
A minimalistic hydrolase based on co-assembled cyclic dipeptides†
Alexander J.
Kleinsmann
a and
Boris J.
Nachtsheim
 *b
*b
aInstitut für Organische Chemie Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
bInstitut für Organische und Analytische Chemie Universität Bremen, Leobener Straße 7, 28359 Bremen, Germany. E-mail: nachtsheim@uni-bremen.de
First published on 26th November 2019
Abstract
The self-assembly of small peptides into larger aggregates is an important process for the fundamental understanding of abiogenesis. In this article we demonstrate that blends of cyclic dipeptides (2,5-diketopiperazines – DKPs) bearing either histidine or cysteine in combination with a lipophilic amino acid form highly stable aggregates in aqueous solution with esterase-like activity. We demonstrate that the catalytic activity is based on an intermolecular cooperative behavior between histidine and cysteine. A high control of the molecular arrangement of the peptide assemblies was gained by C–H-π interactions between Phe and Leu or Val sidechains, resulting in a significant increase in catalytic activity. These interactions were strongly supported by Hartree–Fock calculations and finally confirmed via1H-NMR HRMAS NOE spectroscopy.
Introduction
The transition of simple small molecular building blocks, in particular fatty-, amino- and nucleic acids, into self-replicating systems with an autonomous metabolism is the critical step for the emergence of the first living cells with a minimalistic genotype and phenotype.1 The initial manifestation of small peptides as enzyme precursors that could have provided important catalytic properties for autonomous self-replicating systems is still underexplored.2,3 Here, self-assembly processes that form higher ordered aggregates from spontaneously formed small oligopeptides through intermolecular H-bonding interactions is believed to be an important initial step.2,4a–dArtificial enzymes, in particular esterases based on the self-assembly of short oligopeptides have been described frequently.5 Gazit and co-workers recently demonstrated that even single amino acids, in particular phenylalanine, in the presence of zinc can form supramolecular amyloid-like structures that render a potent esterase activity.6 However, usually, lipophilic tripeptides, amphiphilic oligopeptides or amyloid-forming peptides are necessary to generate self-assembled nanostructures with esterase-like activity.7 Catalytically active aggregates can also be formed based on artificial dendrimers, by fixation of a peptide onto nanoparticles,8 or the generation of other amino acid-derived hybrids.9,10 The relevance of these approaches in abiogenesis is questionable due to the artificial nature of the underlying molecular building blocks.
In this regard, cyclic dipeptides (2,5-diketopiperazines – DKPs) are observed frequently as undesired side-products during peptide formation and under prebiotic conditions,11 in particular as degradation products of small oligopeptides.12 They form spontaneously upon thermal treatment of linear dipeptides in the solid state.13 In addition, they have been found on the Yamato-791198 and Murchison carbonaceous chondrites.14 We recently demonstrated that a variety of Phe-containing DKPs form highly stable aggregates in aqueous solutions.15 Their self-aggregation is the result of strong H-bonding interactions between the cyclic amides and additional π–π– or C–H-π-interactions between the Phe sidechains. This was further revealed by fundamental investigations of Govindaraju and co-workers on cyclic dipeptides containing Phe- or PhG side chains or Fmoc-protected lysine-derived DKPs.16,17 Intermolecular hydrogen bond patterns have also been observed in solid state structures of DKPs, for example by Marchesan and co-workers very recently.18 For proving the relevance of DKPs in the context of abiogenesis, their catalytic properties must be elucidated. So far this has only been demonstrated by Lipton and co-workers in solution for the asymmetric Strecker reaction as well as in epoxidations in multiphase systems as shown by Voyer and coworkers.19,20 Presuming their high tendency to aggregate in water into a defined molecular arrangement, we proposed that simple blends of two DKPs composed of proteinogenic α-amino acids with lipophilic side chains and differing “functional” side chains should render enzyme-like catalytic activity in the co-assembled state through intermolecular cooperative effects.
Results and discussion
To verify this working hypothesis, we generated a minimalistic hydrolase mimic (Fig. 1a). | ||
| Fig. 1 (a) DKP-based mimic of a catalytic dyade (b) chemical structure of investigated DKPs. | ||
In the catalytically active side of hydrolases, imidazoles of His-residues are in close proximity to Ser, Cys or Asp sidechains as the structural basis for catalytic dyads or triades. To the best of our knowledge simple dipeptides without non-natural synthetic modifications are not known as minimalistic esterase mimics. Based on our recent findings towards the outstanding self-aggregation properties of DKPs, we combined His-DKP 1 and Cys-DKPs (2, 3 and 4) as shown in Fig. 1b. These three different blends [1+2], [1+3] and [1+4] should give co-assembled nanostructures with a putative esterase activity.21 We first investigated the principle co-aggregation properties of all three blends. Co-assembly was verified through hydrogel formation and subsequent investigation of the freeze-dried hydrogels via SEM (Fig. 2). All three blends formed stable hydrogels through a simple heating/cooling cycle in pure water at concentrations between 80 and 106 mM. SEM and TEM images showed the appearance of nanofibers with varying average diameters ([1+2]: 12.3 nm, [1+3]: 32.6 nm and [1+4]: 21.4 nm) (for detailed analysis of representative SEM-images see ESI†).
