrug-resistant phenotypes. Thus, proving PTT superiority over drug therapy or optimizing combined PTT-drug therapies should be pursued in both non-resistant and resistant 3D models. In the aforementioned study, spheroid size was a determinant parameter, but as mentioned earlier, unlike normal organs, cancers can make solid tumors with low vasculature and complex tumor microenvironment—i.e., stroma, extracellular matrix, and immune environment—which can affect or hamper drug transport and distribution, inducing resistance.129 More 3D studies are needed to further investigate the mechanisms and parameters that lead to drug resistance.
Shi et al.159 modeled the ECM by using a 3D system with a matrix of collagen I. The main goal of the study was to improve NP-based drug delivery and cytotoxic effects compared to the free drug. NP penetrability was based on their composition: the chitosan and ε-caprolactone (CS-g-PCL)-5-fluorouracil (5-FU)-loaded NPs triggered drug release under acid pH values. The researchers tested the nanosystem in 2D and 3D colorectal cancer models, both of which contained two different cell lines, HCT8 and HCT116, and their multi-drug variants, HCT8/VCR and HCT116/L-OHP. In contrast with the free 5-FU, the results indicated that 5-FU/NPs enhanced cytotoxicity toward HCT116/L-OHP or HCT8/VCR cells by decreasing the IC50. Interestingly, the IC50 of the drug-loaded NPs on the cell lines resistant to drug (HCT8/VCR and HCT116/L-OHP) in 2D and their 3D model on HCT8 and HCT116, present almost the same resistance to the 5-FU/NPs, with a sensitive more resistance in 3D HCT8 vs. 2D HCT8/VCR than 3D HCT116 vs. 2D HCT116/L-OHP, related to the ability of the HCT8 to form larger and highly densify spheroids compare to HCT116. Additionally, quantification of drug release by HPLC tracking indicated that peak saturation was reached after 8 hours of incubation for the 2D model, while saturation was never reached in the 3D model, indicating two different drug release profiles.
In a 3D pancreatic cancer model made by seeding cells in collagen I/agarose, Shen et al.160 showed the versatility of RGD targeting by designing polymeric NPs made of mPEG–PLGA–PLL, loaded with PTX, and surface-functionalized with a final size ∼130 nm. In this study, the focus was on the ability of Pluronic F-127 to make a thermoactivated gel to mimic PTX release and diffusion in tissue through a 3D model system. This kind of model developed with a thermoactive gel (>37 °C) might be highly significant in PTT therapy, especially if the gel can be injected directly within the tumor site and the nanoparticles dispersed in a gel can enhance the PTT and locally deliver the drug. It could allow fast PTT activation and slow release of the drug over an extended time to eliminate photothermal-resistant cancer cells and metastasis. Jiang et al.161 designed an integrin-mediated, PEG-coated, poly(trimethylene carbonate) nanosystem (∼70 nm) loaded with PTX and analyzed its internalization by human glioma cells through various endocytic pathways. The nanosystem's tumor penetration, specificity, and anticancer efficacy were studied in an in vitro (3D glioma spheroids) and in vivo (intracranial glioma mouse). The fluorescent-labeled nanosystem was incubated for 12 hours on 7-day-old spheroids and compared with the following controls: untreated spheroid, NP/PTX, and free PTX. The targeted, drug-functionalized NP system showed the highest penetration and accumulation (throughout the whole spheroids; only in the peripheral region on the controls), resulting in the highest growth inhibition. Additionally, in vivo, this nanosystem had the lowest concentration in the liver and spleen and the highest fluorescence intensity in the brain 24 hours post injection (vs. non-targeted NPs). Here, integrin-mediated nanoparticles showed significant positive results for cellular interaction, stimulating various endocytic pathways, but also showed the ability to diffuse and penetrate inside the 3D structure, allowing higher homogeneity of drug delivery, which provides for better treatment, as shown by in vivo data.
In general, optimization of nanoparticles for drug release and 3D culture is done by varying the parameters we have discussed, including composition, size, shape, surface chemistry, and different triggers, such as pH or temperature. For additional information on the investigation of drug delivery by nanoparticles in 3D systems, readers can refer to already published reviews.20,162
IC50 and drug resistance can be underestimated or misinterpreted between 2D and 3D models. Use of NPs for drug delivery, which could help overcome cancer resistance, can be simulated in a 3D cancer model. This combination of techniques, especially with PTT, might support higher drug dose delivery by causing a triggering effect (pH and heat), increasing cell sensitivity to drugs, or enabling blood diffusion/flow of the drug by heat effect. Because ablative PTT can induce negative side effects, a lower power density must be used to reduce the thermal zone, but this decreases PTT's effect on cancer cells located far from the treatment site. Investigating combining PTT with drug release might answer the following question: could the drug reach cells outside of the thermal zone produced by the PTT? Can the drug be delivered to a higher number of cells, resulting in lower metastasis and/or CTC (circulating tumor cells) generation, or avoid tumor recurrence?
More studies on the combination of PTT with drugs need to be done to fully understand the mechanism of drug-resistance regulation and or reversion in 3D models.
• Gene silencing combination approach
Another chemotherapy alternative that has been tested in 3D cell culture models is gene therapy, which involves RNA interference, cytokines, and apoptosis compound delivery. The success of gene therapy depends primarily on the transportation of genetic materials to targeted tissues or organs while avoiding healthy tissue. Zero premature release from any gene delivery nanocarrier, whether by enteral, parental, or topical administration, is permissible in order to avoid high cytotoxicity in healthy tissues. However, biological barriers and tumor hindrance can retain the nano-gene-carrier and cause negative side effects. A novel approach to nanocarrier functionalization for passive (EPR effect) or active targeting with various targeting ligands such as peptides, antibodies, and aptamers was investigated and showed good distribution and delivery to the cancerous site.163
RNA interference (RNAi) therapy is a new class of cancer gene therapy in which siRNA (small interfering ribonucleic acid or silencing RNA) “interferes” with protein translation by binding to messenger RNAs (mRNA) via a complementary base and promoting their degradation and/or blocking protein synthesis. The goal of the therapy is to target, inhibit, and/or avoid gene overexpression in cancer.164,165 It is very complex to predict and provide efficient delivery of siRNA to tumor sites because the siRNA can easily be destroyed during blood circulation before it reaches the tumor. Protective encapsulation and theranostic applications for siRNA must be improved to determine its biodistribution and cellular uptake. The loading, protection, and diffusion of siRNA by nanocarriers to ensure deep penetration and delivery within tumor regions despite biological barriers could be the key to the success of this therapy.166 Additionally, good protection will also allow the siRNA to be used during PPT, avoiding thermal degradation. Recently, nano-enhanced RNAi therapy was investigated in a 3D model to determine if a suitable model with biological constraints can help optimize penetration and diffusion. To promote efficient delivery and understand if an siRNA-loaded nanostructure can penetrate a complex 3D system, Jung et al.167 developed flexible nanocarriers (FNCs) made of siRNA/PLL and covered in a liposomal membrane. Then, they evaluated how functionalizing the FNCs (∼170 nm, non-spherical) impacted penetrability in a 3D culture of skin melanoma, as well as how it influences RNA interference effects. Lastly, they functionalized the system with PEG and the opener peptide AT1002. Significant RNA interference effects were seen in 2D, comparable to Lipofectamine 2000. Penetrability into 3D cancer spheroids was evaluated by laser scanning microscopy after 24 hours of incubation. The PEG and AT1002 improved the penetration, but the system did not reach deeper than the peripheral region of the spheroids. Penetration optimization was also investigated by Wei et al.,168 who actively functionalized core–shell-siRNA through layer-by-layer assembly of protamine/chondroitin sulfate/siRNA/cationic liposomes. Using this nanosystem, the researchers were able to target the receptor T7 by peptide modification (T7-LPC/siRNA NPs, with LPC: liposome-protamine-chondroitin) in 3D glioma spheroids. 2D in vitro cellular uptake experiments on brain microvascular endothelial cells and glioma cells indicated that the T7-LPC/siRNA NPs caused higher siRNA intracellular fluorescence intensity than the PEG-LPC/siRNA NPs. In the co-culture model, the major down-regulation of EGFR protein expression after treatment with the T7-LPC/siEGFR NPs and T7-LPC/siRNA NPs indicated better penetration into a deeper tumor spheroid region than the PEG-LPC/siRNA NPs. The next study highlights of the combination of gene delivery with photothermal therapy. Chen et al.169 load siRNA on a NIR-to-UV up-conversion spatiotemporally controlled, gene silencing, nanocarrier. They designed azobenzene (Azo)-tagged siRNAs, loaded onto NaYF4:Yb/Tm/Er (sodium yttrium fluoride/thulium/erbium or-lanthanide-based up-conversion nanoparticles (UCNPs)) and functionalized with β-cyclodextrin (CD). The researchers also decorated the UCNP-siRNA complexes with PEG (UCNP-(CD/Azo)-siRNA/PEG NPs), targeting ligands (EGFR-specific GE11 peptide), acid-activated cell-penetrating peptides (TH peptide), and imaging probes (Cy5 fluorophore). The use of UCNP-(CD/Azo)-siRNA/PEG NPs with both GE11 and TH peptides showed significant cellular uptake and endosomal/lysosomal escape abilities. In both 2D and 3D spheroids, NIR-controlled spatiotemporal knockdown of GFP expression occurred.
As for drug-PTT combinatorial effect, associate PTT with gene therapy might be beneficial to enhanced cancer therapeutic effects and minimal side effects. Well investigated in 2D and in vivo170–172 it is still missing state of art in 3D system and present an opportunity of investigation novelty.
