
Understanding the roles of amorphous domains and oxygen-containing groups of nitrogen-doped carbon in oxygen reduction catalysis: toward superior activity†
Jasper
Biemolt
 a,
Gadi
Rothenberg
a and
Ning
Yan
*ab
a,
Gadi
Rothenberg
a and
Ning
Yan
*ab
aVan't Hoff Institute for Molecular Sciences (HIMS), University of Amsterdam, Science Park 904, Amsterdam, The Netherlands. E-mail: n.yan@uva.nl
bSchool of Physics and Technology, Wuhan University, No. 299 Bayi Rd., Wuhan, China
First published on 25th October 2019
Abstract
Nitrogen-doped carbons are promising candidates for replacing platinum catalysts in fuel cell electrodes. They typically contain graphitic and amorphous domains, both of which contribute to the oxygen reduction reaction (ORR) activity. Here, we aim at revealing the catalytic functions of each domain as well of the surface functional groups, and ultimately at maximizing the catalytic performance of the materials in the ORR. We develop a sequential oxidative-pyrolytic treatment to remove the amorphous domains and to alter the surface functionalities. The effectiveness of this approach is evidenced by various techniques. Our electrochemical results show a positive correlation between the ORR activity and the degree of graphitization/oxygen functional group removal. While the oxidation-induced oxygen-containing group impairs the ORR, the amorphous domains seem facilitate the 2-electron transfer ORR, lack electrical conductivity and are rich in oxygen-containing groups. The ORR activity of the optimized sample increases considerably in the alkaline electrolyte, giving a half-wave potential >0.85 V (vs. a reversible hydrogen electrode) that is comparable to that of commercial platinum on carbon.
1. Introduction
Fuel cells are power sources that convert chemical energy into electricity.1,2 They are much more efficient than internal combustion engines (currently 60% vs. 22–45%, respectively).3 Hydrogen-powered fuel cells have an additional advantage that their only effluent is water vapor (the hydrogen itself must be produced somewhere, but the control of centralized emissions is much simpler). Furthermore, unlike batteries, which carry their entire “fuel” supply within them, fuel cells allow fast refill. All these advantages make fuel cells an attractive part of the sustainable energy economy.But there is yet a catch. The cells must oxidize the fuel on one electrode and reduce the oxygen on the other. This so-called oxygen reduction reaction (ORR) is a catalytic bottleneck, often requiring a platinum containing electrocatalyst.4,5 At over 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 US$ per kg, platinum is simply too expensive for large-scale usage. Many ingenious concepts for reducing Pt in fuel cell electrodes have been published recently,6–11 but an ideal solution would be avoiding any platinum-group metals entirely. Earlier, we, and others, showed that this can be done using nitrogen-doped carbons (NCs). These materials, which are inexpensive, readily produced and scalable, show good ORR activity and stability.12–17 Their active sites are ascribed to pyridinic and graphitic nitrogens.18–25
000 US$ per kg, platinum is simply too expensive for large-scale usage. Many ingenious concepts for reducing Pt in fuel cell electrodes have been published recently,6–11 but an ideal solution would be avoiding any platinum-group metals entirely. Earlier, we, and others, showed that this can be done using nitrogen-doped carbons (NCs). These materials, which are inexpensive, readily produced and scalable, show good ORR activity and stability.12–17 Their active sites are ascribed to pyridinic and graphitic nitrogens.18–25
N-doped carbons reported in the literature, in many cases, contain both graphitic and amorphous domains. The active sites in these domains might behave differently. Undoubtedly, graphitic domains show a high catalytic performance: using a well-defined graphene substrate, Guo et al. demonstrated the significance of pyridinic nitrogen for the ORR activity.18 Other similar studies also showed a positive correlation between the pyridinic nitrogen and the ORR performance.19 However, it is unclear whether the same nitrogen moiety shows comparable ORR activity in the amorphous domains. Indeed, Chen et al. and Cao et al. reported good ORR activity in disordered amorphous nitrogen-doped carbons.26,27 Nonetheless, these amorphous carbons also accommodated a relatively large portion of graphitic domains, making it difficult to differentiate the performance between the two domains. In theory, both types of domains have pros and cons: the graphitic domains give high conductivity and contain orderly pyridinic nitrogens as the active sites, the vital characteristics for electrocatalysts. In contrast, disordered domains have more intrinsic defects, which generally function as active sites in catalysis in addition to the N-induced extrinsic defects.28,29 As a downside, the disorder also generates more labile carbon bonds, which are prone to carbon corrosion.30–33
Therefore, we still neither know how the amorphous phase contributes to ORR catalysis nor have a rational approach for optimizing the carbon catalyst in terms of the two domains. Here, starting from a well-characterized series of nitrogen-doped carbons with good ORR activity, we developed a rational and simple approach for selective removal of the amorphous phase and the surface oxygen-containing groups (OCGs). Our results show that the new carbon outperforms nearly all the N-doped carbons today and exhibits comparable activity to that of the commercial 20 wt% Pt/C in an alkaline electrolyte.