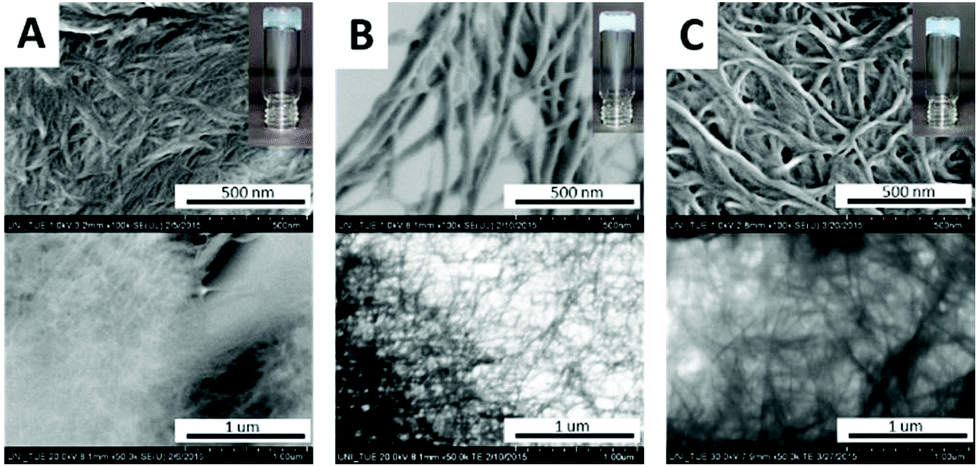 | ||
| Fig. 2 SEM- and TEM-images of co-assembled DKP-blends A: [1 + 2]; B: [1 + 3]; C: [1 + 4]. | ||
To investigate esterase-like activity of the co-aggregates, the hydrolysis of sodium 4-acetoxy-3-nitrobenzenesulfonate (ANBS, Fig. 1a), a water-soluble derivative of the common model compound 2,4-dinitrophenyl acetate (DNPA), was chosen as the model reaction. A solution of ANBS was added on top of the preformed hydrogel and reaction kinetics were monitored by UV/Vis. Initial Job's plot analysis revealed a maximum initial rate constant v0 at X = 0.4 for blend [1+2] and X = 0.5 for blends [1+3] and [1+4] (Fig. 3A). We then investigated the pH-dependency of the ester hydrolysis (Fig. 3B). While with [1+2]v0 reaches a maximum at pH = 7.50, co-assemblies of [1+3] and [1+4] reached explicit maxima at slightly lower pH-values (7.25 and 7.38).
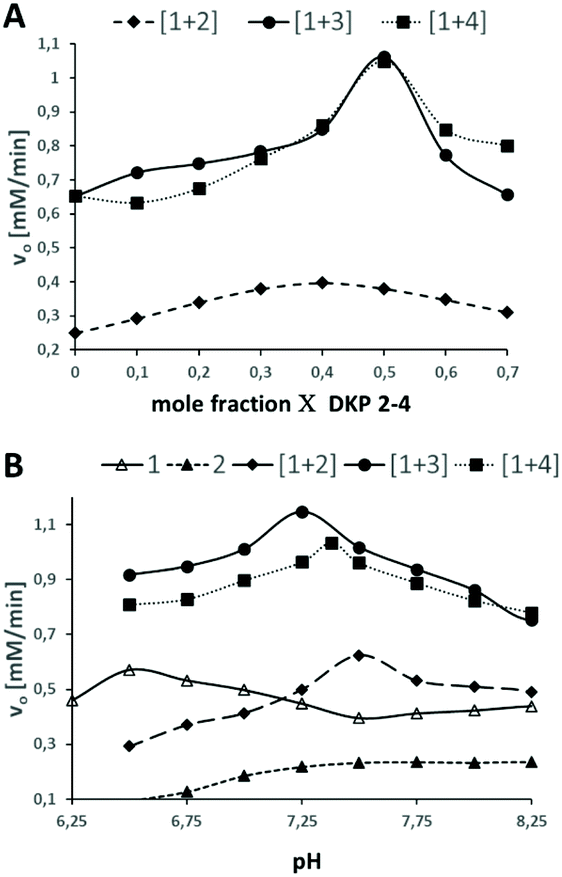 | ||
| Fig. 3 A: Job's plot analysis of DKP-blends [1+2], [1+3] and [1 + 4]. B: pH-dependency of the initial rate constants. | ||
In sharp contrast, v0 of pure self-assembled 1 has a maximum at a pH of 6.50 which corresponds well to the pKa-value of His. For self-assembled DKP 2v0 increases until pH 7.5 and reaches a plateau. The broad maximum of Job's plot analysis for blend [1+2] together with the slight shift from the theoretical optimal ratio of both DKPs from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 as observed for [1+3] and [1+4] is indicative for a random distribution of 1 and 2 within the fibrous network (Fig. 4A). The sharp maxima at X = 0.5 for [1+3] and [1+4] on the other hand indicate a highly defined co-assembly of both DKPs (Fig. 4B).
