
The first total synthesis of rebaudioside R†
Guo-En
Wen
,
Zhi
Qiao
,
Hui
Liu
,
Zhi-Yong
Zeng
,
Yuan-Hong
Tu
,
Jian-Hui
Xia
,
Qing-Ju
Zhang
 * and
Jian-Song
Sun
*
* and
Jian-Song
Sun
*
National Research Centre for Carbohydrate Synthesis, Jiangxi Normal University, 99 Ziyang Avenue, Nanchang 330022, China. E-mail: zhangq@jxnu.edu.cn; jssun@jxnu.edu.cn
First published on 27th November 2019
Abstract
With easily available monosaccharides and steviol as starting materials, the first total synthesis of rebaudioside R with a xylosyl core in the C13–OH linked sugar chain was accomplished via two distinct approaches. The first approach features the stepwise installation of branch-sugar residues via an order of C2–OH first and then C3–OH of the xylosyl core, laying a firm foundation for the synthesis of analogues with different branch sugars, while the second route features the introduction of the C13 trisaccharide sugar chain via a convergent strategy, securing the overall synthetic efficiency. Through the synthetic study, the effect of protecting groups (PGs) at the vicinal hydroxy group on the reactivity of OH acceptors was illustrated.
Introduction
Steviol glycosides, isolated from the leaves of Stevia rebaudiana Bertoni, have been used as substitutes to sucrose worldwide. In comparison to other sweeteners, steviol glycosides are preferred by the consumers because of their evident advantages of natural origin, high sweetness, and reliable safety.1 The wide acceptance of steviol glycosides as sweeteners promotes the development of the stevioside industry rapidly, which is further boosted by the acceptance of the statement that steviol glycosides (rebaudienosides, Fig. 1) are “Generally Recognized As Safe” (GRAS) by the FDA in 2010.2 However, factors impeding the advancement of the stevioside industry still exist, among which the unpleasant bitter after-taste of the existing stevioside-type sweeteners and the difficulty associated with the precise quality control of the crude extract of S. rebaudiana, the feedstock of steviol glycoside-type sweeteners, are prominent. To tackle these problems, easy access to steviol glycosides with a defined chemical structure is urgently required, which will not only expedite the searching process of ideal sweeteners by offering various testing substrates but also improve the quality control of the crude extract of S. rebaudiana by providing reference compounds. Furthermore, with the discovery of the beneficial effect of steviol glycosides on human health,3 the potential pharmaceutical application of these compounds is drawing more and more attention both from chemists and from pharmacologists.4 The search for effective lead compounds with promising medicinal use also call for efficient access to steviosides.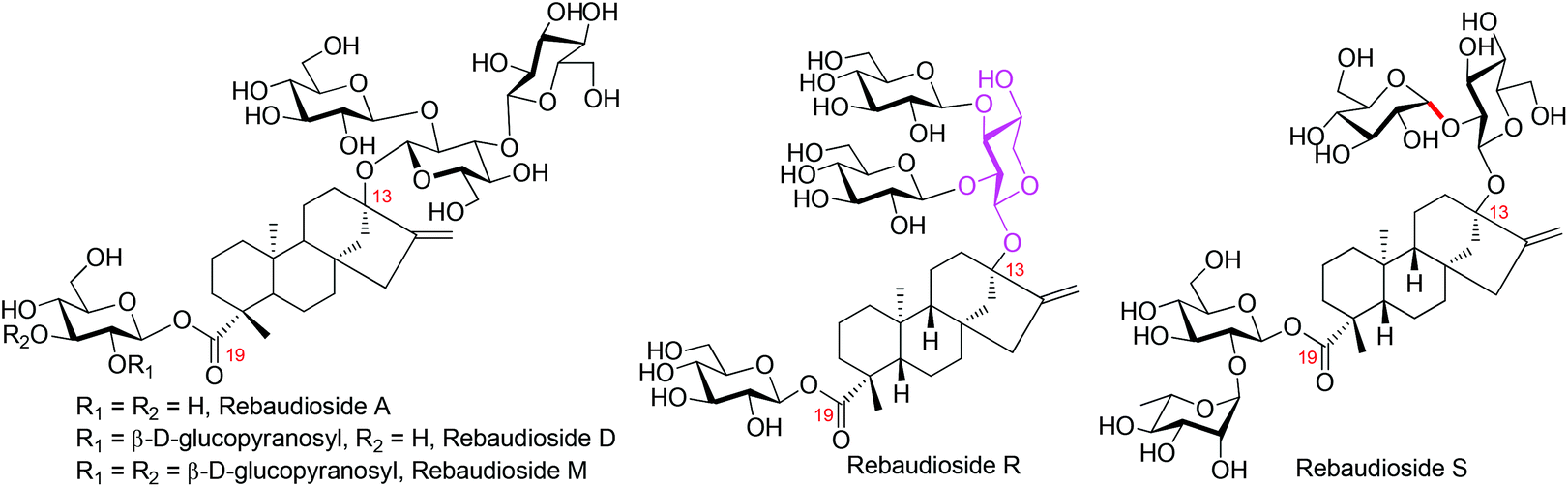 | ||
| Fig. 1 The chemical structures of representative rebaudiosides. | ||
At present, the acquisition of pure steviosides is highly dependent on direct isolation from natural sources.5,6 Due to the high microheterogeneity of naturally occurring steviosides, the separation of stevioside individuals from natural sources is tedious and challenging, so it is only suited to members with high natural content. With easily accessible pure steviosides as starting materials, the enzymatic synthesis can also provide stevioside derivatives. However, the complicated separation process cannot be avoided because the multiple reaction sites existing in the starting materials lead to mixtures of products.7 As such, the chemical synthesis becomes a reliable approach to provide pure and ample amounts of steviosides. Unfortunately, the chemical synthesis of steviol glycosides has only been sporadically reported, and the overall efficiency is far from satisfactory owing to the high acid-sensitivity of the steviol aglycan.8,9
So far, more than 50 steviol glycosides have been isolated and characterized from S. rebaudiana,9 and the number is still increasing rapidly.10 In 2016, McChesney and co-workers characterized two novel steviosides, the rebaudioside R and S.11 With a xylosyl moiety as the core sugar of the C13 sugar chain, rebaudioside R strikingly distinguishes itself from other congeners. Because of the extremely low natural content of Reb-R (below 1%), no taste screenings and biological tests were conducted. Considering that the presence of a xylosyl core may bring about dramatic changes in both taste and bioactivity in comparison to the well investigated Reb-A, D, and M (Fig. 1),12 the synthesis of Reb-R was conducted, culminating with its first total synthesis.12
Results and discussion
Based on the experience accumulated during the synthesis of Reb-A, D, and M and to improve the overall synthetic efficiency,12 the branch sugars in the C13 sugar chain of Reb-R were envisioned to be installed in a single glycosylation step with glucosyl trichloroacetimidate (TCAI) 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 as the donor. In turn, the incorporation of a xylosyl residue into steviol aglycon 212 was planned to be achieved by the xylosyl TCAI donor 3 with its C2,3 hydroxy groups that are different from the levulinoyl (Lev) groups. With regard to the mono-glucosyl residue at the C19 carboxylic group, the phase-transfer conditions (PTC) of the glycosylation protocol was preferred because of its proven reliable efficiency.12,14 Thus, in the forward sense, the target molecule 1 could be assembled by building blocks 2, 3, 4, and 515 (Scheme 1).
13 as the donor. In turn, the incorporation of a xylosyl residue into steviol aglycon 212 was planned to be achieved by the xylosyl TCAI donor 3 with its C2,3 hydroxy groups that are different from the levulinoyl (Lev) groups. With regard to the mono-glucosyl residue at the C19 carboxylic group, the phase-transfer conditions (PTC) of the glycosylation protocol was preferred because of its proven reliable efficiency.12,14 Thus, in the forward sense, the target molecule 1 could be assembled by building blocks 2, 3, 4, and 515 (Scheme 1).
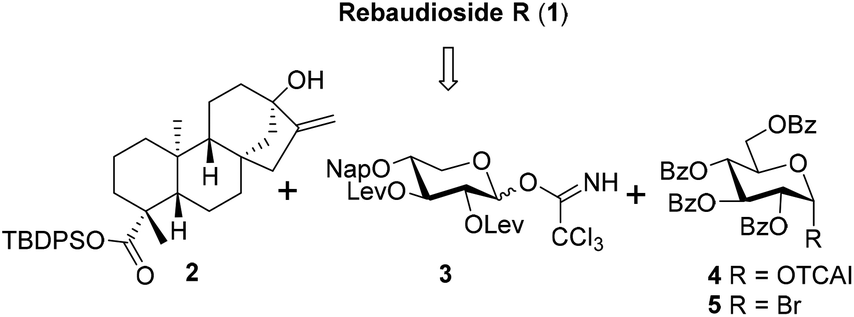 | ||
| Scheme 1 Retrosynthetic analysis of Reb-R. | ||
The synthetic endeavor commenced with the synthesis of the core xylosyl TCAI donor 3 (Scheme 2). 2-Naphthylmethylation of 616 under standard conditions provided 7 (76%), which was then subjected to acid conditions to generate diol 8 (87%). The C2,3 hydroxy groups were re-blocked with Lev groups (9, 99%) before anomeric methoxyphenyl (MP) group removal and TCAI introduction, furnishing donor 3 (67%, 2 steps).
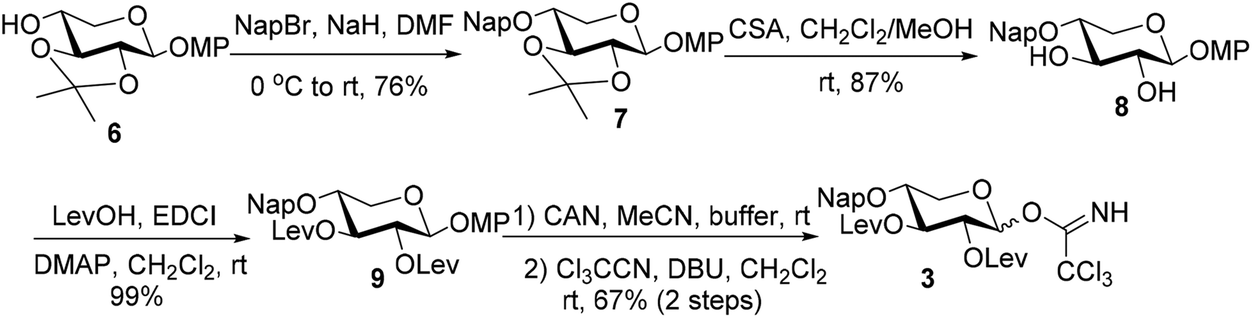 | ||
| Scheme 2 Synthesis of donor 3. | ||
With donor 3 in hand, its coupling with 2 was carried out under the previously optimized conditions (0.2 eq. TMSOTf, −40 °C),12 and, pleasantly, the monosaccharide glycoside 10 was isolated with a good 87% yield, devoid of a double bond migration by-product (Scheme 3). The cleavage of NH2NH2·HOAc-mediated Lev groups converted 10 to 11 (82%), which is ready for the introduction of branch-sugars at C2,3 hydroxy groups of the central xylosyl residue. To avoid the possible C16–C17 double bond migration, the following glycosylation was conducted at further decreased temperature (−50 °C). Under otherwise identical conditions, the condensation between donors 4 and 11 proved to be inefficient, and the desired trisaccharide glycoside 12 was isolated only in 11% yield, accompanied by 53% yield of 1,3-linked disaccharide glycoside 12a. Attempt was then made to improve the yield of 12 by increasing the amounts of donor 4 from 3.0 to 5.0 equivalents (to the acceptor 11). However, no evident increase in the yield of 12 was observed. Instead, with the increase in the donor amounts, the reaction mixture became more complex because of the decomposition of the excess of donors, complicating the isolation of 12 and 12a from the reaction system. This result implies that the desired trisaccharide 12 is presumably not derived from 12avia the introduction of branch-sugars of C3–OH in a step-wise manner first and then the C2-OH of the central sugar residue, which has been determined during the syntheses of rebaudioside A, D, M, and oleanane-type saponins.17 This assumption was verified by resubjecting the isolated 12a to glycosylation conditions identical to those mediating the condensation between 4 and 11 in the presence of 4, and not even trace amounts of 12 were detected. Thus, a reversed order of the introduction of branch-sugars, that is C2–OH prior to C3–OH, presumably should be responsible for the formation of 12, which was tentatively tested with 13, derived from the regioselective levulinoylation of the C3-OH of the xylosyl residue in 11 (62%). However, under the catalysis of TMSOTf at −50 °C, no glycosylation product 14 was detected from the coupling between 13 and 4. The failure of condensation was attributed to the deactivation effect of the C3 levulinoyl group on the free C2′–OH of the xylosyl moiety.18
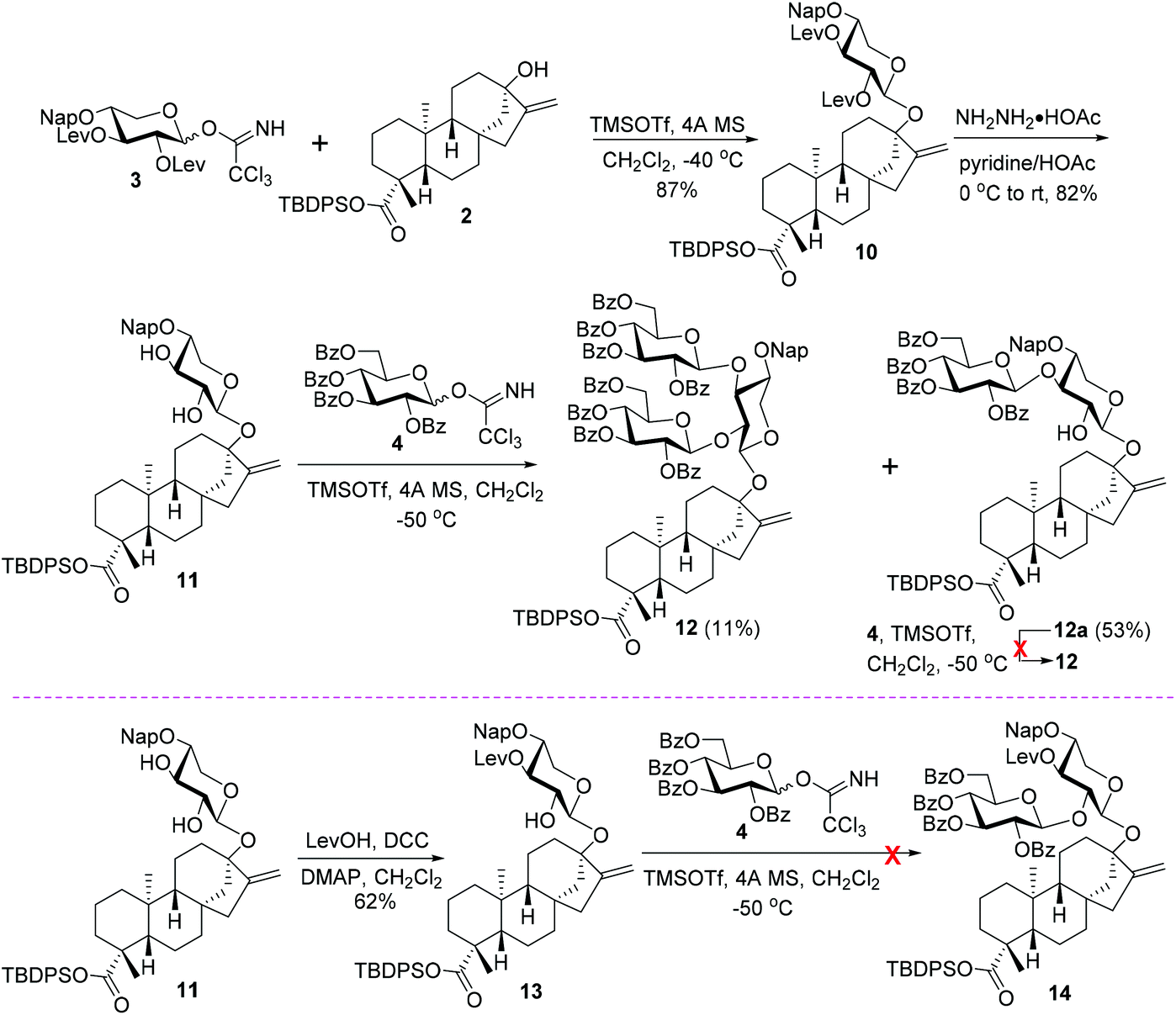 | ||
| Scheme 3 Attempts to fabricate the C13 trisaccharide sugar chain via different branch-sugar introduction sequences. | ||
To further check the feasibility of C13 sugar chain assembly via a sequence of C2–OH first and then C3–OH of the xylosyl core, different xylosyl donors 19, 23, and 28 were synthesized, with the C3–OH protected as a 2-naphthylmethyl (Nap) ether while the C2–OH blocked with a Lev group (Scheme 4). The introduction of a Nap group into the C3-OH of xylosyl donors could eliminate the deactivation effect of an ester protecting group (PG) on the reactivity of the C2–OH group in the following branch-sugar introduction step, while the incorporation of various PGs with distinct electronic and steric properties into C4–OH could facilitate the evaluation of their effect on the reactivity of the C3–OH acceptor for branch-sugar installation.
 | ||
| Scheme 4 Synthesis of xylosyl donors with different protecting groups at the C-4 hydroxy group. | ||
The synthesis of 19 with a benzoyl group at C4–OH commenced with diol 15, which was subjected to regioselective acetylation under the effect of Ac2O/Yb(OTf)3 to afford 16 (54%).19 The remaining free hydroxy group in 16 was then protected with a benzoyl group (17, 97%) before switching the PG at C2–OH from an acetyl (Ac) group to a Lev group via a sequence of selective Ac removal in the presence of 4-O-Bz20 and dehydrative levulinoylation to yield 18 (69%, 2 steps). Then, thioglycoside 18 was converted to xylosyl trichloroacetimidate 19 through thioglycoside hydrolysis and trichloroacetimidate formation (43%, 2 steps).
The synthesis of 23 bearing an electron-donating tert-butyldimethylsilyl (TBS) at C4–OH started with thioglycoside 20.19 The silylation of 20 was followed by replacing the C2-O-benzoyl group with a Lev group to deliver 22via21 (76%, 3 steps). Following the similar procedures to those used for the transformation of 18 to 19, compound 22 was converted to donor 23 smoothly (54%, 2 steps).
Similarly, with 20 as the starting material, the synthesis of donor 28 carrying an allyl (All) group at C4–OH was also achieved (Scheme 4). As the conditions for thioglycoside hydrolysis could not reconcile with the All group, the anomeric STol substituent had to be adjusted to OMP before the installation of the C4-O-All group. This transformation was realized by thioglycoside hydrolysis, trichloroacetimidate formation, and condensation with p-methoxyphenol (MPOH) (25, 49% over 3 steps), following the acetylation of 20 to 24 (89%). Selective cleavage of the C4-O-Ac group in the presence of C2-O-Bz was followed by allylation under neutral conditions with allyl ethyl carbonate as the allylating reagent,21 furnishing 26 (86%, 2 steps). With 26 in hand, the remaining steps required for donor 28 synthesis entailed the exchange of C2-O-Bz with a Lev group (27, 86%, 2 steps) and switching the anomeric MP group to TCAI (28, 52% over 2 steps).