• Photodynamic therapy (PDT) combination approach
PDT uses light irradiation to kill tumors by generating reactive oxygen molecules. Three parameters are required for PDT to result in cell death: an excitation light, a photosensitizer (or light-sensitive drug), and the generation of reactive oxygen species (ROS). Under the appropriate wavelength, the photosensitizer's energy level increases and is transmitted to oxygen to generate ROS in the biological system (type II photosensitizer) or in direct interaction with a specific biomolecule to generate ROS (type I photosensitizer). The photosensitizer may need to be metabolized to become active (ex.: pro-drugs). This ROS generation and reactivity disturb the normal functions of tumor cells, resulting in DNA, lysosomal, and/or mitochondria membrane damage and death by necrosis, apoptosis, or both.174 Thus, PDT is a promising cancer treatment with low systemic toxicity (the pro-drugs are inactive without laser irradiation), limited side effects, and no considerable drug resistance.173 However, limitations still hamper its development, including light penetration and its highly localized action. NIR can be used to optimize tissue penetration, but long exposure to NIR light can harm normal tissue. Additionally, a higher photosensitizer concentration is needed at the cancer site, but because many photosensitizers are water insoluble and cannot travel within the bloodstream, this cannot always be achieved.174
To overcome some of these limitations, photosensitizer delivery by NPs has been investigated in 2D and in vivo,175 and 3D culture investigations have begun recently. In early 2018, Mohammad et al.21 reviewed the use of 3D cancer models with PDT and nano-delivery systems. They mentioned the work of Lee et al.,176 who worked to overcome the issue of hydrophobic photosensitizer delivery by engineering lipophilic/hydrophilic NPs, specifically fusogenic liposomes (MFLs). MFLs or exosomes loaded with two fluorescent dyes were studied in terms of penetration efficiency in comparison with conventional cationic liposomes in two different cancer cell lines (HeLa and CT26) on 3D spheroids and in vivo tumor models. They showed that MFLs can be delivered to the outer layer of the spheroids but also to the core by successive rounds of cellular uptake inducing membrane vesicles production, rather than spheroid penetration. They also studied the toxicity induced by MFLs loaded with zinc phthalocyanine under 600 nm laser excitation and demonstrated, comparatively, that only MFL-type NPs treated with a laser were able to reduce the spheroid integrity by more than 50%. Lipophilic NPs for photosensitizer delivery were also investigated by Hung et al.,177 who used EtNBS (5-ethylamino-9-diethylaminobenzo[a]phenothiazinium chloride) encapsulated in poly(lactic-co-glycolic acid) to create the NPs (PLGA-EtNBS). The release of the photosensitizer before laser exposure can induce non-specific cell death through other mechanisms besides ROS generation and is an undesired side effect called “dark toxicity”. The PLGA encapsulation allowed for the photosensitizer to only be released in the presence of laser irradiation after deep penetration into the acidic/hypoxic core in a 3D spheroid model. Nano-photosensitizer performance and characterization in 3D culture were also investigated by Hinger et al.178 with two different sizes of lipodots (50 and 120 nm), carrying m-tetrahydroxyphenylchlorin (mTHPC) in a 3D tongue carcinoma model. mTHPC encapsulated into 50 nm particles showed higher performance in cancer spheroids to induce disorganization of the volume, structure, and apoptotic markers, compared to free mTHPC. Concerning the functionalization/surface chemistry and composition, three other nanoemulsion formulations of mTHPC named Foscan® (standard), Foslip® (non-PEGylated liposome), Fospeg® (PEGylated liposome) were investigated on avascular 3D cervical cancer model by Gaio et al.179 TEM analysis showed evidence of photodamage in the spheroids, even inside the core. Interestingly, the three formulations have similar cytotoxicity after laser exposure but the liposomal forms possess a better photosensitizer delivery with better cellular uptake, a higher impact on the integrity of the spheroids (disorganization of the structure) and a significant reduced “dark toxicity” than the standard formulation Foscan®.
PDT was also investigated in a 3D culture with a dynamic flow system by Yang et al.180 They designed a microfluidic device for breast cancer with circulation-like dynamic medium flow, using human breast cancer cells and primary adipose-derived stromal cells to control the 3D microenvironment for breast cancer tissue formation. Using this system, they delivered drug supplements via Au NPs and monitored tissue progression in real-time. Active targeting by nanoparticles NPs for PDT and therapeutic monitoring was tested by Jin et al.181 in a 3D model of TNBC model with MDA-MB-231 cell line, which lacks estrogen receptors, progesterone receptors, and HER2. The researchers synthesized cRGD-MEH-PPV composed of cyclic RGD (cRGD) peptide conjugated on polymeric nanoparticles (DSPE-PEG-maleimide) encapsulating the photosensitizer, poly[2-methoxy-5-(2-ethyl-hexyloxy)-1,4-phenylenevinylene]. This type of conjugation and functionalization was used to avoid aggregation, responsible for the decrease/reduction of ROS generation. 3D system was made on microwell arrays and cultured to reach a diameter of 600 μm before to be incubated with cRGD-MEH-PPV nanoparticles then light irradiated (∼100 mW cm−2). Compared to the 2D model experimentation, showing 100% cell death after 2 min irradiation, 3D TNBC showed 24% cancer death. Adjustment of the time of irritation, indicate consequent cells death, >70%, after 30 min light exposure. Using the same targeting, same polymeric nanoparticle and the same 3D cancer model, they also tested the performance of another photosensitizer, called 2-(2,6-bis((E)-4-(phenyl(40-(1,2,2-triphenylvinyl)-[1,10-biphenyl]-4-yl)amino)styryl)-4H-pyran-4-ylidene) malononitrile or TTD, to by-pass aggregation ROS reduction. This time, aggregation was used to enhance the PDT performance using up-conversion nanoparticles (UCNPs) with aggregation-induced emission (AIEgen). UCNP@TTD-cRGD NPs incubated with the 3D model and irritated for 30 min showed >60% cell viability. Apoptosis/necrosis followed 12, 24 and 48 h after irradiation showed respective rates of 20.5%/12.5%, 54.5%/24.2%, and 12.2%/78.9%.182 This last study is very interesting because compared to the first one trying to avoid aggregation, using the same kind of nanoparticle but modifying the photosensitizer, researchers were able to obtain similar result in terms of toxicity. The second study develops here adaptation of the therapy to biological features that cannot be avoided especially in for 3D model culture, where penetration, accumulation and especially aggregation under normal physiological conditions might occur.
As shown in 2D and in vivo studies,170–172 the surface area and optical properties of the NPs make them suitable for combining their therapeutic: drug delivery, gene therapy, PDT with PTT. However, state of art of these combinatorial effects are in 3D system still at theirs early beginning.
NP penetration is an important parameter for enhanced drug release as their targeting effect is a dominant factor for gene delivery and the biological light absorption hamper optimal PDT. In another hand, hyperthermia and PTT can regulate the immune function by activating immune effector cells, increase secretion of cytokines and upregulate the expression of heat shock proteins.183 Since all these parameters might depend of the tumor microenvironment, it is important to evaluate them under 3D culture systems. In our opinion, more thorough studies are needed to investigate how PTT can influence the limiting parameters of the other therapies, especially in function of the 3D model used (type of cells, matrix, thickness and stiffness of the general 3D structures, type of fabrication technique, etc.). Optimizing treatments through NPs allowing combination of therapies might avoid recurrence and metastasis of cancers.
Evaluation of nanoparticle interaction in more complex, specific, and multi-interactive 3D cancer models: an opportunity for PTT advancement
When NPs have been analyzed for cancer therapies using 2D models, the results have typically been encouraging. However, when NPs are incubated with monolayer cell cultures, their interactions are hampered by the model's physical limitations. Likewise, in vivo, the biological barriers or ECM of the tumorous system drastically hamper NPs’ mobility, penetration, and diffusion. Additionally, 2D cultures are limited to studying tumor cells and the complex interactions with other cells (immune cells, stromal cells or cancer-associated cells, extracellular matrix, and protein over-expression). Today, we know that effective cancer therapies must overcome cancer cells’ adaptive systems that introduce hypoxia, pH environment, avasculature or exacerbated angiogenesis, and/or drug resistance. With our increasing knowledge about these considerations, cancer therapies need to be able to interact with the entire tumor system. Though still a new concept, 3D cancer models can be designed to mimic part of the tumor microenvironment, including certain mechanisms of action, or even to discover mechanisms that have not been observed yet. These points need to be considered in future nanoparticle designs for PPT.ECM interaction and reorganization
In vivo, the ECM is a primary element of connective tissue; in solid tumors (breast, prostate, pancreas, liver), the cancer cells grow and multiply inside a structural ECM, which can be overproduced by the connective tissue around it (e.g. stroma). 3D models that incorporate ECM materials—such as collagen, laminin, fibronectin, and other beta-actin—provide a structural cellular organization that mimics the tumor microenvironment, which can reduce and/or inhibit therapeutic capabilities and NP penetration, modify cell signaling, and generate hypoxia or gradient in nutritional supplementation. Lack of nutrition and/or hypoxia in cancer does not mean “death” but “adaptation,” allowing one system to have different phenotypic cells at different stages, including cancer progression, angiogenesis, metastasis, and resistance to therapy (Fig. 2A).184To mimic the ECM of a tumor system, researchers have created simplified models, generally using scaffolds, microbeads, or polymeric support. Charoen et al.185 studied the influence of the size of spheroids embedded inside collagen I matrix, in association with the matrix stiffness, on two cancer models, breast (MDA-MB-231) and bone (U-2 OS). They showed that an ECM surrounding a multicellular spheroid can impact the size of the final spheroids formed. Focusing on their developed 3D MDA-MB-231 model, the researchers compared PTX delivered as practiced in clinical settings with PTX delivered by an expandable nanoparticle, Pax-eNP, made of polymethylmethacrylate. After 24 hours of incubation, penetration in a 3D model was evaluated by fluorescence tracking of the rhodamine-labeled Pax-eNP. 3D and in vivo murine xenograft models showed beneficial results for the NP, while the 2D culture showed no significant difference between the conventional PTX nanoparticle and the expandable nanoparticle. In another study using collagen I, Le et al.186 studied the efficiency and cytotoxicity of NP-based drug delivery (5-FU) by tracking the penetration and diffusion of coumarin fluorescence-loaded NPs in HCT-116, 95-D, and U-87 cell lines, introducing the collagen as a scaffold during the formation of the spheroids to closely mimic the dimensional obstacle found in vivo. The study showed that micelle NPs of >100 nm in this matrix system had lower penetration than smaller molecules, such as free drug, and presented lower cytotoxicity.
Collagen is a dominant component of the ECM, but ECM's total composition is a mix of multiple proteins and polysaccharides. One of them, hyaluronic acid (HA), is found in the enriched tumor-associated stroma, such as those seen in pancreatic or prostate cancer.187 Xu et al.86 evaluated the potential of their engineered model—which included a hydrogel scaffold made of an HA derivative—for anti-cancer drug screening and delivery. They based the model on prostate cancer, with LNCaP prostate cancer cells embedded inside acrylate and thiolate-HA to form a cylindrical model with a height of 1.8 mm and diameter of 12 mm. After 7 days of 3D formation, the cells started to present a drug-resistant phenotype with high expression of MRP1 (multidrug resistance-associated protein 1 also called ABCC1) and lipoprotein receptor-related protein 1 markers, compared to these levels for the same cells in a 2D culture. Polymeric PEG-b-P(CL-ran-TSU) NPs with an average size of ∼50 nm were loaded with Dox, which acts as both a therapeutic agent and a fluorescent tracker. The Dox-NPs were able to penetrate entire spheroids, confirming penetration of the system. However, the drug was localized mainly inside the cytoplasm of cells within the 3D model, though it was found in the nucleus in 2D. Additionally, in the 3D model, a higher IC50 and a lower apoptotic level were observed for free drug treatment compared to the values seen in the 2D model, which can be explained by the drug resistance marker evaluated and by the fact that Dox is actively functional on proliferative and dividing cells (3D culture cells have, on average, decreased proliferative capacity compared to 2D); this property reduces the drug's potency. More surprisingly, the IC50 value for Dox-NPs was similar to that of the free Dox in 3D. Hydrophobic drugs with a low molecular weight diffused easily, suggesting that the cellular uptake occurred via a different pathway. This kind of scaffold mimicking HA-stromal expression might interact well with CD44 or HA-functionalized nanoparticles. However, to our knowledge, no research was done to characterize in-depth the interaction and penetration of NPs in such a 3D system.
Natural polymers can also mimic the thickness, porosity, and/or mechanical properties of the ECM. In 2013, Godugu et al.188 investigated the ability of AlgiMatrix™ (alginate) scaffolds to form a size dispersion of 3D lung cancer tumoroids in order to study the efficiency, penetration, and diffusion of polymeric NPs loaded with different drugs, such as docetaxel, Dox, cisplatin, gemcitabine, 5-fluorouracil, and camptothecin. The researchers showed that, on average, the IC50 of these nanosystems was under-evaluated in 2D models compared to 3D (higher IC50 were found in 3D AlgiMatrix™ model); the phenomenon of drug resistance was also observed inside the alginate-ECM matrix.
