
Enhancing the chemotherapeutic efficacy of platinum prodrug nanoparticles and inhibiting cancer metastasis by targeting iron homeostasis†
Fang
Ding
 abc,
Lingpu
Zhang
bd,
Hao
Chen
bd,
Haiqin
Song
*ef,
Shiguo
Chen
*a and
Haihua
Xiao
*bc
abc,
Lingpu
Zhang
bd,
Hao
Chen
bd,
Haiqin
Song
*ef,
Shiguo
Chen
*a and
Haihua
Xiao
*bc
aNanshan District Key Lab for Biopolymers and Safety Evaluation, Shenzhen Key Laboratory of Polymer Science and Technology, Guangdong Research Center for Interfacial Engineering of Functional Materials, College of Materials Science and Engineering, Shenzhen University, Shenzhen, 518060, P. R. China. E-mail: csg@szu.edu.cn
bBeijing National Laboratory for Molecular Sciences, State Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China. E-mail: hhxiao@iccas.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
dCollege of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, P. R. China
eDepartment of General Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, P. R. China
fShanghai Minimally Invasive Surgery Center, Shanghai, 200025, P. R. China. E-mail: shq604shq@163.com
First published on 16th April 2020
Abstract
Iron plays important roles in tumor growth and metastasis, and iron depletion has become a new therapeutic strategy for iron overload cancers. Cisplatin is widely applied in the clinical therapy of various malignancies, but it has no inhibitory effect on cancer metastasis. In the present study, we found that the combination of cisplatin and iron chelator Dp44mT resulted in enhanced cell apoptosis as well as attenuated cell mobility and migration in vitro. Next, we developed a nano-carrier system to promote intracellular drug accumulation and reduce the side effects in cancer cells. Results showed that the as-synthesized nanoparticles (NPs) exhibited excellent antitumor efficiency when combined with Dp44mT. In breast tumor-bearing mice, the combination of the NPs and Dp44mT dramatically prevented orthotopic mammary tumor growth and inhibited metastasis via downregulation of VEGFα, MMP2 and Vimentin. In conclusion, as a versatile nano-platform for the combination of chemotherapy and iron chelators, the current design holds great potential for metastasis-inhibited cancer therapy.
New conceptsIron depletion is a new therapeutic strategy for iron overload cancers. Cisplatin can induce cell apoptosis via the formation of platinum–DNA adducts; it also disrupts cellular iron metabolism and eventually leads to cell death. In this study, we investigated the combination therapy using iron chelator Dp44mT with cisplatin prodrug nanoparticles (NPs). The as-synthesized NPs exhibited excellent antitumor efficiency in the presence of Dp44mT. Moreover, the combination of the NPs and Dp44mT simultaneously prevented the metastasis of 4T1 orthotopic mammary tumors in mice. As the first report about a versatile nano-platform for the combination of chemotherapy and iron chelators, our study holds great potential for metastasis-inhibited cancer therapy. |
1. Introduction
Iron is an essential element and plays important roles in cell growth, replication and energy metabolism.1,2 Neoplastic cells require more iron to maintain the active status of DNA synthesis and cell proliferation, and excessive iron leads to tumor initiation, growth and metastasis.3–5 Therefore, a variety of iron chelators have been explored as possible therapeutic interventions for various cancer treatments, since Fe deprivation may effectively prevent tumor growth.6–8 Currently, deferoxamine (DFO) is widely applied for clinical therapy of iron overload diseases, and exhibits remarkable antitumor activity.9,10 However, as a standard iron chelator, the poor membrane permeability and short half-life period of DFO seriously limited its clinical application.11 Moreover, DFO displayed no reduction in tumor growth and metastasis of breast cancer due to the fact that DFO could conversely enhance iron uptake in triple-negative breast cancer cells by activating the IL-6/PI3K/AKT pathway.12 In contrast, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT), a novel thiosemicarbazone chelator with more effective Fe-binding ligands and better membrane permeability, holds great potential to fight against cancers.13,14 It is verified that the growth inhibitory effect of Dp44mT on human oral squamous cell carcinoma might be partly attributed to the induction of NDRG1 and NDRG3 and repression of cyclin D1.15 More importantly, Dp44mT can significantly inhibit the metastasis of cancer cells with high metastatic potential, such as osteosarcoma and hepatocellular carcinoma cells.16,17Over the past few decades, great efforts have been made towards treating cancers. Surgery, radiotherapy and chemotherapy are the main strategies for cancer therapy.18–20 Owing to their high apoptosis-inducing capacity, platinum (Pt)-based drugs are widely applied in the clinical therapy of various malignancies, such as testicular, breast, ovarian and lung cancers.21–23 Among them, cisplatin is the most frequently applied Pt drug as the model for a curable neoplasm in the therapy of many cancers.24 It can interact with DNA via the formation of Pt–DNA adducts, leading to the shutdown of gene replication and transcription and eventually inducing cell apoptosis.25,26 Besides, the Pt–DNA adducts can inhibit RNA polymerase II initiation and elongation. More importantly, cisplatin can disrupt the function of iron regulatory protein 2 (the master regulator of iron metabolism, IRP2) by adducting to Cys512 and Cys516, which leads to sustained cellular iron deficiency and eventually cell death.27 These findings expand our understanding of the anticancer mechanism of cisplatin, and broaden the clinical application of cisplatin with iron chelators.
Although cisplatin has shown a broad spectrum of anticancer activities, its clinical use is always accompanied by serious side effects including dose-limiting toxicities as well as intrinsic or acquired drug resistance.28,29 Moreover, the chronic administration of cisplatin will increase the risk of metastasis, which greatly limits its clinical application.30 Nanotechnology is one of the innovative strategies to overcome the above shortcomings through the EPR effect and the stabilization of the drugs encapsulated in the nanoparticles.31,32 When compared to small-molecule based chemotherapeutic drugs, nanoscale drug delivery systems notably enhanced their cytotoxic performances.33,34 To date, a variety of nanoplatforms have been developed to enhance the efficacy of Pt drugs and decrease adverse effects, whose axial ligands can be modified to carry favorable pharmacological properties in the prodrugs.35–37 It is found that the combination of an alkylated cisplatin prodrug and paclitaxel using nanoformulations improved the outcomes of ovarian and breast cancer treatments.38 Moreover, pulmonary delivery of cisplatin and doxorubicin co-loaded nanoparticles displayed promoted therapeutic efficacy and reduced systemic toxicity.39 Recent studies have verified that co-treatment with free cisplatin and iron chelator DFO led to enhanced anticancer activity via augmented iron depletion in cancer cells.27 Nevertheless, no information is available about the anticancer and anti-metastatic activities of Pt prodrugs with iron chelators.
In this study, we designed a Pt prodrug loaded cancer theranostic nanoplatform by targeting iron homeostasis to promote chemotherapeutic efficacy and inhibit cancer metastasis. In particular, the cisplatin prodrug (denoted as Pt(IV) hereafter) was prepared by the oxidation of cisplatin in H2O2, which was attached with a long lipid chain and morpholine at the two axial positions, respectively. Our previous work revealed that attachment of a morpholine moiety to cisplatin could yield significantly higher antiproliferative activity against varieties of cancer cells (unpublished data). Subsequently, an oxidation-sensitive polymer (namely poly(PSDE-co-LDI)) was synthesized by using ROS sensitive monomers that can be broken down and thus release Pt drugs into the tumor microenvironment (denoted as P1 hereafter).40 Finally, the as-synthesized Pt(IV) and P1 were encapsulated into the nanoparticle (denoted as NP hereafter) via hydrophobic and hydrophilic interactions (Scheme 1). With satisfactory drug delivery efficiency, the nanoparticles could facilitate its tumor-targeting ability and induce serious apoptosis. Meanwhile, the iron chelator Dp44mT was co-delivered into tumor tissues, and was not encapsulated into the NPs due to its high cytotoxicity (IC50 less than 100 nM in most cell lines) and low water solubility. We found that the combination of NPs and Dp44mT could severely induce cell apoptosis and suppress cancer metastasis in vitro and in vivo. Our study would advance the clinical application of platinum complexes with iron chelators.
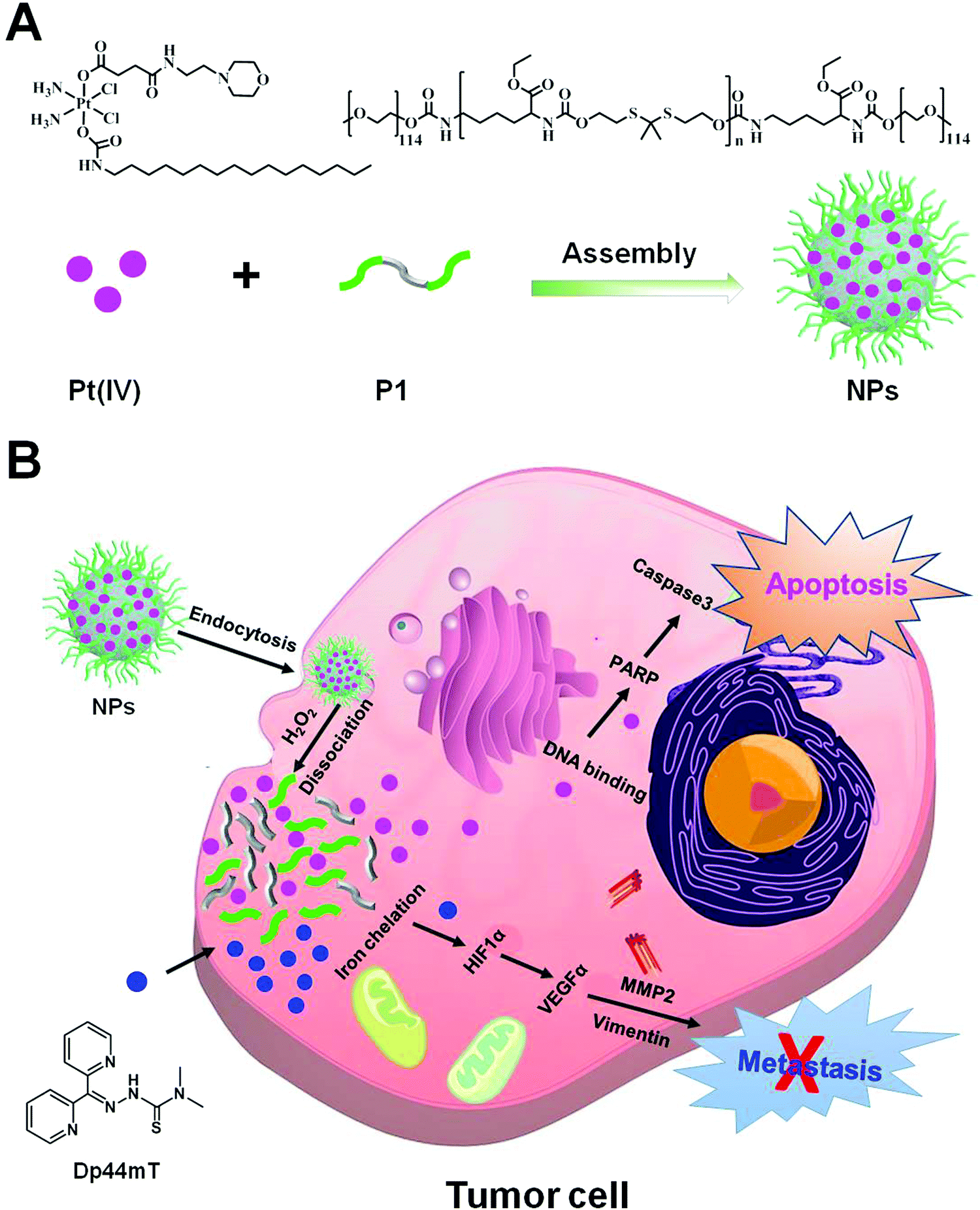 | ||
| Scheme 1 Schematic illustration of the preparation of NPs and the cancer theranostic mechanism with Dp44mT. (A) Preparation of NPs by assembling the Pt(IV) prodrug and P1. (B) The anti-cancer and anti-metastatic mechanisms of NPs and iron chelator Dp44mT, respectively. | ||
2. Materials and methods
2.1 Materials
Tetrahydrofuran (THF), hexane, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), methyl sulfoxide (DMSO), ethanol, wortmannin, sodium azide (NaN3), genistein and cyclohexane were purchased from Sigma-Aldrich (Shanghai, China). N,N-Dimethylformamide (DMF), acetonitrile and hydrogen peroxide were purchased from Beijing Chemical Works (Beijing, China). Cisplatin (purity 99%) was purchased from Shandong Boyuan Chemical Company (Shandong, China). All reagents and solvents were of analytical grade and used without further purification. 2-(4-Amidinophenyl)-1H-indole-6-carboxamidine (DAPI), a DNA extraction kit and an Annexin-V-FITC apoptosis detection kit were purchased from Beyotime Biotechnology (Beijing, China).2.2 General measurements
Dynamic light scattering (DLS) was performed using a Malvern Zetasizer NanoZS90 (Malvern Instruments, Malvern, UK). Inductively Coupled Plasma (ICP) analysis was done using an Inductively Coupled Plasma Optical Emission Spectrometer (ICP-OES, iCAP 6300, Thermo Scientific, USA). Transmission electron microscopy (TEM) was performed using a JEM-1011 electron microscope. Flow cytometry was performed using a Cytomics FC500 Flow Cytometer (Beckman Coulter Ltd). Confocal laser scanning microscopy (CLSM) was accomplished using a ZEISS LSM880. 1H NMR spectra were recorded using a Unity-400 MHz NMR spectrometer (Bruker). Inductively coupled plasma mass spectrometry (ICP-MS) was performed using an Agilent Technologies 7700 series. All OD values were recorded using a SpectraMax M3.2.3 Cell lines and animals
Established lung cancer cell lines A549 (cisplatin sensitive) and A549DDP (cisplatin acquired resistant) were obtained from the American Type Culture Collection (ATCC, USA) and cultured in DMEM supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin/streptomycin (HyClone, USA). 4T1 murine breast cancer cells were purchased from the Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China), and were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. All these cells were incubated in a humidified incubator at 37 °C with 5% CO2.Female Kunming (KM) mice and female Balb/c mice (6–8 weeks old, 18–22 g) were obtained from Vital River Laboratory Animal Technology Co. (Beijing, China). All animals were raised under specific-pathogen-free conditions and housed in a barrier facility with a 12 h light/dark cycle. All animal experimental procedures were performed in accordance with the ethical guidelines issued by the Institutional Animal Care and Use Committee of Shenzhen University.