With donors 19, 23, and 28 in hand, the stage was now set for the subsequent study of the fabrication of the C13 trisaccharide chain via the branch-sugar incorporation sequence of C2–OH first and then C3–OH of the xylosyl core (Scheme 5). Thus, under the previously optimized conditions, the condensations between donors 19, 23, and 28 with acceptor 2 proceeded uneventfully, providing monosaccharide glycosides 29–31 with good to excellent yields (29: 92%, 30: 94%, 31: 77%). The subsequent C2–OH branch-sugar installation of the xylosyl core entailed the glycosylation of 32–34, derived from 29–31 by selective removal of the Lev group, with donor 4 at −50 °C, which uniformly furnished the desired disaccharide glycosides 35–37 with good efficiency (above 75% yields), demonstrating the profound effect of C3–OH PGs on the reactivity of C2–OH (cf. the coupling between 13 and 4 in Scheme 3). The subsequent C3–OH release was effected by DDQ to cleave the Nap group to give acceptors 38–40, which were ready for the C3 branch sugar introduction. When 38 with an electron-withdrawing Bz at C4–OH of the xylosyl residue was chosen as the acceptor, no desired trisaccharide product was detected when coupled with 4 under identical conditions as above, as with the condensation between 39 bearing a TBS group at C4–OH and 4. Surprisingly, when 40 with an All group at C4–OH was selected as the acceptor, the free 3-OH of 40 proved to be reactive enough to condense with donor 4 under the same conditions, furnishing the coveted trisaccharide 41 in a reasonable 67% yield. The only difference for acceptors 38–40 lies in the C4–OH protection pattern of the xylosyl residue, with 38 bearing an electron-withdrawing Bz group of moderate steric demand while 39 carrying a strongly electron-donating TBS group with high steric hindrance and 40 bringing a moderately electron-donating All group with moderate steric demand. These divergences in the electronic and steric properties of PGs at C4 should also be responsible for the reactivity discrepancy between acceptors 38–40. The electron-withdrawing effect of the C4 benzoyl group in 38 and the deleterious steric effect of the TBS group in 39 deactivate the reactivity of the vicinal C3–OHs resulting in futile glycosylation attempts. Although the allyl group in 40 cannot compete with the TBS group in terms of the electron-donating effect, the moderate steric demand compensates for the diminished electron-donating capacity leading to a viable acceptor for trisaccharide glycoside synthesis.
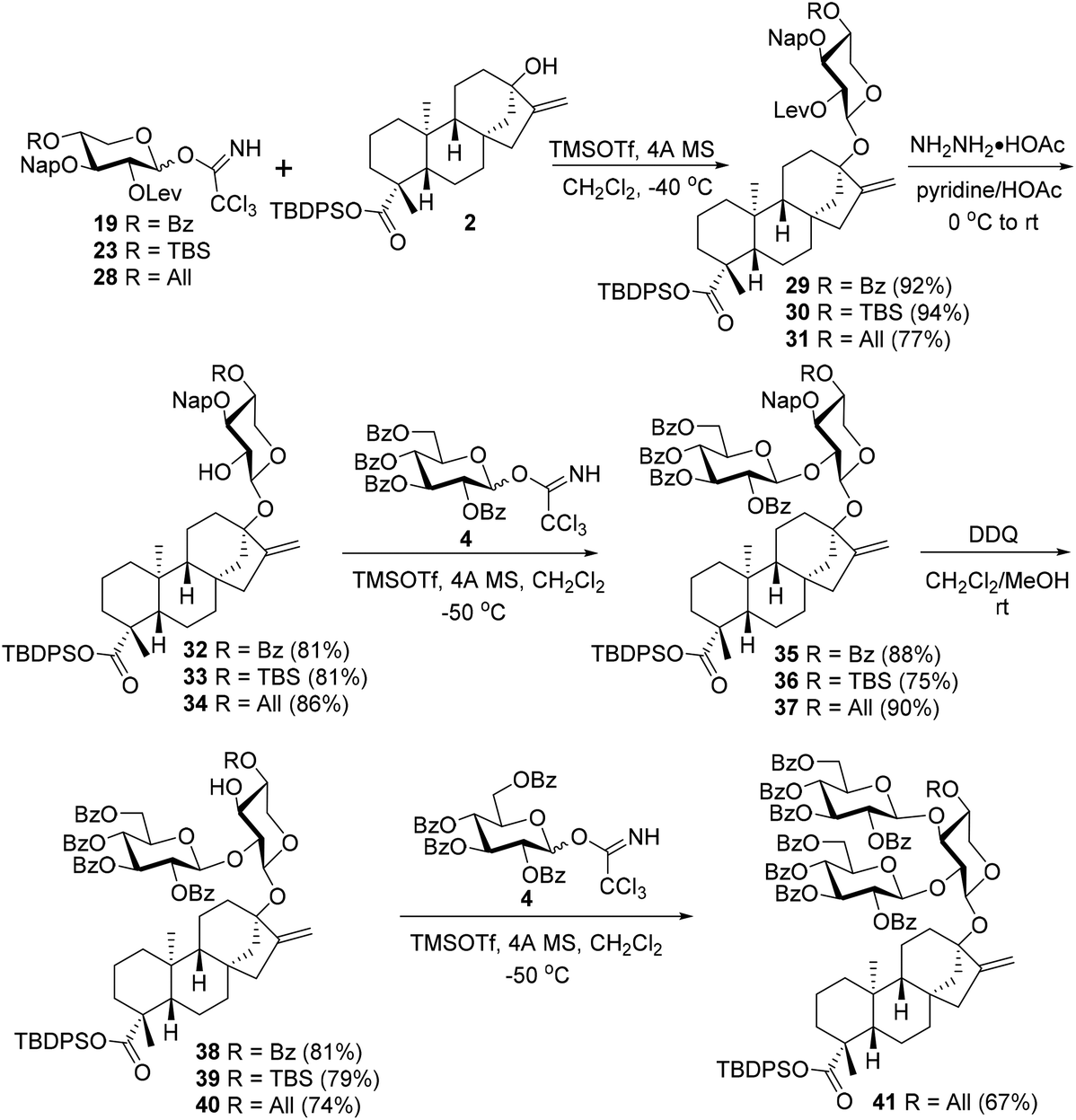 | ||
| Scheme 5 Attempts to assemble the C13 trisaccharide chain via a branch-sugar introduction sequence of C2–OH first and then C3–OH of the xylosyl core. | ||
Eventually, an efficient route to prepare the key intermediate 41 was achieved via a stepwise branch-sugar introduction strategy, which is also applicable to the synthesis of Reb-R analogues with different branch sugars. The branch sugar-introduction order of the core xylosyl residue that is contrary to that of the corresponding glucosyl moiety is tentatively attributed to the conformational flexibility of the xylosyl moiety.22
Considering that Reb-R has two identical branch sugars, the incorporation of the two branch glucosyl residues simultaneously in one glycosylation step is highly desirable owing to the concise synthetic steps. As the inherent reactivity hierarchy of C2,3–OHs (3-OH > 2-OH) of the xylosyl core with a steviol residue at the anomeric position conflicts with the established branch-sugar installation sequence (2-OH prior to 3-OH), the introduction of two branch glucosyl residues through one step glycosylation with C2′,3′-diol of steviol 13-O-xyloside as the acceptor is not feasible, as exemplified by the condensation between 11 and 4 (cf.Scheme 3). Thus, a more convergent strategy, wherein the trisaccharide chain was incorporated into 2 as a whole, was examined at the risk of the comprised stereoselectivity due to the absence of a neighboring group participation effect (Scheme 5).
With diol 8 as the acceptor, the glycosylation with 4 was carried out under the optimized conditions (TMSOTf, −40 °C) to give trisaccharide 42 with a good 77% yield. In comparison to the disappointing result obtained in coupling between 4 and 11, the profound effect of the anomeric substituents on the activity of C2–OH of the core xylosyl moiety is also illustrated. The anomeric MP group of 42 was removed and the resulting hemiacetal was then transformed to trichloroacetimidate 43 (54%) and o-alkynylbenzoate 43′ (88%), respectively. The Schmidt donor2343 was used first to condense with 2, and, surprisingly, the α trisaccharide glycoside 12′ was formed exclusively and even donor 43 was equipped with a bulky perbenzoylated glucosyl residue at C2–OH of the xylosyl core (85%).24 To our delight, the α-stereoselectivity propensity of the coupling was finally reversed with Yu donor2543′ under the catalysis of Ph3PAuNTf2 with the aid of a nitrile solvent effect,26 delivering 12 as the predominating isomer (12: 53%, 12′: 21%). At this junction, the two synthetic approaches converged, offering the key intermediate 12 jointly. Compounds 12, obtained by the linear and convergent approaches, and 12′, easily separated from 12, were then advanced to Reb-R 1 as well as its analogue 1′ separately. C-19 carboxylic groups in 12 and 12′ were liberated with TBAF, furnishing the carboxylic acid intermediates 44 (95%) and 44′ (97%), which were then subjected to PTC glycosylation conditions in the presence of glucosyl bromide 5 to give tetrasaccharide glycosides 45 (83%) and 45′ (94%), respectively. Finally, the global deprotection of 45 and 45′via DDQ-mediated Nap cleavage and NaOMe-promoted saponification furnished 1 (86%) and its analogue 1′ (82%) efficiently. The NMR spectroscopy data of synthetic 1 match well with those recorded for the natural product.27
In fact, the establishment of conditions for the convergent synthesis of 12 (cf.Scheme 6) was achieved by systematic investigations (Table 1). Although only 12′ was detected with 43 as the donor and TMSOTf (0.2 eq.) as the catalyst, a promising result was recorded when B(C6F5)328 was applied as the catalyst under otherwise identical conditions, and the glycosylation product was isolated as a 1:1 α/β-mixture (entries 1 and 2). The combination of B(C6F5)3-catalysis and nitrile effect was thus examined, but, unfortunately, no reaction was observed even at elevated temperature (entry 3). Interestingly, with 43′ as the donor, an α/β ratio of 3.7:1 was obtained in CH2Cl2 under the catalysis of Ph3PAuNTf2 at room temperature (57% yield, entry 4). The yield and the α/β ratio were ameliorated by adjusting the reaction temperature to −40 °C to rt (entry 5, α/β = 2.9:1, 86%). Further optimization of donor 43′ included applying acetonitrile as a solvent, leading to 12 as the major product in a 74% combined yield (entry 6).
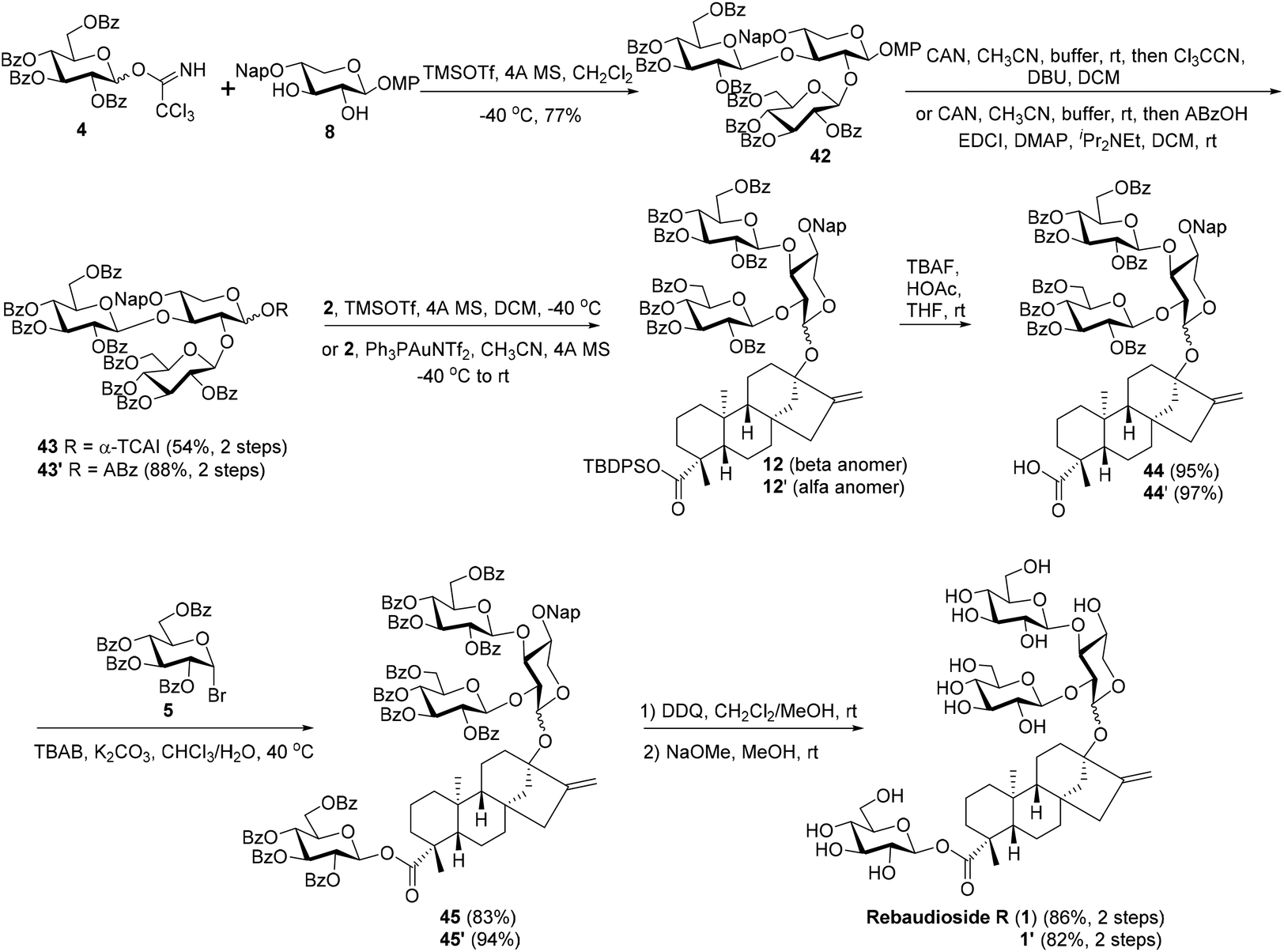 | ||
| Scheme 6 The assembly of C13 branched trisaccharide via a convergent strategy as well as the completion synthesis of rebaudioside R (1) and its analogue 1′. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Donor | Catalyst (eq.) | Solvent | Temperature (°C) | Resulta |
| a Isolated yield. | |||||
| 1 | 43 | TMSOTf (0.2) | CH2Cl2 | −40 °C | 85% (12′ only) |
| 2 | 43 | B(C6F5)3 (0.1) | CH2Cl2 | −40 °C | 85% (12/12′ = 1:1) |
| 3 | 43 | B(C6F5)3 (0.1) | CH3CN | −40 °C to rt | NR |
| 4 | 43′ | Ph3PAuNTf2 (0.2) | CH2Cl2 | rt | 57% (12/12′ = 1:3.7) |
| 5 | 43′ | Ph3PAuNTf2 (0.2) | CH2Cl2 | −40 °C to rt | 86% (12/12′ = 1:2.9) |
| 6 | 43′ | Ph3PAuNTf2 (0.2) | CH3CN | −40 °C to rt | 74% (12/12′ = 2.5:1) |
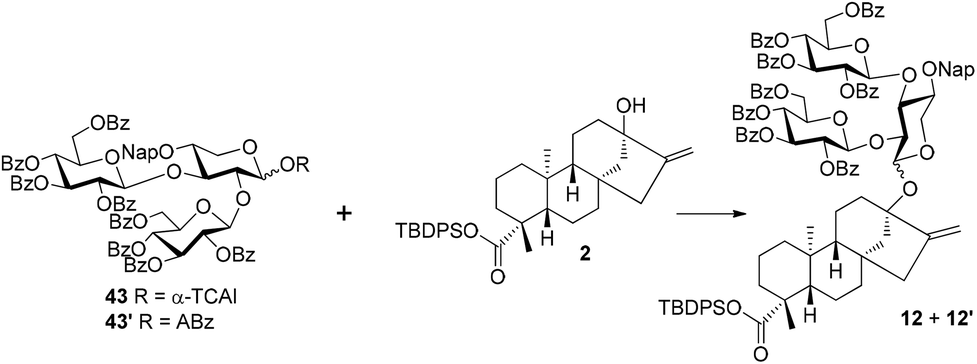
Conclusion
In summary, the first total synthesis of rebaudioside R, featuring a core xylosyl residue in a C13-appended branched sugar chain, was accomplished via two different approaches. In the first approach, the branch sugars were introduced into the core xylosyl moiety via a stepwise manner under C2–OH first and then C3–OH order. Although contrary to the previously established branch sugar-incorporation sequence, the stepwise strategy enables the synthesis of the target molecule Reb-R, and is potentially applicable to the synthesis of Reb-R analogues with distinct branch sugar residues. In the second approach, the branched C13 trisaccharide chain was fabricated prior to the steviol aglycon installation, and the satisfactory β-stereoselectivity in the ensuing glycosylation step with the trisaccharide donor bearing a C2–OH branch was secured by the nitrile solvent effect. Furthermore, through the synthetic investigations, the effect of PGs at vicinal OHs on the reactivity of OH acceptors was illustrated, which will provide reference to the synthesis of a C2,3-branched sugar chain with a conformationally flexible xylosyl core. The synthetic studies toward Reb-R and its analogues can provide not only reference compounds for the precise quality control of stevioside feedstock production but also testing compounds for promising lead compounds that could be used either as ideal sweeteners or in pharmaceuticals.Experimental section
4-Methoxyphenyl 2,3-O-isopropylidene-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (7)
To a stirred solution of 6 (7 g, 23.6 mmol) in dry DMF (25 mL), NaH (1.14 g, 28.3 mmol) was added in batches at 0 °C. After stirring for 20 min under an N2 atmosphere, NapBr (6.26 g, 28.3 mmol) was added to the reaction mixture, which was allowed to stir for another 3 h at the same temperature. The reaction mixture was quenched with water, diluted with ethyl acetate, washed with water and brine, and then dried over anhydrous Na2SO4. Filtration was followed by concentration under reduced pressure to yield the crude product, which was further purified by silica gel column chromatography (PE/EA = 10:1) to afford 7 (7.8 g, 17.8 mmol, 76%) as a light yellow syrup: [α]25D = −34.1 (c 1.4, CHCl3); 1H NMR (400 MHz, acetone-d6) δ 7.92–7.87 (m, 4H), 7.54–7.47 (m, 3H), 7.06–7.00 (m, 2H), 6.89–6.84 (m, 2H), 5.37 (d, J = 7.2 Hz, 1H), 4.95 (d, J = 12.0 Hz, 1H), 4.80 (d, J = 12.4 Hz, 1H), 4.21 (dd, J = 12.4, 4.8 Hz, 1H), 4.00–3.96 (m, 1H), 3.93 (dd, J = 9.6, 8.4 Hz, 1H), 3.74 (s, 3H), 3.68 (dd, J = 9.6, 7.2 Hz, 1H), 3.62 (dd, J = 12.4, 6.0 Hz, 1H), 1.48 (s, 3H), 1.47 (s, 3H); 13C NMR (100 MHz, acetone-d6) δ 156.2, 151.7, 137.1, 134.3, 134.0, 128.9, 128.8, 128.6, 127.1 (2C), 126.8 (2C), 119.0, 115.4, 112.4, 101.1, 81.2, 77.5, 77.3, 72.2, 66.4, 55.9, 27.3, 27.1; HRMS (ESI-TOF) calcd for C26H28O6K [M + K]+ 475.1517, found 475.1514.