Thus, studies indicate that adding an extracellular component to a 3D model to embed cells or create a scaffold is a fast, repeatable, and controlled way to analyze how the components and properties of the ECM can affect drug delivery, efficiency, and resistance. However, we also know that some cells overproduce the ECM in response to inflammation or injury, making this system constantly evolving and non-static. In addition, it is rather rare to find a tumor composed of only one type of cell, and the extracellular matrix for cell–cell interaction, especially when the cells are completely different in nature, can be difficult to synthetically engineer. This complexity makes cell co-culture inside a 3D model highly interesting. While tumors in their natural state cannot be absolutely controlled and investigated, a co-cultured 3D model's complex micro-environment can indicate the limitations of NPs, forcing us to optimize their internal and surface chemistry. Studies have demonstrated that tumor cells grown in co-culture with endothelial cells have more aggressive properties when implanted in vivo, including increased metastatic capability, which suggests that obtaining knowledge of treatment efficacy against co-cultured spheroids may be significantly more clinically relevant when a 3D co-cultured model is used rather than monolayer cell cultures.189 Specific to nanomaterials, in 2015, Sethi et al.190 designed a complex 3D TNBC model made of tumor cells, endothelial cells, and fibroblasts as color-coded murine tumor tissue analogs (TTA) to characterize the impact of the microenvironment/milieu on TNBC treated with nanosized chemotherapy and radiation. Implantation of these 3D systems in immuno-compromised mice led to enhanced growth and aggressive metastasis; the in vivo tumors were used to test preferential endothelial tumor cell targeting via radiation-induced stromal enrichment of galectin-1.
The ECM should also be considered when designing NPs-based active targeting. Recently, it was shown that tumor ECMs can overexpress matrix metalloproteinase (MMP) markers. MMPs are enzymes that are responsible for protein degradation in the ECM. They can be categorized according to the class of proteins that they degrade—collagenases, gelatinases, stromelysins, matrilysins, etc.191 Because their upregulated expression plays an important role in cancer invasion and angiogenesis, and because of their enzymatic peptide activation/recognition, these markers can be used as active targeting agents for drug delivery. One of the major problems with excess ECM (around blood vessel or interstitial space) is its ability to shield the tumor site, resulting in blocked drug delivery or even trapped NPs within the ECM, never reaching the tumor cells. To counteract this shielding effect, 3D models are being explored that will help researchers answer the following questions: Can NPs be delivered by targeting MMPs to enhance enzymatic degradation of the extracellular matrix? Can we stimulate the production of MMPs by targeting specific cells with NPs to accelerate the degradation of the ECM? Can drug delivery be increased with degradable peptide structures and tested in a 3D cancer model in which the ECM shields the tumor cells?
In 2013, Gu et al.192 developed a low molecular weight protamine (ALMWP) loaded on PEG-co-PCL nanoparticles (poly(ethylene glycol)–poly(ε-caprolactone) block copolymer); this system was able to act as a drug carrier (here, PTX) and be recognized by the cells only in the presence of a matrix metalloproteinase (MMP) receptor (here, MMP2 and MMP9). This strategic synthesis of cell-penetrating peptides (CPP) is based on combining them with a sequence of polycationic CPP and an MMP-sensitive peptide linker. The MMP2/9 present in glioblastomas destroyed the linker, allowing the NPs to release the polycationic portion (ALMWP) and adhere to the cells. Testing in vitro C6 tumor glioma spheroids (with a highly similar expression of MMP 2 and 9 to real cancer tissue) showed that the ALMWP-NPs can penetrate the system, as evidenced by fluorescence tracking of their distribution and accumulation. MMP's enzymatic degradation led to the development of a synergistic drug release/active targeting strategy, in which the drug can be released from the NPs only when they reach the active enzyme site of the MMP, where the linker is then cut and the drug released inside the cells. Based on this strategy, in 2014, Kulkarni et al.193 designed multilayer nanosized vesicles functionalized with 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-choline (POPC) and cholesteryl-hemisuccinate to encapsulate the drug (gemcitabine) and anchor/activate the MMP9 site, as well as the synthesized reduction sensitive, PEGylated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine lipid (POPE–SS–PEG5000), to allow better blood circulating time and protection for the MMP9 linker (glutathione levels in cells will destroy the –SS– bonding to expose the MMP9 linker to the enzymatic site). The researchers utilized 3D pancreatic cancer spheroids to analyze the release of compounds from the targeted MMP9 carboxyfluorescein-encapsulated nanovesicles, showing that only lipid-functionalized NPs were internalized. Successful in vivo tests in mice followed (Fig. 10).
 | ||
| Fig. 10 (A) Schematic of the synthesis of nanovesicles with MMP9 substrate lipopeptides and reduction-sensitive POPE–SS–PEG, as well as their degradation via GSH (glutathione) and MMP action. (B) Pictures of the PANC-1 cell spheroids with fluorescent tracking of the released carboxyfluorescein, treated with (B) MMP9-responsive nanovesicles or nanovesicles without the MMP9-responsive lipopeptide (A). Figure and caption reproduced with permission.193 Copyright 2014, American Chemical Society. | ||
In conclusion, ECM-rich 3D systems can benefit NP-based PTT by allowing characterization of the following parameters:
(1) Interaction of NPs with the ECM, specifically diffusion and penetration, as a function of NP size, shape, and surface functionalization and characterization. NPs’ interactive effects—concentration gradient and aggregation state—may influence their heat conversion performance. It will also be interesting to see future studies elucidating if ECM influences only the diffusion of the NPs by modulating the interstitial space or by modifying the cellular activity of uptake, such as endocytosis.
(2) Impact of NP–PTT treatment not only on cancer cells but also on the ECM, chiefly, the impact of heat therapy on the ECM by evaluating protein denaturation and mechanical and physical properties
(3) Long-term influence of PTT on ECM-rich 3D spheroids: evaluation of the role of the ECM on mechanisms of cancer resistance, heat resistance, and protein heat shock production, recovery, or laser/heat shielding.
Metastasis, stem cells, and proliferation
Metastasis and cancer reappearance can occur in patients several years after they have undergone primary tumor ablation or therapy. Early dissemination of cancer cells from a primary site followed by a period of dormancy suggests that metastatic stem cells infiltrate the target organ in its functional state. The start of the dormant period and the later reactivation are controlled by intrinsic mechanisms and nursed by specialized extracellular matrix niches.60,194 Functional characterization of dormant tumor cells in a 3D model is key for overcoming the limitations of 2D models in order to better understand the clinical dormancy phenomenon.195 While such investigations in tissue cultures have obvious limitations, 3D culture systems made from basement membranes have been able to more accurately reflect in vivo ECM components. To our knowledge, the first investigation of a 3D dormant/activated cancer model was reported in 2017 by Grandhi et al.196 with a co-culture of bladder cancer cells and stromal cells (NIH3T3 murine fibroblasts). The design of the 3D model accounted for dormancy and reactivation by reconstituting the different stages (arrest in the G0/G1 phase of the cell cycle) inside a specific microenvironment that induces drug resistance, facilitated by an aminoglycoside-derived hydrogel, Amikagel. This hydrogel supported the proliferation, migration, and formation of metastasis-nodule sites, confirming that this model can be used to test novel drug discoveries. The researchers showed that NP (liposome)-mediated calcium delivery significantly sped up ER stress-mediated 3D spheroid cell death, but seeding treated spheroids inside weaker, adhesive Amikagels led to selective reactivation of a dormant sub-population of N-cadherin-deficient cells.New therapeutic systems are needed that can treat the primary site while also considering the metastatic site. Fitzgerald et al. studied metastatic prostate cancer cell lines derived from prostate metastasis in bone and compared its phenotype and genotype with another lymph node metastatic cell line, exposing these cells in a co-culture with human fetal osteoblast cell lines (hFOB 1.19 cells) on a collagen-nHA scaffold (representative of the in vivo bone microenvironment). Compared to a monoculture, the growth rate was slower in the co-culture model, which is most likely more similar to in vivo growth conditions. Additionally, the level of MMP9 in co-culture, 2D or 3D, increased significantly. This simulated 3D in vitro co-culture model of prostate cancer bone metastasis was then used to analyze the following gene delivery therapy: NP-mediated siRNA delivery and gene knockdown, cationic cyclodextrin, and commercially available Lipofectamine 2000.197
The interaction of NPs with a 3D breast cancer metastasis phenotype198 or 3D stem cells targeted to prevent migration107 has also been investigated. Interestingly, studies showed that the 3D culture can mimic the metastasis by itself, enabling it to treat cancer when it is implanted inside a host organ, where the tissue and function are different from the cancer organ of origin. Subia et al.199 found this to be true when they tested a potential nanotherapy in a 3D model made of breast cancer and bone cells to reconstitute metastatic breast cancer tissue in bone. Similar results were also seen by Alonso-Nocelo et al.,200 who simulated the ability of breast cancer to invade the lymph nodes by co-culturing lung cancer cells with lymphocytes in 3D scaffolds. They studied this system under flow by placing it in a bioreactor coupled to an automatic syringe pump, and the NP-PEGylated polyglutamic acid nanocapsules, fluorescence-tagged by DiD or DiD-DCX (lipophilic carbocyanine fluorescent dyes conjugated NPs), perfused under dynamic conditions to mimic the lymphatic flow.
In conclusion, studying NP-enhanced PTT in 3D systems allows metastasis and stem cell behavior to be analyzed, which aids the evaluation of therapeutic efficacy. The ability of 3D systems to be cultured for a long period of time and be more resistant to therapies might be an advantage for this type of study. 3D models may also help characterize PTT behavior in ECM-rich environments and evaluate the synergistic effects of PTT/NPs on metastasis. This can be done by identifying the impact of PTT on the potential of cancer cells to become metastatic/resistant, determining their influence on the dormancy-reactivation mechanism.
Brain cancer and the blood–brain-barrier
When designing 3D models with highly interactive micro-environments and protective mechanisms, the challenges of treating brain tumors should be considered. 3D models could simulate the natural biological barrier that hampers the efficiency of brain tumor therapies. The BBB is an active physical cellular interface between circulating blood and the brain. It is composed of endothelial cells that form the cerebral vessels and capillaries in the brain and it is one of the most important obstacles to drug delivery and therapeutic efficacy, limiting the distribution and accumulation of substances in the tumor tissue. Because of the BBB's complex selectivity, the study of chemotherapy distribution in brain tumors needs to be aided by a suitable biomimetic platform that can represent both the functions of the BBB—barrier, and carrier. The reproducibility and permeability of in vitro brain tumor models is a major challenge: triple co-culture (culture of brain endothelial cells in the presence of pericytes and astrocytes), transwell systems, and microfluidic models are available, but all possess different advantages and disadvantages.201 Because NP penetration can be highly variable from one system to another, a 3D in vitro system should mimic, as much as possible, the real effect of the BBB in order to help optimize NP surface chemistry modifications that improve cell discrimination, penetration, diffusion, and accumulation. Multicellular BBB spheroids with a core composed mainly of astrocytes, brain endothelial cells, and pericytes at the surface are a new kind of 3D model that takes into consideration in vivo concerns such as high protein expression, tight junction proteins, permeability with VEGF dependence, efflux pump activity, and receptor-mediated transcytosis.202Targeting is the key to overcoming the BBB and treating tumors while limiting the side effects of drugs as they penetrate, accumulate, and diffuse through the complex BBB-glioma system. Therefore, it is essential to develop 3D models that allow testing of targeted nanosystems in vitro. Zong et al.85 investigated the effect of multiple targeting by loading liposomal NPs with (1) transferrin T7 (HAIYPRH), a specific marker expressed on the surface of glioma and BBB cells, to enhance active targeting, and (2) a nonspecific marker, TAT peptide (AYGRKKRRQRRR), to enhance passive targeting through the BBB and increase accumulation inside tumor tissue. The goal was to improve the efficacy of Dox by enabling it to cross the BBB. The study had 3 phases: 2D to evaluate the concentration and toxicity; 3D to quantify penetration, diffusion, and accumulation (NPs tracked by couramin-6 fluorescence); and in vivo in mice to confirm the 2D and 3D results and assess general toxicity throughout the whole body.