2.4 Synthesis of Pt(IV)-OH
Pt(IV)-OH was prepared according to a previous report with minor modifications.41 In brief, 300 mg cisplatin was suspended in H2O2 (30% w/v, 36 mL). The mixture was placed at 55 °C with continuous stirring for 4 h, then the temperature was increased to 100 °C until the solution became clear. Subsequently, the above mixture was placed at 4 °C overnight after cooling down to room temperature, then a large amount of yellow-green crystal was precipitated. The product was washed with acetone, ether and H2O in turn and dried in a vacuum oven.2.5 Preparation of C16-Pt(IV)-Suc
The above-synthesized Pt(IV)-OH (400 mg) was suspended in 200 mL anhydrous DMF. Next, succinic anhydride (133 mg) was added to the above mixture with continuous stirring for 48 h at room temperature. The reaction solution was filtered and dried under vacuum, which was further precipitated in ether and dried to obtain a white solid product, namely Pt(IV)-Suc.Next, 200 mg Pt(IV)-Suc and 122 mg hexadecyl isocyanate were dissolved in 10 mL anhydrous DMF, followed by vigorous stirring at 75 °C overnight. The product (denoted C16-Pt(IV)-Suc) was then separated with MeOH and dried under vacuum as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 6.51 (m, 7H), 2.87 (q, 2H), 2.35 (m, 4H), 1.23 (m, 28H), 0.85 (t, 3H). ESI-MS (negative mode), calcd for C21H45Cl2N3O6Pt−, 700.2; found, 700.1.
2.6 Preparation of C16-Pt(IV)-Mor, Pt(IV)
The above-synthesized C16-Pt(IV)-Suc (200 mg) was dissolved in 10 mL anhydrous DMF with the addition of EDCl and NHS. After stirring for 2 h, 2-morpholinoethan-1-amine (100 mg) was added into the mixture. The above reaction continued for 5 days at room temperature, and the final product C16-Pt(IV)-Mor (namely Pt(IV)) was collected with MeOH as a yellow solid. 1H NMR (400 MHz, DMSO-d6): δ 7.73 (t, 1H), 6.54 (d, 7H), 3.60–3.51 (m, 4H), 3.15 (d, 2H), 2.88 (d, 2H), 2.44 (d, 2H), 2.33 (d, 6H), 2.30–2.20 (m, 2H), 1.29 (d, 28H), 0.85 (t, 3H); ESI-MS, calcd for C27H58Cl2N5O6Pt+ [M + H]+, 813.3; found, 813.3.2.7 Synthesis of 2,2′-(propane-2,2-diylbis(sulfanediyl))bis(ethan-1-o1), PSDE
Mercaptoacetic acid (16 mL) and acetone (40 mL) were placed in a three-necked bottle followed by continuous injection of dry HCl. The mixture was stirred at room temperature for 2 h. Thereafter, the reaction solution was filtered to obtain a light yellow solid, and washed with ethyl acetate and water, respectively. A white product was obtained after being dried under vacuum.The above-synthesized white product (5 g) was suspended in anhydrous THF, and cooled in an ice bath for 15 min. Lithium aluminum hydride (2.5 g) was then slowly added to the mixture and heated to 50 °C for 8 h, which was quenched by the addition of saturated sodium hydroxide solution. The organic phase of the above solution was separated by ethyl acetate extraction and dried with anhydrous magnesium sulfate. The mixture was further filtered and steamed, and finally, a yellow viscous product, namely PSDE, was obtained via column chromatography.
2.8 Synthesis of poly(PSDE-co-LDI)-PEG, P1
PSDE (0.1 mM) and L-lysine diisocyanate (LDI, 0.1 mM) were placed in a 50 mL round-bottom flask, then 10 mL DMF was added into the flask with continuous stirring for 24 h. Subsequently, mPEG5k-OH (0.01 mmol) was added for the purpose of the end-capping of the polymer. After stirring overnight, the final product poly(PSDE-co-LDI)-PEG (namely P1) was collected through dialysis and dried under vacuum.2.9 Preparation of nanoparticles, NPs
The as-synthesized polymer P1 (10 mg) was initially dissolved in DMF, then the solution was dispersed in 10 mL de-ionized water with continuous stirring. Pt(IV) (1 mg) was dissolved in 1 mL DMF, which was added dropwise into the above mentioned aqueous solution. After vigorous stirring for 15 min, the mixture was collected and dialyzed using a dialysis bag (MWCO: 3500 Da) overnight. The nanoparticles (denoted as NPs hereafter) were then separated by centrifugation and washed twice with de-ionized water.2.10 Dissociation kinetics of NPs
The dissociation kinetics of NPs was performed according to a previous report with minor modifications.42 In brief, P1 (10 mg) and the Pt(IV) prodrug (1 mg) were dissolved in 1 mL DMF. 10 μL of Nile red stock solution (0.05 mg mL−1) in THF were added into the above mixture. Hereafter, with gentle stirring, 9 mL deionized water were added dropwise to prepare new nanoparticles. The changes in the fluorescence curves were recorded at an excitation wavelength of 550 nm and emission wavelengths from 620 to 720 nm by adding 10 mM H2O2 at different time points. The half-life time (t1/2) was calculated based on the fluorescence measurements.2.11 Drug release of NPs
The in vitro release behaviors of platinum from NPs at different pH values were investigated by the dynamic analysis method.43 In brief, 0.5 mL NPs was dispersed in 9.5 mL PBS at pH 7.4 and pH 5.0. The solution was then immersed in a pre-swelled dialysis bag with a molecular weight cutoff of 3.5 kDa and placed in 100 mL 0.1 M PBS (pH 7.4 and pH 5.0) in a horizontal shaker at 37 °C. At different time intervals, 1.5 mL release medium were withdrawn from the incubation medium for Pt concentration measurement by ICP-MS. Meanwhile, an equal volume of fresh PBS was immediately added into the incubation medium after sampling. The Pt released from the micelles was expressed as the percentage of cumulative Pt outside the dialysis bag to the total Pt in the micelles.Similar procedures were carried out in the presence of 10 mM H2O2 to detect Pt release under oxidic conditions.
2.12 In vitro cytotoxicity
The in vitro cytotoxicity profiles of cisplatin and Dp44mT as well as the NPs were evaluated by the MTT method against A549 and A549DDP cells.44 Briefly, cells were seeded into 96-well plates at a density of 5 × 103 cells per well and incubated in 5% CO2 at 37 °C. After overnight culture, cells were incubated with various concentrations of cisplatin (0–60 μM), Pt(IV) (0–60 μM), Dp44mT (0.1 μM, 0.25 μM and 0.5 μM) and NPs (0–40 μM) dispersed in the culture medium for 48 h. Thereafter, 10 μL MTT solution (5 mg mL−1) was added into the wells for another 4 h incubation. Next, 10% SDS was added to dissolve the product crystals for 12 h, and the absorbance of each well was measured using a microplate reader.2.13 Apoptotic detection
Cell apoptosis was detected according to early reports with minor modifications.41 Briefly, A549DDP or 4T1 cells were seeded into 6-well plates at a density of 3 × 105 cells per well and grew overnight. The cells were then exposed to Dp44mT (0.25 μM), cisplatin, Pt(IV) and NPs (the Pt concentration in the culture medium was regulated to 2.5 μM) at 37 °C for 24 h. For apoptosis assay, the cells were harvested using trypsin and washed twice with PBS and resuspended. The cells were then incubated with Annexin-V-FITC and PI for 15 min protected from light at 4 °C. After incubation, cell apoptosis was analyzed using flow cytometry (Beckman, USA).2.14 Colony formation assay
For colony formation assay, a total of 1000 A549 cells were seeded into 35 mm dishes and maintained in RPMI 1640 medium with 10% FBS at 37 °C for two weeks. During the growth stage, cells were subjected to the following treatments: cisplatin (0.1 μM), Dp44mT (0.01 μM), Pt(IV) (0.1 μM), cisplatin (0.1 μM) + Dp44mT (0.01 μM) and Pt(IV) (0.1 μM) + Dp44mT (0.01 μM). With two weeks of incubation, cells grew into visible colonies, which were washed with PBS and fixed with 70% ethyl alcohol for 15 min. Subsequently, the colony cells were stained with 0.5% crystal violet for another 15 min and the colonies were photographed with a camera. The number of colonies of each treatment was counted for statistical analysis.2.15 Cellular uptake of the NPs
In order to track the entry of NPs into the cells, fluorescent dye Cy5.5 was incorporated into the aforementioned Pt(IV) and polymer P1 to assemble new nanoparticles (denoted Cy5.5@NPs). Cell slides were placed at the bottom of the wells in advance, then 4T1 cells at a density of 2 × 104 per well were seeded and incubated at 37 °C overnight. The cells were exposed to Cy5.5@NPs with a final concentration of 2 μg ml−1 of Cy5.5 for 2 h and 5 h. After that, cells were washed with cold PBS and fixed with paraformaldehyde. For cell colocalization, the nuclei were stained with DAPI (blue) and filamentous actin cytoskeletons were stained with Phalloidin (green). Finally, images were immediately captured by confocal laser scanning microscopy (CLSM) at a 100× magnification.The intracellular uptake studies of the NPs were also performed by flow cytometry. In particular, 4T1 cells were seeded into 6-well plates at a density of 2 × 105 cells per well. After incubation overnight, cells were exposed to the above synthesized Cy5.5@NPs for another 2 h, 5 h and 8 h at 37 °C. Next, cells were washed with PBS and harvested with trypsin, and the mean fluorescence intensity (MFI) of the cells was detected by flow cytometry.
In order to further investigate the cellular uptake mechanism of Cy5.5@NPs, 4T1 cells were pretreated with various endocytosis inhibitors for 2 h as follows: NaN3 (120 mM), wortmannin (Wort, 0.2 μM) and genistein (Gen, 200 μM). Subsequently, the cells were washed with cold PBS and incubated in fresh medium containing Cy5.5@NPs with a final concentration of 2 μg mL−1 of Cy5.5. After incubation for another 3 h at 37 °C, cells were collected and the MFIs of cells treated with the various aforementioned inhibitors were analyzed by flow cytometry. Cells without any inhibitor treatment were set as the control group.