4-Methoxyphenyl 4-O-(2-naphthylmethyl)-β-D-xylopyranoside (8)
To a stirred solution of 7 (5.7 g, 13.1 mmol) in a mixed solvent of MeOH (10 mL) and DCM (10 mL), camphor-10-sulfonic acid (CSA, 304 mg, 1.31 mmol) was added at room temperature. The reaction mixture was allowed to stir for 2 h at the same temperature and TLC indicated the complete disappearance of the starting material. After being quenched by Et3N, the solvent was removed under reduced pressure to yield the crude product, which was further purified by silica gel column chromatography (PE/EA = 2:1) to afford 8 (4.5 g, 11.35 mmol, 87%) as a white solid: [α]25D = −39.3 (c 1.0, CHCl3); 1H NMR (400 MHz, acetone-d6) δ 7.89–7.83 (m, 4H), 7.55 (dd, J = 8.4, 1.6 Hz, 1H), 7.50–7.44 (m, 2H), 7.02–6.97 (m, 2H), 6.85–6.79 (m, 2H), 4.98 (d, J = 12.0 Hz, 1H), 4.87 (d, J = 12.4 Hz, 1H), 4.82 (d, J = 7.6 Hz, 1H), 4.03 (ddd, J = 11.6, 5.2, 2.4 Hz, 1H), 3.71 (s, 3H), 3.62–3.56 (m, 1H), 3.53 (dd, J = 8.8, 7.2 Hz, 1H), 3.42–3.36 (m, 1H); 13C NMR (100 MHz, acetone-d6) δ 154.3, 150.8, 135.8, 132.6, 132.2, 127.1, 127.0, 126.8, 125.4, 125.3, 125.2, 125.0, 117.4, 113.6, 101.8, 76.8, 75.0, 72.8, 71.8, 62.7, 54.2; HRMS (ESI-TOF) calcd for C23H25O6 [M + H]+ 397.1646, found 397.1651.
4-Methoxyphenyl 2,3-di-O-levulinoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (9)
To a stirred solution of 8 (1 g, 2.52 mmol) in dry CH2Cl2 (10 mL), LevOH (2.34 g, 19.82 mmol), DMAP (1.23 g, 10.07 mmol), DIPEA (3.34 mL, 19.2 mmol), and EDCI (3.8 g, 19.82 mmol) were added successively at 0 °C under a nitrogen atmosphere. The reaction mixture was allowed to warm up to room temperature and stirring was continued for another 12 h at room temperature. Then the reaction mixture was diluted with ethyl acetate, washed successively with water, saturated aqueous NaHCO3, and brine, and then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel column chromatography to afford 9 (1.49 g, 2.514 mmol, 99%) as a colorless syrup: [α]25D = −11.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.84–7.81 (m, 3H), 7.76 (s, 1H), 7.50–7.47 (m, 2H), 7.45 (dd, J = 8.4, 1.6 Hz, 1H), 6.97–6.92 (m, 2H), 6.83–6.77 (m, 2H), 5.29 (t, J = 8.8 Hz, 1H), 5.12 (t, J = 8.0 Hz, 1H), 4.91 (d, J = 7.2 Hz, 1H), 4.81 (dd, J = 15.0 Hz, J = 12.4 Hz, 2H), 4.07 (dd, J = 12.0, 5.2 Hz, 1H), 3.79–3.69 (m, 4H), 3.44 (dd, J = 11.6, 9.2 Hz, 1H), 2.81–2.48 (m, 8H), 2.14 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 206.4, 206.3, 172.0, 171.6, 155.7, 151.1, 135.3, 133.3, 133.2, 128.4, 128.0, 127.8, 126.8, 126.3, 126.2, 125.8, 118.8, 114.7, 100.7, 74.8, 73.4, 73.3, 71.2, 63.8, 55.8, 55.7, 37.8, 29.8, 28.1, 28.0; HRMS (ESI-TOF) calcd for C33H37O10 [M + H]+ 593.2381, found 593.2382.2,3-Di-O-levulinoyl-4-O-(2-naphthylmethyl)-α/β-D-xylopyranosyl trichloroacetimidate (3)
To a stirred solution of 9 (600 mg, 1.01 mmol) in a mixed solvent of acetonitrile (28.5 mL) and buffer (6.91 mL, pH = 7.0), CAN (2.77 g, 5.05 mmol) was added at 0 °C. The reaction mixture was stirred at the same temperature for 15 min before ethyl acetate was added. The resulting solution was washed successively with water, saturated aqueous NaHCO3, and brine, and then dried over anhydrous Na2SO4. Filtration was followed by concentration to yield the crude product, which was further purified by silica gel column chromatography (PE/EA = 1:1) to afford the hemiacetal intermediate (410 mg, 0.84 mmol), which was used directly in the next step without detailed characterization.
To a solution of the above-obtained hemiacetal intermediate in dry CH2Cl2 (5 mL), Cl3CCN (0.34 mL, 3.4 mmol) and DBU (0.05 mL, 0.33 mmol) were added at room temperature. The mixture was stirred for another 1 hour at the same temperature, and then the solvent was evaporated in vacuo to yield a residue, which was subjected to silica gel column chromatography (PE/EA = 3:1) to afford 3 (430 mg, α/β = 1:0.3, 0.68 mmol, 67% for 2 steps). An aliquot of pure α-anomer was obtained for detailed characterization: [α]25D = +49.1 (c 3.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 0.3H), 8.61 (s, 1H), 7.85–7.74 (m, 5.2H), 7.52–7.41 (m, 3.9H), 6.36 (d, J = 4.0 Hz, 1H), 5.89 (d, J = 6.4 Hz, 0.3H), 5.61 (dd, J = 10.4, 8.8 Hz, 1H), 5.30 (t, J = 7.8 Hz, 0.3H), 5.14 (dd, J = 8.0, 6.4 Hz, 0.3H), 5.03 (dd, J = 10.0, 3.6 Hz, 1H), 4.82–4.75 (m, 2.6H), 4.13 (dd, J = 12.0, 4.4 Hz, 0.3H), 3.91–3.60 (m, 3H), 2.77–2.44 (m, 10.4H), 2.17–2.09 (m, 7.8H); 13C NMR (100 MHz, CDCl3) δ 206.5, 206.3, 172.0, 171.9, 161.1, 135.1, 133.3, 133.2, 128.5, 128.0, 127.8, 127.0, 126.4, 126.3, 125.9, 93.6, 91.0, 74.8, 73.6, 73.0, 71.3, 70.2, 62.4, 37.8, 37.7, 29.9 (4C), 28.1, 27.8; HRMS (ESI-TOF) calcd for C28H31Cl3NO9 [M + H]+ 630.1059, found 630.1068.
13-O-[2,3-Di-O-levulinoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (10)
To a solution of 3 (247 mg, 0.39 mmol) and 2 (145 mg, 0.26 mmol) in dry CH2Cl2 (5 mL), activated powdered 4 Å MS (400 mg) was added at room temperature under an N2 atmosphere. The suspension was stirred at the same temperature for 30 min and was then cooled to −40 °C, to which TMSOTf (14.5 μL, 0.08 mmol) was added. After being stirred at −40 °C for another 1 h, Et3N was added to quench the reaction. Filtration was followed by concentration under reduced pressure, and the obtained residue was purified by silica gel column chromatography (PE/EA = 4:1) to yield 10 (233 mg, 0.227 mmol, 87%) as a colorless foam: [α]25D = −52.1 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.82 (d, J = 8.8 Hz, 3H), 7.74 (s, 1H), 7.69–7.66 (m, 4H), 7.49–7.35 (m, 9H), 5.20 (t, J = 9.6 Hz, 1H), 4.92 (s, 1H), 4.87 (dd, J = 9.6, 7.6 Hz, 1H), 4.80 (s, 1H), 4.75 (s, 2H), 4.60 (d, J = 7.6 Hz, 1H), 3.94 (dd, J = 12.0, 5.6 Hz, 1H), 3.68–3.62 (m, 1H), 3.25 (dd, J = 11.6, 10.0 Hz, 1H), 2.70–2.54 (m, 8H), 2.26 (d, J = 13.6 Hz, 1H), 2.14 (s, 3H), 2.11 (s, 3H), 2.00–1.92 (m, 2H), 1.87–1.69 (m, 5H), 1.63–1.28 (m, 7H), 1.25 (s, 3H), 1.14 (s, 9H), 1.04–0.77 (m, 2H), 0.96–0.78 (m, 3H), 0.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.6, 206.3, 176.8, 172.2, 171.4, 152.3, 135.7, 135.4, 133.3, 133.2, 132.2 (2C), 130.1 (2C), 128.4, 130.0, 127.8, 127.7, 126.9, 126.3, 126.2, 125.9, 104.9, 96.4, 86.0, 75.2, 74.0, 73.3, 71.9, 63.8, 57.1, 53.8, 47.5, 45.3, 43.9, 42.5, 41.4, 40.7, 39.5, 38.7, 37.8, 37.7, 37.1, 29.9 (2C), 29.4, 28.2, 28.0, 27.3, 22.2, 20.4, 19.4, 16.4; HRMS (ESI-TOF) calcd for C62H76O11SiK [M + K]+ 1063.4788, found 1063.4790.
13-O-[4-O-(2-Naphthylmethyl)-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (11)
To a solution of 10 (233 mg, 0.227 mmol) in a mixed solvent of pyridine (23 mL) and HOAc (15 mL), the freshly prepared NH2NH2·HOAc (0.314 mL, prepared by mixing NH2NH2 and HOAc with a volume ratio of 1:3.28) was added at 0 °C. The reaction mixture was allowed to warm up to room temperature, and stirring was continued for another 30 min at the same temperature. Then the mixture was quenched with acetone and diluted with ethyl acetate. The resulting solution was washed successively with water, saturated aqueous NaHCO3, and brine, and then dried over anhydrous Na2SO4. Filtration was followed by concentration to yield the crude product, which was further purified by silica gel column chromatography (PE/EA = 3:1) to afford 11 (154 mg, 0.186 mmol, 82%): [α]25D = −58.7 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.86–7.82 (m, 3H), 7.79 (s, 1H), 7.71–7.68 (m, 4H), 7.50–7.36 (m, 9H), 4.98 (s, 1H), 4.89 (d, J = 4.0 Hz, 1H), 4.85 (s, 1H), 4.84 (s, 2H), 4.09 (dd, J = 12.4, 3.2 Hz, 1H), 3.91 (t, J = 5.6 Hz, 1H), 3.60–3.50 (m, 3H), 2.26 (d, J = 12.8 Hz, 1H), 2.18–2.08 (m, 2H), 2.02 (d, J = 16.8 Hz, 1H), 1.89–1.1.28 (m, 12H), 1.27 (s, 3H), 1.16 (s, 9H), 1.05–1.03 (m, 2H), 0.95–0.73 (m, 2H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.7, 153.0, 135.8, 135.7, 135.1, 133.3, 133.2, 132.2, 132.2, 130.1, 128.6, 128.0, 127.8, 126.9, 126.4, 126.2, 125.8, 104.5, 96.9, 86.1, 76.7, 72.4, 71.0, 70.6, 59.7, 57.1, 53.8, 47.5, 45.3, 43.8, 42.5, 41.5, 40.7, 39.5, 38.7, 37.0, 29.8, 29.3, 27.3, 22.2, 20.4, 19.4, 16.3; HRMS (ESI-TOF) calcd for C52H65O7Si [M + H]+ 829.4494, found 829.4487.
13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-β-D-xylopyranoside]steviol tert-butyldiphenylsilyl ester (12)
To a solution of 4 (80.5 mg, 0.109 mmol) and 11 (30 mg, 0.036 mmol) in dry CH2Cl2 (2 mL), activated powdered 4 Å MS (200 mg) were added at room temperature under an N2 atmosphere. The suspension was stirred at the same temperature for 30 min and then cooled to −50 °C, to which TMSOTf (4 μL, 0.022 mmol) was added. After being stirred at −50 °C for another 1 h, the reaction mixture was quenched with Et3N, followed by 4 Å MS removal by filtration. The filtrate was concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (PE/EA = 3:1) to afford 12 (8 mg, 0.004 mmol, 11%) as a white foam and 12a (27 mg, 0.019 mmol, 53%) as a white foam. For 12: [α]25D = +24.9 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.28 (t, J = 7.2 Hz, 4H), 7.93–7.85 (m, 12H), 7.81–7.64 (m, 12H), 7.57–7.52 (m, 4H), 7.47–7.28 (m, 23H), 7.22 (t, J = 7.6 Hz, 2H), 5.89–5.80 (m, 2H), 5.62 (dd, J = 10.0, 8.0 Hz, 1H), 5.56–5.46 (m, 3H), 4.97 (s, 1H), 4.94 (d, J = 12.8 Hz, 1H), 4.87 (d, J = 7.6 Hz, 1H), 4.78 (d, J = 7.6 Hz, 1H), 4.77 (d, J = 12.8 Hz, 1H), 4.58 (s, 1H), 4.50 (d, J = 6.4 Hz, 1H), 4.24–4.17 (m, 4H), 3.94 (t, J = 8.8 Hz, 1H), 3.70–3.62 (m, 2H), 3.54–3.48 (m, 1H), 3.10 (dd, J = 11.6, 9.6 Hz, 1H), 2.64–2.51 (m, 2H), 2.27 (d, J = 13.2 Hz, 1H), 2.04–1.97 (m, 1H), 1.94 (d, J = 10.8 Hz, 1H), 1.86–1.76 (m, 5H), 1.65–1.61 (m, 2H), 1.48–1.40 (m, 4H), 1.35–1.25 (m, 2H), 1.23 (s, 3H), 1.15 (s, 9H), 1.03–0.97 (m, 2H), 0.88–0.75 (m, 2H), 0.84 (d, J = 7.2 Hz, 1H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.1 (2C), 166.0, 165.8, 165.2, 165.0 (2C), 153.3, 136.2, 135.8, 135.7, 133.6 (3C), 133.4 (2C), 133.3, 133.1 (2C), 132.3, 132.2, 130.3, 1230.2, 130.1 (2C), 130.0, 129.8 (2C), 129.7 (2C), 129.6, 129.5 129.2, 129.1, 129.0, 128.5 (2C), 128.4, 128.3, 128.2, 128.0, 127.8, 127.7, 126.6, 126.1, 125.9, 125.8, 104.5, 100.4 (2C), 97.0, 85.0, 82.2, 79.4, 77.4, 75.1, 73.6, 73.1, 73.0, 72.8, 72.7, 71.5, 71.4, 69.7, 69.6, 63.7, 63.0, 62.8, 57.1, 54.0, 47.0, 45.3, 43.6, 42.8, 41.4, 40.7, 39.5, 38.8, 36.3, 29.4, 27.3, 22.2, 20.4, 19.5, 19.4, 16.5; HRMS (ESI-TOF) calcd for C120H116O25SiNa [M + Na]+ 2008.7499, found 2008.7461.
13-O-[3-O-Levulinoyl-4-O-(2-naphthylmethyl)-β-D-xylopyranosilyl]-steviol tert-butyldiphenylsilyl ester (13)
To a stirred solution of compound 11 (100 mg, 0.121 mmol), 1,3-dicyclohexylcarbodiimide (DCC, 50 mg, 0.242 mmol), and 4-dimethylaminopyridine (DMAP, 1.46 mg, 0.012 mmol) in dry DCM (2.5 mL), levulinic acid (LevOH, 13.5 μL, 0.132 mmol) was added dropwise at room temperature. After the addition was completed, stirring was continued at the same temperature for another 15 min before methanol was added to quench the reaction. Filtration through a pad of Celite removed the white solid. After the solid was thoroughly washed with EA/DCM (10/1), the filtrate was concentrated in vacuo to afford a residue, which was further purified by silica gel column chromatography (PE/EA = 3:1) to afford 13 (69 mg, 0.074 mmol, 62%) as a white solid: [α]25D = −19.1 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.85–7.80 (m, 3H), 7.75 (s, 1H), 7.70–7.65 (m, 4H), 7.49–7.35 (m, 9H), 5.12 (t, J = 8.4 Hz, 1H), 5.00 (s, 1H), 4.82 (s, 1H), 4.76 (s, 2H), 4.58 (d, J = 6.4 Hz, 1H), 3.95 (dd, J = 12, 4.8 Hz, 1H), 3.60 (td, J = 8.8, 4.4 Hz, 1H), 3.46 (dd, J = 8.4, 6.4 Hz, 1H), 3.28 (dd, J = 12.0, 8.8 Hz, 1H), 2.73 (t, J = 6.8 Hz, 2H), 2.58 (t, J = 6.8 Hz, 2H), 2.24 (d, J = 13.2 Hz, 1H), 2.19–2.11 (m, 4H), 2.04–1.29 (m, 14H), 1.25 (s, 3H), 1.14 (s, 9H), 1.06–0.77 (m, 4H), 0.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.9, 176.8, 172.5, 153.0, 135.8, 135.7, 135.4, 133.3, 133.2, 132.2, 132.1, 130.1, 128.4, 128.0, 127.8, 127.7, 126.8, 126.3, 126.2, 125.8, 104.5, 97.9, 86.0, 77.4, 74.8, 74.7, 72.9, 71.9, 62.8, 57.1, 53.8, 47.6, 45.3, 43.8, 42.4, 41.4, 40.7, 39.5, 38.7, 38.2, 37.5, 33.7, 30.0, 29.4, 28.3, 27.3, 25.6, 24.9, 22.2, 20.4, 19.4, 16.3; HRMS (ESI-TOF) calcd for C57H71O9Si [M + H]+ 927.4862, found 927.4854.
4-Methylphenyl 2-O-acetyl-3-O-(2-naphthylmethyl)-1-thio-β-D-xylopyranoside (16)
To a solution of diol 15 (100 mg, 0.252 mmol) and Yb(OTf)3 (15 mg, 0.024 mmol) in dioxane (1 mL), Ac2O (27 μL, 0.29 mmol) was added dropwise at room temperature under an N2 atmosphere. After the addition was completed, stirring was continued for 10 h at the same temperature. The reaction mixture was quenched with MeOH and the solvent was evaporated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (PE/EA = 3:1) to give 16 (60 mg, 0.137 mmol, 54%) as a white solid: [α]25D = −42.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.86–7.80 (m, 4H), 7.50–7.46 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 5.15 (t, J = 6.0 Hz, 1H), 4.96 (d, J = 11.6 Hz, 1H), 4.89 (d, J = 6.0 Hz, 1H), 4.80 (d, J = 12.0 Hz, 1H), 4.39 (dd, J = 12.0, 4.0 Hz, 1H), 3.79–3.74 (m, 1H), 3.61 (t, J = 6.0 Hz, 1H), 3.46 (dd, J = 12.0, 6.8 Hz, 1H), 2.46 (d, J = 6.8 Hz, 1H), 2.34 (s, 3H), 2.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.5, 138.1, 135.2, 133.4, 133.2, 132.6, 130.6, 129.9, 128.6, 128.1, 127.8, 126.9, 126.4, 126.2, 125.8, 87.2, 79.4, 73.8, 71.0, 68.6, 66.0, 21.3; HRMS (ESI-TOF) calcd for C25H27O5S [M + H]+ 439.1574, found 439.1577.