2. Results and discussion
Nitrogen-doped carbons can be prepared in many ways, from chemical synthesis to biomass pyrolysis.34–36 Here, we emphasize the importance of working with a well-defined system. This is essential if one wants to have reproducible results. We started from our previously reported material, which contains both amorphous and graphitic domains.37 In a typical synthesis (Fig. 1a), multi-gram scale samples were prepared by dissolving nitrilotriacetic acid (NTA) and magnesium carbonate in water (see the Experimental section in the ESI† for detailed procedures). The precursors contain no ORR-active element namely Fe, Co, or Ni, excluding their interference in the reaction.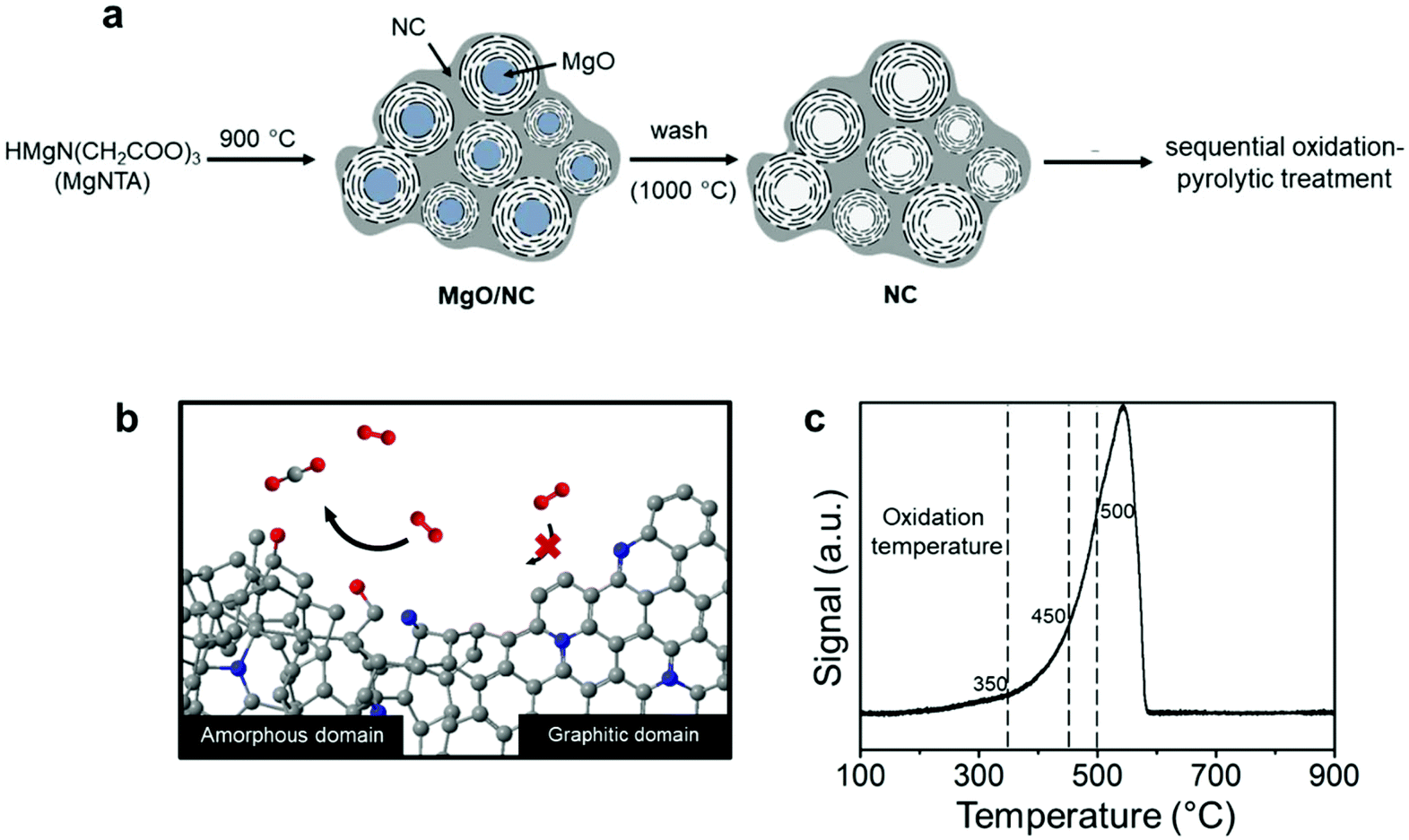 | ||
| Fig. 1 (a) The schematic procedure of synthesizing NC from NTA; (b) the schematic of selective oxidation of the amorphous domains over the graphitic domains via the sequential oxidation-pyrolytic treatment. (c) TPO of the pristine NC material, measured from 100 to 900 °C with a ramp rate of 2 °C min−1 and a flow of 2 mL min−1 under 5% O2/He. The dashed lines indicate the temperatures used for the oxidations. | ||
The fact that this NC has both amorphous and graphitic domains makes it an excellent starting point. We then used a thermal treatment to gradually burn away the amorphous parts (Fig. 1b). Previous studies on charcoal and graphite showed that amorphous carbon reacts readily with reactive oxygen species, while graphitic carbon reacts much more slowly.38,39 However, this difference in oxidation kinetics only applies at the right temperature. To pinpoint this temperature, we measured the reactivity of the NC surface with oxygen using temperature programmed oxidation (TPO, see Fig. 1c). The oxidation profile showed a single peak between 300 °C and 580 °C, in agreement with the results reported for activated carbon.40 This single peak indicates a fast transition between the amorphous and the graphitic carbon oxidation. That said, the changing slope of the oxidation profile shows that the oxidation kinetics changes as the temperature is increased. To understand this behavior, we