Dual targeting enhancement was also investigated by Xin et al.,87 who used PTX, a drug with clinically relevant results to treat malignant glioma and metastasis that has highly limited access to the brain as a “free” drug but great potential when loaded on NPs.203–205 They studied the ability to increase the delivery of PEG-NPs by functionalizing the surface of the nanostructures with Agiopep-2, which can transport the NP in brain endothelial cells through low-density lipoprotein receptor-related protein-1-mediated transcytosis, as the protein is highly expressed on the BBB and in human glioma cells. Avascular 3D glioma spheroids were used in the preliminary study of this treatment to evaluate the efficacy of the NPs. Results showed higher penetration by ANG-PEG-NP after 24 hours of incubation (noted by fluorescence tracking of RBITC-labeled NPs) as well as better diffusion and accumulation than plain PEG–PCL NPs (PEG-NP). Paclitaxel ANG-PEG-NP incubated in spheroids also had higher toxicity and growth inhibition by relative spheroid volume compared to a treated control (∼60% diminution), Taxol (free drug), and PEG-NP (no targeted, no drug).
Combination of targeting with aptamer and peptide was investigated by Gao et al.90 for dual targeting of glioma both to treat the tumor and penetrate through the BBB. They functionalized methoxy PEG–poly(ε-caprolactone) NPs with a phage-displayed TGN peptide (TGNYKALHPHNG) and an AS1411 aptamer, which are specific targeting ligands of the BBB and cancer cells (bEnd.3 and C6 cells), respectively. In 3D tumor spheroid penetration studies with a C6 monoculture, the uptake of the aptamer-NP was highest, and the non-functionalized NP had the lowest uptake. However, the spheroids cultured with additional bEnd.3 monolayers significantly decreased the uptake of the aptamer-NP, making it lower than that of the peptide-NP. The peptide-NPs improved the movement of NPs thought the bEnd.3 monolayer but did not improve uptake and penetration, leading only to a surface distribution. These results showed that dual targeting with AS1411 and TGN allowed successful transport of the NPs across the biological barriers as well as uptake by the cancer spheroids. In vivo tracking and fluorescence imaging of the brain indicated that the aptamer-peptide-NPs had the best tumor spreading, as well as the highest and tumor-to-normal brain ratio discrimination. Loading docetaxel on the dual-targeting NP resulted in significantly improved survival for glioma-bearing mice. Here, the use of co-culture in 3D shows the ability of nanoparticles to be dual-targeting to overcome certain biological barriers. It highlights that it is important to actively target not only cancer cells but also some components of the microenvironment (here BBB) to achieve complete delivery and optimal therapy.
In 2017, studying a different biological pathway for penetration, Kou et al.101 loaded PTX on PLGA NPs to determine the benefit of the expression of the organic cation/carnitine transporter 2 (OCTN2) for mediating the b-oxidation of fatty acids in the mitochondria, which is present on the surface of both brain capillary endothelial cells and glioma cells. The researchers actively functionalized and conjugated the NPs with the targeting agent L-carnitine (LC) (attachment antibody/ligand optimized with PEG anchor of different lengths). The BBB endothelial cell line hCMEC/D3 and a glioma cell line were used in a 2D culture to verify the accumulation of NPs (tracked by fluorescence image of couramin-6 loaded on the PLGA) and in 3D spheroid models to confirm the efficacy of the paclitaxel-LC-PLGA treatment by tracking the morphology modification (relative size modification) of the spheroids: −250 μm diminution size in 9 days of therapy. In vivo results with mice followed the predicted results. The L-carnitine active targeting was also investigated for drug-nanodelivery by the same team in 3D colorectal cancer the same year.100
While penetration through the BBB is essential for optimal delivery, a malignant glioma can also develop its own barrier—the brain tumor barrier (BTB). The BTB is a kind of defective version of the BBB that has been invaded by tumor cells to divert its function and allow the progression, nutrition, and invasion of cancer.206 Miao et al.207 investigated how the coexistence of the BBB and BTB can hinder the accumulation of chemotherapeutic agents in a brain tumor. To improve diffusion through a potentially defective BBB, they investigated the efficacy of dual-targeting the BBB/BTB and glioma with two biomarkers: lactoferrin (Lf), whose receptor is overexpressed on brain endothelial cells and glioma cells, and the tumor-homing peptide tLyP-1, which can facilitate penetration in brain tissue through the NRP-1 internalization pathway. These biomarkers were co-functionalized on the surface of PEG-poly(lactic acid) NPs. In both BCEC and C6 cells (2D model), interaction was facilitated and a considerable increase in cytotoxicity was seen when the dual-targeting NPs were loaded with PTX. Deeper penetration was achieved by the Lf NPs in avascular 3D glioma spheroids after 4 hours of incubation. An in vivo comparative study in rats also showed a better concentration of the drug in blood circulation and better accumulation in the brain just 2 hours after injection of the dual-targeting NPs.
A 3D culture model that can mimic the BBB's function will be extremely advantageous for PPT and cancer therapies; in fact, it was shown that the permeability of the BBB can be disrupted under thermotherapy.208 Consequently, a model that mimics patient tumor configuration without extensive use of animal models might be essential for optimizing bimodal NPs for enhanced local treatment (PPT), drug delivery, and deeper tumor tissue treatment.
Conclusion, limitations, and future directions
Throughout this review, we highlight examples that demonstrate the superiority of 3D cell culture models over 2D cell cultures, particularly for analyzing nanoparticle functionalization and conjugation strategies, as well as drug delivery and cellular interaction and penetration/diffusion in tissues. The tunability of NPs makes them suitable to aid many cancer therapies, including drug, protein, and gene delivery; PTT enhancement; and even a combination of therapies. Studies have shown the relevance of nanomedicine in cancer applications using complex 3D models, allowing validation before pre-clinical (animals) or clinical (human) trials. These 3D culture models quantify and visualize the complex interactions of NPs and cancer, guiding biological and/or chemical adaptions to optimize them for the application at hand.Advanced experimentations are starting to use 3D models because they can simulate micro-environment constraints, enabling more effective design of novel nanoparticles and potential configurations for drug delivery, targeting, and tumor monitoring and imaging. For example, in the context of hypoxia, Nichols et al.209 highlighted the barrier to deep drug penetration and imaging caused by a lack of oxygen and our inability to evaluate this amount of hypoxia. Based on studying a multicellular 3D system, they designed a 3D ovarian cancer model and tested a novel dendrimer NP based on click-assembled oxygen sensing nanoconjugate, composed of a sequential click-based ligation of poly(amidoamine)-like subunits. Using NIR confocal phosphorescence microscopy, the researchers were able to penetrate ∼100 μm into the spheroids, correlating NP sensitivity to oxygen changes throughout the nodule to support more direct calibration and quantitative oxygen mapping in the cellular environment. 3D cultures also enable us to explore how NPs can help discover new cancer mechanisms and treat and prevent the disease. In early 2018, Westmeier et al.32 investigated a very complex gastric 3D model by treating H. pylori with different NPs then exposing them to a 3D gastric cancer cell, aiming to determine if NP attachment on bacteria can disturb their pathobiological effect and, consequently, their ability to interact with cancer cells. Results showed that the interaction of NPs with bacteria did not modify their attachment and interaction with cancer cells—without the need for a large number of animal experiments.
PTT has shown impressive clinical results for cancer therapy, and NPs enhance its targeting, toxicity, and efficiency. Based on the current research and clinical applications, the basic criteria for the NPs selection for PTT in 3D culture will be (1) absorption capacity in the NIR regions (700–1350 nm); (2) less than 100 nm in size, which allows for maximum absorption by tissue; (3) enhanced absorption cross-section, and (4) good biocompatibility as well as low toxicity.56 As discussed in this review, evaluating NPs in 2D cultures can lead to misunderstandings and false results, such as underestimated nanoparticle concentration, time of laser exposure, or even an NP's light-to-heat conversion ability. The study of NPs for PTT in 3D models is becoming important because it allows researchers to do the following:
(1) Determine the corresponding IC50 or NP concentration inside a multidirectional system that can be modulated in size, density, and composition
(2) Analyze NP retention and penetration inside a system to determine NP surface chemistry, size, or shape modifications that might be needed in a real microenvironment to improve tumor-NP compatibility without hampering their plasmonic properties
(3) Evaluate the potential of therapies in a system that mimics the microenvironment, including an external ECM (collagen, chitosan, alginate, other potential cells (stromal, immune))
(4) Combine PTT with drug delivery or PDT and evaluate the real contribution of each
(5) Possibly reduce the amount of expensive in vivo experimentation needed to determine key parameters of PTT, such as the time of laser exposure, power density, and wavelength conversion and nanoparticle specifications such as efficient concentration and “dark-toxicity”
(6) Allow faster discrimination of NPs suitable for PTT by providing more complex evaluations than just the study of the elevation of temperature in a solution in an in vitro model that is very close to the final in vivo application
(7) Determine the impact of PTT on cancer cells and normal cells with a co-culture of cancer cells/normal cells and design targeting NPs that can discriminate and reduce therapy's impact on the normal cells
(8) Evaluate the impact of PTT and immunotherapy, as indicated by the evolution of nanoparticle targeting the TAMs by delivery to macrophages for PTT ablation. It can also help investigate the reaction of NPs combined with PTT and other therapeutic techniques to mimic long-term curative effects with induced anti-tumor immune response.
It is obvious, then, those 3D cultures are an advantageous model for in vitro cancer studies, though it must be acknowledged that the lack of vasculature in 3D models remains a major need. However, in solid tumor growth, there are many regions that are nearly devoid of vasculature, and these partial volumes are where therapy such as nanomedicine needs to be designed to penetrate. The current, non-vascularized 3D spheroid model is generally highly relevant for this therapeutic hurdle. Even with the current lack of vascularization, the micro-engineering field is evolving to mimic and image vasculature genesis in 3D210 (as in heart bioprinting), which may indicate the possibility of mimicking angiogenesis in cancer and the BBB in brain cancer.
There are major potential benefits to widespread 3D model use. To improve the incorporation of NPs in cancer therapy, we need to study their penetration, diffusion, and retention in systems that more closely simulate in vivo architectures. Additionally, the ability of 3D models to reconstitute different phases of cancer, progression, hypoxia, resistance, metastasis, and stem cell metastasis, such as breast/bone and prostate/bone, makes them suitable to test adaptive therapies for primary and metastatic sites. Consequently, 3D cultures can help researchers design and functionalize NPs to be more adaptive to the real cancer environment and metastases.