4-Methylphenyl 2-O-acetyl-3-O-(2-naphthylmethyl)-4-O-benzoyl-1-thio-β-D-xylopyranoside (17)
To a solution of 16 (250 mg, 0.57 mmol) in dry pyridine (3 mL), BzCl (110 μL, 0.95 mmol) was added dropwise at 0 °C under an N2 atmosphere. After the addition was completed, the reaction mixture was gradually warmed up to room temperature and stirring was continued for another 4 h before MeOH was added to quench the reaction. Ethyl acetate was added to dilute the reaction mixture and the resulting solution was washed successively with 1 M HCl, saturated aqueous NaHCO3, and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration in vacuo to afford the crude product, which was further purified by silica gel column chromatography (PE/EA = 7:1) to yield 17 (300 mg, 0.553 mmol, 97%) as a white solid: [α]25D = −61.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.02 (d, J = 6.8 Hz, 2H), 7.82–7.76 (m, 4H), 7.56 (t, J = 7.6 Hz, 1H), 7.49–7.39 (m, 7H), 7.13 (d, J = 7.6 Hz, 2H), 5.22–5.17 (m, 2H), 5.08 (d, J = 5.2 Hz, 1H), 4.95 (d, J = 12.0 Hz, 1H), 4.88 (d, J = 12.0 Hz, 1H), 4.63 (dd, J = 12.4, 3.2 Hz, 1H), 3.92 (t, J = 5.6 Hz, 1H), 3.66 (dd, J = 12.4, 5.6 Hz, 1H), 2.34 (s, 3H), 1.99 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 169.7, 165.6, 138.0, 135.0, 133.5, 133.3, 133.1, 132.5, 130.7, 129.8, 129.7, 128.5, 128.4, 128.1, 127.8, 126.8, 126.2, 126.1, 125.8, 86.6, 75.6, 73.5, 70.2, 69.5, 62.5, 21.3, 21.2, 21.1 (2C); HRMS (ESI-TOF) calcd for C32H31O6S [M + H]+ 543.1836, found 543.1838.
4-Methylphenyl 2-O-levulinoyl-3-O-(2-naphthylmethyl)-4-O-benzoyl-1-thio-β-D-xylopyranoside (18)
To a solution of 17 (300 mg, 0.553 mmol) in a mixed solvent of dry CH3OH/CH2Cl2 (8 mL/8 mL), AcCl (0.8 mL) was added. The mixture was stirred at room temperature for 24 h, at which time TLC indicated that the reaction was completed. The reaction mixture was then neutralized with Et3N, and the volatile solvent was removed under reduced pressure. The resulting residue was passed through a silica gel column (PE/EA = 5:1) to give a 2-OH intermediate (220 mg, 80%), which was used directly in the next step without detailed characterization. The above-obtained 2-OH intermediate (220 mg, 0.44 mmol), EDCI (337 mg, 1.76 mmol), DMAP (107 mg, 0.88 mmol), LevOH (204 mg, 1.76 mmol) and DIPEA (0.29 mL, 1.76 mmol) were dissolved in dry CH2Cl2 (4.0 mL), and the resulting mixture was stirred at room temperature for 4 h. Ethyl acetate was added to dilute the reaction mixture, and the obtained solution was washed successively with water, saturated aqueous NaHCO3, and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel column chromatography (PE/EA = 3:1) to afford 18 (230 mg, 0.429 mmol, 69% for 2 steps) as a colorless syrup: [α]25D = −59.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.13 (d, J = 6.8 Hz, 1H), 8.04 (d, J = 7.2 Hz, 2H), 7.81–7.76 (m, 4H), 7.63–7.54 (m, 1H), 7.50–7.39 (m, 6H), 7.14 (d, J = 7.6 Hz, 2H), 5.24–5.21 (m, 2H), 5.07 (d, J = 5.6 Hz, 1H), 4.98 (d, J = 12.0 Hz, 1H), 4.90 (d, J = 12.0 Hz, 1H), 4.61 (dd, J = 12.4, 4.0 Hz, 1H), 3.96 (t, J = 6.0 Hz, 1H), 3.65 (dd, J = 12.4, 6.0 Hz, 1H), 2.68–2.47 (m, 4H), 2.35 (s, 3H), 2.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.3, 171.6, 171.5, 165.5, 138.0, 135.1, 133.7, 133.4, 133.2, 133.0, 132.6, 130.3, 130.2, 129.8 (2C), 129.6, 129.4, 128.5, 128.4, 128.2, 128.0, 127.7, 126.6, 126.1, 126.0, 125.8, 86.5, 76.2, 73.6, 70.6, 69.7, 62.9, 37.7, 29.8, 28.0, 21.2; HRMS (ESI-TOF) calcd for C35H34O7SK [M + K]+ 637.1657, found 637.1659.
2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-benzoyl-β-D-xylopyranosyl trichloroacetimidate (19)
To a solution of 18 (200 mg, 0.334 mmol) in a mixed solvent of acetone and H2O (2 mL, v/v = 9:1), NBS (178 mg, 1.0 mmol) was added portionwise at 0 °C. After the reaction mixture was stirred at the same temperature for another 20 min, ethyl acetate was added to dilute the reaction mixture. The obtained solution was washed successively with water, saturated aqueous NaHCO3, and brine, and was then dried over Na2SO4. Filtration was followed by concentration in vacuo to give the crude product, which was further purified by silica gel column chromatography (PE/EA = 1:1) to afford the hemiacetal intermediate (135 mg, 82%). The hemiacetal intermediate was used in the next step without further characterization. To a solution of the above obtained hemiacetal intermediate in dry CH2Cl2 (3 mL), Cl3CCN (0.13 mL, 1.3 mmol) and DBU (18 μL, 0.13 mmol) were added at room temperature. The mixture was stirred at the same temperature for another 1 h, and then the solvent was evaporated in vacuo to give the crude product, which was purified by silica gel column chromatography (PE/EA = 3:1) to afford 19 (91 mg, 0.208 mmol, α:β = 5:1, 43% for 2 steps) as a colorless syrup. [α]25D = +2.2 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 0.2H), 8.65 (s, 1H), 8.23 (d, J = 8.0 Hz, 0.2H), 8.01 (s, 0.2H), 7.97–7.88 (m, 2.4H), 7.76–7.63 (m, 4H), 7.56–7.31 (m, 7.2H), 6.52–6.47 (m, 1.2H), 5.40–5.31 (m, 1.2H), 5.18 (dd, J = 9.6, 3.6 Hz, 1H), 4.93 (s, 2H), 4.37 (t, J = 9.2 Hz, 0.2H), 4.29 (t, J = 9.2 Hz, 1H), 4.13 (dd, J = 11.2, 5.6 Hz, 1.2H), 3.82 (t, J = 10.8 Hz, 1.2H), 2.70–2.47 (m, 4.8H), 2.13 (s, 0.6H), 2.11 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.1, 171.9, 165.5, 160.9, 135.3, 133.5, 133.2, 133.1, 129.8 (2C), 129.3, 128.6, 128.4, 128.3, 128.0, 127.8, 126.9, 126.2, 126.1 (2C), 93.9, 91.1, 76.2, 75.1 (2C), 72.3, 70.8, 61.5, 37.8, 29.9, 27.8 (2C); HRMS (ESI-TOF) calcd for C30H27Cl3NO8 [M − H]− 634.0808, found 634.0823.
4-Methylphenyl 2-O-benzoyl-3-O-(2-naphthylmethyl)-4-O-tert-butyldimethylsilyl-1-thio-β-D-xylopyranoside (21)
Compound 20 (500 mg, 1.0 mmol) was dissolved in dry DMF (3 mL), to which imidazole (343 mg, 5.0 mmol) and TBSCl (545 mg, 3.6 mmol) were added at room temperature under an N2 atmosphere. The reaction mixture was stirred at the same temperature for another 15 min, at which time TLC indicated the completion of reaction. The addition of methanol was followed by continuous stirring for another 15 min. Concentration at reduced pressure gave a residue and it was further purified by silica gel column chromatography (PE/EA = 40:1) to afford 21 (600 mg, 0.976 mmol, 98%) as a white solid: [α]25D = +63.8 (c 1.0, CHCl3); 1H NMR (400 MHz, chloroform-d) δ 7.92 (d, J = 6.8 Hz, 2H), 7.69–7.53 (m, 2H), 7.60 (s, 1H), 7.55–7.50 (m, 2H), 7.42–7.26 (m, 7H), 7.09 (d, J = 8.0 Hz, 2H), 5.24 (t, J = 8.8 Hz, 1H), 4.95 (d, J = 11.6 Hz, 1H), 4.81 (d, J = 11.6 Hz, 1H), 4.76 (d, J = 9.2 Hz, 1H), 4.08 (dd, J = 11.2, 4.8 Hz, 1H), 3.97–3.91 (m, 1H), 3.70 (t, J = 8.0 Hz, 1H), 3.35 (dd, J = 11.6, 9.6 Hz, 1H), 2.32 (s, 3H), 0.94 (s, 9H), 0.12 (d, J = 4.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 165.3, 138.1, 135.5, 133.1 (2C), 133.0, 132.9, 129.9, 129.8, 129.7, 129.4, 128.3, 128.1, 127.9, 127.7, 126.8, 126.1, 125.9, 125.7, 87.7, 83.2, 75.2, 71.9, 71.3, 69.8, 25.9, 21.2, 18.0, −4.5, −4.6; HRMS (ESI-TOF) calcd for C36H42O5SSiK [M + K]+ 653.2154, found 653.2154.
4-Methylphenyl 2-O-levulinoyl-3-O-(2-naphthylmethyl)-4-O-tert-butyldimethylsilyl-1-thio-β-D-xylopyranoside (22)
K2CO3 (78.6 mg, 0.569 mmol) was added to a solution of 21 (350 mg, 0.569 mmol) in a mixed solvent of THF and CH3OH (9 mL, v/v = 1:2) at room temperature, and then the suspension was stirred at the same temperature for 24 h, at which time TLC indicated that the reaction was completed. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel column chromatography (PE/EA = 25:1) to afford the C2–OH intermediate (240 mg), which was used directly in the next step without detailed characterization. To a solution of the above-obtained C2–OH intermediate (240 mg, 0.47 mmol), 1,3-dicyclohexylcarbodiimide (DCC, 194 mg, 0.94 mmol), and 4-dimethylaminopyridine (DMAP, 5.7 mg, 0.047 mmol) in DCM (3 mL), levulinic acid (LevOH, 58 μL, 0.56 mmol) was added portionwise at room temperature under an N2 atmosphere. After the addition was completed, stirring was continued at the same temperature for another 15 min, at which time TLC indicated the completion of the reaction. To the reaction mixture methanol was added, and stirring was continued for additional 15 min. Filtration through a pad of Celite was followed by thorough washing of the solid with EA/DCM (10/1). The filtrate was concentrated in vacuo and the resulting residue was purified by silica gel column chromatography (PE/EA = 20:1) to afford 22 (270 mg, 0.443 mmol, 78% for 2 steps) as a colorless syrup: [α]25D = +27.6 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.84–7.80 (m, 3H), 7.74 (s, 1H), 7.49–7.41 (m, 3H), 7.36 (d, J = 8.0 Hz, 2H), 7.10 (d, J = 8.0 Hz, 2H), 4.95 (t, J = 11.6 Hz, 2H), 4.80 (d, J = 12.0 Hz, 1H), 4.57 (d, J = 9.6 Hz, 1H), 4.00 (dd, J = 11.6, 5.2 Hz, 1H), 3.86–3.80 (m, 1H), 3.52 (t, J = 8.4 Hz, 1H), 3.25 (dd, J = 11.6, 9.6 Hz, 1H), 2.60–2.42 (m, 3H), 2.34–2.26 (m, 4H), 2.05 (s, 3H), 0.9 (s, 9H), 0.08 (d, J = 3.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 206.2, 171.4, 138.0, 135.9, 133.2, 132.9, 132.8, 129.7, 129.3, 128.0, 127.9, 127.7, 126.5, 126.1, 126.0, 125.9, 87.5, 83.6, 75.2, 71.6, 71.1, 69.8, 37.7, 29.8, 28.0, 25.8, 21.2, 18.0, −4.6, −4.7; HRMS (ESI-TOF) calcd for C34H44O6SSiK [M + K]+ 647.2260, found 647.2262.
2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-tert-butyldimethylsilyl-α-D-xylopyranosyl trichloroacetimidate (23)
Similar procedures to those used for the synthesis of 19 were adopted to convert 22 (420 mg, 0.69 mmol) to 23 (243 mg, 0.376 mmol, 54% for 2 steps) as a white oil: [α]25D = +70.2 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 7.84–7.78 (m, 4H), 7.50–7.44 (m, 3H), 6.38 (d, J = 3.2 Hz, 1H), 5.06–4.98 (m, 2H), 4.87 (d, J = 12.0 Hz, 1H), 3.91 (d, J = 4.8 Hz, 2H), 3.78–3.70 (m, 2H), 2.53–2.24 (m, 4H), 2.01 (s, 3H), 0.9 (s, 9H), 0.13 (d, J = 6.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 205.9, 172.0, 161.0, 136.0, 133.3, 133.0, 128.0, 127.9, 127.7, 126.5, 126.2, 126.1, 125.9, 94.2, 91.2, 79.6, 75.6, 72.1, 71.0, 64.9, 37.6, 29.7, 27.7, 25.9, 18.1, −4.5, −4.6; HRMS (ESI-TOF) calcd for C29H38Cl3NO7SiNa [M + Na]+ 668.1375, found 668.1370.4-Methylphenyl 2-O-benzoyl-3-O-(2-naphthylmethyl)-4-O-acetyl-1-thio-β-D-xylopyranoside (24)
To a solution of 20 (500 mg, 1 mmol) in dry pyridine (4 mL), AcCl (0.107 mL, 1.5 mmol) was added dropwise at 0 °C under an N2 atmosphere. Then the mixture was allowed to gradually warm up to room temperature and stirring was continued at the same temperature for 4 h. Then the reaction mixture was quenched with MeOH, and diluted with ethyl acetate. The resulting solution was washed successively with 1 M HCl, saturated aqueous NaHCO3 and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford a residue, which was further purified by silica gel column chromatography (PE/EA = 8:1) to give 24 (480 mg, 0.885 mmol, 89%) as a white solid: [α]25D = −29.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.06 (d, J = 6.8 Hz, 2H), 7.80–7.73 (m, 4H), 7.58 (t, J = 7.6 Hz, 1H), 7.48–7.39 (m, 7H), 7.11 (d, J = 7.6 Hz, 2H), 5.40 (t, J = 5.2 Hz, 1H), 5.17 (d, J = 5.2 Hz, 1H), 5.01–4.97 (m, 1H), 4.94 (d, J = 12.0 Hz, 1H), 4.86 (d, J = 12.0 Hz, 1H), 4.57 (dd, J = 12.4, 3.2 Hz, 1H), 3.90 (t, J = 5.6 Hz, 1H), 3.60 (dd, J = 12.0, 5.6 Hz, 1H), 2.33 (s, 3H), 2.00 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.0, 165.2, 138.0, 135.0, 133.5, 133.3, 133.1, 132.6, 130.6, 129.9, 129.8 (2C), 128.5, 128.3, 128.0, 127.8, 126.8, 126.2, 126.0, 125.8, 86.6, 75.5, 73.5, 70.5, 69.4, 62.4, 21.2, 21.1; HRMS (ESI-TOF) calcd for C32H30O6SK [M + K]+ 581.1395, found 581.1394.
4-Methoxyphenyl 2-O-benzoyl-3-O-(2-naphthylmethyl)-4-O-acetyl-β-D-xylopyranoside (25)
To a solution of 24 (3.25 g, 5.99 mmol) in a mixed solvent of acetone and H2O (20 mL, v/v = 9:1), NBS (3.2 g, 17.9 mmol) was added portionwise at 0 °C. After the reaction mixture was stirred at the same temperature for another 20 min, ethyl acetate was added to dilute the reaction mixture. The resulting solution was washed successively with water, saturated aqueous NaHCO3, and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel chromatography (PE/EA = 1:1) to afford the hemiacetal intermediate (1.8 g, 70%), which was used in the next step without further characterization. To a solution of the above-obtained hemiacetal intermediate (1.8 g, 4.12 mmol) in dry CH2Cl2 (5 mL), Cl3CCN (2.6 mL, 26 mmol) and DBU (0.6 mL, 4 mmol) were added at room temperature, and the mixture was stirred at the same temperature for another 1 h. Then the solvent was evaporated in vacuo to give the crude product, which was purified by silica gel column chromatography (PE/EA = 4:1) to afford the Schmidt donor (1.8 g, 75%), which was used for the next step without detailed characterization. In a flame dried flask, 4 Å molecular sieves (600 mg), 4-methoxyphenol (1.16 g, 9.3 mmol), anhydrous DCM (10 mL), and the above-obtained Schmidt donor (1.8 g, 3.1 mmol) were charged sequentially. The mixture was stirred at room temperature for 30 min, and then cooled to 0 °C. After being stirred at 0 °C for 5 min, TMSOTf (114 μL, 0.63 mmol) was added at the same temperature. The resulting mixture was allowed to gradually warm up to room temperature and stirring was continued for another 5 h at the same temperature. After being quenched with Et3N, the MS were removed by filtration through a pad of Celite. The filtrate was concentrated under vacuum, and the residue was purified by silica gel column chromatography (PE/EA = 3:1) to afford 25 (1.6 g, 2.95 mmol, 49%, 3 steps) as a white solid: [α]25D = +0.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.0 Hz, 2H), 7.79–7.71 (m, 4H), 7.58 (t, J = 7.6 Hz, 1H), 7.48–7.41 (m, 5H), 6.97 (d, J = 8.8 Hz, 2H), 6.80 (d, J = 8.8 Hz, 2H), 5.52 (dd, J = 6.4, 4.8 Hz, 1H), 5.26 (d, J = 4.8 Hz, 1H), 5.12–5.08 (m, 1H), 4.95 (d, J = 12.0 Hz, 1H), 4.90 (d, J = 12.0 Hz, 1H), 4.36 (dd, J = 12.4, 4.0 Hz, 1H), 3.96 (t, J = 6.0 Hz, 1H), 3.75 (s, 3H), 3.57 (dd, J = 12.4, 6.0 Hz, 1H), 2.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.1, 165.2, 155.4, 150.9, 135.2, 133.5, 133.3, 133.1, 129.9, 129.6, 128.5, 128.3, 128.0, 127.8, 126.7, 126.2, 126.0, 125.8, 118.6, 114.7, 99.2, 75.8, 73.3, 70.4, 69.9, 61.0, 55.7, 21.1; HRMS (ESI-TOF) calcd for C32H31O8 [M + H]+ 543.2013, found 543.2012.