2.16 Platinum uptake in the cells
To determine the ratios of cellular uptake, 4T1 cells were seeded into 6-well plates at a density of 3 × 105 cells per well and grew overnight. The cells were then exposed to cisplatin and NPs with the platinum concentration in the culture medium regulated to10 μM. After incubation at 37 °C for 2 h or 5 h, cells were washed with PBS three times and lysed with cell lysis buffer. The platinum content in the cell lysis solution was determined by ICP-MS.2.17 Pt–DNA adducts in the cells
A549 and A549/DDP cells were seeded into 6-well plates at a density of 1 × 106 per well and incubated at 37 °C for 12 h. Cisplatin and NPs were added to the wells at a final concentration of 20 μM Pt. After incubation for 2 h and 5 h at 37 °C, cells were washed three times with cold PBS, and DNA was extracted using a DNA extraction kit (Beyotime, Beijing). DNA yields were evaluated using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA) and Pt contents were determined via ICP-MS. The DNA–Pt adduct amounts were expressed as pg Pt per μg DNA.2.18 Western blot assay
Western blot was performed as previously described.45 In this study, the following primary antibodies were used: anti-Caspase3 (#9662, Cell Signaling Technology, CST), anti-c-Caspase3 (#9661, CST), anti-PARP (#9542, CST), anti-c-PARP (#9548, CST), anti-VEGFα (#ab46154, abcam), anti-HIF1a (#36169, CST) and GAPDH (#5174, CST). Typically, A549 or A549DDP cells were seeded into 6-well plates and incubated with free cisplatin (2.5 μM), Pt(IV) (2.5 μM), Dp44mT (0.25 μM) and NPs (2.5 μM) for 24 h separately. Cells were harvested and the lysates were homogenized in RIPA lysis buffer followed by centrifugation (Beyotime, Beijing, China). A total of 30 μg protein was separated by SDS-PAGE and transferred onto a PVDF membrane. Following incubation with the primary antibodies overnight at 4 °C, the membranes were washed with TBST and subsequently incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit secondary antibody for 1 h. Bands of interest were analyzed using an image analysis system (Bio-Rad, USA) according to the manufacturer's instructions. The lysates from the xenografts with different treatments were also homogenized in RIPA for western blot assay. GAPDH was used as the internal control.2.19 Wound-healing assay
A549DDP cells were seeded into 35 mm dishes at a density of 6 × 105 cells per well and incubated for 12 h to grow a monolayer. Next, a linear wound was generated across the middle of the well surface with a p200 pipette tip, and cells were incubated with different drugs in serum-free media for another 24 h. Cell images were captured subsequently under a microscope (Olympus, Japan) and the wound healing closure was calculated using ImageJ software.2.20 Cell migration assay
For cell migration assay, a total of 5 × 105 A549DDP cells in 100 μL serum-free medium were plated in the top chambers of Transwell plates, and 500 μL medium containing 20% FBS was added in the lower chambers as the chemotactic factor. For both chambers, the following treatments were performed: PBS (control), Pt(IV) (2.5 μM), Dp44mT (0.25 μM), Pt(IV) (2.5 μM) + Dp44mT (0.25 μM), NPs (2.5 μM) and NPs (2.5 μM) + Dp44mT (0.25 μM). After incubation for 24 h, cells that did not migrate through the Transwell membrane were gently removed with a cotton swab, while the migrating cells on the lower surface were then stained with 0.5% crystal violet for 15 min. The migrating cells were photographed under a microscope at ×100 magnification. To quantify the cell migration ratio, 33% acetic acid was added to dissolve the crystal violet absorbed by the cells, and the absorbance was determined at 570![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm using a microplate reader.
nm using a microplate reader.
2.21 In vivo toxicity evaluation
In vivo toxicity evaluation was performed as per a previous report.46 Healthy female KM mice (weighing 18–22 g) were randomly assigned to six groups and injected via the tail vein with PBS, cisplatin (2.5 mg Pt per kg body weight), Dp44mT (3 mg per kg body weight), cisplatin (2.5 mg Pt per kg body weight) + Dp44mT (3 mg per kg body weight), NPs (2.5 mg Pt per kg body weight) and NPs (2.5 mg Pt per kg body weight) + Dp44mT (3 mg per kg body weight) twice a week for two weeks, respectively (n = 5). The mice were weighed every other day and sacrificed 14 days post the first administration. In order to assess the potential toxicity of the above drugs, histological analysis was performed on the major organs including the heart, spleen, liver, kidneys and lungs. Tissues from these organs were collected, fixed with 4% paraformaldehyde solution, and sliced for hematoxylin and eosin (H&E staining) assay. Meanwhile, blood biochemistry assays were performed to quantitatively evaluate the potential kidney, heart and liver function as follows: nitrogen (BUN) and creatinine (CR) (indicators for kidney function), creatine kinase (CK) and creatine kinase MB (CK-MB) (indicators for heart function), and aminotransferase (ALT) and aminotransferase (AST) (indicators for hepatic function).2.22 Tissue distribution and tumor accumulation in mice with breast cancer
As reported previously, the in vivo distribution of NPs was investigated in female mice bearing 4T1 breast cancer, which was initiated by subcutaneous injection of 1 × 106 4T1 cells on the right mammary gland.47 To monitor the NPs in vivo, Cy7.5, a near-infrared dye, was loaded into the NPs (denoted as Cy7.5@NPs hereafter). When the tumors grew to 100 mm3, the mice were randomly divided into two groups and injected via the tail vein with PBS (control group) and Cy7.5@NPs, respectively. At designated time points post injection, the mice were imaged using an IVIS in vivo imaging system (PerkinElmer, Ex/Em = 780 nm/810 nm, Waltham, USA) to detect the tumor accumulation. After 24 h of injection, the mice were sacrificed and the main organs including the heart, liver, lungs, kidneys, spleen and tumors were isolated and used for ex vivo imaging.2.23 In vivo anticancer and antimetastatic effects in mice with breast cancer
For an antitumor experiment, a female Balb/c mice 4T1 xenograft tumor model was established according to a literature report with some modifications.48 Typically, tumors were allowed to grow to reach a size of about 100 mm3, then mice were randomly divided into 6 groups with the following treatments (n = 6): cisplatin (2.5 mg Pt per kg body weight), Dp44mT (0.3 mg Pt per kg body weight), cisplatin (2.5 mg Pt per kg body weight) + Dp44mT (0.3 mg Pt per kg body weight), NPs (2.5 mg Pt per kg body weight), NPs (2.5 mg Pt per kg body weight) + Dp44mT (0.3 mg Pt per kg body weight) and control (0.9% saline) twice a week for four weeks. During the experimental period, the body weights and tumor volumes were measured every other day using the following equation: tumor volume (V) = length × width2 × 0.5.At the end of the experiment, all the mice were sacrificed and the tumor weights were recorded. Meanwhile, the primary lungs and livers were taken out and photographed with a camera, and the histological sections of the lungs and livers were stained with H&E for histopathological examination. For IHC staining assay, primary tumor sections were separately incubated with anti-MMP2 and anti-Vimentin rabbit antibodies (CST, USA) at 4 °C overnight, and then blotted with the corresponding HRP-conjugated secondary antibodies for 1 h at room temperature. Thereafter, the sections were stained with diaminobenzidine solution for about 5 min and counterstained with hematoxylin.
2.24 Statistical analysis
The results are presented as means ± standard deviation (S.D.). The data were subjected to ANOVA testing and means were assessed for significance using Student's t-test. Differences are considered to be significant for p values less than 0.05 (*p < 0.05) and very significant for p values less than 0.01 (**p < 0.01). Unless specifically mentioned, all assays were performed in triplicate in three independent experiments. All statistical analyses were performed using SPSS.3 Results and discussion
3.1 In vitro cytotoxicity of cisplatin and Dp44mT
To investigate the in vitro cytotoxicity of cisplatin and Dp44mT, MTT assay was conducted to evaluate the cytotoxicity of cisplatin and Dp44mT in A549 and A549DDP cells after incubation for 48 h. As shown in Fig. 1A, compared with the control group, both cisplatin and Pt(IV) can significantly inhibit the proliferation ability of A549 and A549DDP cells in a dose-dependent manner. Notably, a higher inhibition efficiency was observed for the combined treatment with Dp44mT and cisplatin, whose IC50 values decreased over 10 times that of cisplatin alone (Table S1, ESI†). Similar results were observed with Dp44mT and Pt(IV), demonstrating that the combination of the Pt prodrug and Dp44mT could induce augmented cytotoxicity (Table S2, ESI†). It should be noted that the co-treatment using Dp44mT with carboplatin or oxaliplatin also resulted in improved cytotoxicity to cancer cells, which further confirms that iron chelator Dp44mT facilitates the anti-proliferative effect of chemotherapeutic drugs (Fig. S3, ESI†). Next, the cell apoptotic profiles of the above drugs against A549DDP cells were evaluated by flow cytometric analysis. They reveal that cisplatin or Pt(IV) alone resulted in gentle apoptosis of the cells with apoptotic rates of 19.3% and 9.31%, respectively (Fig. 1B and C). However, when the cells were co-treated with Dp44mT, the apoptotic rates of the cells exposed to cisplatin and Pt(IV) increased to 26.92% and 32.8%, respectively, suggesting enhanced cell apoptosis. Besides, colony formation assay was performed to explore the effects of these drugs on the clonogenicity of A549DDP cells (Fig. 1D and E). After two weeks of incubation, there were 118 colonies in the control group, while cisplatin or Pt(IV) treatment alone depleted the cells to form only 80 and 62 colonies, respectively. However, this effect was further exacerbated in the presence of Dp44mT, leading to less than 40 colonies available. This suggests that the combination of Dp44mT and cisplatin (especially Pt(IV)) could aggravate the inhibitory effect on the colony formation ability of cancer cells. At last, we explored the mechanism of the enhanced cell apoptosis, which is partly through the activation of apoptosis signaling pathways in cancer cells. Therefore, the expression profiles of apoptosis-related proteins were detected by western blot. They reveal that cisplatin or Pt(IV) alone can hardly induce the expression of apoptosis markers such as c-PARP and c-caspase3. However, obvious up-regulation of c-PARP and c-caspase3 was detected upon co-treatment using cisplatin or Pt(IV) with Dp44mT, demonstrating promoted apoptosis (Fig. 1F).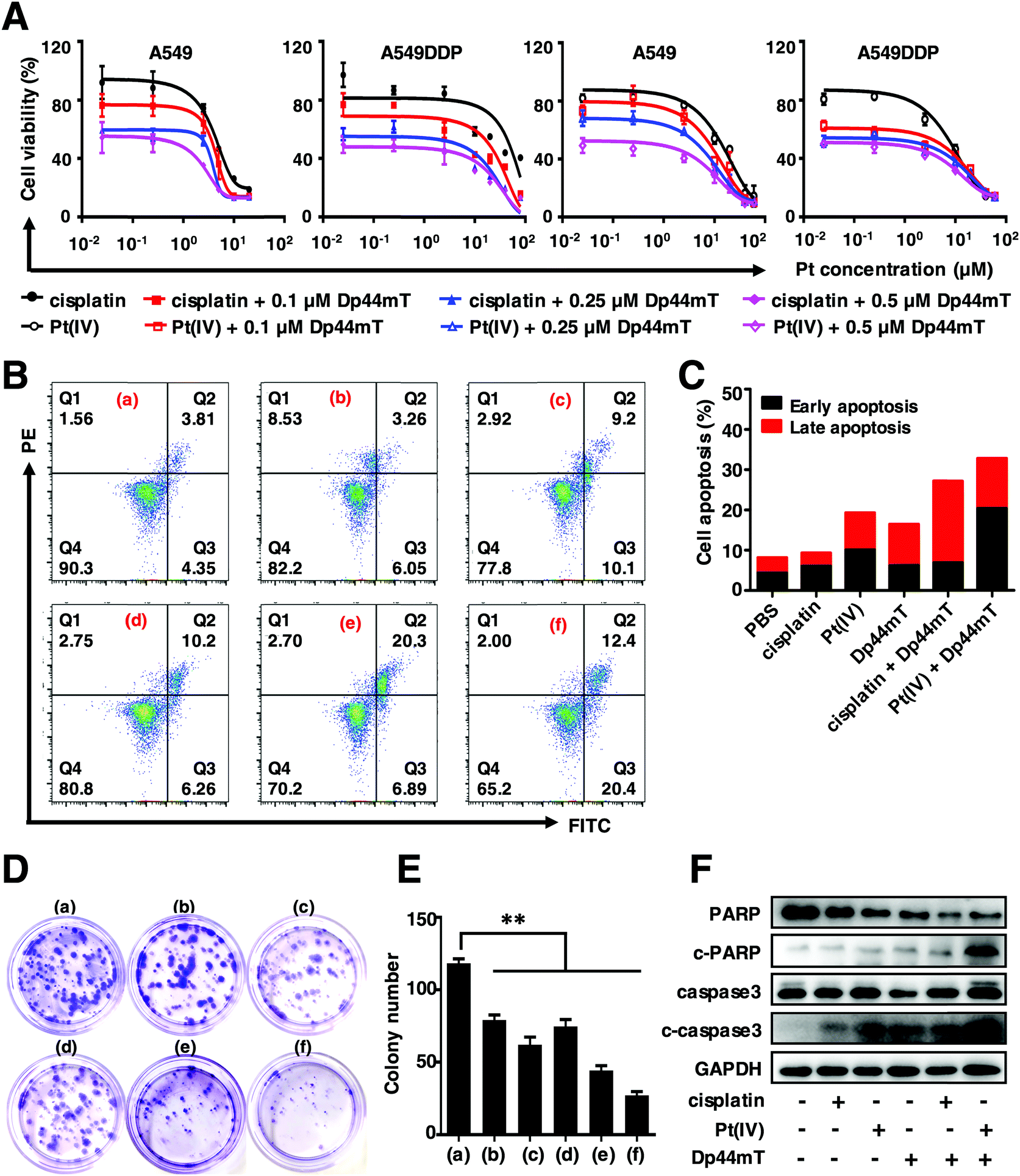 | ||
| Fig. 1 In vitro cytotoxicity of free cisplatin or Pt(IV) with Dp44mT. (A) The relative cell viabilities of A549 and A549DDP cells exposed to cisplatin or Pt(IV) in the presence of different concentrations of Dp44mT for 48 h, respectively. (B) Cell apoptosis results and (C) the quantitative analysis of A549DDP cells with various treatments for 24 h as indicated below: (a) PBS, (b) 2.5 μM cisplatin, (c) 2.5 μM Pt(IV), (d) 0.25 μM Dp44mT, (e) 2.5 μM cisplatin + 0.25 μM Dp44mT and (f) 2.5 μM Pt(IV) + 0.25 μM Dp44mT. (D) Clonogenicity (E) and the corresponding statistical analysis of A549DDP cells with the above treatments for 2 weeks. **p < 0.01 for comparison with the control group. (F) Expression patterns of key apoptotic proteins of A549DDP cells after various treatments as mentioned above for 24 h by western blot. | ||
Taken together, the above results indicated that cisplatin or Pt(IV) can suppress cell viability, inhibit the colony formation capacity and induce cell apoptosis. However, the addition of Dp44mT could significantly enhance the cytotoxicity of cisplatin and Pt(IV). Noteworthily, in all the above experiments, the Pt(IV) showed higher toxicity than free cisplatin, which might be attributed to the attachment of the morpholine core at the axial position. Our study is in good accordance with previous reports that compounds containing morpholine exhibit broad biological activities such as antimicrobial, anti-oxidant, antidiabetic and antitumor activities.49,50 In view of this, Pt(IV) is used to further explore the anticancer mechanism with Dp44mT in the subsequent experiments.