3D cultures not only enable the study of more complex systems, but they also support the design of more complex nanoparticles. In the future, NPs should be more interactive with the tumor microenvironment to up/down regulate the ECM and/or the immune system. In addition, if nanoparticles could modulate some of their properties, such as size or surface charge, in function with their environment, this would enable them to flow in avascular systems in different cellular systems or ECM, allowing higher penetration/diffusion in 3D structures for optimal PTT. More and more studies are designing NPs (especially polymeric ones) that modify or adapt their shape in function with their environment to increase cellular delivery. For example, Takechi-Haraya et al.84 used atomic force microscopy to investigate if NP surface or bending membrane rigidity (such as liposomes) can impact its interaction with a 3D culture of cervix cells. Results indicated that optimal penetration occurred with the NP whose membrane bending modulus (Kc) reached ≥3.3 × 10−19 J, followed by saturation or plateau of the penetration at higher Kcs. This data contradicted the study's theoretical prediction that they would see more efficient endocytic cellular uptake if the liposomal bending modulus to cellular membrane bending modulus ratio increased. The actual results indicated that spheroid penetration by the liposomes is influenced not just by endocytosis but by other cell parameters as well.
3D models can also help us develop and analyze NP-enhanced multi-imaging and multi-therapeutics methods, lead to a better understanding of the holistic impact of each of them. Because 3D models are denser and more compact than 2D cultures, they can aid in the discovery of new analytical techniques and imaging methods, with or without the use of NPs. Finally, 3D cultures could both reduce the cost of research and expand the research capabilities of laboratories that do not have easy access to in vivo units. This type of cell culture may be incredibly valuable to improve the design of personalized, patient-specific therapeutic approaches, as well as to test the efficacy of targeted therapy and, in the near future, of custom-made therapeutic NPs ex vivo.
However, the use of 3D cultures with NPs is still a relatively immature field. 3D multicellular spheroid tumor models made of multicellular entities with different metabolic and proliferative states need to be further understood in order to increase their impact and relevance. The newness of this field makes it extremely difficult to compare between studies, especially considering the variations in spheroid preparation techniques, size, shape (index of sphericity), cell nature, time of formation, and exposure times with NPs of various sizes and compositions. For example, it has been reported that the size and shape of spheroids may be the cause of the variability observed in studies and can make them respond differently to the same treatment.211 Homogeneous spheroids may provide more accurate results with commercially available cytotoxicity tests. However, this is not always true—for example, liquid overlay in a 96-well plate can form one single spheroid or multi-spheres. This possibility might not be mentioned in the protocol and method descriptions of publications, and only a picture allows the reader to note the difference. In that case, can we consider these two approaches similar, and can we still consider “spheroids” individual if multiple spheres are created the same way? Furthermore, which model is the most suitable to represent a real tumorous system? Finally, what is the impact on the data (penetration, drug efficiency, functionalization of NPs) when in a well of the same size, NPs may have been incubated with a single spheroid or with multi-spheres?
Additionally, the terminology of the 3D model field is not yet consistent. For example, the terms “organoid,” “spheroid,” and “tumoroid” are difficult to use correctly because no clear definitions have been established—organoids can be defined as a more complex and accurate structure than spheroids due to the natural matrix components and/or complexity of its ECM or as the combination of two or more cells, such as cancer cells cultured with mesenchymal cells and/or stem cells.18 Consequently, some papers use the term organoids to define spheroids or the term spheroids for a 3D cluster formed in a scaffold. Some research uses organoids because they are using one type of cancer cells from an isolated biopsied primary tumor, while other research calls a 3D sphere composed of 2 to 3 different cell lines a spheroid. To provide crucial clarity on these terms, publishers and reviewers must begin asking for better terminology and definitions of the 3D culture model.
Another consistency issue relates to the procedures used to make spheroids—they need to be presented in detail. For example, stating that an ultra-low attachment well plate was used can be misleading: for some spheroids, a round-bottom Corning 4515 well plate is considered an ultra-low attachment plate when it includes a hydrogel with ultra-low properties. However, publications do not always mention if they used the ultra-low attachment plate from Corning without the hydrogel (category liquid overlay on low attachment plate) or with it (liquid overlay on a gel). Can we assume that NP interaction—influenced by charge, size, functionalization—will be the same with a spheroid generated on a liquid overlay on an ultra-low attachment as it is with one generated on a liquid overlay with gel? Also, the inability to routinely image/detect NPs in 3D other than by fluorescence makes it difficult to confirm the accuracy of penetration and diffusion data, as these results are based on unreliable variables, like the indirect fluorescent signal. Better evidence and protocols for characterization of 3D culture studies, such as MRI, X-ray, CT scan, photoacoutic and photothermal microscopies, and Raman spectroscopy, are critically needed. It is imperative to cross-link these techniques to confirm and identify the limitations of each (ability of signal penetration in bio-tissue), especially when spheroids with diameters over 400 μm are used. It is also important to mention that when fluorescence is the only way to track the NPs, data might lack accuracy. Nothing can confirm or contradict whether the fluorescence is released solely from the nanoparticles, especially in the case of drug-release models. Thus, combining NP imaging with the techniques mentioned above is critically needed.
Finally, due to the complexity of the organ-on-a-chip development, the number of publications using microfluidics and bioprinting to screen nanoparticles in 3D cultures is very low.212 However, in the future, microfluidic chip-mimicking 3D systems might allow researchers to overcome some 3D limitations by mimicking vasculature, microtissue functionalities, and cell reactions and functionalities to biophysical stimuli.213 Flow, accumulation, and retention of NPs in such systems might indirectly impact the efficiency of PTT therapy, and the evolution of 3D cultures for these dynamic systems could help improve their design and development, as well as define and support adaptive photothermal parameters such as exposure time, laser wavelength, and power.
Conflicts of interest
The authors report no conflicts of interest.Acknowledgements
We are very grateful to Emily Davis and Dr Daniel Casciano for the review and assistance with the technical editing of this manuscript. We acknowledge partial support from the Center for Advanced Surface Engineering, under the National Science Foundation Grant No. IIA-1457888 and the Arkansas EPSCoR Program, ASSET III.Notes and references
- L. Zou, H. Wang, B. He, L. Zeng, T. Tan, H. Cao, X. He, Z. Zhang, S. Guo and Y. Li, Theranostics, 2016, 6, 762–772 CrossRef CAS.
- A. M. Pekkanen, M. R. DeWitt and M. N. Rylander, J. Biomed. Nanotechnol., 2014, 10, 1677–1712 CrossRef CAS PubMed.
- S. Govindaraju and K. Yun, 3 Biotech, 2018, 8, 113 CrossRef PubMed.
- D. Jaque, L. Martinez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martin Rodriguez and J. Garcia Sole, Nanoscale, 2014, 6, 9494–9530 RSC.
- H. Yan, C. Teh, S. Sreejith, L. Zhu, A. Kwok, W. Fang, X. Ma, K. T. Nguyen, V. Korzh and Y. Zhao, Angew. Chem., Int. Ed., 2012, 51, 8373–8377 CrossRef CAS.
- Q. Chen, C. Wang, L. Cheng, W. He, Z. Cheng and Z. Liu, Biomaterials, 2014, 35, 2915–2923 CrossRef CAS.
- R. Vankayala and K. C. Hwang, Adv. Mater., 2018, 30, e1706320 CrossRef PubMed.
- C. Yue, P. Liu, M. Zheng, P. Zhao, Y. Wang, Y. Ma and L. J. B. Cai, Biomaterials, 2013, 34, 6853–6861 CrossRef CAS PubMed.
- Y. Hu, S. Mignani, J.-P. Majoral, M. Shen and X. Shi, Chem. Soc. Rev., 2018, 47, 1874–1900 RSC.
- D. A. Nedosekin, T. Fahmi, Z. A. Nima, J. Nolan, C. Cai, M. Sarimollaoglu, E. Dervishi, A. Basnakian, A. S. Biris and V. P. Zharov, Photoacoustics, 2017, 6, 16–25 CrossRef.
- E. Darrigues, Z. A. Nima, W. Majeed, K. B. Vang-Dings, V. Dantuluri, A. R. Biris, V. P. Zharov, R. J. Griffin and A. S. Biris, Drug Metab. Rev., 2017, 49, 212–252 CrossRef CAS.
- E. Darrigues, V. Dantuluri, Z. A. Nima, K. B. Vang-Dings, R. J. Griffin, A. R. Biris, A. Ghosh and A. S. Biris, Drug Metab. Rev., 2017, 49, 253–283 CrossRef CAS.
- Y. Liu, Z. Chang, H. Yuan, A. M. Fales and T. Vo-Dinh, Nanoscale, 2013, 5, 12126–12131 RSC.
- L. Pompili, M. Porru, C. Caruso, A. Biroccio and C. Leonetti, J. Exp. Clin. Cancer Res., 2016, 35, 189 CrossRef.
- Y. Fang and R. M. Eglen, SLAS Discovery, 2017, 22, 456–472 CAS.
- M. W. Pickup, J. K. Mouw and V. M. Weaver, EMBO Rep., 2014, 15, 1243–1253 CrossRef CAS PubMed.
- J. Hoarau-Véchot, A. Rafii, C. Touboul and J. Pasquier, Int. J. Mol. Sci., 2018, 19, 181 CrossRef PubMed.
- S. Alhaque, M. Themis and H. Rashidi, Philos. Trans. R. Soc., B, 2018, 373, 20170216 CrossRef.
- M. C. Cox, L. M. Reese, L. R. Bickford and S. S. Verbridge, ACS Biomater. Sci. Eng., 2015, 1, 877–894 CrossRef CAS.
- D. T. Leong and K. W. Ng, Adv. Drug Delivery Rev., 2014, 79, 95–106 CrossRef.
- L. Mohammad-Hadi, A. J. MacRobert, M. Loizidou and E. Yaghini, Nanoscale, 2018, 10, 1570–1581 RSC.
- H. Lu and M. H. J. S. Stenzel, Small, 2018, 14, 1702858 CrossRef.
- B.-W. Huang and J.-Q. Gao, J. Controlled Release, 2017, 270, 248–259 Search PubMed.
- S. Sheng, M. M. Bernardo, S. H. Dzinic, K. Chen, E. I. Heath and W. A. Sakr, Cancer Metastasis Rev., 2018, 37, 655–663 CrossRef.
- F. Pampaloni, E. G. Reynaud and E. H. Stelzer, Nat. Rev. Mol. Cell Biol., 2007, 8, 839–845 CrossRef CAS.
- R. Edmondson, J. J. Broglie, A. F. Adcock and L. Yang, Assay Drug Dev. Technol., 2014, 12, 207–218 CrossRef CAS.
- Y. Imamura, T. Mukohara, Y. Shimono, Y. Funakoshi, N. Chayahara, M. Toyoda, N. Kiyota, S. Takao, S. Kono, T. Nakatsura and H. Minami, Oncol. Rep., 2015, 33, 1837–1843 CrossRef CAS.
- S. L. Chia, C. Y. Tay, M. I. Setyawati and D. T. Leong, Small, 2015, 11, 702–712 CrossRef CAS.
- A. M. Privalova, S. V. Uglanova, N. R. Kuznetsova, N. L. Klyachko, Y. I. Golovin, V. V. Korenkov, E. L. Vodovozova and E. A. Markvicheva, J. Nanosci. Nanotechnol., 2015, 15, 4806–4814 CrossRef CAS.
- A. W. Du, H. Lu and M. H. Stenzel, Biomacromolecules, 2015, 16, 1470–1479 CrossRef CAS.
- G. Huang, M. Li, Q. Yang, Y. Li, H. Liu, H. Yang and F. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 1155–1166 CrossRef CAS PubMed.