:1 as observed for [1+3] and [1+4] is indicative for a random distribution of 1 and 2 within the fibrous network (Fig. 4A). The sharp maxima at X = 0.5 for [1+3] and [1+4] on the other hand indicate a highly defined co-assembly of both DKPs (Fig. 4B).
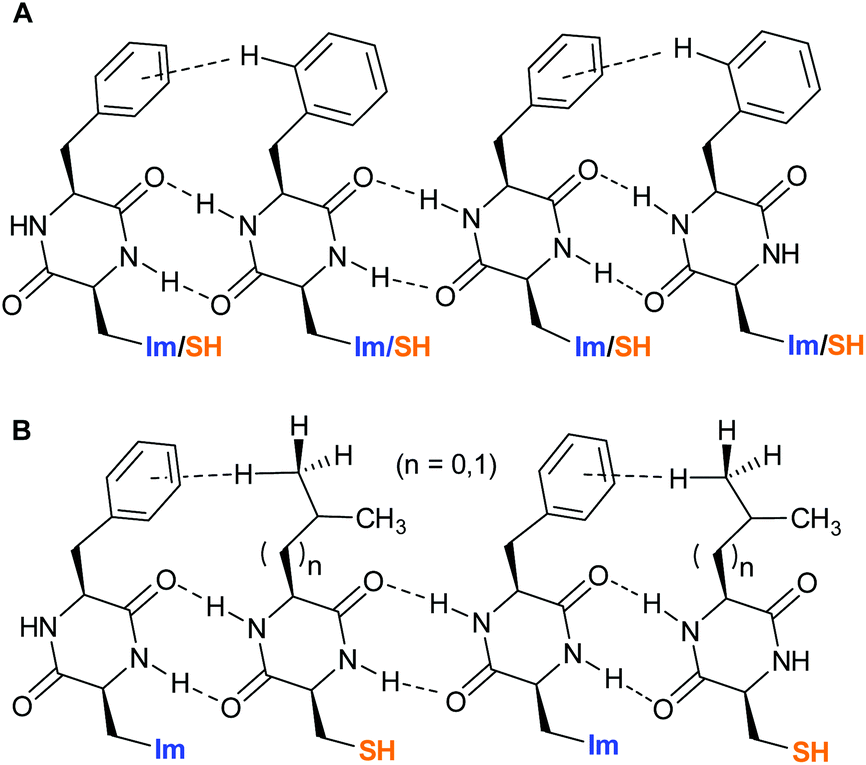 | ||
| Fig. 4 A: Random distribution of DKPs 1 and 2 within the co-assembly. B: Alternating distribution of DKPs 1 and 3 or 4 within the co-assembly. | ||
This defined alternating co-assembly should also result in a better catalytic performance of blends [1+3] and [1+4], as already indicated by the significantly higher v0-values. Next, we wanted to compare v0 of the DKPs between the co-assembled hydrogel state and a solution by disturbing the co-assembly process through DMF addition. In general, v0 should be reduced for the hydrogels since substrate availability is initially strongly limited by diffusion processes. In addition, the accessibility of the catalytically active His and Cys residues should be strongly limited in the self-assembled hydrogel state through intermolecular interactions of individual strands to form the three-dimensional network. As an initial control experiment, we tested the catalytic activity of pure His-DKP 1 in solution (Fig. 5A, dotted line). Even though 1 accelerated ANBS hydrolysis, it cannot be defined as catalyst. The solution exhibits a fast initial reaction turnover in the first 10 minutes and finally approaches asymptotically a substrate conversion that matches the total DKP concentration. Hence, only one turnover is observed. With the same absolute molarity, the corresponding hydrogel of 1 shows an inferior substrate conversion, also with a strongly decelerating slope finally converging to an overall conversion close to the DKP concentration (Fig. 5A, solid line). This diminished reactivity strongly indicates the lower accessibility of the His-residues in the aggregated state. The observed saturation in both the solution and the gel state of 1 indicates a quick N-acetylation of the His-residue followed by a very slow deacetylation, excluding catalytic behaviour.