4-Methoxyphenyl 2-O-benzoyl-3-O-(2-naphthylmethyl)-4-O-allyl-β-D-xylopyranoside (26)
AcCl (1 mL, 14.0 mmol) was added dropwise to a solution of 25 (1.0 g, 1.8 mmol) in a mixed solvent of dry CH3OH/CH2Cl2 (10 mL/10 mL) at room temperature. The resulting mixture was stirred at the same temperature for 24 h, at which time TLC (PE/EA = 3:1) indicated that the reaction was completed. Et3N was added to quench the reaction, and the volatile materials were removed under reduced pressure. The obtained residue was passed through a silica gel column (PE/EA = 3:1) to give the C4–OH intermediate (800 mg, 89%), which was used directly in the next step without detailed characterization. To a solution of the above-obtained C4–OH intermediate (800 mg, 1.6 mmol) and allyl ethyl carbonate (0.64 mL, 4.87 mmol) in degassed THF (3.2 mL), a mixture of Pd2(dba)3 (37 mg, 0.04 mmol) and dppb (133 mg, 0.31 mmol) in degassed THF (3.2 mL) was added at room temperature. Then the reaction mixture was heated to 65 °C and stirring was continued for 6 h at the same temperature. After the reaction mixture was cooled down to room temperature, the solvent was evaporated and the crude product was purified by silica gel column chromatography (PE/EA = 7:1) to afford 26 (837 mg, 1.55 mmol, 86%, 2 steps) as a white solid: [α]25D = +64.4 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.0 Hz, 2H), 7.71–7.69 (m, 1H), 7.66–7.62 (m, 2H), 7.54 (t, J = 8.4 Hz, 2H), 7.43–7.30 (m, 5H), 6.89 (d, J = 9.2 Hz, 2H), 6.75 (d, J = 9.2 Hz, 2H), 5.99–5.89 (m, 1H), 5.44 (dd, J = 8.0, 6.8 Hz, 1H), 5.32 (dd, J = 17.6, 1.6 Hz, 1H), 5.22 (dd, J = 10.0, 1.6 Hz, 1H), 4.98 (t, J = 12.0 Hz, 2H), 4.88 (d, J = 12.0 Hz, 1H), 4.26–4.15 (m, 3H), 3.84–3.75 (m, 2H), 3.73 (s, 3H), 3.45–3.40 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 165.4, 155.5, 151.2, 135.5, 134.6, 133.2 (2C), 133.0, 129.9, 129.8, 128.4, 128.2, 127.9, 127.7, 126.9, 126.2, 126.0, 125.8, 118.8, 117.7, 114.6, 101.0, 80.0, 77.4, 74.5, 72.5, 72.2, 63.6, 55.7; HRMS (ESI-TOF) calcd for C33H33 O7 [M + H]+ 541.2221, found 541.2224.
4-Methoxyphenyl 2-O-levulinoyl-3-O-(2-naphthylmethyl)-4-O-allyl-β-D-xylopyranoside (27)
To a solution of 26 (650 mg, 1.20 mmol) in a mixed solvent of MeOH (4 mL) and CH2Cl2 (4 mL), the freshly prepared NaOMe (in absolute MeOH, 1 mL) was added at room temperature. The reaction mixture was stirred at the same temperature for 1 h before Amberlite (H+) was added to adjust the pH value of the reaction mixture to around 7. Filtration was followed by concentration under reduced pressure to give the crude product, which was further purified by silica gel column chromatography (PE/EA = 5:1) to afford the C2–OH intermediate (450 mg, 86%), which was used directly in the next step without detailed characterization. The above-obtained C2–OH intermediate (450 mg, 1.03 mmol), EDCI (790 mg, 4.12 mmol), DMAP (251 mg, 2.05 mmol), LevOH (478.4 mg, 4.12 mmol), and DIPEA (0.68 mL, 3.9 mmol) were dissolved in dry CH2Cl2(5.0 mL) at room temperature, and the resulting mixture was stirred at the same temperature for another 4 h before ethyl acetate was added. The obtained solution was washed successively with water, saturated aqueous NaHCO3, and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel column chromatography (PE/EA = 3:1) to afford 27 (550 mg, 1.03 mmol, 86%, 2 steps) as a white solid: [α]25D = +31.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.88–7.83 (m, 4H), 7.52–7.46 (m, 3H), 6.99 (d, J = 9.2 Hz, 2H), 6.83 (d, J = 8.8 Hz, 2H), 5.98–5.88 (m, 1H), 5.32 (dd, J = 17.2, 1.6 Hz, 1H), 5.24–5.20 (m, 2H), 5.02 (d, J = 12.0 Hz, 1H), 4.92 (d, J = 12.0 Hz, 1H), 4.87 (d, J = 7.2 Hz, 1H), 4.23–4.12 (m, 3H), 3.74 (s, 3H), 3.72–3.67 (m, 2H), 3.39–3.34 (m, 1H), 2.66–2.42 (m, 4H), 2.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.1, 171.4, 155.4, 151.1, 135.8, 134.5, 133.2, 132.9, 128.0, 127.9, 127.6, 126.6, 126.0 (2C), 125.8, 118.6, 117.4, 114.5, 100.8, 80.3, 77.1, 74.4, 72.3, 72.0, 63.4, 55.5, 37.6, 29.7, 27.9; HRMS (ESI-TOF) calcd for C31H35O8 [M + H]+ 535.2326, found 535.2325.
2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-allyl-α/β-D-xylopyranosyl trichloroacetimidate (28)
Similar procedures to those used for the synthesis of 3 from 9 were applied to convert 27 (180 mg, 0.337 mmol) to 28 (100 mg, 0.174 mmol, 52%, 2 steps): [α]25D = +42.3 (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3δ 8.58 (s, 1H), 7.82 (d, J = 10.4, 4H), 7.50–7.44 (m, 3H), 6.38 (d, J = 3.6 Hz, 1H), 5.97–5.86 (m, 1H), 5.31 (dd, J = 17.2, 1.6 Hz, 1H), 5.22 (dd, J = 10.4, 1.6 Hz, 1H), 5.04 (d, J = 11.6 Hz, 1H), 5.01 (dd, J = 10.0, 3.6 Hz, 1H), 4.92 (d, J = 11.6 Hz, 1H), 4.27–4.13 (m, 2H), 4.02 (dd, J = 10.0, 8.4 Hz, 1H), 3.93 (dd, J = 10.4, 4.8 Hz, 1H), 3.78–3.66 (m, 2H), 2.62–2.38 (m, 4H), 2.06 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.1, 172.0, 161.0, 135.9, 134.5, 133.3, 133.0, 128.1, 128.0, 127.7, 126.7, 126.2, 126.0, 117.8, 94.0, 78.5, 77.2, 75.4, 72.8, 72.1, 62.6, 37.7, 29.8, 27.8; HRMS (ESI-TOF) calcd for C26H27Cl3NO7 [M − H]− 570.0857, found 570.0877.13-O-[2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-benzoyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (29)
A similar procedure to that used for the synthesis of 10 from 3 and 2 was applied to mediate the coupling between 19 (91 mg, 0.14 mmol) and acceptor 2 (53 mg, 0.095 mmol) to provide 29 (90 mg, 0.087 mmol, 92%) as a white foam: [α]25D = −38.0 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 6.8 Hz, 2H), 7.77–7.65 (m, 8H), 7.56 (t, J = 7.6 Hz, 1H), 7.45–7.32 (m, 11H), 5.29–5.22 (m, 1H), 5.13 (dd, J = 8.4, 6.4 Hz, 1H), 5.01 (s, 1H), 4.91 (d, J = 12.0 Hz, 1H), 4.85 (d, J = 12.0 Hz, 2H), 4.76 (d, J = 6.4 Hz, 1H), 4.23 (dd, J = 11.6, 4.8 Hz, 1H), 3.93 (t, J = 7.6 Hz, 1H), 3.35 (dd, J = 11.6, 8.0 Hz, 1H), 2.64–2.49 (m, 4H), 2.26 (d, J = 13.2 Hz, 1H), 2.20–2.13 (m, 1H), 2.10–1.98 (m, 5H), 1.89–1.72 (m, 6H), 1.57–1.38 (m, 5H), 1.26 (s, 3H), 1.13 (s, 9H), 1.06–0.93 (m, 3H), 0.86–0.79 (m, 1H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.3, 176.8, 171.4, 165.5, 152.6, 135.7 (2C), 135.6, 133.4, 133.3, 133.0, 132.2, 130.0, 129.8, 129.7, 128.5, 128.2, 128.0, 127.8, 127.6, 126.7, 126.1, 126.0, 125.9, 104.8, 95.8, 85.8, 78.0, 73.6, 72.5, 71.2, 61.8, 57.0, 53.8, 47.4, 45.3, 43.8, 42.5, 41.4, 40.7, 39.5, 38.7, 37.8, 37.1, 29.9, 29.3, 28.1, 27.2, 22.2, 20.4, 19.4 (2C), 16.4; HRMS (ESI-TOF) calcd for C64H75O10Si [M + H]+ 1031.5124, found 1031.5116.13-O-[2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-tert-butyldimethylsilyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (30)
A similar procedure to that used for the synthesis of 10 from 3 and 2 was adopted to mediate the coupling between 23 (243 mg, 0.37 mmol) and 2 (139 mg, 0.25 mmol) to provide 30 (246 mg, 0.236 mmol, 94%) as a white solid: [α]25D = +2.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.88–7.83 (m, 3H), 7.79 (s, 1H), 7.73–7.70 (m, 4H), 7.52–7.37 (m, 9H), 5.03 (d, J = 12.4 Hz, 1H), 4.97 (dd, J = 9.6, 8.0 Hz, 1H), 4.95 (s, 1H), 4.85 (d, J = 12.0 Hz, 1H), 4.82 (s, 1H), 4.54 (d, J = 8.0 Hz, 1H), 3.92–3.83 (m, 2H), 3.54 (t, J = 9.2 Hz, 1H), 3.18 (t, J = 9.6 Hz, 1H), 2.49–2.25 (m, 5H), 2.20–2.11 (m, 1H), 2.04–1.96 (m, 5H), 1.91–1.69 (m, 6H), 1.65–1.38 (m, 5H), 1.35–1.30 (m, 1H), 1.28 (s, 3H), 1.17 (s, 9H), 1.09–1.00 (m, 2H), 0.96 (s, 9H), 0.94–0.91 (m, 1H), 0.86–0.79 (m, 1H), 0.77 (s, 3H), 0.13 (d, J = 7.3 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 206.3, 176.7, 171.2, 152.4, 136.2, 135.6 (2C), 133.3, 132.9, 132.1 (2C), 130.0, 128.0 (2C), 127.7, 127.6, 126.5, 126.1, 126.0, 125.8, 104.7, 96.7, 85.5, 82.9, 77.4, 75.0, 73.3, 71.6, 66.1, 56.9, 53.6, 47.2, 45.2, 44.0, 42.5, 41.3, 40.6, 39.4, 38.6, 37.6, 36.7, 29.8, 29.3, 28.0, 27.2, 25.8, 22.1, 20.3, 19.3, 18.0, 16.4, −4.5, −4.7; HRMS (ESI-TOF) calcd for C63H84O9Si2K [M + K]+ 1079.5285, found 1079.5280.13-O-[2-O-Levulinoyl-3-O-(2-naphthylmethyl)-4-O-allyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (31)
A similar procedure to that used for the synthesis of 10 from 3 and 2 was applied to mediate the condensation between 28 (40 mg, 0.0698 mmol) and 2 (26 mg, 0.0466 mmol) to afford 31 (35 mg, 0.036 mmol, 77%) as a colorless foam: [α]25D = +5.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.85–7.79 (m, 4H), 7.67 (d, J = 6.4 Hz, 4H), 7.50–7.35 (m, 9H), 5.95–5.87 (m, 1H), 5.30 (dd, J = 17.2, 1.6 Hz, 1H), 5.20 (dd, J = 10.4, 1.6 Hz, 1H), 5.00–4.86 (m, 4H), 4.80 (s, 1H), 4.52 (d, J = 7.6 Hz, 1H), 4.19 (dd, J = 12.8, 5.6 Hz, 1H), 4.13 (dd, J = 12.8, 5.6 Hz, 1H), 3.98 (dd, J = 12.0, 4.4 Hz, 1H), 3.65–3.56 (m, 2H), 3.13 (dd, J = 12.0, 9.6 Hz, 1H), 2.57–2.52 (m, 2H), 2.43–2.39 (m, 2H), 2.26 (d, J = 13.6 Hz, 1H), 2.14 (d, J = 17.2 Hz, 1H), 2.04 (s, 3H), 2.00–1.95 (m, 2H), 1.85–1.65 (m, 6H), 1.57–1.35 (m, 5H), 1.25 (s, 3H), 1.14 (s, 9H), 1.05–1.00 (m, 2H), 0.93 (d, J = 8.0 Hz, 1H), 0.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 206.4, 176.8, 171.4, 152.6, 136.2 135.7 (2C), 134.8, 133.4, 133.1, 132.2, 130.1, 128.1 (2C), 127.8, 127.7, 126.7, 126.3, 126.1, 125.9, 117.6, 104.8, 96.6, 85.7, 81.4, 77.8, 74.6, 73.4, 72.3, 63.6, 57.1, 53.8, 47.4, 45.3, 44.0, 42.6, 41.4, 40.7, 39.5, 38.8, 37.8, 37.0, 29.9, 29.4, 28.1, 27.2, 22.2, 20.4, 19.5, 19.4, 16.5; HRMS (ESI-TOF) calcd for C60H74O9SiK [M + K]+ 1005.4734, found 1005.4755.13-O-[3-O-(2-Naphthylmethyl)-4-O-benzoyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (32)
A similar procedure to that used for the synthesis of 11 was applied to convert 29 (110 mg, 0.107 mmol) to 32 (81 mg, 0.087 mmol, 81%) as a white foam: [α]25D = −42.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.94 (d, J = 6.8 Hz, 2H), 7.78–7.66 (m, 8H), 7.57 (t, J = 7.2 Hz, 1H), 7.48–7.35 (m, 11H), 5.29–5.23 (m, 1H), 5.12 (s, 1H), 5.10 (d, J = 12.0 Hz, 1H), 5.01 (d, J = 12.0 Hz, 1H), 4.91 (s, 1H), 4.59 (d, J = 7.2 Hz, 1H), 4.16 (dd, J = 11.6, 5.6 Hz, 1H), 3.84 (t, J = 8.8 Hz, 1H), 3.75 (dd, J = 10.4, 7.6 Hz, 1H), 3.27 (dd, J = 11.6, 9.6 Hz, 1H), 2.53 (s, 1H), 2.28 (d, J = 12.8 Hz, 1H), 2.22 (d, J = 17.6 Hz, 1H), 2.06 (d, J = 14.0 Hz, 2H), 1.95–1.75 (m, 6H), 1.60–1.43 (m, 4H), 1.29 (s, 3H), 1.17 (s, 9H), 1.10–0.97 (m, 3H), 0.78 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 165.6, 152.8, 135.8, 135.7 (2C), 133.3, 133.2, 133.0, 132.1 (2C), 130.0, 129.7, 129.6, 128.5, 128.2, 128.0, 127.7, 127.6, 126.8, 126.1, 126.0, 125.8, 104.7, 98.3, 86.1, 80.4, 77.4, 77.3, 74.4, 74.3, 71.5, 62.7, 57.0, 53.7, 47.6, 45.2, 43.9, 42.3, 41.4, 40.7, 39.5, 38.7, 37.6, 29.3, 27.2, 22.2, 20.4, 19.4, 16.3; HRMS (ESI-TOF) calcd for C59H69O8Si [M + H]+ 933.4756, found 933.4765.13-O-[3-O-(2-Naphthylmethyl)-4-O-tert-butyldimethylsilyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (33)
A similar procedure to that used for the synthesis of 11 was adopted to convert 30 (120 mg, 0.115 mmol) to 33 (88 mg, 0.093 mmol, 81%) as a white solid: [α]25D = −21.0 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.84 (d, J = 8.4 Hz, 4H), 7.73–7.69 (m, 4H), 7.55 (d, J = 8.8 Hz, 1H), 7.50–7.37 (m, 8H), 5.08 (s, 1H), 5.06 (d, J = 12.0 Hz, 1H), 5.01 (d, J = 12.0 Hz, 1H), 4.88 (s, 1H), 4.55 (d, J = 6.8 Hz, 1H), 3.90 (dd, J = 11.6, 4.8 Hz, 1H), 3.85–3.80 (m, 1H), 3.58 (dd, J = 8.4, 6.8 Hz, 1H), 3.48 (t, J = 8.0 Hz, 1H), 3.21 (dd, J = 11.6, 9.2 Hz, 1H), 2.27 (d, J = 13.2 Hz, 1H), 2.20 (d, J = 17.2 Hz, 1H), 1.92–1.74 (m, 6H), 1.68–1.42 (m, 6H), 1.29 (s, 3H), 1.17 (s, 9H), 1.09–1.02 (m, 2H), 0.99–0.96 (m, 1H), 0.94 (s, 9H), 0.76 (s, 3H), 0.13 (s, 3H), 0.12 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 153.0, 136.4, 135.8, 135.7, 133.4, 133.0, 132.2, 132.1, 130.0 (2C), 128.1, 128.0, 127.8, 127.6, 126.5, 126.4, 126.0, 125.8, 104.6, 98.2, 85.8, 83.6, 77.4, 74.7, 73.5, 70.9, 65.8, 57.0, 53.7, 47.5, 45.2, 44.0, 42.4, 41.4, 40.7, 39.5, 38.7, 37.4, 29.3, 27.3, 25.9, 22.2, 20.4, 19.4, 18.1, 16.3, −4.5, −4.6; HRMS (ESI-TOF) calcd for C58H78O7Si2K [M + K]+ 981.4918, found 981.4925.13-O-[3-O-(2-Naphthylmethyl)-4-O-allyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (34)