studied samples treated at different temperatures: 350, 450 and 500 °C (dashed lines in Fig. 1c). Intuitively, one would imagine that such a treatment will also introduce oxygen-containing groups as well as surface amorphization. Thus, we also subjected each sample to a pyrolytic treatment under an inert atmosphere at 900 °C to prohibit their possible interference. This cascade oxidative-pyrolytic treatment guaranteed the removal of the amorphous domains while introducing few OCGs.
The effectiveness of this treatment was analyzed with Raman spectroscopy. Generally, this technique is used to monitor the ratio between ordered (G-band at 1580 cm−1) and disordered graphite planes (D-band at 1360 cm−1). Another band for amorphous carbon is located at 1500 cm−1.41,42 Herein, we refer to these three bands as G, D1 and D2, respectively. Fig. 2a shows the deconvolution of the Raman spectrum of pristine NC with the D1, D2 and G bands. Empirically, the decrease in the trough shown by the arrow reflects the ratio decrease in D2. This is supported by the complete deconvolution of a set of Raman spectra in Fig. S2.† Therefore, we simply used the depth of this trough as a easier indicator of the ratio of amorphous carbon in the samples. In general, the ratio between the D1 and G peaks did not change across the set of samples (Fig. 2b–d, where oxidized samples are denoted as Ox-temp, and pyrolyzed samples as Ox-temp-P). This means that defects in the graphite lattice were hardly introduced or removed by the treatments. The trough did decrease after the treatments, confirming the removal of amorphous carbon. The largest loss in amorphous carbon occurred at 450 °C. Increasing the temperature to 500 °C resulted in the surface oxidation of the graphitic carbon. However, the amount of such oxidation-introduced amorphous carbon decreased after pyrolysis (see the spectrum of Ox-500-P in Fig. 2d), as expected.
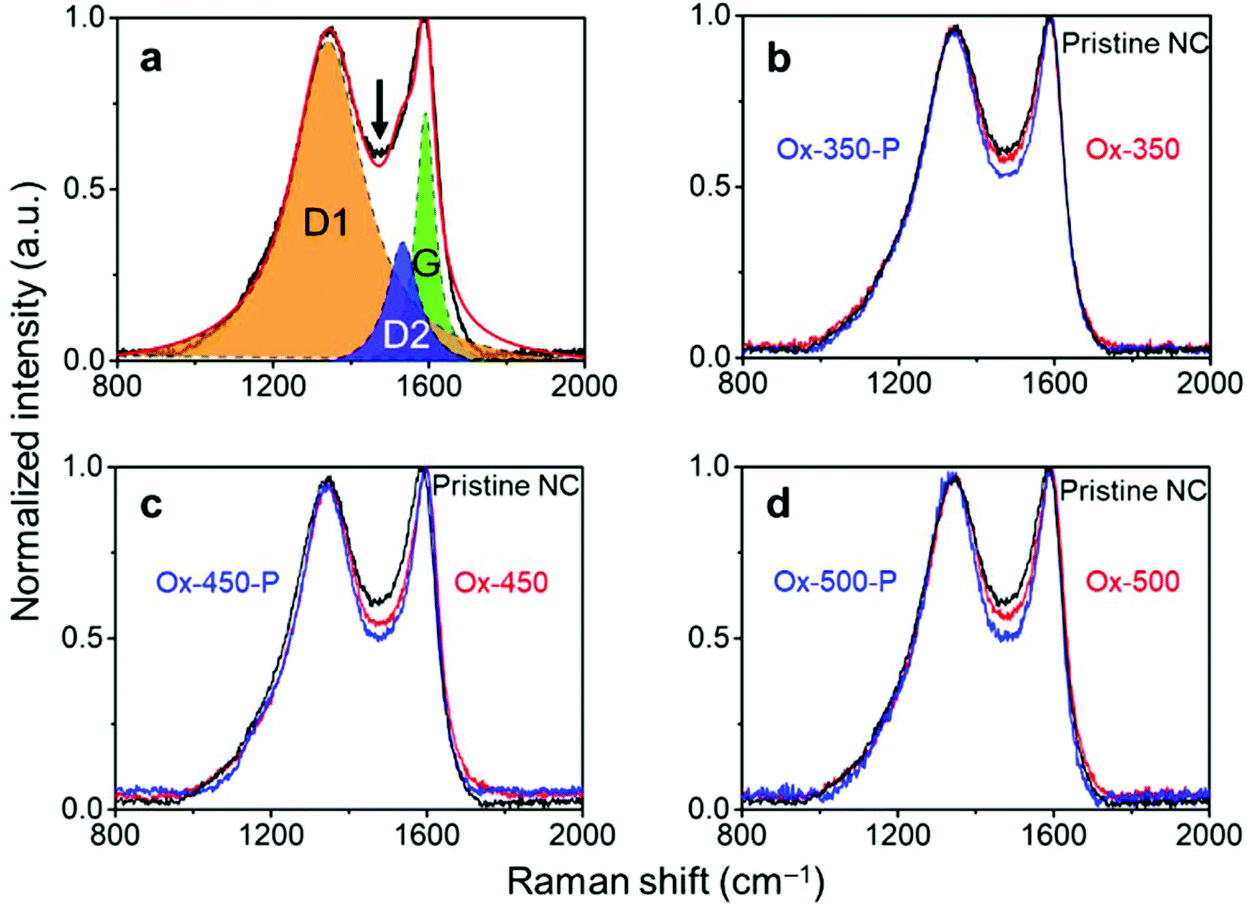 | ||