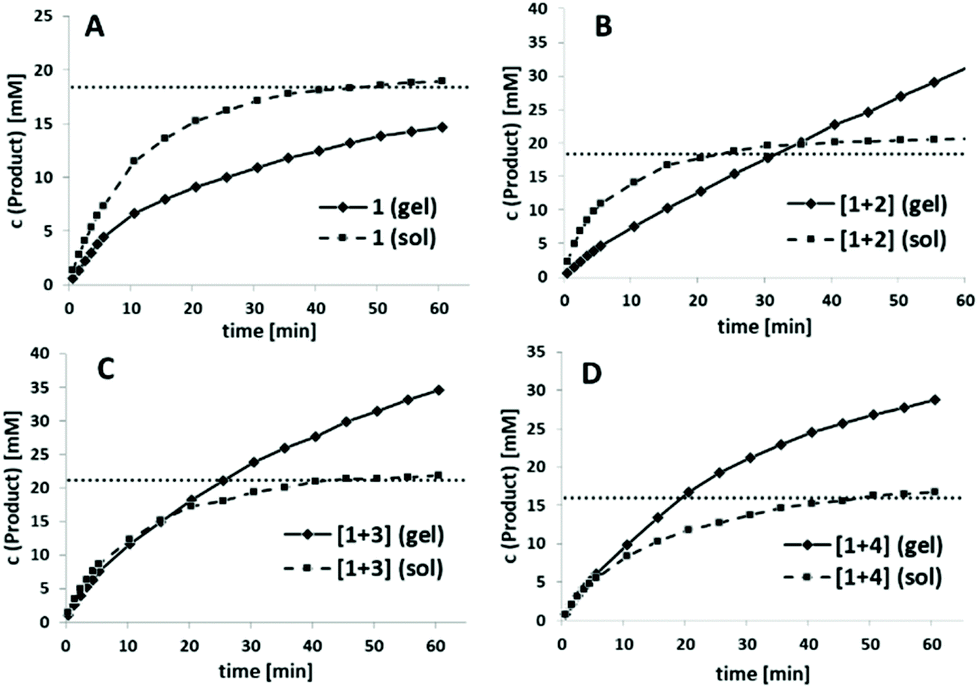 | ||
| Fig. 5 Product formation in ANBS hydrolysis, c (ANBS) = 60 mM; solid lines: reaction was performed in the self-assembled hydrogel (gel); dashed lines: reaction was performed in solution (sol) (HEPES:DMF = 1:1, V = 1.25 ml); dotted horizontal lines: total DKP concentration referenced to the total volume; A: 1, pH = 6.50, c (1-hydrogel) = 92 mM; B: [1+2] (1.5:1), pH = 7.50, c = 92 mM; C: [1+3] (1:1), pH = 7.25, c = 106 mM; D: [1+4] (1:1), pH = 7.38, c = 80 mM. Product conversion was detected via UV/Vis at λ = 406 nm. In all experiments background hydrolysis of ANBS was measured in the corresponding buffers at the same pH with identical substrate concentration and subtracted. | ||
Next we investigated the corresponding blends (Fig. 5B–D). In all cases the self-assembled blended DKPs were compared with the corresponding DKPs kept in solution as a control. As already observed for pure 1, all blended solutions, even though accelerating ester hydrolysis, provided only one turnover. Catalytic behaviour is only observed for blended hydrogels. For [1+2] total conversion of the solution again converges to the initial DKP-concentration while in the co-assembled state product concentration exceeds DKP-concentration after 35 min (Fig. 5B).
A similar catalytic behaviour was observed for [1+3] and [1+4], although, as already indicated in Fig. 3A, ANBS hydrolysis was throughout faster, exceeding the initial DKP concentration after 20–25 min. In sharp contrast to pure 1 and [1+2], initial hydrolysis rates using the co-assembled blends [1+3] and [1+4] were comparable to the solution phase experiments (Fig. 5C and D). Overall, the co-assembled blend [1+4] shows the best results in direct comparison with the corresponding solution phase and in direct comparison to the other blends.
For a more precise comparison of their catalytic efficiency, v0 was investigated in dependence of the substrate concentration at the optimal pH and ratio for each blend. The Michaelis–Menten enzyme kinetics model was used to calculate the rate constants for all co-assembled hydrogels. In all blends, catalyst turnover became the rate-limiting step at very high substrate concentrations, a typical behaviour for enzyme-catalysed reactions (see ESI – Table S2†). Michaelis Menten constants (KM), rate constants (Kcat) as well as the catalytic efficiencies (Kcat/KM) are given in Table 1.
The highest substrate-affinity and the highest catalytic efficiency was once again observed for blend [1+4] (KM = 6.81).
K cat values between [1+3] and [1+4] differ only insignificantly but KM is twofold higher for [1+3]. This is indicative for a significantly weaker substrate affinity and might be the result of sterically more favourable or multiple C–H-π-interactions between 1 and 4 which subsequently leads to a closer proximity of the imidazole and thiol functionalities at the catalytic active site of the dipeptide.