A similar procedure to that used for the synthesis of 11 was applied to convert 31 (180 mg, 0.186 mmol) to 34 (140 mg, 0.161 mmol, 86%) as a white solid: [α]25D = −15.0 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.88–7.83 (m, 4H), 7.73–7.70 (m, 4H), 7.57 (d, J = 8.4 Hz, 1H), 7.51–7.37 (m, 8H), 5.99–5.90 (m, 1H), 5.32 (dd, J = 17.2, 1.6 Hz, 1H), 5.21 (dd, J = 10.4, 1.6 Hz, 1H), 5.09 (s, 1H), 5.07 (d, 11.6 Hz, 1H), 5.03 (d, J = 12.0 Hz, 1H), 4.88 (s, 1H), 4.56 (s, 1H), 4.24–4.14 (m, 2H), 4.03 (dd, J = 12.4, 3.6 Hz, 1H), 3.59 (s, 3H), 3.24–3.19 (m, 1H), 2.57 (bs, 1H), 2.28 (d, J = 13.2 Hz, 1H), 2.24–2.16 (m, 1H), 2.07–2.02 (m, 2H), 1.93–1.74 (m, 6H), 1.70–1.39 (m, 6H), 1.29 (s, 3H), 1.18 (s, 9H), 1.09–1.00 (m, 2H), 0.98 (d, J = 8.0 Hz, 1H), 0.87–0.80 (m, 1H), 0.77 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 153.0, 136.3, 135.7 (2C), 134.7, 133.4, 133.1, 132.1 (2C), 130.0, 128.2, 128.0, 127.8, 127.6, 126.6, 126.1, 125.8, 117.4, 104.6, 98.1, 85.8, 82.2, 77.4, 74.5, 73.3, 72.0, 63.0, 57.0, 53.8, 47.6, 45.2, 43.9, 42.4, 41.4, 40.7, 39.5, 38.7, 37.5, 29.3, 27.2, 22.2, 20.4, 19.4, 16.3; HRMS (ESI-TOF) calcd for C55H69O7Si [M + H]+ 869.4807, found 869.4802.13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-3-O-(2-naphthylmethyl)-4-O-benzoyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (35)
A similar procedure to that used for the synthesis of 12 was applied to mediate the coupling between 32 (35 mg, 0.0375 mmol) and 4 (35 mg, 0.0472 mmol) to provide 35 (50 mg, 0.0334 mmol, 88%) as a colorless foam: [α]25D = −18.7 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.03 (d, J = 6.8 Hz, 2H), 7.92 (d, J = 7.2 Hz, 2H), 7.87 (d, J = 6.8 Hz, 2H), 7.81 (d, J = 6.8 Hz, 2H), 7.70–7.64 (m, 8H), 7.45–7.16 (m, 26H), 5.88 (t, J = 9.6 Hz, 1H), 5.76 (t, J = 10.0 Hz, 1H), 5.63 (dd, J = 9.6, 8.0 Hz, 1H), 5.34 (d, J = 8.0 Hz, 1H), 5.15 (s, 1H), 5.13–5.07 (m, 1H), 4.84 (d, J = 12.0 Hz, 1H), 4.81 (d, J = 6.4 Hz, 1H), 4.75 (s, 1H), 4.61 (d, J = 12.0 Hz, 1H), 4.57 (dd, J = 12.4, 2.8 Hz, 1H), 4.47 (dd, J = 12.0, 4.4 Hz, 1H), 4.08–4.02 (m, 2H), 3.94 (dd, J = 8.8, 6.4 Hz, 1H), 3.82 (t, J = 8.4 Hz, 1H), 3.22 (dd, J = 12.0, 7.6 Hz, 1H), 2.25 (d, J = 13.2 Hz, 1H), 2.14–2.04 (m, 2H), 1.97–1.63 (m, 6H), 1.60–1.33 (m, 6H), 1.25 (s, 3H), 1.12 (s, 9H), 1.04–1.00 (m, 2H), 0.91 (d, J = 7.6 Hz, 1 H), 0.83–0.76 (m, 1H), 0.72 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.9, 166.3, 165.9, 165.6, 165.2 (2C), 153.5, 135.8, 135.7, 135.6, 133.5, 133.3, 133.2, 133.1 (2C), 133.0, 132.2 (2C), 130.0, 129.9 (3C), 129.7, 129.5, 129.4, 128.9 (2C), 128.5 (3C), 128.4, 128.3, 128.2, 128.0, 127.8, 127.6 (2C), 126.4, 126.1, 125.8 (2C), 104.6, 101.1, 97.1, 85.3, 81.5, 79.6, 77.4, 75.1, 73.4, 72.8, 72.3 (2C), 69.5, 63.1, 62.2, 57.1, 53.9, 47.2, 45.3, 44.0, 42.9, 41.4, 40.7, 39.5, 38.8, 36.2, 29.8, 29.4, 27.2, 22.2, 20.4, 19.5, 19.4, 16.4; HRMS (ESI-TOF) calcd for C93H94O17SiNa [M + Na]+ 1534.6185, found 1534.6207.13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-3-O-(2-naphthylmethyl)-4-O-tert-butyldimethylsilyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (36)
A similar procedure to that used for the synthesis of 12 was applied to mediate the condensation between 33 (70 mg, 0.074 mmol) and 4 (70 mg, 0.094 mmol) to furnish 36 (84.4 mg, 0.055 mmol, 75%) as a white solid: [α]25D = +25.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.02–7.91 (m, 5H), 7.85 (d, J = 8.4 Hz, 4H), 7.80–7.77 (m, 3H), 7.72–7.68 (m, 4H), 7.55–7.32 (m, 18H), 7.26–7.17 (m, 3H), 5.84 (t, J = 9.6 Hz, 1H), 5.72 (t, J = 10.0 Hz, 1H), 5.58 (dd, J = 9.6, 8.0 Hz, 1H), 5.45 (d, J = 8.0 Hz, 1H), 5.16 (s, 1H), 4.91 (d, J = 11.2 Hz, 1H), 4.73 (d, J = 11.2 Hz, 1H), 4.69 (s, 1H), 5.56 (d, J = 7.6 Hz, 1H), 4.54 (dd, J = 7.6, 4.8 Hz, 1H), 4.48 (dd, J = 12.0, 4.4 Hz, 1H), 4.05–4.01 (m, 1H), 3.84–3.71 (m, 3H), 3.46 (t, J = 8.8 Hz, 1H), 3.10 (t, J = 11.6 Hz, 1H), 2.27 (d, J = 13.2 Hz, 1H), 2.14–2.04 (m, 2H), 1.96–1.83 (m, 5H), 1.64–1.36 (m, 7H), 1.27 (s, 3H), 1.16 (s, 9H), 1.06–1.02 (m, 2H), 0.92 (d, J = 7.6 Hz, 1H), 0.83 (s, 9H), 0.78 (s, 3H), −0.01 (s, 3H), −0.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.9, 166.3, 165.8, 165.2 (2C), 153.2, 136.1, 135.8, 135.7, 133.5, 133.4, 133.2, 133.1 (2C), 133.0, 132.3, 132.2, 130.0 (3C), 129.9 (2C), 129.8 (2C), 129.4, 129.0, 128.4 (2C), 128.3, 128.2, 128.1, 127.9, 127.7, 126.2, 126.0, 125.9, 125.7, 104.8, 100.8, 97.4, 85.6, 85.3, 78.7, 77.4, 75.2, 73.4, 72.9, 72.2, 71.9, 69.7, 66.0, 63.3, 57.1, 53.9, 47.1, 45.3, 44.0, 42.9, 41.5, 40.7, 39.6, 38.8, 36.2, 29.4, 27.3, 25.8, 22.2, 20.5, 19.5, 19.4, 18.0, 16.5, −4.5; HRMS (ESI-TOF) calcd for C92H104O16Si2Na [M + Na]+ 1543.6759, found 1543.6755.13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-3-O-(2-naphthylmethyl)-4-O-allyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (37)
A similar procedure to that used for the synthesis of 12 was applied to mediate the coupling between 34 (140 mg, 0.161 mmol) and 4 (152.3 mg, 0.205 mmol) to provide 37 (210 mg, 0.145 mmol, 90%) as a white solid: [α]25D = +24.3 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.04 (d, J = 6.4 Hz, 2H), 7.97–7.81 (m, 11H), 7.73 (d, J = 8.0 Hz, 4H), 7.56–7.25 (m, 20H), 5.93 (t, J = 9.6 Hz, 1H), 5.85–5.75 (m, 2H), 5.64 (dd, J = 9.6, 7.6 Hz, 1H), 5.48 (d, J = 7.6 Hz, 1H), 5.22–5.16 (m, 2H), 5.12 (dd, J = 10.0, 1.6 Hz, 1H), 4.88 (d, J = 11.2 Hz, 1H), 4.78 (d, J = 11.2 Hz, 1H), 4.74 (s, 1H), 4.63 (d, J = 7.2 Hz, 1H), 4.59 (dd, J = 12.0, 2.8 Hz, 1H), 4.51 (dd, J = 12.4, 4.4 Hz, 1H), 4.10–4.06 (m, 1H), 4.01 (d, J = 5.6 Hz, 2H), 3.93 (dd, J = 11.6, 4.8 Hz, 1H), 3.84 (t, J = 8.4 Hz, 1H), 3.58–3.48 (m, 2H), 3.12 (dd, J = 11.6, 9.2 Hz, 1H), 2.31 (d, J = 13.2 Hz, 1H), 2.16–2.08 (m, 2H), 1.99–1.71 (m, 7H), 1.63–1.40 (m, 6H), 1.29 (s, 3H), 1.19 (s, 9H), 1.12–0.95 (m, 2H), 0.94 (d, J = 7.6 Hz, 1H), 0.88–0.81 (m, 1H), 0.80 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.2, 165.9, 165.2 (2C), 153.3, 136.2, 135.7 (2C), 134.6, 133.5, 133.4, 133.2, 133.1 (2C), 133.0, 132.2 (2C), 130.0, 129.9 (2C), 129.8 (2C), 129.7, 129.4, 128.9, 128.4 (2C), 128.3 (2C), 128.2, 128.1, 127.8, 127.6, 126.4, 126.2, 126.0, 125.9, 117.4, 104.6, 100.9, 97.2, 85.2, 84.2, 79.1, 78.2, 77.4, 75.2, 73.4, 72.8, 72.2, 71.8, 69.6, 63.2, 63.1, 57.1, 53.9, 47.2, 45.3, 44.0, 42.8, 41.4, 40.7, 39.5, 38.7, 36.3, 29.3, 27.2, 22.1, 20.4, 19.4 (2C), 16.4; HRMS (ESI-TOF) calcd for C89H95O16Si [M + H]+ 1447.6384, found 1447.6405.13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-benzoyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (38)
To a solution of 35 (72 mg, 0.048 mmol) in a mixed solvent of CH2Cl2 (3.8 mL) and CH3OH (1.09 mL), DDQ (46.54 mg, 0.205 mmol) was added at room temperature. The suspension was stirred at the same temperature for 6 h before ethyl acetate was added to dilute the reaction mixture. The resulting mixture was washed with saturated aqueous NaHCO3 and brine, and the organic phase was then dried over Na2SO4. Filtration and concentration under reduced pressure gave a residue, which was purified by silica gel column chromatography (PE/EA = 4:1) to give 38 (53 mg, 0.0386 mmol, 81%) as a white solid: [α]25D = +1.5 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.01 (d, J = 8.4 Hz, 2H), 7.93 (d, J = 11.2 Hz, 2H), 7.89–7.86 (m, 4H), 7.81 (d, J = 6.8 Hz, 2H), 7.66–7.63 (m, 4H), 7.57–7.25 (m, 21H), 5.89 (t, J = 9.6 Hz, 1H), 5.75 (t, J = 9.6 Hz, 1H), 5.58 (dd, J = 9.6, 7.6 Hz, 1H), 5.30 (d, J = 8.0 Hz, 1H), 5.06 (s, 1H), 4.92–4.87 (m, 2H), 4.78 (s, 1H), 4.59 (dd, J = 12.4, 2.8 Hz, 1H), 4.46 (dd, J = 12.4, 4.0 Hz, 1H), 4.18–4.08 (m, 2H), 3.86 (t, J = 7.6 Hz, 1H), 3.75 (dd, J = 7.6, 5.6 Hz, 1H), 3.35 (dd, J = 12.4, 6.4 Hz, 1H), 2.23 (d, J = 13.6 Hz, 1H), 2.12–2.06 (m, 2H), 1.95 (d, J = 18.0 Hz, 1H), 1.86–1.57 (m, 6H), 1.52–1.31 (m, 6H), 1.23 (s, 3H), 1.12 (s, 9H), 1.02–0.96 (m, 2H), 0.89–0.87 (m, 1H), 0.80–0.69 (m, 1H), 0.66 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.3 (2C), 165.9, 165.6, 165.2, 153.5, 135.8, 135.7, 133.5, 133.4, 133.3, 133.2, 132.2 (2C), 130.0, 129.9 (4C), 129.6 (2C), 129.2, 128.9 (2C), 128.5 (2C), 128.4, 127.7 (2C), 104.5, 102.1, 96.2, 85.6, 80.9, 77.4, 73.2, 72.7, 72.6, 72.4, 69.3, 63.0, 61.1, 57.1, 53.8, 47.1, 45.3, 44.3, 42.8, 41.4, 40.7, 39.5, 38.7, 35.9, 29.8, 29.4, 27.3, 22.2, 20.3, 19.4, 16.4; HRMS (ESI-TOF) calcd for C82H90NO17Si [M + NH4]+ 1388.5973, found 1388.5970.
13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-tert-butyldimethylsilyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (39)
A similar procedure to that used for the synthesis of 38 was applied to convert 36 (84 mg, 0.055 mmol) to 39 (60 mg, 0.0434 mmol, 79%) as a white solid: [α]25D = +1.6 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 6.8 Hz, 4H), 7.85 (dd, J = 14.8, 6.8 Hz, 4H), 7.69–7.66 (m, 4H), 7.54–7.28 (m, 18H), 5.89 (t, J = 10.0 Hz, 1H), 5.75 (t, J = 10.0 Hz, 1H), 5.56 (dd, J = 10.0, 8.0 Hz, 1H), 5.30 (d, J = 7.6 Hz, 1H), 5.11 (s, 1H), 4.74 (s, 1H), 4.57 (dd, J = 12.4, 3.2 Hz, 1H), 4.51–4.45 (m, 2H), 4.16–4.12 (m, 1H), 3.66 (dd, J = 11.6, 5.2 Hz, 1H), 3.51 (dd, J = 8.8, 7.6 Hz, 1H), 3.45–3.39 (m, 1H), 3.32 (t, J = 8.8 Hz, 1H), 3.00 (t, J = 11.6 Hz, 1H), 2.24 (d, J = 13.2 Hz, 1H), 2.08 (d, J = 16.8 Hz, 1H), 2.01 (d, J = 10.0 Hz, 1H), 1.92 (d, J = 16.8 Hz, 1H), 1.86–1.31 (m, 12H), 1.25 (s, 3H), 1.13 (s, 9H), 1.06–0.0.97 (m, 2H), 0.88–0.84 (m, 1H), 0.78 (s, 9H), 0.76–0.71 (m, 1H), 0.67 (s, 3H), −0.06 (s, 3H), −0.07 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.9, 166.3, 165.9, 165.6, 165.2, 153.5, 135.8, 135.7, 133.5, 133.3, 133.1, 132.2, 132.1, 130.0, 129.9 (3C), 129.8 (2C), 129.7, 129.0 (2C), 128.5 (2C), 128.4 (2C), 127.6, 104.5, 102.0, 97.2, 85.4, 81.6, 77.4, 73.2, 73.0, 72.3, 71.1, 69.4, 65.6, 63.0, 57.1, 53.7, 47.1, 45.3, 44.2, 42.8, 41.4, 40.7, 39.5, 38.7, 36.0, 29.4, 27.3, 25.8, 22.2, 20.3, 19.4 (2C), 18.1, 16.4, −4.7 (2C); HRMS (ESI-TOF) calcd for C81H100NO16Si2 [M + NH4]+ 1398.6575, found 1398.6588.13-O-[2-O-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-allyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (40)
A similar procedure to that used for the synthesis of 38 was adopted to convert 37 (87 mg, 0.06 mmol) to 40 (58 mg, 0.0443 mmol, 74%) as a white solid: [α]25D = +0.05 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.02–7.97 (m, 4H), 7.87 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 7.2 Hz, 2H), 7.67 (d, J = 6.8 Hz, 4H), 7.51–7.26 (m, 18H), 5.89 (t, J = 10.0 Hz, 1H), 5.83–5.71 (m, 2H), 5.55 (dd, J = 9.6, 7.6 Hz, 1H), 5.32 (d, J = 7.6 Hz, 1H), 5.19–5.09 (m, 3H), 4.73 (s, 1H), 4.60–4.55 (m, 2H), 4.47 (dd, J = 12.0, 4.0 Hz, 1H), 4.16–4.12 (m, 1H), 3.99 (dd, J = 5.6, 1.6 Hz, 2H), 3.86 (dd, J = 11.6, 4.8 Hz, 1H), 3.60–3.50 (m, 2H), 3.28–3.23 (m, 1H), 3.03 (dd, J = 11.6, 9.6 Hz, 1H), 2.25 (d, J = 13.2 Hz, 1H), 2.11–2.00 (m, 2H), 1.92 (d, J = 16.0 Hz, 1H), 1.85–1.72 (m, 5H), 1.64–1.59 (m, 1H), 1.53–1.31 (m, 6H), 1.24 (s, 3H), 1.14 (s, 9H), 1.02–0.98 (m, 2H), 0.88–0.78 (m, 1H), 0.80–0.70 (m, 1H), 0.68 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.3, 165.9, 165.6, 165.2, 153.5, 135.7 (2C), 134.6, 133.5, 133.3 (2C), 133.1, 133.2, 132.1, 130.0, 129.9 (2C), 129.8, 129.7, 129.5, 128.9, 128.5 (2C), 128.4 (2C), 127.6, 117.7, 104.5, 101.7, 96.9, 85.4, 81.2, 77.0, 75.5, 73.2, 72.9, 72.2, 71.7, 69.4, 63.0, 62.8, 57.1, 53.8, 47.1, 45.3, 44.1, 42.8, 41.4, 40.6, 39.4, 38.7, 36.1, 29.4, 27.2 (2C), 22.1, 20.3, 19.4 (2C), 16.4; HRMS (ESI-TOF) calcd for C78H86O16SiNa [M + Na]+ 1329.5577, found 1329.5568.13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-allyl-β-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (41)
A similar procedure to that used for the synthesis of 12 was adopted to mediate the coupling between 40 (31 mg, 0.024 mmol) and 4 (44 mg, 0.059 mmol) to provide 41 (30 mg, 0.016 mmol, 67%) as a white solid: [α]25D = +41.8 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.25 (t, J = 8.8 Hz, 4H), 7.94–7.87 (m, 11H), 7.69–7.62 (m, 8H), 7.57–7.27 (m, 27H), 5.86–5.71 (m, 3H), 5.56–5.45 (m, 4H), 5.15 (dd, J = 17.2, 2.0 Hz, 1H), 5.01(s, 1H), 4.96 (dd, J = 10.4, 1.6 Hz, 1H), 4.78 (dd, J = 18.0, 7.6 Hz, 2H), 4.62 (s, 1H), 4.49 (d, J = 6.4 Hz, 1H), 4.26–4.15 (m, 5H), 4.01 (dd, J = 13.2, 6.0 Hz, 1H), 3.84–3.77 (m, 2H), 3.65–3.60 (m, 1H), 3.44–3.38 (m, 1H), 3.05 (dd, J = 12.0, 9.6 Hz, 1H), 2.63–2.53 (m, 2H), 2.26 (d, J = 13.2 Hz, 1H), 2.06–1.96 (m, 2H), 1.89–1.28 (m, 13H), 1.26 (s, 3H), 1.15 (s, 9H), 1.07–0.98 (m, 2H), 0.91–0.83 (m, 2H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.1, 166.0 (2C), 165.2, 165.1, 165.0, 153.4, 135.8, 135.7, 135.2, 133.6, 133.4, 133.2, 133.1, 132.3, 132.2, 130.3, 130.2, 130.1, 129.9 (2C), 129.8, 129.7 (2C), 129.5, 129.2 (2C), 129.0, 128.5 (2C), 128.4 (2C), 127.7, 116.5, 104.6, 100.4, 100.3, 97.1, 85.1, 82.3, 79.4, 75.5, 73.1, 72.8, 72.6, 71.5, 71.3, 69.6, 63.6, 62.9, 57.2, 54.1, 47.2, 45.4, 43.6, 42.8, 41.5, 40.7, 39.5, 38.8, 36.3, 29.8, 29.4, 27.3, 22.8, 22.2, 20.4, 19.5, 19.4, 16.5, 14.3; HRMS (ESI-TOF) calcd for C112H112O25SiNa [M + Na]+ 1908.7186, found 1908.7173.4-Methoxyphenyl 2,3-di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-β-D-xylopyranoside (42)