- D. Westmeier, G. Posselt, A. Hahlbrock, S. Bartfeld, C. Vallet, C. Abfalter, D. Docter, S. K. Knauer, S. Wessler and R. H. Stauber, Nanoscale, 2018, 10, 1453–1463 RSC.
- G. Kaushik, M. P. Ponnusamy and S. K. Batra, Stem Cells, 2018, 36, 1329–1340 CrossRef.
- M. E. Katt, A. L. Placone, A. D. Wong, Z. S. Xu and P. C. Searson, Front. Bioeng. Biotechnol., 2016, 4, 12 Search PubMed.
- A. Fatehullah, S. H. Tan and N. Barker, Nat. Cell Biol., 2016, 18, 246 CrossRef.
- G. Huang, F. Li, X. Zhao, Y. Ma, Y. Li, M. Lin, G. Jin, T. J. Lu, G. M. Genin and F. Xu, Chem. Rev., 2017, 117, 12764–12850 CrossRef CAS.
- P. Kaur, M. L. Aliru, A. S. Chadha, A. Asea and S. Krishnan, Int. J. Hyperthermia, 2016, 32, 76–88 CrossRef CAS.
- A. Sohail, Z. Ahmad, O. A. Bég, S. Arshad and L. Sherin, Bull. Cancer, 2017, 104, 452–461 CrossRef PubMed.
- E. Schena, P. Saccomandi and Y. Fong, J. Funct. Biomater., 2017, 8, 19 CrossRef PubMed.
- C. Brace, IEEE Pulse, 2011, 2, 28–38 CAS.
- R. W. Y. Habash, R. Bansal, D. Krewski and H. T. Alhafid, Crit. Rev. Biomed. Eng., 2006, 34, 459–489 CrossRef.
- A. M. Smith, M. C. Mancini and S. Nie, Nat. Nanotechnol., 2009, 4, 710 CrossRef CAS PubMed.
- K. Welsher, Z. Liu, S. P. Sherlock, J. T. Robinson, Z. Chen, D. Daranciang and H. Dai, Nat. Nanotechnol., 2009, 4, 773 CrossRef CAS PubMed.
- E. Hemmer, A. Benayas, F. Légaré and F. Vetrone, Nanoscale Horiz., 2016, 1, 168–184 RSC.
- Y. Lyu, J. Li and K. Pu, Small Methods, 2019, 3, 1900553 CrossRef.
- R. P. Dings, M. L. Loren, Y. Zhang, S. Mikkelson, K. H. Mayo, P. Corry and R. J. Griffin, Int. J. Hyperthermia, 2011, 27, 42–52 CrossRef PubMed.
- C. W. Song, H. Park and R. J. Griffin, Radiat. Res., 2001, 155, 515–528 CrossRef CAS.
- H. D. Suit and L. E. Gerweck, Cancer Res., 1979, 39, 2290–2298 CAS.
- J. Van der Zee, Ann. Oncol., 2002, 13, 1173–1184 CrossRef CAS.
- G. F. Baronzio, A. Gramaglia, A. Baronzio and I. Freitas, Hyperthermia in Cancer Treatment: A Primer, Springer, 2006, pp. 67–91 Search PubMed.
- B. M. Crawford, R. L. Shammas, A. M. Fales, D. A. Brown, S. T. Hollenbeck, T. Vo-Dinh and G. R. Devi, Int. J. Nanomed., 2017, 12, 6259–6272 CrossRef CAS PubMed.
- I. Freestone, N. Meeks, M. Sax and C. Higgitt, Gold Bull., 2007, 40, 270–277 CrossRef CAS.
- M. Faraday, Philos. Trans. R. Soc. London, 1857, 147, 145–181 CrossRef.
- G. Mie, Contributions to the optics of turbid media, particularly of colloidal metal solutions Transl. into ENGLISH from Ann. Phys.(Leipzig), v. 25, no. 3, 1908, p 377-445, 1976, 1, 377-445.
- R. S. Riley and E. S. Day, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2017, 9, e1449 Search PubMed.
- F. Jabeen, M. Najam-ul-Haq, R. Javeed, C. W. Huck and G. K. Bonn, Molecules, 2014, 19, 20580–20593 CrossRef PubMed.
- C. M. Hessel, V. P. Pattani, M. Rasch, M. G. Panthani, B. Koo, J. W. Tunnell and B. A. Korgel, Nano Lett., 2011, 11, 2560–2566 CrossRef CAS PubMed.
- K. Bian, X. Zhang, K. Liu, T. Yin, H. Liu, K. Niu, W. Cao and D. Gao, ACS Sustainable Chem. Eng., 2018, 6, 7574–7588 CrossRef CAS.
- C. Iodice, A. Cervadoro, A. Palange, J. Key, S. Aryal, M. R. Ramirez, C. Mattu, G. Ciardelli, B. E. O’Neill and P. Decuzzi, Opt. Laser Eng., 2016, 76, 74–81 CrossRef.
- J. C. Chang, Medicine, 2016, 95, S20–25 CrossRef CAS.
- D. P. Gonçalves, R. D. Rodriguez, T. Kurth, L. J. Bray, M. Binner, C. Jungnickel, F. N. Gür, S. W. Poser, T. L. Schmidt and D. R. Zahn, Acta Biomater., 2017, 58, 12–25 CrossRef PubMed.
- S. Liang, C. Li, C. Zhang, Y. Chen, L. Xu, C. Bao and X. Wang, Theranostics, 2015, 5, 970–984 CrossRef CAS.
- M. S. Mohamed, S. Veeranarayanan, A. C. Poulose, Y. Nagaoka, H. Minegishi, Y. Yoshida, T. Maekawa and D. S. Kumar, Biochim. Biophys. Acta, Gen. Subj., 2014, 1840, 1657–1669 CrossRef CAS PubMed.
- M. S. Mohamed, S. Veeranarayanan, A. Baliyan, A. C. Poulose, Y. Nagaoka, H. Minegishi, S. Iwai, Y. Shimane, Y. Yoshida and T. Maekawa, Macromol. Biosci., 2014, 14, 1696–1711 CrossRef CAS.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn, 2013, pp. 395–463 Search PubMed.
- A. Li Volsi, C. Scialabba, V. Vetri, G. Cavallaro, M. Licciardi and G. Giammona, ACS Appl. Mater. Interfaces, 2017, 9, 14453–14469 CrossRef CAS PubMed.
- G. Chen, B. Ma, Y. Wang, R. Xie, C. Li, K. Dou and S. Gong, ACS Appl. Mater. Interfaces, 2017, 9, 41700–41711 CrossRef CAS PubMed.
- Z. Chen, X. Feng, C. J. Herting, V. A. Garcia, K. Nie, W. W. Pong, R. Rasmussen, B. Dwivedi, S. Seby and S. A. Wolf, Cancer Res., 2017, 77, 2266–2278 CrossRef CAS.
- J. M. Brown, L. Recht and S. Strober, Clin. Cancer Res., 2017, 23, 3241–3250 CrossRef.
- S. J. Madsen, S.-K. Baek, A. R. Makkouk, T. Krasieva and H. Hirschberg, Ann. Biomed. Eng., 2012, 40, 507–515 CrossRef PubMed.
- C. Christie, S. Madsen, Q. Peng and H. Hirschberg, J. Environ. Pathol., Toxicol. Oncol., 2017, 36, 229–235 CrossRef.
- L. Beqa, Z. Fan, A. K. Singh, D. Senapati and P. C. Ray, ACS Appl. Mater. Interfaces, 2011, 3, 3316–3324 CrossRef CAS.
- X. Bian, Z. L. Song, Y. Qian, W. Gao, Z. Q. Cheng, L. Chen, H. Liang, D. Ding, X. K. Nie, Z. Chen and W. Tan, Sci. Rep., 2014, 4, 6093 CrossRef CAS PubMed.
- U. Dembereldorj, S. Y. Choi, E. O. Ganbold, N. W. Song, D. Kim, J. Choo, S. Y. Lee, S. Kim and S. W. Joo, Photochem. Photobiol., 2014, 90, 659–666 CrossRef CAS.
- B. N. Eldridge, B. W. Bernish, C. D. Fahrenholtz and R. Singh, ACS Biomater. Sci. Eng., 2016, 2, 963–976 CrossRef CAS.
- R. A. Coss, Int. J. Hyperthermia, 2005, 21, 695–701 CrossRef CAS.
- J. W. Fisher, S. Sarkar, C. F. Buchanan, C. S. Szot, J. Whitney, H. C. Hatcher, S. V. Torti, C. G. Rylander and M. N. Rylander, Cancer Res., 2010, 70, 9855–9864 CrossRef CAS PubMed.
- P. Zhang, H. Huang, J. Huang, H. Chen, J. Wang, K. Qiu, D. Zhao, L. Ji and H. Chao, ACS Appl. Mater. Interfaces, 2015, 7, 23278–23290 CrossRef CAS.
- J. Liu, K. Liu, L. Feng, Z. Liu and L. Xu, Biomater. Sci., 2017, 5, 331–340 RSC.
- X. Song, Q. Chen and Z. J. N. R. Liu, Nano Res., 2015, 8, 340–354 CrossRef CAS.
- H. Li, H. Liu, T. Nie, Y. Chen, Z. Wang, H. Huang, L. Liu and Y. Chen, Biomaterials, 2018, 178, 620–629 CrossRef CAS.
- C. G. Alves, R. Lima-Sousa, D. de Melo-Diogo, R. O. Louro and I. J. Correia, Int. J. Pharm., 2018, 542, 164–175 CrossRef CAS.
- S. K. Rajendrakumar, K. Cherukula, H. J. Park, S. Uthaman, Y. Y. Jeong, B. I. Lee and I. K. Park, J. Controlled Release, 2018, 276, 72–83 CrossRef CAS.
- Y. Takechi-Haraya, Y. Goda and K. Sakai-Kato, Mol. Pharmaceutics, 2017, 14, 2158–2165 CrossRef CAS.
- T. Zong, L. Mei, H. Gao, K. Shi, J. Chen, Y. Wang, Q. Zhang, Y. Yang and Q. He, J. Pharm. Sci., 2014, 103, 3891–3901 CrossRef CAS PubMed.
- X. Xu, C. R. Sabanayagam, D. A. Harrington, M. C. Farach-Carson and X. Jia, Biomaterials, 2014, 35, 3319–3330 CrossRef CAS PubMed.
- H. Xin, X. Sha, X. Jiang, W. Zhang, L. Chen and X. Fang, Biomaterials, 2012, 33, 8167–8176 CrossRef CAS.
- B. Wang, L. Lv, Z. Wang, Y. Zhao, L. Wu, X. Fang, Q. Xu and H. Xin, Biomaterials, 2014, 35, 5897–5907 CrossRef CAS.
- H. Gao, J. Qian, Z. Yang, Z. Pang, Z. Xi, S. Cao, Y. Wang, S. Pan, S. Zhang and W. Wang, Biomaterials, 2012, 33, 6264–6272 CrossRef CAS.
- H. Gao, J. Qian, S. Cao, Z. Yang, Z. Pang, S. Pan, L. Fan, Z. Xi, X. Jiang and Q. Zhang, Biomaterials, 2012, 33, 5115–5123 CrossRef CAS PubMed.
- H. Gao, S. Zhang, Z. Yang, S. Cao, X. Jiang and Z. Pang, Int. J. Pharm., 2014, 466, 8–17 CrossRef CAS PubMed.