To verify this hypothesis, gas phase calculations based on the low cost Hartree–Fock/minimal basis set composite method HF-3C which shows excellent performance for noncovalent interactions have been accomplished for dimers of [1+2], [1+3] and [1+4] (Fig. 6).22 Each energy minimized structure confirms two central intermolecular H-bonds between the two cyclic amides with typical O–H-distances ranging from 1.74 to 1.92 Å. H-Bonds between the lipophilic amino acids are significantly longer (1.89–1.92 Å) than the H-bonds between the His and Cys amino acids (1.74–1.76 Å). As predicted, all lipophilic side chains show significant C–H-π-interactions. For [1+2] two C–H-π-interactions of the ortho- and meta protons of the Phe side chain in 2 and the π-system of the Phe side chain in 1 give a disordered T-shape geometry between the two benzene rings with C–H-π-distances of 2.80 and 3.17 Å. In the calculated structure of [1+3] two significant C–H-π-interaction between two C–Hγ protons of the terminal CH3-group of the Val side chain in 3 and the benzene ring in 1 exist. The calculated C–H-π-distances to the centroid of the benzene ring is 3.04 Å and 3.05 Å to the centroid of the C3–C4-π-bond. For [1+4], two C–H-π-interaction are observed with C–H-π-distances of 2.70 and 2.77 Å between C–Hδ protons of both terminal CH3 groups and two distinct C–C-π-bonds of Phe. All distances are in good agreement with typical average distances of C–H-π interactions as observed in solid state protein structures.23 Obviously, the additional methylene group in the side chain of 4 allows a significantly stronger C–H-π-interaction as implicated by shorter C–H-centroid distances.
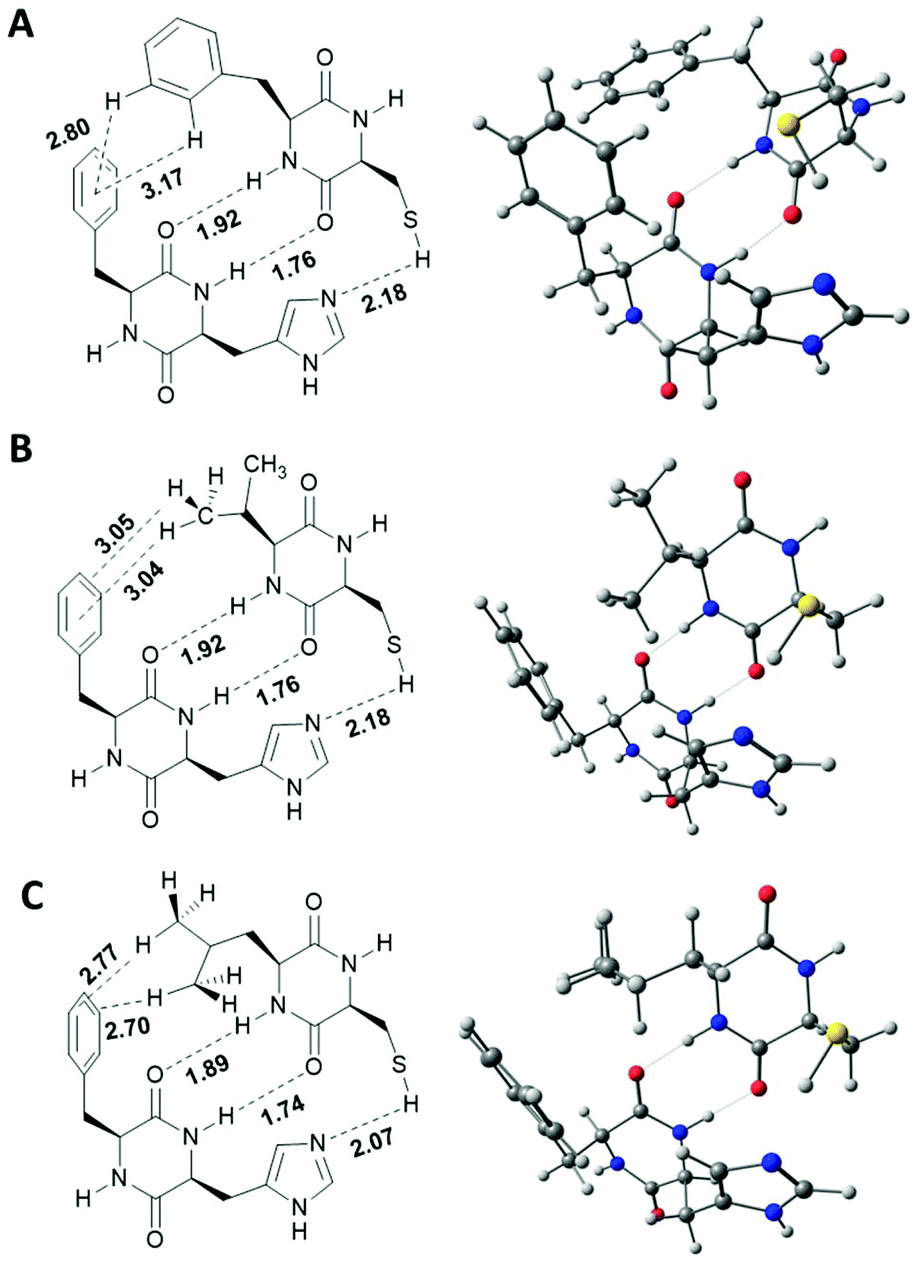 | ||
| Fig. 6 Calculated structures of DKP-dimers. A: [1+2]; B: [1+3]; C: [1+4]. Structures were calculated using the semi-empirical HF-3c functional in the gas phase. | ||
In all blends, combination of the two central amide hydrogen bonds and the additional C–H-π-interaction arranges the functional imidazole and thiol functionalities into close proximity. Calculated S–H–N-distances vary from 2.18 Å in [1+2] and [1+3], and 2.07 Å for [1+4]. It has to be mentioned, that the horizontal dimension of these calculated single-strands (approx. 