A similar procedure to that used for the synthesis of 10 was applied to mediate the coupling between 4 (237 mg, 0.32 mmol) and 8 (50 mg, 0.126 mmol) to provide 42 (150 mg, 0.096 mmol, 77%) as a colorless foam: [α]25D = +70.8 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 7.2 Hz, 2H), 8.26 (d, J = 7.2 Hz, 2H), 7.98–7.28 (m, 43H), 6.85 (d, J = 9.2 Hz, 2H), 6.58 (d, J = 8.8 Hz, 2H), 5.87 (q, J = 9.6 Hz, 2H), 5.66–5.53 (m, 4H), 5.01 (d, J = 12.4 Hz, 1H), 4.92 (d, J = 8.0 Hz, 2H), 4.83 (dd, J = 12.0, 7.6 Hz, 2H), 4.35–4.26 (m, 3H), 4.18 (dd, J = 12.0, 3.6 Hz, 1H), 4.08 (t, J = 8.8 Hz, 1H), 3.90–3.84 (m, 2H), 3.67–3.63 (m, 4H), 3.31 (dd, J = 12.0, 9.2 Hz, 1H), 2.81–2.77 (m, 1H), 2.73–2.69 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 166.0 (2C), 165.8, 165.2, 165.1, 165.0 (2C), 155.0, 151.3, 136.0, 133.8, 133.6 (2C), 133.5, 133.4, 133.3, 133.1 (2C), 133.0, 130.3, 130.2, 129.8 (3C), 129.7 (2C), 129.6, 129.5 (2C), 129.4 (2C), 129.0 (2C), 128.9, 128.8, 128.5 (2C), 128.4 (2C), 128.3, 128.2, 128.0, 127.8, 126.7, 126.1, 125.9, 125.8, 117.8, 114.5, 100.7, 100.4, 100.2, 81.2, 79.7, 77.4, 74.7, 73.6, 73.1, 72.8, 72.7, 72.6, 71.6, 71.4, 69.7, 69.0, 63.9, 62.7, 62.3, 55.6; HRMS (ESI-TOF) calcd for C91H80NO24 [M + NH4]+ 1570.5065, found 1570.5138.2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-α-D-xylopyranosyl trichloroacetimidate (43)
Similar procedures to those used for the synthesis of 3 were applied to convert 42 (642 mg, 0.41 mmol) to 43 (350 mg, 0.22 mmol, 54%, 2 steps) as a colorless foam: [α]25D = +89.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.35–8.29 (m, 4H), 7.98–7.28 (m, 43H), 6.20 (d, J = 3.6 Hz, 1H), 5.80 (t, J = 10.0 Hz, 1H), 5.74 (t, J = 10.0 Hz, 1H), 5.63 (dd, J = 10.0, 7.6 Hz, 1H), 5.55–5.48 (m, 3H), 5.11 (d, J = 12.0 Hz, 1H), 4.91 (d, J = 12.0 Hz, 1H), 4.85 (d, J = 7.6 Hz, 1H), 4.59 (d, J = 7.6 Hz, 1H), 4.43 (dd, J = 12.4, 3.2 Hz, 1H), 4.36 (dd, J = 12.0, 3.2 Hz, 1H), 4.29–4.22 (m, 3H), 3.37 (dd, J = 9.2, 3.6 Hz, 1H), 3.70 (d, J = 8.4 Hz, 2H), 3.63 (t, J = 8.0 Hz, 1H), 2.77–2.72 (m, 1H), 2.63–2.58 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 166.0, 165.9, 165.8, 165.2, 165.0, 164.9, 164.5, 161.0, 135.7, 133.7 (2C), 133.6, 133.4, 133.3, 133.2, 133.1 (2C), 130.2, 130.0, 129.8 (3C), 129.7, 129.6 (2C), 129.5 (2C), 129.4, 129.0, 128.9, 128.8, 128.7, 128.6, 128.5 (2C), 128.4 (2C), 128.3, 128.0, 127.8, 126.9, 126.1, 126.0, 125.9, 100.3 (2C), 95.8, 91.1, 78.6, 77.4, 74.1, 74.0, 73.2, 72.6, 72.2, 72.1, 72.0, 71.5, 69.4, 69.3, 62.8, 62.2; HRMS (ESI-TOF) calcd for C86H74Cl3N2O23 [M + NH4]+ 1607.3782, found 1607.3783.2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-α/β-D-xylopyranosyl o-cyclopropylethynylbenzoate (43′)
To a stirred solution of 42 (600 mg, 0.386 mmol) in a mixed solvent of acetonitrile (10.7 mL) and buffer (2.7 mL, pH 7.0), CAN (636 mg, 1.16 mmol) was added at 0 °C. The reaction mixture was stirred at the same temperature for another 15 min, and then ethyl acetate was added. The solution was washed successively with saturated aqueous NaHCO3, water, and brine, and was then dried over anhydrous Na2SO4. Filtration was followed by concentration to afford the crude product, which was further purified by silica gel column chromatography (PE/EA = 2:1) to afford the hemiacetal intermediate (500 mg, 89%), which was used directly in the next step without detailed characterization. The above-obtained hemiacetal intermediate (500 mg, 0.34 mmol) was dissolved in dry CH2Cl2 (3 mL), to which ABzOH (96.5 mg, 0.518 mmol), iPr2NEt (0.172 mL, 1.0 mmol), DMAP (84 mg, 0.69 mmol), and EDCI (198 mg, 1.03 mmol) were added at 0 °C under an N2 atmosphere. Then the reaction mixture was allowed to warm up to room temperature and stirring was continued at the same temperature for 6 h. Ethyl acetate was added to dilute the reaction mixture, and the resulting solution was washed with water, brine, and then dried over anhydrous Na2SO4. Filtration was followed by concentration to give a residue which was further purified by silica gel chromatography (PE/EA = 3:1) to deliver 43′ (550 mg, 0.34 mmol, 88%, 2 steps) as a mixture of α/β anomers (α/β = 3:1). An aliquot of pure α-isomer was isolated for detailed characterization: [α]25D = +63 (c 2.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 7.2 Hz, 4H), 8.05 (d, J = 7.6 Hz, 1H), 7.96–7.70 (m, 20H), 7.66–7.28 (m, 24H), 7.15 (d, J = 8.0 Hz, 2H), 6.29 (d, J = 3.6 Hz, 1H), 5.77 (t, J = 10.0 Hz, 1H), 5.67–5.40 (m, 5H), 5.07 (d, J = 12.0 Hz, 1H), 4.84 (d, J = 12.0 Hz, 1H), 4.79 (d, J = 7.6 Hz, 1H), 4.58 (d, J = 7.6 Hz, 1H), 4.38 (dd, J = 12.0, 3.6 Hz, 1H), 4.29–4.20 (m, 4H), 3.79 (dd, J = 9.2, 3.6 Hz, 1H), 3.71–3.58 (m, 3H), 2.71–2.67 (m, 1H), 2.60–2.55 (m, 1H), 1.59–1.52 (m, 1H), 0.87–0.84 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 166.0, 165.9 (3C), 165.2, 165.0 (2C), 164.7, 164.6, 135.9, 134.1, 133.8 (2C), 133.6, 133.4 (2C), 133.3, 133.2, 133.1 (2C), 131.8, 131.7, 131.2, 130.2, 130.1, 129.9 (2C), 129.8, 129.6 (2C), 129.5 (2C), 129.1, 129.0, 128.9, 128.8, 128.6, 128.5 (3C), 128.3, 128.2, 128.0, 127.8, 127.3, 126.8, 126.2, 126.0, 125.9, 124.9, 100.5 (2C), 99.7, 92.6, 79.1, 77.4, 77.1, 74.5, 74.4, 74.1, 73.2, 72.7, 72.3, 72.2, 72.1, 71.3, 69.5 (2C), 62.7, 62.5, 62.1, 9.1, 9.0, 0.9; HRMS (ESI-TOF) calcd for C97H79O26 [M + HCOO]− 1659.4854, found 1659.4876.
13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-α-D-xylopyranosyl]-steviol tert-butyldiphenylsilyl ester (12′)
A similar procedure to that used for the synthesis of 10 was applied to mediate the coupling between 43 (86 mg, 0.054 mmol) and 2 (25 mg, 0.045 mmol) to provide 12′ (76 mg, 0.0383 mmol, 85%) as a colorless foam: [α]25D = +42.0 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.38–8.34 (m, 4H), 8.00–7.66 (m, 24H), 7.64–7.27 (m, 29H), 5.84 (t, J = 9.6 Hz, 1H), 5.78 (t, J = 9.6 Hz, 1H), 5.65 (dd, J = 9.6, 7.6 Hz, 1H), 5.64–5.57 (m, 2H), 5.50 (t, J = 9.6 Hz, 1H), 5.44 (s, 1H), 5.16 (d, J = 12.0 Hz, 1H), 5.03 (s, 1H), 4.97–4.92 (m, 3H), 4.63 (d, J = 8.0 Hz, 1H), 4.44–4.29 (m, 4H), 4.12 (dd, J = 12.4, 4.4 Hz, 1H), 3.72 (t, J = 10.4 Hz, 1H), 3.57–3.48 (m, 3H), 2.81–2.77 (m, 1H), 2.60–2.55 (m, 1H), 2.27 (d, J = 13.2 Hz, 1H), 1.90–1.79 (m, 7H), 1.66–1.43 (m, 7H), 1.28 (s, 3H), 1.16 (s, 9H), 1.06–0.91 (m, 3H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 176.8, 166.0, 165.9 (2C), 165.8, 165.1 (2C), 165.0, 164.7, 150.1, 136.2, 135.7, 135.6, 133.6 (3C), 133.5, 133.4, 133.3, 133.2, 133.1, 133.0, 132.1, 132.0, 130.2 (2C), 130.0, 129.9, 129.8 (2C), 129.7 (2C), 129.6 (3C), 129.5, 129.4, 129.0 (2C), 128.9, 128.8, 128.6, 128.4 (2C), 128.3, 128.2, 128.1, 127.8, 127.6, 126.9, 126.1, 126.0, 125.8, 105.8, 100.5, 100.0, 93.4, 85.6, 78.2, 78.0, 77.4, 75.7, 74.1, 73.5, 72.8, 72.5, 72.1, 71.8, 71.3, 69.5, 69.4, 62.8, 61.9, 59.7, 57.1, 53.6, 47.6, 45.2, 43.7, 41.6, 41.3, 40.7, 39.5, 39.2, 38.7, 29.8, 29.3, 27.2 (2C), 22.2, 20.0, 19.4, 19.3, 16.2; HRMS (ESI-TOF) calcd for C120H116O25SiNa [M + Na]+ 2007.7473, found 2007.7467.13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-β-D-xylopyranosyl]-steviol (44)
To a solution of 12 (128 mg, 0.064 mmol) in dry THF (5 mL), HOAc (7.3 μL, 0.128 mmol) and TBAF (1 N in THF, 71.5 μL) were added at room temperature. After the reaction mixture was stirred at the same temperature for 30 min, ethyl acetate was added to dilute the reaction mixture. The resulting solution was washed successively with water and brine and was then dried over anhydrous Na2SO4. Filtration was followed by concentration in vacuo to give the crude product, which was further purified by silica gel column chromatography (PE/EA = 2:1) to afford 44 (107 mg, 0.061 mmol, 95%) as a colorless foam: [α]25D = +15.8 (c 1.0, CHCl3); 1H NMR (400 MHz, acetone-d6) δ 8.32 (dd, J = 7.6, 2.8 Hz, 4H), 8.01–7.89 (m, 16H), 7.73–7.67 (m, 6H), 7.63–7.48 (m, 13H), 7.42–7.35 (m, 8H), 6.08–5.98 (m, 2H), 5.84–5.67 (m, 4H), 5.19–5.13 (m, 3H), 5.01 (d, J = 12.4 Hz, 1H), 4.88 (d, J = 12.4 Hz, 1H), 4.71 (s, 1H), 4.62 (d, J = 6.4 Hz, 1H), 4.46–4.35 (m, 4H), 4.06 (t, J = 8.8 Hz, 1H), 3.88–3.79 (m, 2H), 3.66–3.58 (m, 1H), 3.28–3.24 (m, 1H), 3.18–3.06 (m, 2H), 2.21 (t, J = 12.4 Hz, 2H), 2.11–1.87 (m, 6H), 1.80 (d, J = 12.4 Hz, 1H), 1.71–1.38 (m, 7H), 1.24 (s, 3H), 1.12–0.98 (m, 5H), 0.94 (d, J = 6.8 Hz, 1H), 0.86–0.76 (m, 1H); 13C NMR (100 MHz, acetone-d6) δ 179.0, 166.5, 166.2, 166.0, 165.8 (2C), 165.5, 165.4, 153.5, 137.4, 134.5, 134.4 (2C), 134.3, 134.1, 133.8 (2C), 130.7, 130.6, 130.5 (3C), 130.4 (3C), 130.3 (3C), 130.1 (2C), 129.9, 129.8 (2C), 129.7, 129.4 (2C), 129.3 (2C), 129.2, 128.8, 128.7 (2C), 128.5, 127.1, 126.9, 126.8, 126.6, 104.9, 101.0, 100.8, 97.6, 86.4, 82.5, 79.9, 75.4, 73.9, 73.8, 73.7, 73.6, 73.3, 72.3, 70.6, 70.4, 64.0, 63.8, 63.5, 57.4, 54.6, 48.1, 44.4, 44.0, 42.7, 42.1, 41.3, 40.1, 38.8, 38.2, 22.7, 20.8, 19.9, 16.4; HRMS (ESI-TOF) calcd for C104H98O25Na [M + Na]+ 1769.6301, found 1769.6289.
13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-α-D-xylopyranosyl]-steviol (44′)
A similar procedure to that used for the synthesis of 44 was applied to convert 12′ (200 mg, 0.1 mmol) to 44′ (170 mg, 0.097 mmol, 97%) as a colorless foam: [α]25D = +33.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.33–8.28 (m, 4H), 7.96–7.21 (m, 43H), 5.78–5.70 (m, 2H), 5.61–5.53 (m, 3H), 5.35 (td, J = 9.6, 3.2 Hz, 1H), 5.28 (s, 1H), 5.07 (d, J = 12.0 Hz, 1H), 4.99 (d, J = 3.2 Hz, 1H), 4.96 (s, 1H), 4.86 (dd, J = 10.0, 2.0 Hz, 2H), 4.54 (d, J = 6.8 Hz, 1H), 4.38 (dd, J = 12.0, 3.2 Hz, 1H), 4.32–4.23 (m, 3H), 4.16–4.12 (m, 1H), 3.74 (t, J = 10.4 Hz, 1H), 3.50–3.41 (m, 3H), 2.82–2.78 (m, 1H), 2.53–2.49 (m, 1H), 2.18 (d, J = 13.2 Hz, 1H), 2.08–1.74 (m, 8H), 1.73–1.50 (m, 5H), 1.47–1.33 (m, 2H), 1.24 (s, 3H), 1.11–0.97 (m, 2H), 0.94 (s, 3H), 0.91–0.75 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 182.1, 166.0, 165.9, 165.8, 165.6, 165.1, 165.0, 164.7, 151.4, 136.2, 133.9, 133.6 (2C), 133.5, 133.4 (3C), 133.2, 133.1, 133.0, 130.3, 130.2, 130.1, 129.9 (2C), 129.8 (2C), 129.7, 129.6 (2C), 129.5, 129.4, 129.1, 129.0, 128.8 (2C), 128.6, 128.5 (2C), 128.3, 128.2, 128.1, 127.8 126.9, 126.2, 126.0, 125.8, 105.4, 100.6, 100.1, 93.5, 85.8, 78.4, 77.4, 75.5, 74.0, 73.5, 72.9, 72.6, 72.0, 71.8, 71.3, 70.2, 69.4, 62.7, 62.3, 60.0, 56.9, 53.7, 47.6, 44.1, 43.7, 41.8, 41.4, 40.8, 39.6, 38.0, 29.0, 22.0, 20.2, 19.2, 15.7; HRMS (ESI-TOF) calcd for C104H98O25Na [M + Na]+ 1769.6298, found 1769.6289.13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-β-D-xylopyranosyl]-steviol (2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl) ester (45)
To a solution of 44 (107 mg, 0.061 mmol) in a mixed solvent of CHCl3 and H2O (3 mL, v/v = 1:1), K2CO3 (25.3 mg, 0.18 mmol) and TBAB (39.4 mg, 0.122 mmol) were added at room temperature. After the mixture was stirred at the same temperature for 10 min, perbenzoylated glucosyl bromide 5 (80.6 mg, 0.122 mmol) was added. After being stirred at 40 °C for another 14 h, the reaction mixture was cooled to room temperature. Ethyl acetate was added to dilute the reaction mixture, and the resulting solution was washed successively with water and brine and was dried over anhydrous Na2SO4. Filtration and evaporation in vacuo gave the crude product, which was further purified by silica gel column chromatography (PE/THF = 2.5:1) to deliver 45 (118 mg, 0.051 mmol, 83%) as a colorless foam: [α]25D = +56.6 (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.30 (dd, J = 7.6, 5.2 Hz, 4H), 8.06 (d, J = 7.6 Hz, 2H), 8.00–7.19 (m, 61H), 6.17 (d, J = 8.0 Hz, 1H), 5.97 (t, J = 9.6 Hz, 1H), 5.90–5.76 (m, 4H), 5.66 (dd, J = 9.6, 8.0 Hz, 1H), 5.59–5.50 (m, 3H), 5.02 (s, 1H), 4.95 (d, J = 12.1 Hz, 1H), 4.90 (d, J = 7.6 Hz, 1H), 4.83 (d, J = 7.6 Hz, 1H), 4.79 (d, J = 12.1 Hz, 1H), 4.66 (dd, J = 12.3, 3.6 Hz, 2H), 4.57–4.53 (m, 2H), 4.30–4.18 (m, 5H), 3.97 (t, J = 8.8 Hz, 1H), 3.76 (dd, J = 11.6, 5.6 Hz, 1H), 3.69 (dd, J = 9.2, 6.4 Hz, 1H), 3.59–3.53 (m, 1H), 3.17 (dd, J = 11.6, 9.2 Hz, 1H), 2.62–2.57 (m, 2H), 2.23 (d, J = 13.2 Hz, 1H), 2.07–1.99 (m, 2H), 1.88–1.71 (m, 5H), 1.62–1.54 (m, 1H), 1.51–1.25 (m, 9H), 0.99 (s, 3H), 0.95–0.81 (m, 4H), 0.76 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 175.2, 166.1, 166.0 (2C), 165.9, 165.7 (2C), 165.2, 165.1, 165.0, 164.8, 152.8, 136.2, 133.6, 133.5, 133.4 (4C), 133.3, 133.2, 133.0 (2C), 130.2, 129.9, 129.8 (2C), 129.7 (2C), 129.6 (3C), 129.4, 129.1, 129.0, 128.9, 128.8 (2C), 128.6, 128.7 (2C), 128.5 (2C), 128.4 (3C), 128.3, 128.2, 128.1, 128.0, 127.7, 126.5, 126.0, 125.9, 125.8, 104.6, 100.4, 100.2, 96.8, 91.6, 85.4, 82.1, 79.2, 77.4, 75.0, 73.5, 73.1, 73.0, 72.8, 72.7, 72.6, 71.5, 71.3, 71.0, 69.6, 69.3, 63.8, 62.9, 62.6, 57.2, 54.0, 47.4, 44.1, 43.5, 42.5, 41.3, 40.4, 39.4, 37.8, 36.9, 32.0, 29.8, 28.8, 22.8, 21.5, 20.2, 19.3, 16.3, 14.2; HRMS (ESI-TOF) calcd for C138H124O34Na [M + Na]+ 2347.7874, found 2347.7866.