3.2 Preparation and characterization of NPs
As mentioned above, in this study Dp44mT was not packaged with the Pt(IV) prodrug into the nano-carrier to assemble the NPs because of the low water solubility and high cytotoxicity of Dp44mT (IC50 less than 100 nM against most cells). Indeed, few reports are available about the fabrication of Dp44mT-loaded nanoparticles via poly(lactic-co-glycolic acid) (PLGA).51 The Pt(IV) prodrug was obtained by the oxidation of cisplatin in H2O2, a long lipid chain and a morpholine group were subsequently attached to the axial positions, respectively (Scheme S1, ESI†). The polymer drug carrier poly(PSDE-co-LDI) was prepared to encapsulate the Pt(IV), which could be broken down and release Pt drugs into the tumor microenvironment (Scheme S2, ESI†). The as-synthesized PSDE and polymer P1 were characterized by 1H-NMR (Fig. S1 and S2, ESI†). To optimize the nanoparticles, various ratios of Pt(IV) to the polymer (ranging from 0.05 to 1) were studied by DLS. It is verified that the Pt(IV) to polymer ratio of 0.1 would yield a satisfactory nanoparticle diameter and PDI.To characterize the as-synthesized nanoparticles, dynamic light scattering (DLS) was performed. As shown in Fig. 2A, the hydrodynamic diameter of the NPs is approximately 156 nm with a narrow and monomodal size distribution (PDI = 0.124). Moreover, the stability of the NPs was monitored during 5 days of storage at room temperature by DLS. As expected, no significant changes in size were observed, confirming that the current nano-system is stable enough (Fig. 2B). In order to study the degradation kinetics of the polymer P1, the fluorescence spectra of NPs labelled with Nile red were determined after H2O2 treatment. They reveal that the fluorescence intensity of the NPs gradually decreased as time went by (Fig. 2C). Besides, the half-life (t1/2) of 1.62 h demonstrated the rapid dissociation of the NPs (Fig. 2D). To further investigate the in vitro Pt release profile of NPs, we studied the kinetic behavior of drug release in the absence and presence of H2O2 under different pH conditions by ICP-MS. In this study, pH 5.0 and pH 7.4 were used to mimic normal tissue and the intravascular circulation environment, respectively. As depicted in Fig. 2E, less than 25% Pt was released at pH 7.4 and pH 5.0. However, in the presence of H2O2 that facilitates the conversion of Pt(IV) to active Pt(II), over 90% Pt was released within 48 h. Obviously, Pt release at pH 5.0 was much higher than that at pH 7.4. These results indicate that the as-synthesized nano-system was sensitive to oxidic as well as acidic conditions.
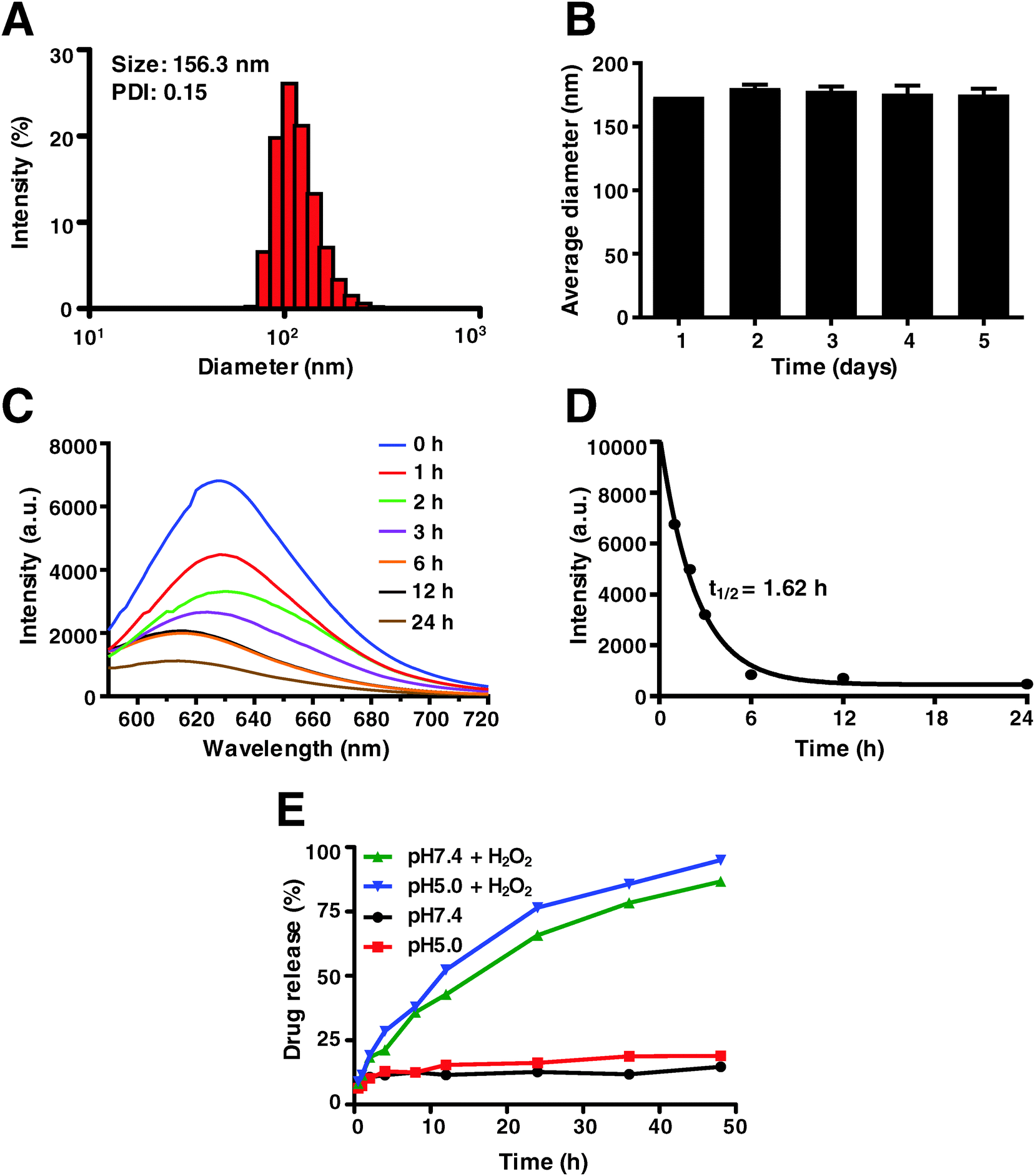 | ||
| Fig. 2 Characterization of NPs. (A) Size distribution of NPs by DLS. (B) Stability of NPs within 5 days monitored by DLS. (C) The changes in the fluorescence intensity curves of the NPs (3 mg mL−1 P1, 50 μL) labelled with Nile red after being treated with H2O2 (10 mM, 50 μL) for 24 h. (D) The dissociation half-life of the NPs encapsulated with Nile red exposed to H2O2. (E) Cumulative Pt release profiles of NPs at pH 5.4 and pH 7.4 in the presence of 10 mM H2O2. | ||
3.3 Cellular uptake of NPs
To investigate the intracellular internalization pathway of nanoparticles, the above mentioned NPs loaded with hydrophobic fluorescent dye Cy5.5 were prepared (denoted Cy5.5@NPs) and detected using a confocal laser scanning microscope (CLSM). As shown in Fig. 3A, negligible red fluorescence was observed within the cells when exposed to the Cy5.5@NPs for 2 h due to the minor cellular internalization. However, after 5 h of incubation, notable red fluorescence was observed near the cell nucleus, indicating that the cellular uptake of the Cy5.5@NPs is time-dependent. In order to verify this process, we next measured the cellular uptake profiles of the NPs by flow cytometry. Cells were incubated with the Cy5.5@NPs for 2 h, 5 h and 8 h (Fig. 3B and C). Consistently, we noticed that the longer incubation time resulted in much stronger cell internalization of the NPs, as the Mean Fluorescence Intensity (MFI) with 8 h incubation was 3.66-fold that with 2 h incubation. To distinguish the internalization pathway of Cy5.5@NPs, various endocytosis inhibitors were applied to treat 4T1 cells before co-incubation with the NPs, including wortmannin (Wort, clathrin-mediated pathway), genistein (Gen, caveola-dependent pathway) and sodium azide (NaN3, ATP depleting agent).52 It seems that the intracellular internalization of NPs was predominantly inhibited by NaN3, since the MFI was decreased by 2.6 times compared to the control group (Fig. 3B and C). Besides, Wort and Gen also exhibited slight inhibition to the cellular uptake of the NPs, leading to a reduction of the MFI by approximately 1.7-fold. These results demonstrate that the internalization process of the NPs occurs via various pathways, with the ATP dependent endocytosis being the predominant pathway.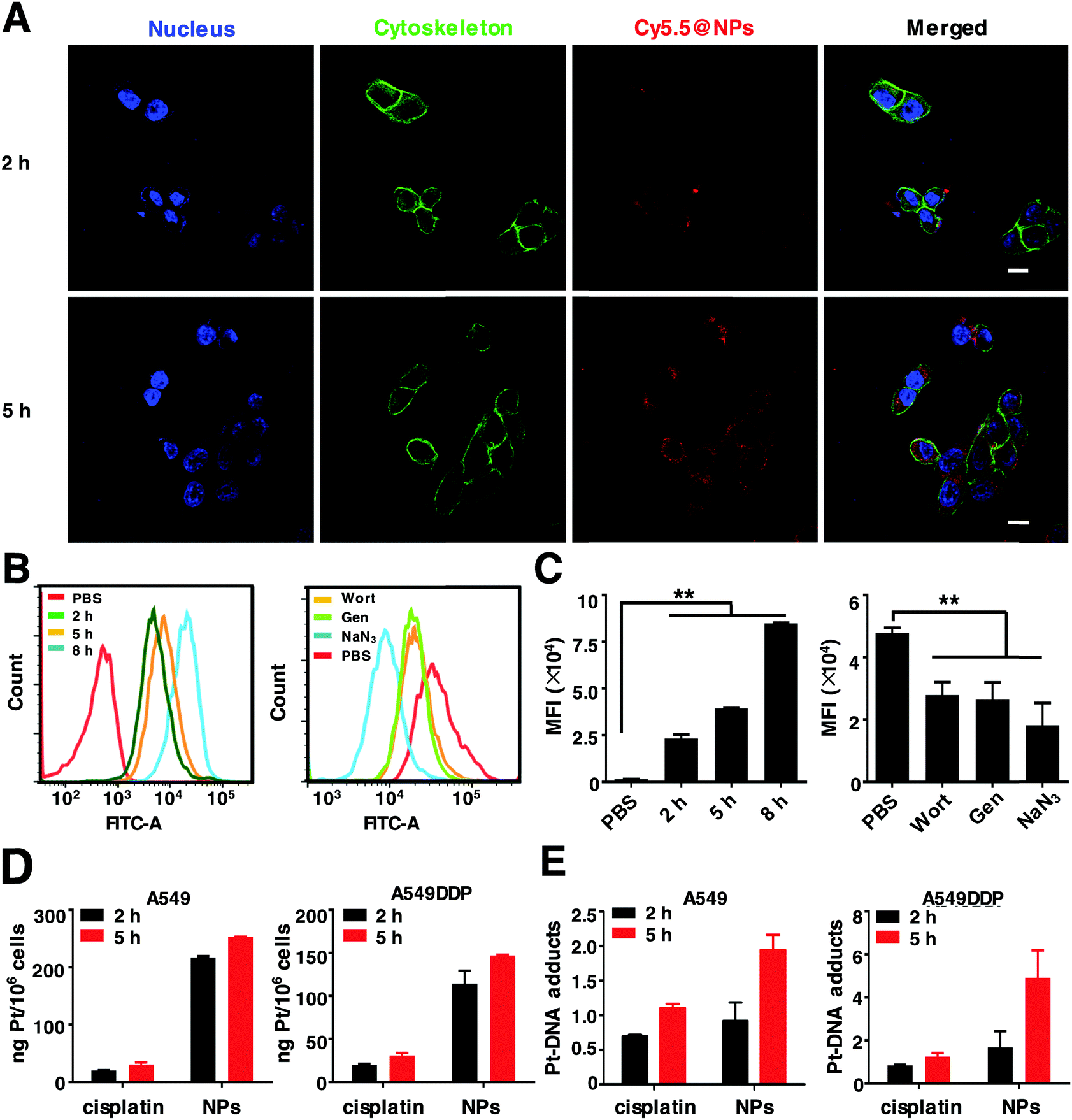 | ||