1 nm) is one dimension below the observed fibre thickness as observed via SEM and TEM. Thus, further inter-strand interactions must be operational which strongly limits the true accessibility of the catalytically active sides in the self-assembled state. Under this premise it is even more surprising that blends [1+3] and [1+4] show similar initial hydrolysis rates in comparison to the corresponding DKPs kept in solution.
To finally verify that the calculated alternating co-assembly in [1+3] (Fig. 7A) and [1+4] (Fig. 7B) is based on C–H-π-interactions, 1H HRMAS NOESY experiments of the co-assembled hydrogels were performed in D2O. Clearly, the strongest NOE correlation was observed between C–Hγ of 3 or C–Hδ of 4 and C–Haryl of 1 supporting the calculated structure of this blend.24
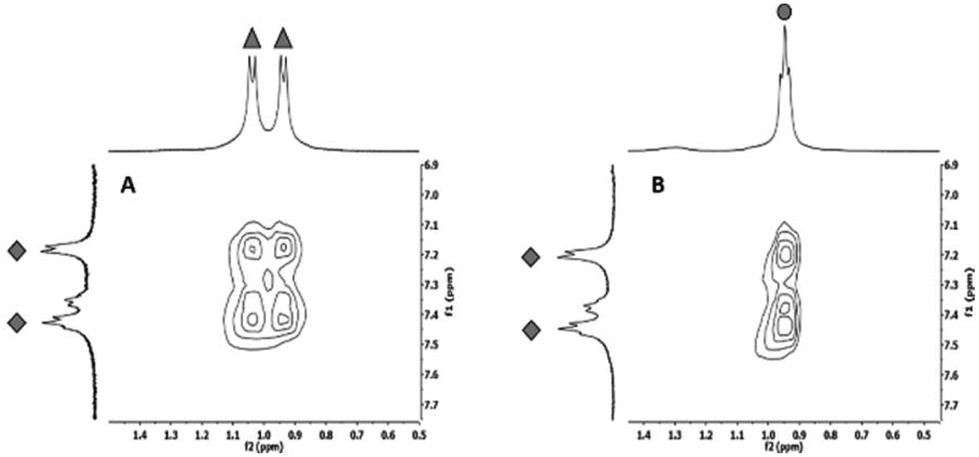 | ||
| Fig. 7 A: Detail magnifications of 1H HRMAS NOE spectra in D2O of A: Hydrogel [1+3] (1:1) and B: Hydrogel [1+4] (1:1); diamonds: aromatic protons of the Phe sidechain of 1, triangles: C–Hγ protons of the Val sidechain of 3, circles: C–Hδ protons of the Leu sidechain of 4. | ||
Conclusions
In summary we described the most minimalistic peptide self-assembly with an enzyme-like activity. It is based on abiogenesis relevant cyclic dipeptides solely build from the proteinogenic amino acids Phe, His, Val, Leu and Cys. A catalytic turnover is exclusively observed in the self-aggregated state for heterologous mixtures (blends) of His- and a Cys-containing cyclic dipeptides. Hartree–Fock calculations as well as HRMAS NOE experiments strongly indicate that C–H-π-interactions as well as intermolecular amide hydrogen bonds are responsible for the heterologous self-aggregation which finally leads to a close proximity of His and Cys side chain to give a catalytic dyade. These findings offer a new understanding toward a potential role of the so far undervalued role of cyclic dipeptides in chemical evolution and further implies the importance of self-assembled peptide aggregates in the pre-Darwin evolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial Support by the Fonds der Chemischen Industrie (FCI) is gratefully acknowledged.References
- T. A. E. Jakschitz and B. M. Rode, Chem. Soc. Rev., 2012, 41, 5484 RSC.