13-O-[2,3-Di-O-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)-4-O-(2-naphthylmethyl)-α-D-xylopyranoside] steviol (2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl) ester (45′)
A similar procedure to that used for the synthesis of 45 was adopted to mediate the coupling between 44′ (80 mg, 0.045 mmol) and 5 (59 mg, 0.09 mmol) to provide 45′ (99 mg, 0.0425 mmol, 94%) as a white solid: [α]25D = +21.4 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 9.2 Hz, 4H), 8.00–7.20 (m, 63H), 6.16 (d, J = 8.4 Hz, 1H), 6.00 (t, J = 9.2 Hz, 1H), 5.84 (t, J = 9.6 Hz, 1H), 5.79–5.73 (m, 3H), 5.66–5.47 (m, 4H), 5.42 (s, 1H), 5.09 (d, J = 12.0 Hz, 1H), 5.02 (s, 1H), 4.97 (d, J = 3.6 Hz, 1H), 4.93 (d, J = 7.6 Hz, 1H), 4.87 (d, J = 12.0 Hz, 1H), 4.66–4.62 (m, 2H), 4.53 (dd, J = 12.4, 6.0 Hz, 1H), 4.43–4.25 (m, 5H), 4.12 (dd, J = 12.4, 4.4 Hz, 1H), 3.76 (t, J = 12.8 Hz, 1H), 3.58–3.51 (m, 3H), 2.80–2.76 (m, 1H), 2.59–2.54 (m, 1H), 2.21 (d, J = 13.2 Hz, 1H), 1.94–1.69 (m, 8H), 1.53–1.19 (m, 7H), 1.01–0.85 (m, 7H), 0.74 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 175.3, 166.0 (2C), 165.9, 165.8 (2C), 165.7, 165.2, 165.1 (2C), 164.9, 164.7, 150.2, 136.2, 133.6 (2C), 133.5, 133.4 (2C), 133.3, 133.2, 133.0, 132.9, 130.2 (2C), 130.0, 129.9 (2C), 129.8 (3C), 129.7, 129.6 (2C), 129.5 (3C), 129.4, 129.0 (2C), 128.9, 128.8 (2C), 128.7, 128.6, 128.5, 128.4 (4C), 128.3, 128.1, 128.0, 127.7, 126.8, 126.1, 126.0, 125.7, 105.9, 100.5, 100.0, 93.5, 91.6, 85.7, 78.2, 78.1, 77.4, 75.6, 73.8, 73.5, 73.2, 73.0, 72.8, 72.5, 72.1, 71.8, 71.3, 71.0, 69.5 (2C), 62.8, 61.9, 59.9, 57.2, 53.7, 47.8, 44.2, 43.8, 41.5, 41.3, 40.5, 39.4, 39.2, 37.8, 29.8, 28.8, 21.6, 20.0, 19.3, 16.0; HRMS (ESI-TOF) calcd for C138H128NO34 [M + NH4]+ 2342.8312, found 2342.8289.Rebaudioside R (1)
To a solution of 45 (118 mg, 0.051 mmol) in CH2Cl2 (2.8 mL) and CH3OH (0.8 mL), DDQ (34.7 mg, 0.153 mmol) was added. The suspension was stirred at room temperature for 6 h before ethyl acetate was added to dilute the reaction mixture. The resulting mixture was washed with saturated aqueous NaHCO3 and brine, and dried over anhydrous Na2SO4. Filtration and concentration under reduced pressure obtained a residue, which was purified by silica gel column chromatography (PE/EA = 1.5:1) to give the C4–OH intermediate (100 mg, 91%), which was not characterized in detail and applied directly in the next step. The thus-obtained C4–OH intermediate was dissolved in absolute MeOH (4 mL), to which the freshly prepared NaOMe (in absolute MeOH, 1 mL) was added at room temperature. The reaction mixture was stirred at the same temperature for 1 h, and then neutralized with Amberlite (H+). Filtration was followed by concentration under reduced pressure to give the crude product, which was purified by using an RP-18 silica gel column (MeOH/H2O = 2:1) to afford rebaudioside R (1) (41 mg, 0.044 mmol, 86% for 2 steps) as a white solid: [α]25D = −30.3 (c 0.2, CH3OH); 1H NMR (400 MHz, pyridine-d5) δ 6.14 (d, J = 8.0 Hz, 1H), 5.58 (s, 1H), 5.53 (d, J = 7.6 Hz, 1H), 5.32 (d, J = 7.6 Hz, 1H), 4.99 (s, 1H), 4.94 (d, J = 7.2 Hz, 1H), 4.56 (dd, J = 11.6, 2.4 Hz, 1H), 4.49 (dd, J = 11.6, 2.8 Hz, 1H), 4.42 (dd, J = 12.0, 2.4 Hz, 1H), 4.36–4.31 (m, 2H), 4.29–4.16 (m, 7H), 4.15–3.87 (m, 9H), 3.45 (dd, J = 11.6, 9.6 Hz, 1H), 2.55 (d, J = 11.2 Hz, 1H), 2.38 (t, J = 12.4 Hz, 2H), 2.22–1.84 (m, 6H), 1.75–1.61 (m, 4H), 1.43–1.30 (m, 3H), 1.25 (s, 3H), 1.23 (s, 3H), 1.02 (d, J = 11.6 Hz, 2H), 0.89 (d, J = 6.8 Hz, 1H), 0.73 (dd, J = 14.4, 10.4 Hz, 1H); 13C NMR (101 MHz, pyridine-d5) δ 177.3, 154.1, 105.4, 105.1, 105.0, 98.7, 96.3, 87.9, 87.2, 80.7, 79.7, 79.3, 79.0, 78.9 (2C), 78.5 76.4, 75.8, 74.4, 72.4, 72.1, 71.3, 70.2, 66.4, 63.6, 62.9, 62.5, 57.8, 54.7, 50.1, 48.5, 44.7, 44.4, 42.7, 42.2, 41.1, 40.2, 38.9, 38.4, 29.0, 22.6, 20.9, 19.9, 16.1; HRMS (ESI-TOF) calcd for C43H72NO22 [M + NH4]+ 954.4540, found 954.4556.
13-O-[2,3-Di-O-β-D-glucopyranosyl-4-O-(2-naphthylmethyl)-α-D-xylopyranoside] steviol (β-D-glucopyranosyl) ester (1′)
To a solution of 45′ (72 mg, 0.031 mmol) in CH2Cl2 (1.68 mL) and CH3OH (0.48 mL), DDQ (21 mg, 0.092 mmol) was added. The suspension was stirred at room temperature for 6 h before ethyl acetate was added to dilute the reaction mixture. The resulting mixture was washed with saturated aqueous NaHCO3 and brine, and dried over anhydrous Na2SO4. Filtration and concentration under reduced pressure obtained a residue, which was purified by silica gel column chromatography (PE/EA = 1.5:1) to give the C4′–OH intermediate (60 mg, 0.0274 mmol, 88%) as a white solid: [α]25D = +24.5 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.32–8.29 (m, 4H), 8.01–7.76 (m, 18H), 7.67 (t, J = 7.6 Hz, 2H), 7.57–7.28 (m, 30H), 6.12 (d, J = 8.4 Hz, 1H), 5.95 (t, J = 9.6 Hz, 1H), 5.76–5.70 (m, 3H), 5.59–5.40 (m, 5H), 5.33 (s, 1H), 4.98 (d, J = 3.6 Hz, 1H), 4.93 (s, 1H), 4.60–4.46 (m, 5H), 4.39 (dd, J = 8.4, 3.2 Hz, 1H), 4.31–4.26 (m, 1H), 4.11 (dd, J = 12.4, 4.4 Hz, 1H), 4.12 (dd, J = 12.0, 4.4 Hz, 1H), 3.87 (t, J = 8.4 Hz, 1H), 3.69–3.59 (m, 4H), 2.93–2.88 (m, 1H), 2.63–2.59 (m, 1H), 2.18 (d, J = 13.2 Hz, 1H), 1.92 (d, J = 11.2 Hz, 1H), 1.83–1.68 (m, 6H), 1.58–1.35 (m, 8H), 0.96–0.86 (m, 5H), 0.74 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 175.3, 166.0 (2C), 165.9, 165.8 (2C), 165.3, 165.2, 165.1, 165.0, 165.0, 164.6, 150.2, 133.9, 133.7, 133.6, 133.5, 133.4, 133.3, 133.2, 130.2 (2C), 130.0, 129.9 (3C), 129.8 (3C), 129.6 (3C), 129.5, 129.4, 129.0, 128.9, 128.9 (2C), 128.8 (2C), 128.6 (2C), 128.5 (3C), 128.4, 106.0, 100.3, 99.8, 93.3, 91.6, 86.0, 81.1, 77.4, 76.0, 73.2, 73.0, 72.6 (2), 72.4, 71.9, 71.6, 71.0, 69.4, 69.0, 62.8, 62.1, 61.9, 60.7, 57.3, 53.8, 47.9, 44.2, 43.8, 41.6, 41.4, 40.6, 39.5, 39.2, 37.8, 29.8, 28.9, 21.6, 20.0, 19.4, 16.1, 0.1. HRMS (ESI-TOF) calcd for C127H116O34Na [M + Na]+ 2207.7240, found 2207.7237.
The C4′–OH intermediate (75 mg, 0.343 mmol) was dissolved in absolute MeOH (4 mL), to which the freshly prepared NaOMe (in absolute MeOH, 1 mL) was added at room temperature. The reaction mixture was stirred at the same temperature for 1 h, and then neutralized with Amberlite (H+). Filtration was followed by concentration under reduced pressure to give the crude product, which was purified by using an RP-18 silica gel column (MeOH/H2O = 2:1) to afford 1′ (30 mg, 0.032 mmol, 93%) as a white solid: [α]25D = +4.9 (c 1.0, CH3OH); 1H NMR (400 MHz, pyridine-d5) δ 6.23 (d, J = 8.4 Hz, 1H), 5.72 (d, J = 3.6 Hz, 1H), 5.64 (s, 1H), 5.34 (d, J = 7.6 Hz, 1H), 5.02 (s, 1H), 4.58–4.41 (m, 4H), 4.41–3.97 (m, 17H), 3.93–3.85 (m, 2H), 3.71–3.67 (m, 1H), 2.46 (d, J = 11.2 Hz, 1H), 2.42–2.28 (m, 2H), 2.23–2.10 (m, 1H), 2.00–1.88 (m, 4H), 1.82 (d, J = 11.2 Hz, 1H), 1.76–1.67 (m, 2H), 1.62–1.55 (m, 2H), 1.40–1.34 (m, 2H), 1.31–1.22 (m, 4H), 1.19 (s, 3H), 1.03–0.96 (m, 2H), 0.84 (d, J = 10.4 Hz, 1H), 0.71 (dd, J = 14.8, 10.8 Hz, 1H); 13C NMR (100 MHz, pyridine-d5) δ 179.6, 154.1, 109.0, 108.2, 107.6, 98.7, 97.5, 89.0, 87.8, 82.2, 81.8 (2C), 81.4 (2C), 81.1, 81.0, 78.4, 78.3, 76.9, 74.8, 74.4, 73.8, 73.2, 65.7, 65.2, 64.9, 60.1, 56.7, 50.8, 46.9, 44.6, 12.4, 41.2, 31.4, 24.9, 23.2, 22.4, 18.6; HRMS (ESI-TOF) calcd for C43H69O22 [M + H]+ 937.4275, found 937.4269.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21762024, 21572081, and 21877055) and the Natural Science Foundation of Jiangxi Province (20161ACB20005, 20171BCB23036, 20171BAB203008 and 20181BCB24004). Z.-Y. Z. acknowledges the financial support from the Innovative Fund of Jiangxi Province (YC2018-S181).References
- (a) J. R. Hanson, Sci. Prog., 2016, 99, 413–419 CrossRef CAS PubMed; (b) M. A. A. Gasmalla, R. Yang and X. Hua, Food Eng. Rev., 2014, 6, 150–162 CrossRef CAS; (c) J. M. C. Geuns, V. Bruggeman and J. G. Buyse, J. Agric. Food Chem., 2003, 51, 5162–5167 CrossRef CAS PubMed.
- (a) S. Andress and U.S. Food and Drug Administration, Agency Response Letter GRAS Notice No. GRN 000252, CFSAN/Office of Food Additive Safety, Whole Earth Sweetener Company LLC, Chicago, IL, 2008 Search PubMed; (b) R. S. McQuate and U.S. Food and Drug Administration, Agency Response Letter GRAS Notice No. GRN000304, CFSAN/Office of Food Additive Safety, GRAS Associates, LLC, Bend, OR, 2010 Search PubMed.
- G. Brahmachari, L. C. Mandal, R. Roy, S. Mondal and A. K. Brahmachari, Arch. Pharm. Chem. Life Sci., 2011, 1, 5–9 CrossRef PubMed.
- (a) C. Boonkaewwan, C. Toskulkao and M. Vongsakul, J. Agric. Food Chem., 2006, 54, 758–789 CrossRef PubMed; (b) C. Boonkaewwan and A. Burodom, J. Sci. Food Agric., 2013, 93, 3820–3825 CrossRef CAS PubMed.
- M. Puri, D. Sharma and A. Tiwari, Biotechnol. Adv., 2011, 29, 781–791 CrossRef CAS PubMed.
- G. J. Gerwig, E. M. te Poele, L. Dijkhuizen and J. P. Kamerling, Adv. Carbohydr. Chem. Biochem., 2016, 73, 1–72 CrossRef PubMed.
- (a) V. A. Abelyan, A. M. Balayan, V. T. Ghochikyan and A. A. Markosyan, Appl. Biochem. Microbiol., 2004, 40, 129–134 CrossRef CAS; (b) I. Prakash and V. S. P. Chaturvedula, Molecules, 2014, 19, 20280–20294 CrossRef PubMed.
- (a) T. Ogawa, M. Nozaki and M. Matsui, Tetrahedron, 1980, 36, 2641–2648 CrossRef CAS; (b) S. Esaki, R. Tanaka and S. Kamiya, Agric. Biol. Chem., 1984, 48, 1831–1834 CAS; (c) V. S. P. Chaturvedula, J. Klucik, M. Upreti and I. Prakash, Molecules, 2011, 16, 8402–8409 CrossRef CAS PubMed.
- S. Ceunen and J. M. C. Geuns, J. Nat. Prod., 2013, 76, 1201–1228 CrossRef CAS PubMed.
- W. H. Perera, I. Ghiviriga, D. L. Rodenburg, K. Alves, J. J. Bowling, B. Avula, I. A. Khan and J. D. McChesney, Phytochemistry, 2017, 135, 106–114 CrossRef CAS PubMed.
- M. A. Ibrahim, D. L. Rodenburg, K. Alves, W. H. Perera, F. R. Fronczek, J. Bowling and J. D. McChesney, J. Nat. Prod., 2016, 79, 1468–1472 CrossRef CAS PubMed.
- Z. Qiao, H. Liu, J.-J. Sui, J.-X. Liao, Y.-H. Tu, R. R. Schmidt and J.-S. Sun, J. Org. Chem., 2018, 83, 11480–11492 CrossRef CAS PubMed.
- U. Muhlhausen, R. Schirrmacher, M. Piel, B. Lecher, M. Briegert, A. Piee-Staffa, B. Kaina and F. Rosch, J. Med. Chem., 2006, 49, 263–272 CrossRef PubMed.
- For a review, see: J. Sun, S. Laval and B. Yu, Synthesis, 2014, 1030–1045 Search PubMed.
- S. A. Mitchell, M. R. Pratt, V. J. Hruby and R. Polt, J. Org. Chem., 2001, 66, 2327–2342 CrossRef CAS PubMed.
- J.-I. Tamura, A. Yamaguchi, J. Tanaka and Y. Nishimura, J. Carbohydr. Chem., 2007, 26, 61–82 CrossRef CAS.
- J.-J. Sui, W.-H. Zhou, D.-Y. Liu, M.-Q. Li and J.-S. Sun, Carbohydr. Res., 2017, 452, 43–46 CrossRef CAS PubMed.
- (a) S. van der Vorm, T. Hansen, J. M. A. van Hengst, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Chem. Soc. Rev., 2019, 48, 4688–4706 RSC; (b) S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Angew. Chem., Int. Ed., 2018, 57, 8240–8244 CrossRef CAS PubMed.
- D. Schmidt, F. Schuhmacher, A. Geissner, P. H. Seeberger and F. Pfrengle, Chem. – Eur. J., 2015, 21, 5709–5713 CrossRef CAS PubMed.
- N. E. Byramova, M. V. Ovchinnikov, L. V. Backinwsky and N. K. Kochetkov, Carbohydr. Res., 1983, 124, C8–C11 CrossRef CAS.
- R. Lakhmiri, P. Lhoste and D. Sinou, Tetrahedron Lett., 1989, 30, 4669–4672 CrossRef CAS.
- (a) T.-Y. Huang, M. M. Zulueta and S.-C. Hung, Org. Lett., 2011, 13, 1506–1509 CrossRef CAS PubMed; (b) R. R. Karimov, D. S. Tan and D. Y. Gin, Chem. Commun., 2017, 53, 5838–5841 RSC.
- (a) R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef; (b) X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- N. Hoffmann, J. Pietruszka and C. Soffing, Adv. Synth. Catal., 2012, 354, 959–963 CrossRef CAS.
- (a) Y. Li, Y. Yang and B. Yu, Tetrahedron Lett., 2008, 49, 3604–3608 CrossRef CAS; (b) B. Yu, Acc. Chem. Res., 2018, 51, 507–516 CrossRef CAS PubMed.
- R. R. Schmidt, M. Behrendt and A. Toepfer, Synlett, 1990, 694–696 CrossRef CAS.
- See the ESI.†.
- (a) P. Wang, Y.-J. Kim, M. Navarro-Villalobos, B. D. Rohde and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 3256–3257 CrossRef CAS PubMed; (b) K. B. Mishra, A. K. Singh and J. Kandasamy, J. Org. Chem., 2018, 83, 4204–4212 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ob02422k |
| This journal is © The Royal Society of Chemistry 2020 |