| Fig. 3 The in vitro evaluation of the intracellular uptake of NPs. (A) CLSM images of 4T1 cells treated with Cy5.5@NPs for 2 h and 5 h, respectively. Scale bars: 20 μm. Cell nuclei were stained with DAPI (blue fluorescence). The red fluorescence and green fluorescence come from Cy5.5 and cell skeletons stained with Alexa Fluor™ 488 Phalloidin, respectively. (B) The intracellular uptake of Cy5.5@NPs and the uptake inhibition assay in 4T1 cells at different time points by flow cytometry. (C) Endocytosis statistics of the intracellular uptake and inhibition of 4T1 cells. **p < 0.01 as compared to the control. (D) Mass of Pt and (E) Pt–DNA adduct formation in A549 and A549DDP cells upon treatment with free cisplatin and NPs (the Pt content in the initial solution was 10 μM) at different time points by ICP-MS. | ||
As reported previously, Pt drugs can disrupt DNA duplicates by targeting DNA, form a Pt–DNA adduct and eventually lead to cell apoptosis. Hence, we performed the endocytosis and Pt–DNA adduct formation assay by ICP-MS. It reveals that the Pt uptake amounts of NPs and cisplatin were increased from 2 h to 5 h in a time-dependent manner. Noteworthily, in both A549 and A549DDP cells, the intracellular uptake of NPs was over 5 times higher than that of cisplatin, implying that the NPs could circumvent the intracellular internalization pathway of free cisplatin (Fig. 3D). Similar results were obtained in the Pt–DNA adducts of these cells. The Pt–DNA adduct quantities in the NPs were significantly higher than those of cisplatin (Fig. 3E). In particular, in A549DDP cells, the Pt–DNA adduct contents in the NPs were twice higher than those in cisplatin, demonstrating that the NPs can target cellular DNA and eventually overcome Pt drug resistance.
Taken together, the above results verified the excellent deliverability of the NPs as carriers, which can enhance the drug internalization and promote intracellular drug accumulation in the cells.
3.4 In vitro cytotoxicity of NPs and Dp44mT
Encouraged by the enhanced endocytosis and Pt release efficiency of the NPs, we further explored the cytotoxicity of the as-synthesized NPs with Dp44mT against cancer cells. As shown in Fig. 3A, exposure to NPs induced much higher cytotoxicity than that to Pt(IV) against A549, A549DDP and 4T1 cells. Moreover, among all these treatments, cells co-treated with NPs and Dp44mT exhibited the lowest cell viability due to their enhanced cytotoxicity. In particular, the IC50 value of A549DDP cells with NP and Dp44mT co-treatment was the lowest (only 0.169 μM), demonstrating that this co-administration is beneficial to overcoming the cisplatin resistance of lung cancer cells (Table S3, ESI†). Subsequently, the cell apoptotic profiles of A549DDP cells were evaluated by flow cytometry. The results show that the treatment with NPs could induce stronger apoptosis of the cells than free Pt(IV) with apoptotic rates of 23.29% and 27.98%, respectively (Fig. 4B and C). As expected, when in the presence of Dp44mT, the apoptotic rates of the cells exposed to NPs and Pt(IV) increased to even 61.39% and 46.6%, respectively (Fig. 4B and C). Consistently, similar results were obtained in 4T1 cells with the above treatments (Fig. S4, ESI†). These results, consistent with the above MTT assay, suggest that the combination of NPs and Dp44mT would lead to maximal cytotoxicity to cancer cells. To explore the apoptotic mechanisms of these treatments, we detected the expression patterns of apoptosis-related proteins including caspase 3 and PARP. In both the A549 and A549DDP cells, negligible expression levels of c-caspase 3 and c-PARP were detected upon exposure to Pt(IV) alone. However, exposure to NPs could up-regulate the expression of c-caspase 3 and c-PARP (Fig. 4D). More importantly, the combination of NPs and Dp44mT led to the highest expression levels of both c-caspase 3 and c-PARP, implying that the combined administration can induce maximal apoptosis of the cells.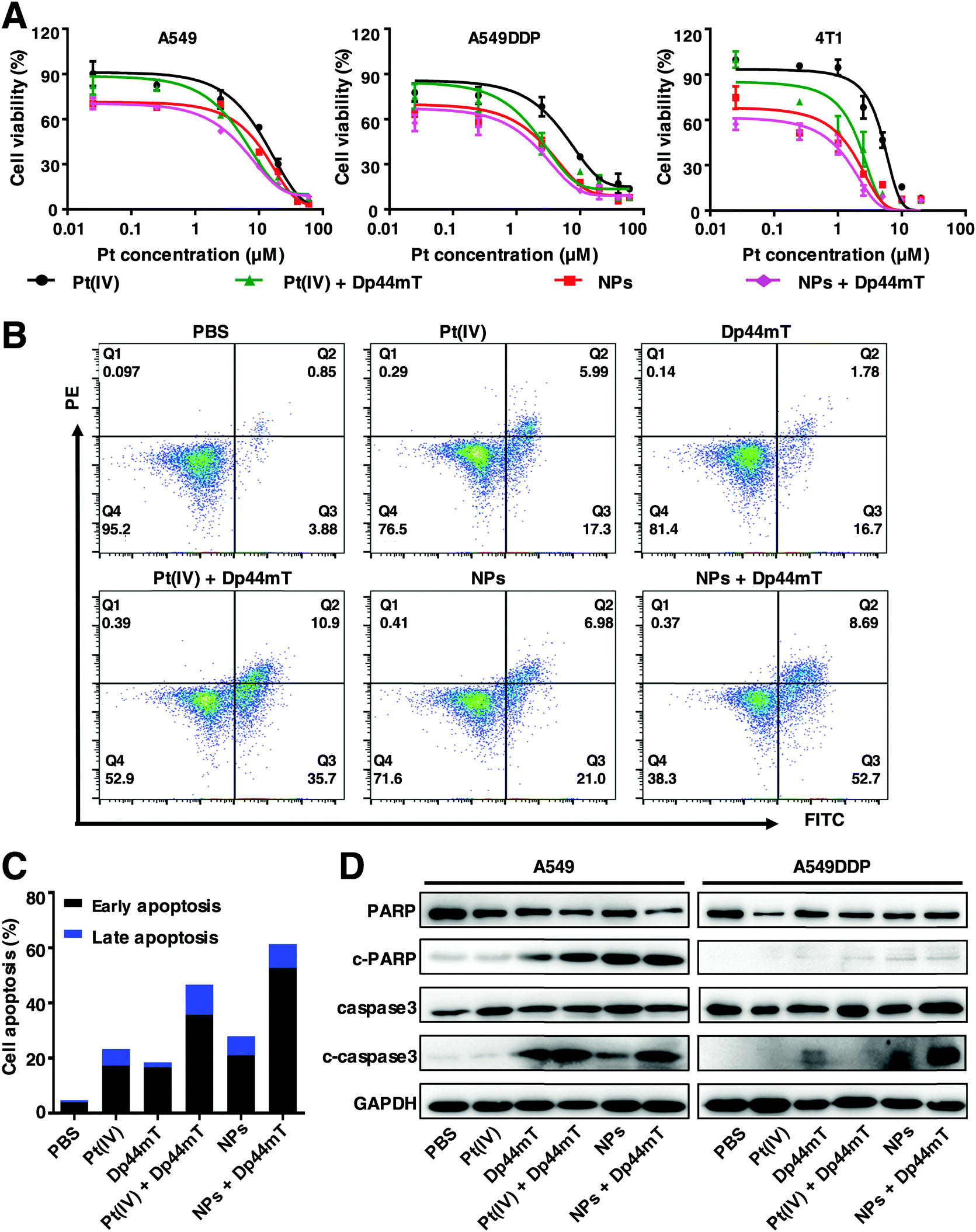 | ||
| Fig. 4 In vitro anticancer effects of the NPs and Dp44mT. (A) Relative cell viabilities of A549, A549DDP and 4T1 cells upon 48 h treatment with NPs, Pt(IV) and Dp44mT, respectively. (B) Apoptosis and (C) the corresponding statistical analysis of A549DDP cells after various treatments for 24 h as indicated below: PBS, 2.5 μM Pt(IV), 0.25 μM Dp44mT, 2.5 μM Pt(IV) + 0.25 μM Dp44mT, 2.5 μM NPs and 2.5 μM NPs + 0.25 μM Dp44mT. (D) Expression levels of apoptosis-related proteins in A549 and A549DDP cells after various treatments as mentioned above for 24 h by western blot. | ||
Based on the above results, it is verified that the NPs exhibit stronger cytotoxicity to cancer cells than Pt(IV) due to their improved endocytosis and Pt release efficiency. Moreover, co-treatment using the NPs with Dp44mT could result in higher cytotoxicity than any single administration method alone, demonstrating the desirable therapeutic effect due to the combination of chemotherapy and the iron chelator. All these results prompt us to further investigate the in vivo antitumor effects of the NPs and Dp44mT in the subsequent experiments.