- H. Gao, Y. Xiong, S. Zhang, Z. Yang, S. Cao and X. Jiang, Mol. Pharmaceutics, 2014, 11, 1042–1052 CrossRef CAS PubMed.
- Y. Chi, S. Zhu, C. Wang, L. Zhou, L. Zhang, Z. Li and Y. Dai, J. Drug Targeting, 2016, 24, 224–232 CrossRef CAS PubMed.
- Y. Yang, A. Roy, Y. Zhao, E. Undzys and S.-D. Li, Bioconjugate Chem., 2017, 28, 1505–1518 CrossRef CAS.
- G. M. Zamora, F. Wang, C.-H. Sun, A. Trinidad, Y. J. Kwon, S. K. Cho, K. Berg, S. J. Madsen and H. Hirschberg, J. Biomed. Opt., 2014, 19, 105009 CrossRef.
- X. Wang, X. Zhen, J. Wang, J. Zhang, W. Wu and X. Jiang, Biomaterials, 2013, 34, 4667–4679 CrossRef CAS.
- X. Wang, H. Tang, C. Wang, J. Zhang, W. Wu and X. Jiang, Theranostics, 2016, 6, 1378–1392 CrossRef CAS.
- Y. Wang, Y. Xie, J. Li, Z.-H. Peng, Y. Sheinin, J. Zhou and D. Oupicky, ACS Nano, 2017, 11, 2227–2238 CrossRef CAS.
- L. B. Sims, M. K. Huss, H. B. Frieboes and J. M. Steinbach-Rankins, J. Nanobiotechnol., 2017, 15, 67 CrossRef.
- L. Kou, Q. Yao, S. Sivaprakasam, Q. Luo, Y. Sun, Q. Fu, Z. He, J. Sun and V. Ganapathy, Drug Delivery, 2017, 24, 1338–1349 CrossRef CAS.
- L. Kou, Y. Hou, Q. Yao, W. Guo, G. Wang, M. Wang, Q. Fu, Z. He, V. Ganapathy and J. Sun, Artif. Cells, Nanomed., Biotechnol., 2017, 1–12, DOI:10.1080/21691401.2017.1384385.
- H.-L. Xu, K.-L. Mao, C.-T. Lu, Z.-L. Fan, J.-J. Yang, J. Xu, P.-P. Chen, D.-L. ZhuGe, B.-X. Shen and B.-H. Jin, Biomaterials, 2016, 107, 44–60 CrossRef CAS.
- D. Chen, B. Li, S. Cai, P. Wang, S. Peng, Y. Sheng, Y. He, Y. Gu and H. Chen, Biomaterials, 2016, 100, 1–16 CrossRef CAS.
- E. Pavlovich, N. Volkova, E. Yakymchuk, O. Perepelitsyna, M. Sydorenko and A. Goltsev, Nanoscale Res. Lett., 2017, 12, 494 CrossRef.
- T. D. Rane and A. M. Armani, PLoS One, 2016, 11, e0167548 CrossRef PubMed.
- S. Yamaguchi, H. Kobayashi, T. Narita, K. Kanehira, S. Sonezaki, Y. Kubota, S. Terasaka and Y. Iwasaki, Photochem. Photobiol., 2010, 86, 964–971 CrossRef CAS.
- J. Wang, J. S. Lee, D. Kim and L. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 39971–39984 CrossRef CAS.
- E. Kim, W. B. Jeon, S. Kim and S.-K. Lee, J. Nanosci. Nanotechnol., 2014, 14, 3356–3365 CrossRef CAS.
- S. Pernal, V. M. Wu and V. Uskokovic, ACS Appl. Mater. Interfaces, 2017, 9, 39283–39302 CrossRef CAS.
- J. S. Basuki, H. T. Duong, A. Macmillan, R. B. Erlich, L. Esser, M. C. Akerfeldt, R. M. Whan, M. Kavallaris, C. Boyer and T. P. Davis, ACS Nano, 2013, 7, 10175–10189 CrossRef CAS.
- A. Hornung, M. Poettler, R. P. Friedrich, B. Weigel, S. Duerr, J. Zaloga, I. Cicha, C. Alexiou and C. Janko, Anticancer Res., 2016, 36, 3093–3101 CAS.
- M. Yu, S. Karmakar, J. Yang, H. Zhang, Y. Yang, P. Thorn and C. Yu, Chem. Commun., 2014, 50, 1527–1529 RSC.
- A. Kienzle, S. Kurch, J. Schlöder, C. Berges, R. Ose, J. Schupp, A. Tuettenberg, H. Weiss, J. Schultze and S. Winzen, Adv. Healthcare Mater., 2017, 6, 1700012 CrossRef.
- G. Jarockyte, D. Dapkute, V. Karabanovas, J. V. Daugmaudis, F. Ivanauskas and R. Rotomskis, Biochim. Biophys. Acta, Gen. Subj., 2017, 1862, 914–923 CrossRef.
- L. Przysiecka, M. Michalska, G. Nowaczyk, B. Peplinska, T. Jesionowski, R. Schneider and S. Jurga, Colloids Surf., B, 2016, 146, 9–18 CrossRef CAS.
- J. Whitney, M. DeWitt, B. M. Whited, W. Carswell, A. Simon, C. G. Rylander and M. N. Rylander, Nanotechnology, 2013, 24, 275102 CrossRef PubMed.
- P. V. Jena, Y. Shamay, J. Shah, D. Roxbury, N. Paknejad and D. A. Heller, Carbon, 2016, 97, 99–109 CrossRef CAS.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS.
- A. S. Mikhail, S. Eetezadi, S. N. Ekdawi, J. Stewart and C. Allen, Int. J. Pharm., 2014, 464, 168–177 CrossRef CAS.
- A. Arranja, A. G. Denkova, K. Morawska, G. Waton, S. Van Vlierberghe, P. Dubruel, F. Schosseler and E. Mendes, J. Controlled Release, 2016, 224, 126–135 CrossRef CAS.
- B. Kim, G. Han, B. J. Toley, C. K. Kim, V. M. Rotello and N. S. Forbes, Nat. Nanotechnol., 2010, 5, 465–472 CrossRef CAS.
- C. G. England, T. Priest, G. Zhang, X. Sun, D. N. Patel, L. R. McNally, V. van Berkel, A. M. Gobin and H. B. Frieboes, Int. J. Nanomed., 2013, 8, 3603–3617 Search PubMed.
- R. Agarwal, P. Jurney, M. Raythatha, V. Singh, S. Sreenivasan, L. Shi and K. Roy, Adv. Healthcare Mater., 2015, 4, 2269–2280 CrossRef CAS.
- J. Bugno, H.-J. Hsu, R. M. Pearson, H. Noh and S. Hong, Mol. Pharmaceutics, 2016, 13, 2155–2163 CrossRef CAS.
- D. L. Priwitaningrum, J.-B. G. Blondé, A. Sridhar, J. van Baarlen, W. E. Hennink, G. Storm, S. Le Gac and J. Prakash, J. Controlled Release, 2016, 244, 257–268 CrossRef CAS.
- C. Corbo, R. Molinaro, A. Parodi, N. E. Toledano Furman, F. Salvatore and E. Tasciotti, Nanomedicine, 2016, 11, 81–100 CrossRef CAS.
- K. Huang, R. Boerhan, C. Liu and G. Jiang, Mol. Pharmaceutics, 2017, 14, 4618–4627 CrossRef CAS.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- R. Nandigama, B. Upcin, B. H. Aktas, S. Ergün and E. Henke, Histochem. Cell Biol., 2018, 150, 631–648 CrossRef CAS PubMed.
- Y. Bi, F. Hao, G. Yan, L. Teng, R. J. Lee and J. Xie, Curr. Drug Metab., 2016, 17, 763–782 CrossRef CAS.
- R. Bazak, M. Houri, S. El Achy, S. Kamel and T. Refaat, J. Cancer Res. Clin. Oncol., 2015, 141, 769–784 CrossRef CAS PubMed.
- D. van den Brand, L. F. Massuger, R. Brock and W. P. Verdurmen, Bioconjugate Chem., 2017, 28, 846–856 CrossRef CAS.
- M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS PubMed.
- R. Ran, A. P. Middelberg and C.-X. Zhao, Colloids Surf., B, 2016, 148, 402–410 CrossRef CAS.
- Y. Wang, J. H. Bahng, Q. Che, J. Han and N. A. Kotov, ACS Nano, 2015, 9, 8231–8238 CrossRef CAS.
- L. B. Sims, L. T. Curtis, H. B. Frieboes and J. M. Steinbach-Rankins, J. Nanobiotechnol., 2016, 14, 33 CrossRef.
- M. Millard, S. Odde and N. Neamati, Theranostics, 2011, 1, 154–188 CrossRef CAS.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–715 CrossRef CAS PubMed.
- D. Ni, H. Ding, S. Liu, H. Yue, Y. Bao, Z. Wang, Z. Su, W. Wei and G. Ma, Small, 2015, 11, 2518–2526 CrossRef CAS PubMed.
- C. L. Waite and C. M. Roth, Bioconjugate Chem., 2009, 20, 1908–1916 CrossRef CAS.
- C. L. Waite and C. M. Roth, Biotechnol. Bioeng., 2011, 108, 2999–3008 CrossRef CAS PubMed.
- F. Wang, Y. Li, Y. Shen, A. Wang, S. Wang and T. Xie, Int. J. Mol. Sci., 2013, 14, 13447–13462 CrossRef PubMed.
- G. L. Zwicke, G. A. Mansoori and C. J. Jeffery, Nano Rev., 2012, 3, 18496 CrossRef CAS PubMed.
- Y. Shen, X. Li, D. Dong, B. Zhang, Y. Xue and P. Shang, Am. J. Cancer Res., 2018, 8, 916 CAS.
- Y. Ma, G. Ai, C. Zhang, M. Zhao, X. Dong, Z. Han, Z. Wang, M. Zhang, Y. Liu, W. Gao, S. Li and Y. Gu, Theranostics, 2017, 7, 1511–1523 CrossRef CAS.
- J. P. Landry, Y. Ke, G.-L. Yu and X. D. Zhu, J. Immunol. Methods, 2015, 417, 86–96 CrossRef CAS.
- H. Attarwala, J. Nat. Sci., Biol. Med., 2010, 1, 53–56 CrossRef PubMed.
- W. Rao, H. Wang, J. Han, S. Zhao, J. Dumbleton, P. Agarwal, W. Zhang, G. Zhao, J. Yu and D. L. Zynger, ACS Nano, 2015, 9, 5725–5740 CrossRef CAS PubMed.
- R. Tammi, K. Rilla, J. P. Pienimaki, D. K. MacCallum, M. Hogg, M. Luukkonen, V. C. Hascall and M. Tammi, J. Biol. Chem., 2001, 276, 35111–35122 CrossRef CAS PubMed.
- J. Lesley, V. C. Hascall, M. Tammi and R. Hyman, J. Biol. Chem., 2000, 275, 26967–26975 CAS.
- M. Takeda, S. Ogino, R. Umemoto, M. Sakakura, M. Kajiwara, K. N. Sugahara, H. Hayasaka, M. Miyasaka, H. Terasawa and I. Shimada, J. Biol. Chem., 2006, 281, 40089–40095 CrossRef CAS PubMed.
- M. H. El-Dakdouki, E. Puré and X. Huang, Nanoscale, 2013, 5, 3895–3903 RSC.