- G. Danger, R. Plasson and R. Pascal, Chem. Soc. Rev., 2012, 41, 5416 RSC.
- (a) A. Brack, Pure Appl. Chem., 1993, 65, 1143 CAS; (b) A. Brack, Chem. Biodiversity, 2007, 4, 665 CrossRef CAS PubMed.
- (a) B. M. Rode, Peptides, 1999, 20, 773 CrossRef CAS PubMed; (b) I. Ghosh and J. Chmielewski, Curr. Opin. Chem. Biol., 2004, 8, 640 CrossRef CAS PubMed; (c) O. Carny and E. Gazit, FASEB J., 2005, 19, 1051 CrossRef CAS PubMed; (d) J. Greenwald, M. P. Friedmann and R. Riek, Angew. Chem., Int. Ed., 2016, 55, 11609 CrossRef CAS PubMed.
- (a) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734 RSC; (b) E. Kuah, S. Toh, J. Yee, Q. Ma and Z. Gao, Chem. – Eur. J., 2016, 22, 8404 CrossRef CAS PubMed; (c) N. Singh, M. Kumar, J. F. Miravet, R. V. Ulijn and B. Escuder, Chem. – Eur. J., 2017, 23, 981 CrossRef CAS PubMed; (d) M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, ACS Catal., 2018, 9, 168 CrossRef; (e) O. Zozulia, M. A. Dolan and I. V. Korendovych, Chem. Soc. Rev., 2018, 47, 3621 RSC.
- P. Makam, S. S. R. K. C. Yamijala, K. Tao, L. J. W. Shimon, D. S. Eisenberg, M. R. Sawaya, B. M. Wong and E. Gazit, Nat. Catal., 2019, 2, 977 CrossRef CAS PubMed.
- (a) K. S. Broo, H. Nilsson, J. Nilsson, A. Flodberg and L. Baltzer, J. Am. Chem. Soc., 1998, 120, 4063 CrossRef CAS; (b) B. Baumeister, N. Sakai and S. Matile, Org. Lett., 2001, 3, 4229 CrossRef CAS PubMed; (c) A. J. Nicoll and R. K. Allemann, Org. Biomol. Chem., 2004, 2, 2175 RSC; (d) M. O. Guler and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 12082 CrossRef CAS PubMed; (e) Z. Huang, S. Guan, Y. Wang, G. Shi, L. Cao, Y. Gao, Z. Dong, J. Xu, Q. Luo and J. Liu, J. Mater. Chem. B, 2013, 1, 2297 RSC; (f) C. Zhang, X. Xue, Q. Luo, Y. Li, K. Yang, X. Zhuang, Y. Jiang, J. Zhang, J. Liu, G. Zou and X.-J. Liang, ACS Nano, 2014, 8, 11715 CrossRef CAS PubMed; (g) C. M. Rufo, Y. S. Moroz, O. V. Moroz, J. Stöhr, T. A. Smith, X. Hu, W. F. DeGrado and I. V. Korendovych, Nat. Chem., 2014, 6, 303 CrossRef CAS PubMed; (h) M. Matsumoto, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2014, 136, 15817 CrossRef CAS PubMed; (i) M. Bélières, N. Chouini-Lalanne and C. Déjugnat, RSC Adv., 2015, 5, 35830 RSC; (j) C. G. Pappas, I. R. Sasselli and R. V. Ulijn, Angew. Chem., Int. Ed., 2015, 54, 8119 CrossRef CAS PubMed; (k) M. P. Friedmann, V. Torbeev, V. Zelenay, A. Sobol, J. Greenwald and R. Riek, PLoS One, 2015, 10, e0143948 CrossRef PubMed; (l) A. J. Burton, A. R. Thomson, W. M. Dawson, R. L. Brady and D. N. Woolfson, Nat. Chem., 2016, 8, 837 CrossRef CAS PubMed; (m) G. Gulseren, M. A. Khalily, A. B. Tekinay and M. O. Guler, J. Mater. Chem. B, 2016, 4, 4605 RSC; (n) Y. Maeda, J. Fang, Y. Ikezoe, D. H. Pike, V. Nanda and H. Matsui, PLoS One, 2016, 11, e0153700 CrossRef PubMed; (o) M. Wang, Y. Lv, X. Liu, W. Qi, R. Su and Z. He, ACS Appl. Mater. Interfaces, 2016, 8, 14133 CrossRef CAS PubMed; (p) Y.-M. Wong, H. Masunaga, J.-A. Chuah, K. Sudesh and K. Numata, Biomacromolecules, 2016, 17, 3375 CrossRef CAS; (q) A. M. Garcia, M. Kurbasic, S. Kralj, M. Melchionna and S. Marchesan, Chem. Commun., 2017, 53, 8110 RSC.