3.5 In vitro anti-metastasis of the NPs and Dp44mT
As previously reported, iron contributes to the metastasis of cancer cells. Therefore, we further investigated the effect of Pt(IV) and Dp44mT on the in vitro metastasis via wound healing and cell migration assays on cancer cells. As shown in Fig. 5A, after 24 h of incubation, A549 cells without treatment exhibited an obvious wound healing ability with 71% wound closure of the former scratching, demonstrating the intense metastatic characteristic of lung cancer cells. Pt(IV) or even the NP treatment alone had a minor inhibitory effect on the wound healing ability in the cells. However, Dp44mT could significantly inhibit the cell motility due to its high efficiency of iron deprival. Notably, cells co-treated with NPs and Dp44mT showed only 13% wound closure with negligible cell motility (Fig. 5C). Next, cell migration assay was carried out to evaluate the effects of Pt(IV) and Dp44mT on the longitudinal motility of A549 cells. As expected, similar results to those of the wound healing assay were observed. For the control group, most of the cells have migrated into the lower surface of the Transwell chamber (Fig. 5B). Pt(IV) or NP treatment alone showed a slight inhibitory effect on the cell migration ability, but Dp44mT could notably suppress cell migration, with fewer migrating cells on the lower surface. In particular, barely cells displayed on the lower surface of the Transwell chamber by the combined treatment with NPs and Dp44mT with an OD value of only 0.24 (less than half of the control group) (Fig. 5D). The above results demonstrate that the combination of cisplatin nanoparticles and iron chelator Dp44mT is beneficial to inhibiting in vitro cell migration.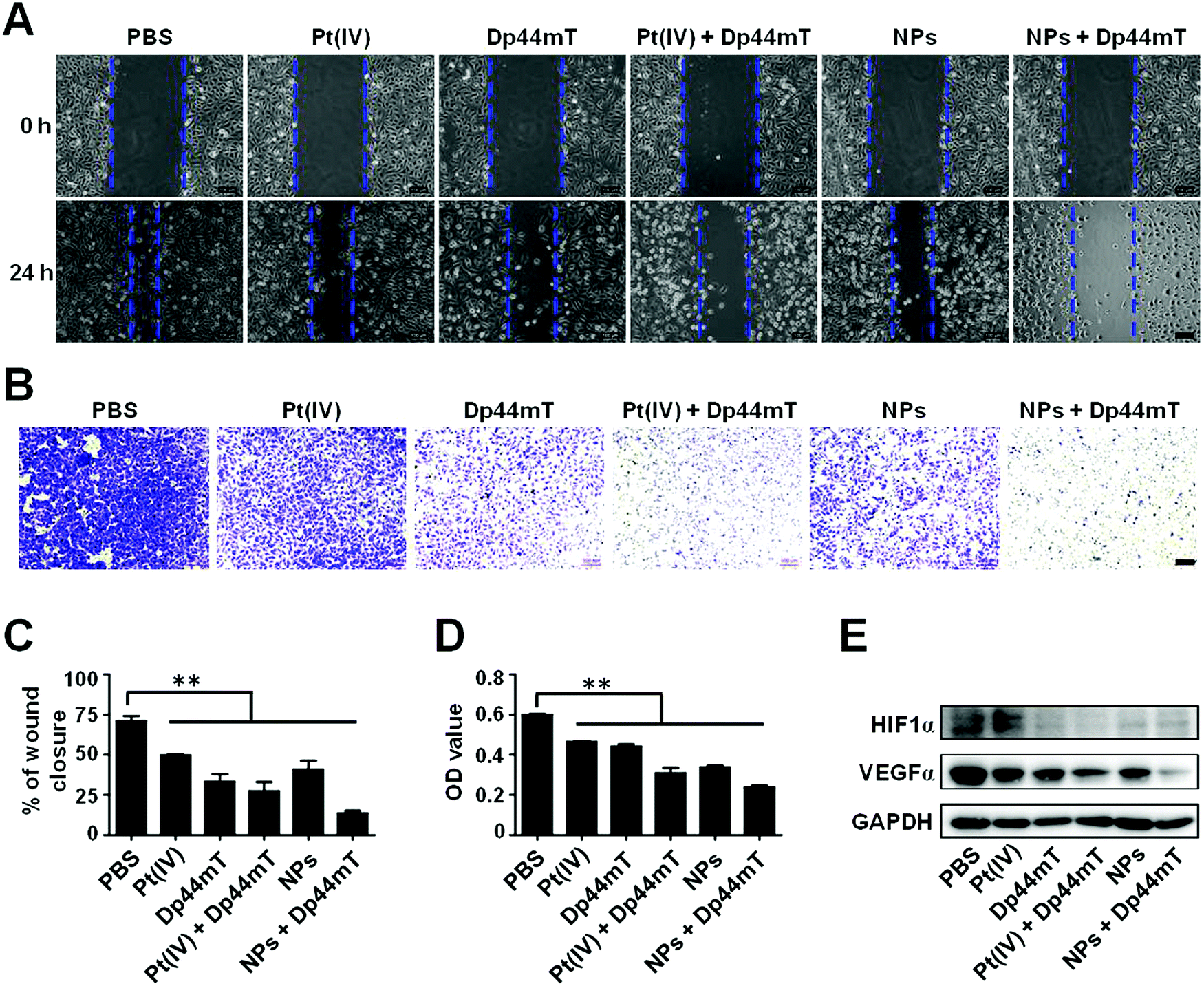 | ||
| Fig. 5 The in vitro anti-metastatic effects of the NPs and Dp44mT. (A) Wound healing, (B) migration, and (C and D) the quantitative assay of A549DDP cells with various treatments for 24 h: PBS, 2.5 μM Pt(IV), 0.25 μM Dp44mT, 2.5 μM Pt(IV) + 0.25 μM Dp44mT, 2.5 μM NPs and 2.5 μM NPs + 0.25 μM Dp44mT. (E) Expression patterns of HIF1α and VEGFα in A549DDP cells exposed to the above treatments for 24 h. Scale bar: 100 μm. **p < 0.01 for comparison with the control. | ||
Next, we investigated the molecular mechanism of NPs and Dp44mT on cell metastasis by western blot. It is well established that vascular endothelial growth factors (VEGFs) play important roles in tumor angiogenesis, which could enhance the proliferation and survival of endothelial cells as well as cell migration.53 HIF1α is up-regulated in primary tumors, and it participates in cell invasion and migration as a major regulator of VEGFα.54 In view of this, we detected the relative expression level of VEGFα and HIF1α in A549DDP cells with the above treatments. Compared to the control group, Pt(IV) or Dp44mT treatment alone resulted in down-regulation of VEGFα expression. The combination of Pt(IV) and Dp44mT caused a much lower expression level of VEGFα, implying the attenuated migration ability of the cells (Fig. 5E). To our surprise, expression of HIF1α showed no significant differences among the above treatments (Fig. 5E). This might be explained as the fact that HIF1α is mostly highly expressed in a hypoxic environment, which could not be activated by the combined treatment of Pt(IV) and Dp44mT in the cancer cells cultured in vitro cultured cancer cell lines.55
3.6 Toxicity evaluation
The prolonged circulation time, improved intratumoral penetration and enhanced tumor accumulation provide us with a strong rationale to explore the therapeutic efficacy of the NPs with Dp44mT on tumor tissue. Healthy KM mice were injected via the tail vein for 2 weeks to assess the potential toxicity of NPs and Dp44mT at a dose of 2.5 mg Pt per kg body weight and 3 mg per kg body weight, respectively. During the treatment period, no apparent changes in the body weight loss were observed among the different groups (Fig. 6A). In addition, no obvious damage was found in the H&E staining of the major organs including the heart, liver, spleen, lungs and kidneys (Fig. 6B). Furthermore, we evaluated the functional toxicities of the major organs of the mice, and no apparent toxicity was seen among the different treatments (Fig. 6C). These results confirm the safety of the current nano-delivery system; it can prevent cisplatin from degradation during circulation and protect the body from its toxic side effects. Meanwhile, the current dose of Dp44mT used for the mice was also tolerable in vivo. As reported previously, cancer cells have high requirements of iron for their rapid growth than normal cells. In this study, the transient iron chelation with Dp44mT might only affect the proliferation and function of tumors rather than the other major organs. Collectively, these results suggest that our nano-formulation and iron chelator with this dose is safe and tolerable in vivo. | ||
| Fig. 6 In vivo toxicity of NPs and Dp44mT. (A) Body weight changes of mice with different treatments during the administration process. (B) H&E staining of the major organs including the heart, liver, spleen, lungs and kidneys of the mice with the different treatments. (C) Hematological analysis for the functional toxicities of the major organs of the mice. BUN and CR: indicators for kidney function; CK and CK-MB: indicators for heart function; ALT and AST: indicators for hepatic function. | ||
3.7 In vivo biodistribution and antitumor effects
In order to evaluate the tumor targeting efficacy of the NPs, Cy7.5@NPs were injected via the tail vein to 4T1-bearing Balb/c mice. At various time points post administration, the mice were imaged using an IVIS in vivo imaging system. The fluorescence intensity gradually increased as time progressed, which doubled from 0 h to 24 h (Fig. 7A, top panel, and Fig. 7B). After 24 h of injection, the mice were sacrificed and the main organs and tumors were isolated for ex vivo imaging. We found intense fluorescence in the tumors, demonstrating that the as-synthesized NPs would effectively accumulate in the tumor sites (Fig. 7A, bottom panel). The quantitative analysis of the fluorescence intensity further confirmed the accumulation of NPs in tumors (Fig. S5, ESI†).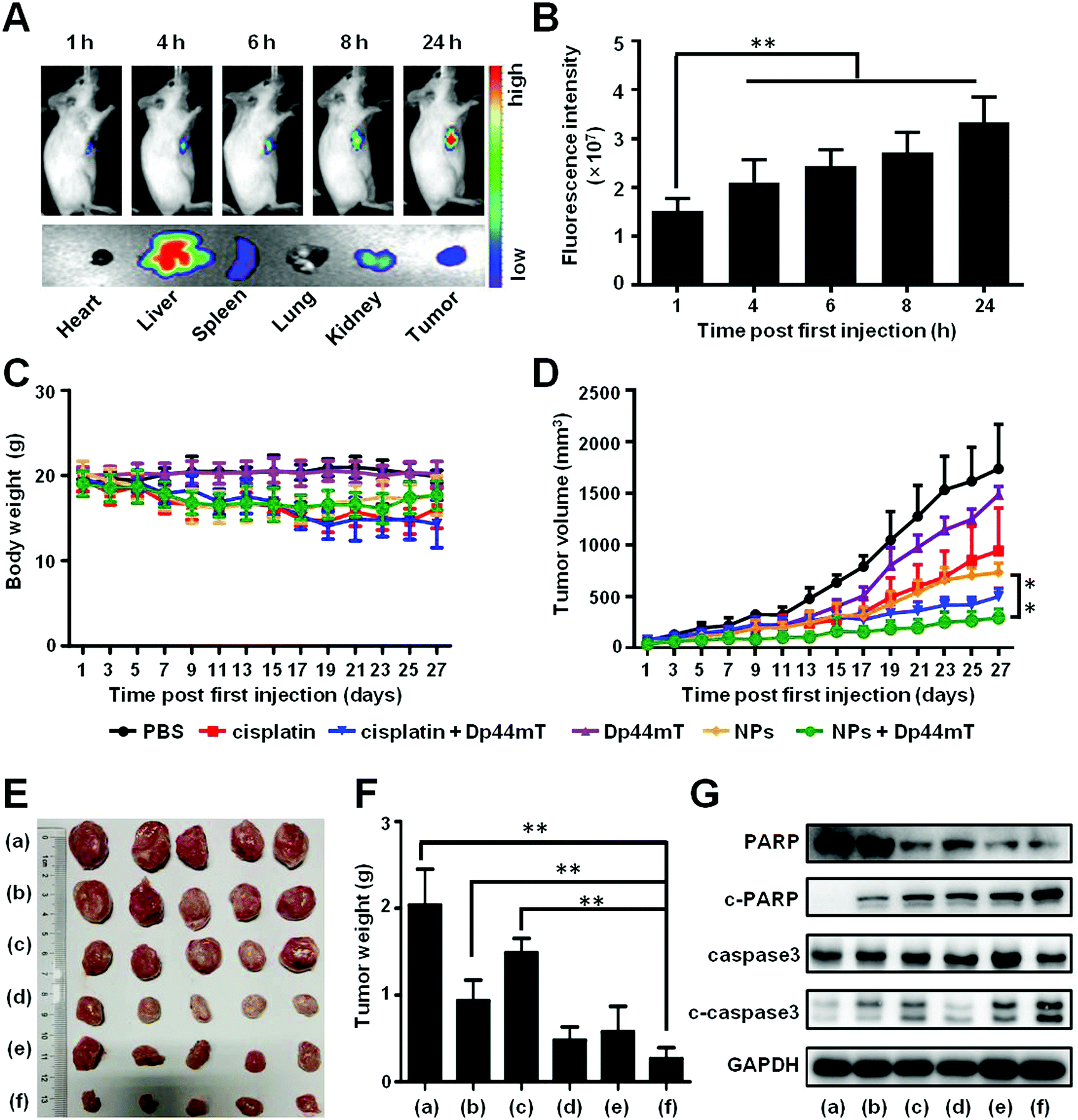 | ||
| Fig. 7 In vivo biodistribution and antitumor effects of the NPs with Dp44mT. (A) In vivo biodistribution of the NPs labelled with Cy7.5 at various time points post injection, and the ex vivo imaging of the main organs in mice bearing 4T1 xenografts 24 h post injection (IVIS, Spectrum CT, PerkinElmer, Ex/Em = 780 nm/810 nm). (B) Pt accumulation in tumors at various time points post injection via the tail vein. (C) Body weight changes and (D) tumor growth curves of the mice during the treatments. (E) Pictures and (F) weights of the tumors removed from the mice at the end of the administration period with different treatments as follows: (a) PBS, (b) cisplatin (2.5 mg Pt per kg body weight), (c) Dp44mT (3 mg Pt per kg body weight), (d) cisplatin (2.5 mg Pt per kg body weight) + Dp44mT (3 mg Pt per kg body weight), (e) NPs (2.5 mg Pt per kg body weight), and (f) NPs (2.5 mg Pt per kg body weight) + Dp44mT (3 mg Pt per kg body weight). (G) Protein expression of tumors with the above-mentioned treatments by western blot. **p < 0.01. | ||
Next, the antitumor efficiency was evaluated on 4T1-bearing Balb/c mice. As depicted in Fig. 7C, the average body weights of the mice treated with free cisplatin or cisplatin with Dp44mT decreased around 4 g, demonstrating that cisplatin itself is toxic to the tumor bearing mice. However, no negligible body weight loss occurred in mice from the other groups, which further confirms that the functionalized NPs as drug carriers could effectively reduce the adverse effect of cisplatin on the mice. Meanwhile, at the end of the administration, the tumor volumes of the mice with cisplatin or Dp44mT injection alone showed a 9-fold and 14-fold increase, respectively (Fig. 7D). However, co-treatment with cisplatin and Dp44mT resulted in a two times decrease of tumor volumes, which were further suppressed by the combination of NPs and Dp44mT. The different therapeutic efficacies were also confirmed by the images and weights of the tumors after the mice were sacrificed at the final stage of the in vivo studies (Fig. 7E and F). Consistent with changes in the tumor growth results, the smallest tumor mass was seen in the mice co-treated with NPs and Dp44mT. What's more, the pathological analysis (H&E staining) of tumor sections was performed. Highly concentrated cytoplasmic with a large number of vacuoles occurred in mice cotreated with NPs and Dp44mT, which displayed obvious necrosis (Fig. S6, ESI†). These results suggest that NPs with Dp44mT could effectively inhibit tumorigenesis with excellent therapeutic efficacy in vivo.