- M. H. El-Dakdouki, E. Puré and X. Huang, Nanoscale, 2013, 5, 3904–3911 RSC.
- P. B. Langevin and J. L. Atlee, Complications in Anesthesia, W.B. Saunders, Philadelphia, 2nd edn, 2007, ch. 30, pp. 110–118 DOI:10.1016/B978-1-4160-2215-2.50035-1.
- M. S. Lee, E. C. Dees and A. Z. Wang, Oncology, 2017, 31, 198–208 Search PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- V. Voliani, G. Signore, R. Nifosí, F. Ricci, S. Luin and F. Beltram, Recent Pat. Nanomed., 2012, 2, 34–44 CrossRef CAS.
- A. Kang, H. I. Seo, B. G. Chung and S.-H. Lee, Nanomedicine, 2015, 11, 1153–1161 CrossRef CAS.
- W. B. Shi, V. M. Le, C. H. Gu, Y. H. Zheng, M. D. Lang, Y. H. Lu and J. W. Liu, J. Pharm. Sci., 2014, 103, 1064–1074 CrossRef PubMed.
- M. Shen, Y.-Y. Xu, Y. Sun, B.-S. Han and Y.-R. Duan, ACS Appl. Mater. Interfaces, 2015, 7, 20530–20537 CrossRef CAS.
- X. Jiang, X. Sha, H. Xin, X. Xu, J. Gu, W. Xia, S. Chen, Y. Xie, L. Chen and Y. Chen, Biomaterials, 2013, 34, 2969–2979 CrossRef CAS.
- M. Millard, I. Yakavets, V. Zorin, A. Kulmukhamedova, S. Marchal and L. Bezdetnaya, Int. J. Nanomed., 2017, 12, 7993–8007 CrossRef CAS.
- S. Nimesh, in Gene therapy, ed. S. Nimesh, Woodhead Publishing, 2013, ch. 6, pp. 113–135 DOI:10.1533/9781908818645.113.
- Y.-J. Ren and Y. Zhang, Expert Opin. Biol. Ther., 2014, 14, 1581–1592 CrossRef CAS.
- S. Jain, K. Pathak and A. Vaidya, Int. J. Biol. Macromol., 2018, 116, 880–892 CrossRef CAS PubMed.
- J.-M. Williford, J. Wu, Y. Ren, M. M. Archang, K. W. Leong and H.-Q. Mao, Annu. Rev. Biomed. Eng., 2014, 16, 347–370 CrossRef CAS PubMed.
- H. Jung, Y. Shimatani, M. Hasan, K. Uno, S. Hama and K. Kogure, Int. J. Pharm., 2017, 516, 258–265 CrossRef CAS PubMed.
- L. Wei, X.-Y. Guo, T. Yang, M.-Z. Yu, D.-W. Chen and J.-C. Wang, Int. J. Pharm., 2016, 510, 394–405 CrossRef CAS PubMed.
- G. Chen, B. Ma, R. Xie, Y. Wang, K. Dou and S. Gong, J. Controlled Release, 2017, 282, 142–155 Search PubMed.
- J. Kim, J. Kim, C. Jeong and W. J. Kim, Adv. Drug Delivery Rev., 2016, 98, 99–112 CrossRef CAS PubMed.
- S. Huang, S. Duan, J. Wang, S. Bao, X. Qiu, C. Li, Y. Liu, L. Yan, Z. Zhang and Y. Hu, Adv. Funct. Mater., 2016, 26, 2532–2544 CrossRef CAS.
- L. Wang, J. Shi, H. Zhang, H. Li, Y. Gao, Z. Wang, H. Wang, L. Li, C. Zhang and C. Chen, Biomaterials, 2013, 34, 262–274 CrossRef CAS PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef.
- M. Sivasubramanian, Y. C. Chuang and L.-W. Lo, Molecules, 2019, 24, 520 CrossRef PubMed.
- J. Lee, J. Kim, M. Jeong, H. Lee, U. Goh, H. Kim, B. Kim and J.-H. Park, Nano Lett., 2015, 15, 2938–2944 CrossRef CAS.
- H. I. Hung, O. J. Klein, S. W. Peterson, S. R. Rokosh, S. Osseiran, N. H. Nowell and C. L. Evans, Sci. Rep., 2016, 6, 33234 CrossRef CAS PubMed.
- D. Hinger, F. Navarro, A. Käch, J.-S. Thomann, F. Mittler, A.-C. Couffin and C. Maake, J. Nanobiotechnol., 2016, 14, 68 CrossRef.
- E. Gaio, D. Scheglmann, E. Reddi and F. Moret, J. Photochem. Photobiol., B, 2016, 161, 244–252 CrossRef CAS PubMed.
- Y. Yang, X. Yang, J. Zou, C. Jia, Y. Hu, H. Du and H. Wang, Lab Chip, 2015, 15, 735–744 RSC.
- G. Jin, R. He, Q. Liu, Y. Dong, M. Lin, W. Li and F. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 10634–10646 CrossRef CAS.
- G. Jin, R. He, Q. Liu, M. Lin, Y. Dong, K. Li, B. Z. Tang, B. Liu and F. Xu, Theranostics, 2019, 9, 246–264 CrossRef CAS.
- X. Hou, Y. Tao, Y. Pang, X. Li, G. Jiang and Y. Liu, Int. J. Cancer, 2018, 143, 3050–3060 CrossRef CAS PubMed.
- B. Muz, P. de la Puente, F. Azab and A. K. Azab, Hypoxia, 2015, 3, 83–92 CrossRef PubMed.
- K. M. Charoen, B. Fallica, Y. L. Colson, M. H. Zaman and M. W. Grinstaff, Biomaterials, 2014, 35, 2264–2271 CrossRef CAS PubMed.
- V. M. Le, M. D. Lang, W. B. Shi and J. W. Liu, Artif. Cells, Nanomed., Biotechnol., 2016, 44, 540–544 CrossRef CAS PubMed.
- V. B. Lokeshwar, S. Mirza and A. Jordan, Advances in cancer research, Elsevier, 2014, vol. 123, pp. 35–65 Search PubMed.
- C. Godugu, A. R. Patel, U. Desai, T. Andey, A. Sams and M. Singh, PLoS One, 2013, 8, e53708 CrossRef CAS PubMed.
- M. Upreti, A. Jamshidi-Parsian, N. A. Koonce, J. S. Webber, S. K. Sharma, A. A. Asea, M. J. Mader and R. J. Griffin, J. Trans. Oncol., 2011, 4, 365–IN363 CrossRef PubMed.
- P. Sethi, A. Jyoti, E. P. Swindell, R. Chan, U. W. Langner, J. M. Feddock, R. Nagarajan, T. V. O’Halloran and M. Upreti, Nanomedicine, 2015, 11, 2013–2023 CrossRef CAS PubMed.
- A. Jablonska-Trypuc, M. Matejczyk and S. Rosochacki, J. Enzyme Inhib. Med. Chem., 2016, 31, 177–183 CrossRef CAS PubMed.
- G. Gu, H. Xia, Q. Hu, Z. Liu, M. Jiang, T. Kang, D. Miao, Y. Tu, Z. Pang and Q. Song, Biomaterials, 2013, 34, 196–208 CrossRef CAS PubMed.
- P. S. Kulkarni, M. K. Haldar, R. R. Nahire, P. Katti, A. H. Ambre, W. W. Muhonen, J. B. Shabb, S. K. Padi, R. K. Singh and P. P. Borowicz, Mol. Pharmaceutics, 2014, 11, 2390–2399 CrossRef CAS PubMed.
- J. J. Campbell, R. D. Hume and C. J. Watson, Mol. Pharmaceutics, 2014, 11, 1971–1981 CrossRef CAS PubMed.
- A. C. Yeh and S. Ramaswamy, Cancer Res., 2015, 75, 5014–5022 CrossRef CAS PubMed.
- T. S. P. Grandhi, T. Potta, R. Nitiyanandan, I. Deshpande and K. Rege, Biomaterials, 2017, 142, 171–185 CrossRef PubMed.
- K. A. Fitzgerald, J. Guo, R. M. Raftery, I. M. Castano, C. M. Curtin, M. Gooding, R. Darcy, F. J. O’Brien and C. M. O’Driscoll, Int. J. Pharm., 2016, 511, 1058–1069 CrossRef CAS PubMed.
- R. K. Delong, M. N. Hurst, S. Aryal and N. K. Inchun, Anticancer Res., 2016, 36, 2097–2103 CAS.
- B. Subia, T. Dey, S. Sharma and S. C. Kundu, ACS Appl. Mater. Interfaces, 2015, 7, 2269–2279 CrossRef CAS PubMed.
- M. Alonso-Nocelo, R. Abellan-Pose, A. Vidal, M. Abal, N. Csaba, M. J. Alonso, R. Lopez-Lopez and M. de la Fuente, J. Nanobiotechnol., 2016, 14, 51 CrossRef PubMed.
- I. Wilhelm and I. n. A. Krizbai, Mol. Pharmaceutics, 2014, 11, 1949–1963 CrossRef CAS.
- C.-F. Cho, J. M. Wolfe, C. M. Fadzen, D. Calligaris, K. Hornburg, E. A. Chiocca, N. Y. Agar, B. L. Pentelute and S. E. Lawler, Nat. Commun., 2017, 8, 15623 CrossRef CAS.
- J. Heimans, J. Vermorken, J. Wolbers, C. Eeltink, O. Meijer, M. Taphoorn and J. Beijnen, Ann. Oncol., 1994, 5, 951–953 CrossRef CAS.
- S. Fellner, B. Bauer, D. S. Miller, M. Schaffrik, M. Fankhänel, T. Spruß, G. Bernhardt, C. Graeff, L. Färber and H. Gschaidmeier, J. Clin. Invest., 2002, 110, 1309–1318 CrossRef CAS.
- E. Nance, C. Zhang, T.-Y. Shih, Q. Xu, B. S. Schuster and J. Hanes, ACS Nano, 2014, 8, 10655–10664 CrossRef CAS PubMed.
- D. R. Groothuis, Neuro-Oncology, 2000, 2, 45–59 CrossRef CAS PubMed.
- D. Miao, M. Jiang, Z. Liu, G. Gu, Q. Hu, T. Kang, Q. Song, L. Yao, W. Li and X. Gao, Mol. Pharmaceutics, 2013, 11, 90–101 CrossRef PubMed.
- A. Rodriguez, S. Tatter and W. Debinski, Pharmaceutics, 2015, 7, 175–187 CrossRef CAS.
- A. J. Nichols, E. Roussakis, O. J. Klein and C. L. Evans, Angew. Chem., Int. Ed., 2014, 53, 3671–3674 CrossRef CAS PubMed.
- G. Kaushik, D. A. Gil, E. Torr, E. S. Berge, C. Soref, P. Uhl, G. Fontana, J. Antosiewicz-Bourget, C. Edington and M. P. Schwartz, Adv. Healthcare Mater., 2019, 8, 1801186 CrossRef PubMed.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS PubMed.
- Q. Yang, Q. Lian and F. Xu, Biomicrofluidics, 2017, 11, 031301 CrossRef PubMed.
- B. Gao, Q. Yang, X. Zhao, G. Jin, Y. Ma and F. Xu, Trends Biotechnol., 2016, 34, 746–756 CrossRef CAS PubMed.
Footnote |
| † E. Darrigues, A. S. Biris and A. Rodriguez are the main authors of this paper. |
| This journal is © The Royal Society of Chemistry 2020 |