- D. Zaramella, P. Scrimin and L. J. Prins, J. Am. Chem. Soc., 2012, 134, 8396 CrossRef CAS PubMed.
- T. Darbre and J.-L. Reymond, Acc. Chem. Res., 2006, 39, 925 CrossRef CAS PubMed.
- N. Singh, M. P. Conte, R. V. Ulijn, J. F. Miravet and B. Escuder, Chem. Commun., 2015, 51, 13213 RSC.
- (a) A. Brack, K. W. Ehler and L. E. Orgel, J. Mol. Evol., 1976, 8, 307 CrossRef CAS PubMed; (b) A. L. Weber and L. E. Orgel, J. Mol. Evol., 1978, 11, 189 CrossRef CAS PubMed; (c) A. L. Weber and L. E. Orgel, J. Mol. Evol., 1979, 13, 185 CrossRef CAS PubMed; (d) A. L. Weber and L. E. Orgel, J. Mol. Evol., 1979, 13, 193 CrossRef CAS PubMed; (e) S. Steinberg and J. L. Bada, Science, 1981, 213, 544 CrossRef CAS PubMed; (f) M. Nagayama, O. Takaoka, K. Inomata and Y. Yamagata, Origins Life Evol. Biospheres, 1990, 20, 249 CrossRef CAS PubMed; (g) J. Ying, R. Lin, P. Xu, Y. Wu, Y. Liu and Y. Zhao, Sci. Rep., 2018, 8, 936 CrossRef PubMed; (h) Y. Furukawa, T. Otake, T. Ishiguro, H. Nakazawa and T. Kakegawa, Origins Life Evol. Biospheres, 2012, 42, 519 CrossRef CAS PubMed.
- (a) K. Kawamura, T. Nishi and T. Sakiyama, J. Am. Chem. Soc., 2005, 127, 522 CrossRef CAS PubMed; (b) K. Kawamura, H. Takeya and T. Kushibe, Adv. Space Res., 2009, 44, 267 CrossRef CAS.
- M. A. Ziganshin, R. A. Larionov, A. V. Gerasimov, S. A. Ziganshina, A. E. Klimovitskii, K. R. Khayarov, T. A. Mukhametzyanov and V. V. Gorbatchuk, J. Pept. Sci., 2019, 25, e3177 CrossRef PubMed.
- A. Shimoyama and R. Ogasawara, Origins Life Evol. Biospheres, 2002, 32, 165 CrossRef CAS PubMed.
- A. J. Kleinsmann and B. J. Nachtsheim, Chem. Commun., 2013, 49, 7818 RSC.
- (a) T. Govindaraju, Supramol. Chem., 2011, 23, 759 CrossRef CAS; (b) T. Govindaraju, M. Pandeeswar, K. Jayaramulu, G. Jaipuria and H. S. Atreya, Supramol. Chem., 2011, 23, 487 CrossRef CAS; (c) S. Manchineella and T. Govindaraju, RSC Adv., 2012, 2, 5539 RSC.
- S. Manchineella and T. Govindaraju, ChemPlusChem, 2017, 82, 88 CrossRef CAS.
- M. Kurbasic, S. Semeraro, A. M. Garcia, S. Kralj, E. Parisi, C. Deganutti, R. de Zorzi and S. Marchesan, Synthesis, 2019, 51, 2829 CAS.
- (a) M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, J. Am. Chem. Soc., 1996, 118, 4910 CrossRef CAS; (b) M. S. Iyer, K. M. Gigstad, N. D. Namdev and M. Lipton, Amino Acids, 1996, 11, 259 CrossRef CAS PubMed.
- C. Bérubé, X. Barbeau, S. Cardinal, P.-L. Boudreault, C. Bouchard, N. Delcey, P. Lagüe and N. Voyer, Supramol. Chem., 2017, 29, 330 CrossRef.
- (a) B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC; (b) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- (a) R. Sure and S. Grimme, J. Comput. Chem., 2013, 34, 1672 CrossRef CAS; (b) J. G. Brandenburg, M. Hochheim, T. Bredow and S. Grimme, J. Phys. Chem. Lett., 2014, 5, 4275 CrossRef CAS PubMed.
- (a) M. Brandl, M. S. Weiss, A. Jabs, J. Sühnel and R. Hilgenfeld, J. Mol. Biol., 2001, 307, 357 CrossRef CAS PubMed; (b) M. Nishio, J. Mol. Struct., 2012, 1018, 2 CrossRef CAS.
- An initial version of this manuscript has been published in the preprint server ChemRxiv: A. Kleinsmann and B. Nachtsheim, A Minimalistic Hydrolase based on Co-Assembled Cyclic Dipeptides, ChemRxiv, 2019 DOI:10.26434/chemrxiv.9919325.v1.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02198a |
| This journal is © The Royal Society of Chemistry 2020 |