Besides, we explored the apoptotic mechanism of the primary tumors with the above treatments by western blot. In accordance with the in vitro results, combination of cisplatin and Dp44mT activated the apoptotic markers including c-caspase3 and c-PARP, whose expression levels were significantly up-regulated (Fig. 7G). Noteworthily, co-treatment with NPs and Dp44mT led to the highest expression levels of these apoptotic related proteins, which further confirms the satisfactory therapeutic effect of chemotherapy and iron chelator combination.
3.8 In vivo anti-metastatic effect
In view of the fact that breast cancer tends to metastasize to other tissues such as the lungs and liver, we further examined the anti-metastasis efficacy of the above treatments. In the current study, we established a 4T1 orthotopic mammary tumor metastasis model. At the end of the administration period, over 15 metastatic nodules were found in both the lung and liver of mice with saline injection, implying severe lung and liver metastases (Fig. 8A). Fortunately, combination therapy with NPs and Dp44mT effectively inhibited the metastasis of the 4T1 orthotopic mammary tumor. Histopathological examination of the lung and liver sections further confirmed the tumor metastasis (Fig. 8B). Compared to the control group mice, cisplatin or Dp44mT administration alone showed a slight anti-metastatic effect on the tumors. However, combined administration of cisplatin and Dp44mT significantly suppressed tumor metastasis with negligible metastatic nodules and a small area of the tumor burden in the tissue slides. Owing to the excellent anti-tumor effects, almost no tumor burden area appeared in the H&E staining images in the mice treated with NPs and Dp44mT, which suggests an attenuated risk of tumor metastasis.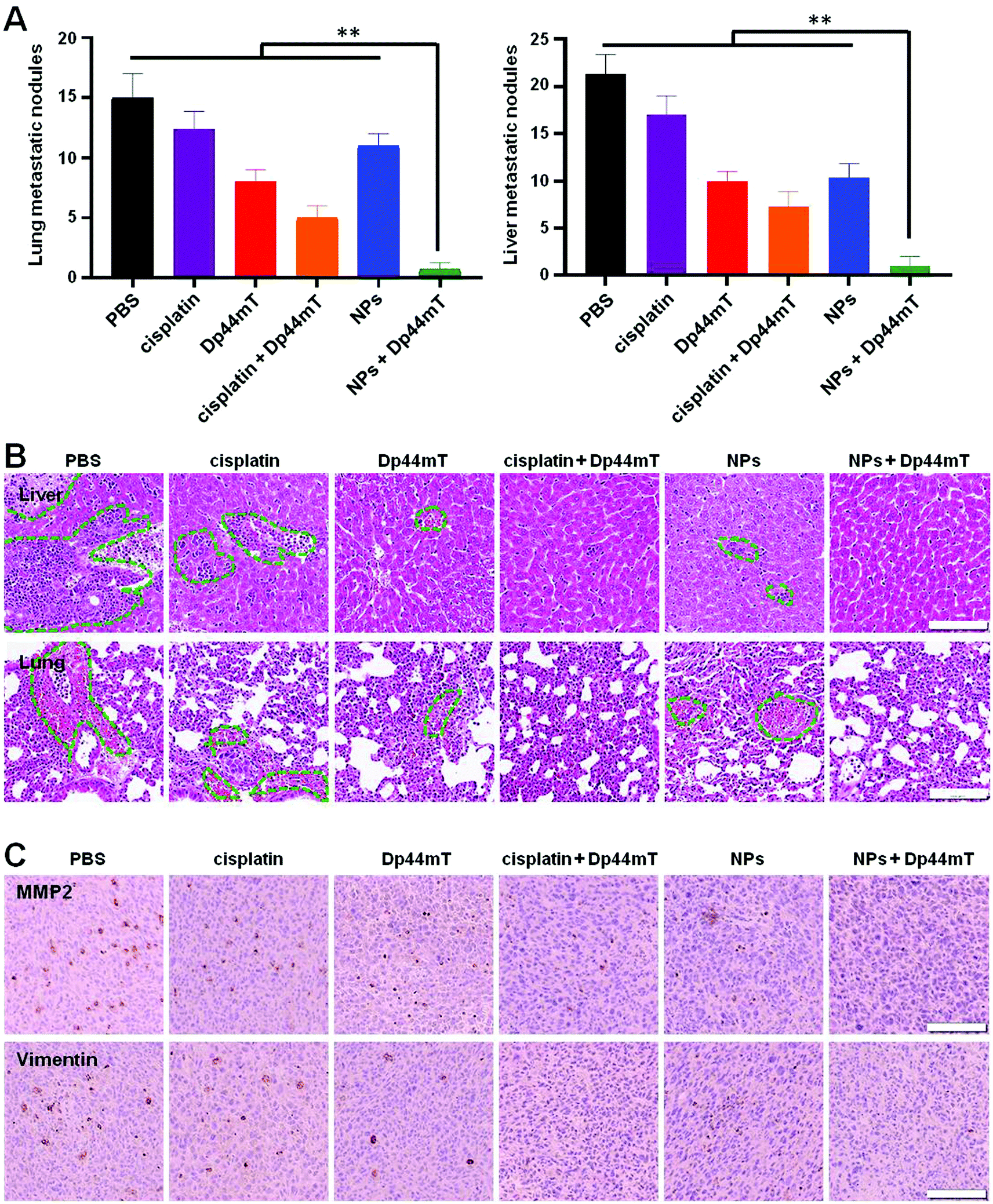 | ||
| Fig. 8 In vivo anti-metastatic effects of NPs and Dp44mT in 4T1 tumor bearing mice. (A) Numbers of lung and liver metastatic nodules with various treatments. **p < 0.01 compared to the NPs + Dp44mT group. (B) Histopathological examination of the liver and lung sections by H&E staining at the end of the administration period, respectively. Green circles represent liver or lung metastatic nodules. (C) IHC staining images with anti-MMP2 and anti-Vimentin in tumor sections with various treatments. Scale bars: 100 μm. | ||
Next, an immunohistochemical (IHC) experiment was carried out to investigate the molecular mechanisms of the above formulations against tumor metastasis. As reported previously, matrix metalloproteinase 2 (MMP2) and Vimentin are overexpressed in tumor tissues with metastatic potential; they are beneficial for cell motility and extracellular matrix (ECM) degradation.45 In the current study, tumor sections of the saline group displayed dark brown staining around the cell nucleus (blue), demonstrating the overexpression of MMP2 and Vimentin in tumors (Fig. 8C). Cisplatin or Dp44mT exposure alone had a minor inhibitory effect on the expression of the two markers. However, co-treatment with cisplatin and Dp44mT significantly suppressed the MMP2 and Vimentin expression. Noteworthily, due to their high accumulation in the tumors, the mice co-treated with NPs and Dp44mT showed inappreciable expression of MMP2 and Vimentin as no brown staining appeared around the cell nucleus. These results demonstrate that combined administration of NPs and Dp44mT exhibited excellent restriction of pulmonary and hepatic metastases via inhibition of the motility of cancer cells and suppressed the degradation of the ECM.
As mentioned before, VEGFα is a key protein in tumor angiogenesis with important roles in the proliferation and survival of endothelial cells as well as cell migration.46 Therefore, we also detected the expression level of VEGFα in tumors with the above treatments by western blot. It seemed that the expression of VEGFα was not down-regulated by the treatment with cisplatin or Dp44mT alone compared to the control group (Fig. S7A, ESI†). However, significant down-regulation of the VEGFα expression level was observed in tumors when co-treated with cisplatin and Dp44mT. In particular, the combination of NPs and Dp44mT resulted in a 5-fold decrease of the VEGFα level (Fig. S7B, ESI†). It is consistent with the in vitro western blot results, which further confirms the inhibitory effects of cisplatin and Dp44mT combination on tumor metastasis.
Taken together, combined use of NPs and Dp44mT could be considered as a promising therapeutic strategy for metastatic breast cancer, based on the effective inhibition of the orthotopic mammary tumor growth and excellent restriction of the liver and lung metastases.
4. Conclusion
In summary, we first confirmed that the combination use of cisplatin and Dp44mT has enhanced antitumor efficiency than each agent alone by inhibiting cancer cell viability and inducing cell apoptosis. Next, we constructed a nanoparticle delivery system to promote drug endocytosis in cancer cells. With excellent delivery capability, the as-synthesized NPs exhibited improved intracellular drug accumulation with less toxic side effects, which lead to excellent antitumor efficiency when co-delivered with Dp44mT. Besides, the combination of cisplatin and Dp44mT showed desirable inhibitory effects on breast cancer metastasis. In conclusion, as a versatile nano-platform for the combination of chemotherapy and iron chelators, the current design holds great potential in clinical treatment of metastatic cancer.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project was financially supported by the National Natural Science Foundation of China (No. 51873218 and No. 51803120), the Beijing Natural Science Foundation (2202071), the Key Research and Development Program of Hunan Province (2019SK2251) and the Guangci Distinguished Young Scholars Training Program (GCQN-2019-A06) of Ruijin Hospital, Shanghai JiaoTong University.References
- L. Wang, X. Li, Y. Mu, C. Lu, S. Tang, K. Lu, X. Qiu, A. Wei, Y. Cheng and W. Wei, J. Trace Elem. Med. Biol., 2019, 56, 131–138 CrossRef CAS PubMed.
- A. R. Bogdan, M. Miyazawa, K. Hashimoto and Y. Tsuji, Trends Biochem. Sci., 2016, 41, 274–286 CrossRef CAS PubMed.
- S. Shinoda, S. Kaino, S. Amano, H. Harima, T. Matsumoto, K. Fujisawa, T. Takami, N. Yamamoto, T. Yamasaki and I. Sakaida, Oncotarget, 2018, 9, 28434 CrossRef PubMed.
- Y. Katsura, T. Ohara, K. Noma, T. Ninomiya, H. Kashima, T. Kato, H. Sato, S. Komoto, T. Narusaka and Y. Tomono, Cancers, 2019, 11, 177 CrossRef CAS PubMed.
- J. L. Buss, B. T. Greene, J. Turner, F. M. Torti and S. V. Torti, Curr. Top. Med. Chem., 2004, 4, 1623–1635 CrossRef CAS PubMed.
- J. L. Hamilton, M. Imran ul-haq, S. Abbina, M. T. Kalathottukaren, B. F. Lai, A. Hatef, S. Unniappan and J. N. Kizhakkedathu, Biomaterials, 2016, 102, 58–71 CrossRef CAS PubMed.
- F. Taghavi, M. Gholizadeh, A. S. Saljooghi and M. Ramezani, MedChemComm, 2017, 8, 1953–1964 RSC.
- L. Wang, X. Li, Y. Mu, C. Lu, S. Tang, K. Lu, X. Qiu, A. Wei, Y. Cheng and W. Wei, J. Trace Elem. Med. Biol., 2019, 56, 131–138 CrossRef CAS PubMed.
- A. Salehabadi, M. Salavati-Niasari and M. Ghiyasiyan-Arani, J. Alloys Compd., 2018, 745, 789–797 CrossRef CAS.
- M. Lazaridou, E. Christodoulou, M. Nerantzaki, M. Kostoglou, D. A. Lambropoulou, A. Katsarou, K. Pantopoulos and D. N. Bikiaris, Pharmaceutics, 2020, 12, 238 CrossRef PubMed.
- J. Lang, X. Zhao, X. Wang, Y. Zhao, Y. Li, R. Zhao, K. Cheng, Y. Li, X. Han and X. Zheng, ACS Nano, 2019, 13, 2176–2189 CAS.
- C. Chen, P. Liu, X. Duan, M. Cheng and L. X. Xu, OncoTargets Ther., 2019, 12, 4359 CrossRef CAS PubMed.
- S. Menezes, Z. Kovacevic and D. Richardson, Free Radical Biol. Med., 2018, 120, 76–77 CrossRef.
- S. Krishan, S. Sahni, L. Leck, P. Jansson and D. Richardson, Biochim. Biophys. Acta, Mol. Basis Dis., 2020, 165657 CrossRef CAS PubMed.
- J.-C. Lee, K.-C. Chiang, T.-H. Feng, Y.-J. Chen, S.-T. Chuang, K.-H. Tsui, L.-C. Chung and H.-H. Juang, Int. J. Mol. Sci., 2016, 17, 1435 CrossRef PubMed.
- P. Li, X. Zheng, K. Shou, Y. Niu, C. Jian, Y. Zhao, W. Yi, X. Hu and A. Yu, Am. J. Transl. Res., 2016, 8, 5370 CAS.
- J. Wang, D. Yin, C. Xie, T. Zheng, Y. Liang, X. Hong, Z. Lu, X. Song, R. Song, H. Yang, B. Sun, N. Bhatta, X. Meng, S. Pan, H. Jiang and L. Liu, Oncotarget, 2014, 5, 8478–8491 Search PubMed.
- H. Xiao, L. Yan, E. M. Dempsey, W. Song, R. Qi, W. Li, Y. Huang, X. Jing, D. Zhou and J. Ding, Prog. Polym. Sci., 2018, 87, 70–106 CrossRef CAS.
- Z. Gu, S. Zhu, L. Yan, F. Zhao and Y. Zhao, Adv. Mater., 2019, 31, 1800662 CrossRef PubMed.
- W. H. Chen, G. F. Luo and X. Z. Zhang, Adv. Mater., 2019, 31, 1802725 CrossRef PubMed.
- R. Zhang, X. Song, C. Liang, X. Yi, G. Song, Y. Chao, Y. Yang, K. Yang, L. Feng and Z. Liu, Biomaterials, 2017, 138, 13–21 CrossRef CAS PubMed.
- T. Wu, J. Liu, M. Liu, S. Liu, S. Zhao, R. Tian, D. Wei, Y. Liu, Y. Zhao and H. Xiao, Angew. Chem., 2019, 58, 14224–14228 CrossRef CAS PubMed.
- L. Shu, L. Ren, Y. Wang, T. Fang, Z. Ye, W. Han, C. Chen and H. Wang, Chem. Commun., 2020, 56, 3069–3072 RSC.
- Z. Ren, S. Sun, R. Sun, G. Cui, L. Hong, B. Rao, A. Li, Z. Yu, Q. Kan and Z. Mao, Adv. Mater., 2020, 32, 1906024 CrossRef CAS PubMed.
- A. Yimit, O. Adebali, A. Sancar and Y. Jiang, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- A. Quarta, M. Amorín, M. J. Aldegunde, L. Blasi, A. Ragusa, S. Nitti, G. Pugliese, G. Gigli, J. R. Granja and T. Pellegrino, Nanoscale, 2019, 11, 23482–23497 RSC.
- M. Miyazawa, A. R. Bogdan and Y. Tsuji, Cell Chem. Biol., 2019, 26, 85–97 CrossRef CAS PubMed.
- N. Wang, Z. Wang, Z. Xu, X. Chen and G. Zhu, Angew. Chem., 2018, 57, 3426–3430 CrossRef CAS PubMed.
- Q. Li, T. Qin, Z. Bi, H. Hong, L. Ding, J. Chen, W. Wu, X. Lin, W. Fu, F. Zheng, Y. Yao, M. L. Luo, P. E. Saw, G. M. Wulf, X. Xu, E. Song, H. Yao and H. Hu, Nat. Commun., 2020, 11, 1456 CrossRef CAS PubMed.
- Y. Zhai, W. Ran, J. Su, T. Lang, J. Meng, G. Wang, P. Zhang and Y. Li, Adv. Mater., 2018, 30, 1802378 CrossRef PubMed.
- Z. Ma, L. Wu, K. Han and H. Han, Nanoscale Horiz., 2019, 4, 1124–1131 RSC.
- J. Li, Y. Liu, X. Li, G. Liang, C. Ruan and K. Cai, Nanoscale Horiz., 2020, 5, 350–358 RSC.
- P. a. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang and Z. Gu, Nano Lett., 2017, 17, 928–937 CrossRef CAS PubMed.
- Z. Zhang, G. Kuang, S. Zong, S. Liu, H. Xiao, X. Chen, D. Zhou and Y. Huang, Adv. Mater., 2018, 30, 1803217 CrossRef PubMed.
- T. Fang, Z. Ye, J. Wu and H. Wang, Chem. Commun., 2018, 54, 9167–9170 RSC.
- Z. Chen, X. Kang, Y. Wu, H. Xiao, X. Cai, S. Sheng, X. Wang and S. Chen, Chem. Commun., 2019, 55, 4781–4784 RSC.
- C. Yang, K. Tu, H. Gao, L. Zhang, Y. Sun, T. Yang, L. Kong, D. Ouyang and Z. Zhang, Biomaterials, 2020, 232, 119751 CrossRef CAS PubMed.
- X. Wan, J. J. Beaudoin, N. Vinod, Y. Min, N. Makita, H. Bludau, R. Jordan, A. Wang, M. Sokolsky and A. V. Kabanov, Biomaterials, 2019, 192, 1–14 CrossRef CAS PubMed.
- C. Xu, Y. Wang, Z. Guo, J. Chen, L. Lin, J. Wu, H. Tian and X. Chen, J. Controlled Release, 2019, 295, 153–163 CrossRef CAS PubMed.
- J. Li, D. Cui, Y. Jiang, J. Huang, P. Cheng and K. Pu, Adv. Mater., 2019, 31, 1905091 CrossRef CAS PubMed.
- X. Kang, Y. Yu, Z. Chen, Y. Wu, D. Wei, Y. Zhao, F. Wang and H. Xiao, J. Mater. Chem. B, 2019, 7, 3346–3350 RSC.
- Y. Lu, Z. Yue, J. Xie, W. Wang, H. Zhu, E. Zhang and Z. Cao, Nat. Biomed. Eng., 2018, 2, 318–325 CrossRef CAS PubMed.
- W. Zhang, X. Hu, Q. Shen and D. Xing, Nat. Commun., 2019, 10, 1–14 CrossRef PubMed.
- M. Yu, D. Su, Y. Yang, L. Qin, C. Hu, R. Liu, Y. Zhou, C. Yang, X. Yang and G. Wang, ACS Appl. Mater. Interfaces, 2018, 11, 176–186 CrossRef PubMed.
- L. Liu, F. Hu, H. Wang, X. Wu, A. S. Eltahan, S. Stanford, N. Bottini, H. Xiao, M. Bottini and W. Guo, ACS Nano, 2019, 13, 5036–5048 CrossRef CAS PubMed.
- L. Rao, G. T. Yu, Q. F. Meng, L. L. Bu, R. Tian, L. S. Lin, H. Deng, W. Yang, M. Zan and J. Ding, Adv. Funct. Mater., 2019, 29, 1905671 CrossRef CAS.
- W. Ma, S. N. Sha, P. L. Chen, M. Yu, J. J. Chen, C. B. Huang, B. Yu, Y. Liu, L. H. Liu and Z. Q. Yu, Adv. Healthcare Mater., 2019, 1901100 Search PubMed.
- X. Ma, Y. Wang, X.-L. Liu, H. Ma, G. Li, Y. Li, F. Gao, M. Peng, H. M. Fan and X.-J. Liang, Nanoscale Horiz., 2019, 4, 1450–1459 RSC.
- F. Arshad, M. F. Khan, W. Akhter, M. M. Alam, L. M. Nainwal, S. K. Kaushik, M. Akhter, S. Parvez, S. M. Hasan and M. Shaquiquzzaman, Eur. J. Med. Chem., 2019, 167, 324–356 CrossRef CAS PubMed.
- K. Ohui, E. Afanasenko, F. Bacher, R. L. X. Ting, A. Zafar, N. R. Blanco-Cabra, E. Torrents, O. Dömötör, N. R. V. May and D. Darvasiova, J. Med. Chem., 2018, 62, 512–530 CrossRef PubMed.
- C. Holley, S. Alkhalifah and S. Majd, 40th Annual International Conference of the IEEE EMBC, 2018, 5733-5736.
- J. Sun, L. Zhang, J. Wang, Q. Feng, D. Liu, Q. Yin, D. Xu, Y. Wei, B. Ding and X. Shi, Adv. Mater., 2015, 27, 1402–1407 CrossRef CAS PubMed.
- Y. Yang, Y. Zhang, H. Iwamoto, K. Hosaka, T. Seki, P. Andersson, S. Lim, C. Fischer, M. Nakamura and M. Abe, Nat. Commun., 2016, 7, 12680 CrossRef CAS PubMed.
- C.-S. Devignes, Y. Aslan, A. Brenot, A. Devillers, K. Schepers, S. Fabre, J. Chou, A.-J. Casbon, Z. Werb and S. Provot, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 992–1001 CrossRef PubMed.
- C.-W. Lin, L.-K. Wang, S.-P. Wang, Y.-L. Chang, Y.-Y. Wu, H.-Y. Chen, T.-H. Hsiao, W.-Y. Lai, H.-H. Lu and Y.-H. Chang, Nat. Commun., 2016, 7, 1–16 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00148a |
| This journal is © The Royal Society of Chemistry 2020 |