| Fig. 2 (a) Raman spectra showing curves fitted with the ordered graphitic carbon (G band), disordered graphitic carbon (D1 band), and amorphous carbon (D2 band). The arrow indicates the trough. (b) Raman spectra of the NC samples oxidized at 350 °C. (c) Raman spectra of the NC samples oxidized at 450 °C. (d) Raman spectra of the NC samples oxidized at 500 °C. Oxidized samples are denoted as Ox-temp, and the pyrolyzed ones as Ox-temp-P. | ||
The removal of amorphous carbon hardly changed the morphology of the material. Earlier, we showed that this nitrogen-doped carbon comprised micro-, meso- and macro-pores.37,43 The relatively low-temperature selective oxidation of amorphous carbon did not significantly alter the porosity of the material. This was confirmed by nitrogen adsorption studies of the oxidized samples (Fig. S3 and Table S1 in the ESI†). The Brunauer–Emmett–Teller (BET) specific surface area (SSA) remained essentially intact after oxidation treatment at 350 °C and 450 °C (∼1300 m2 g−1), yet increased to >1700 m2 g−1 at 500 °C. In the meantime, the total pore volume after the oxidative treatment increased slightly, which was attributable to the removal of the hydrogenated carbon layer on the surface, transforming closed-pores into open pores. These changes are even visible in the scanning electron microscopy (SEM) micrographs (Fig. 3 and Fig. S4†). The surface of the pristine NC is smooth and has fully covered carbon layers. Hence, the mesopores are barely observable (Fig. 3a). This surface layer was removed after the oxidation, creating large visible mesopores (Fig. 3b–d). High-resolution transmission electron microscope (HRTEM) measurement was also carried out (see Fig. S5†), the micrographs confirmed that more open pores have formed after the oxidation treatment.
 | ||
| Fig. 3 The SEM micrographs of (a) the pristine NC, (b) Ox-350, (c) Ox-450 and (d) Ox-500. | ||
We then analyzed the ORR activity of these carbons using linear sweep voltammetry (LSV, Fig. 4). These experiments were all performed in an oxygen-saturated 0.1 M KOH electrolyte. The measured onset potential (Vonset), half-wave potential (Vhalfwave) and electron transfer number (n), derived from the Koutecký–Levich plot,44 are summarized in Table 1. All potentials are reported against the reversible hydrogen electrode (RHE).
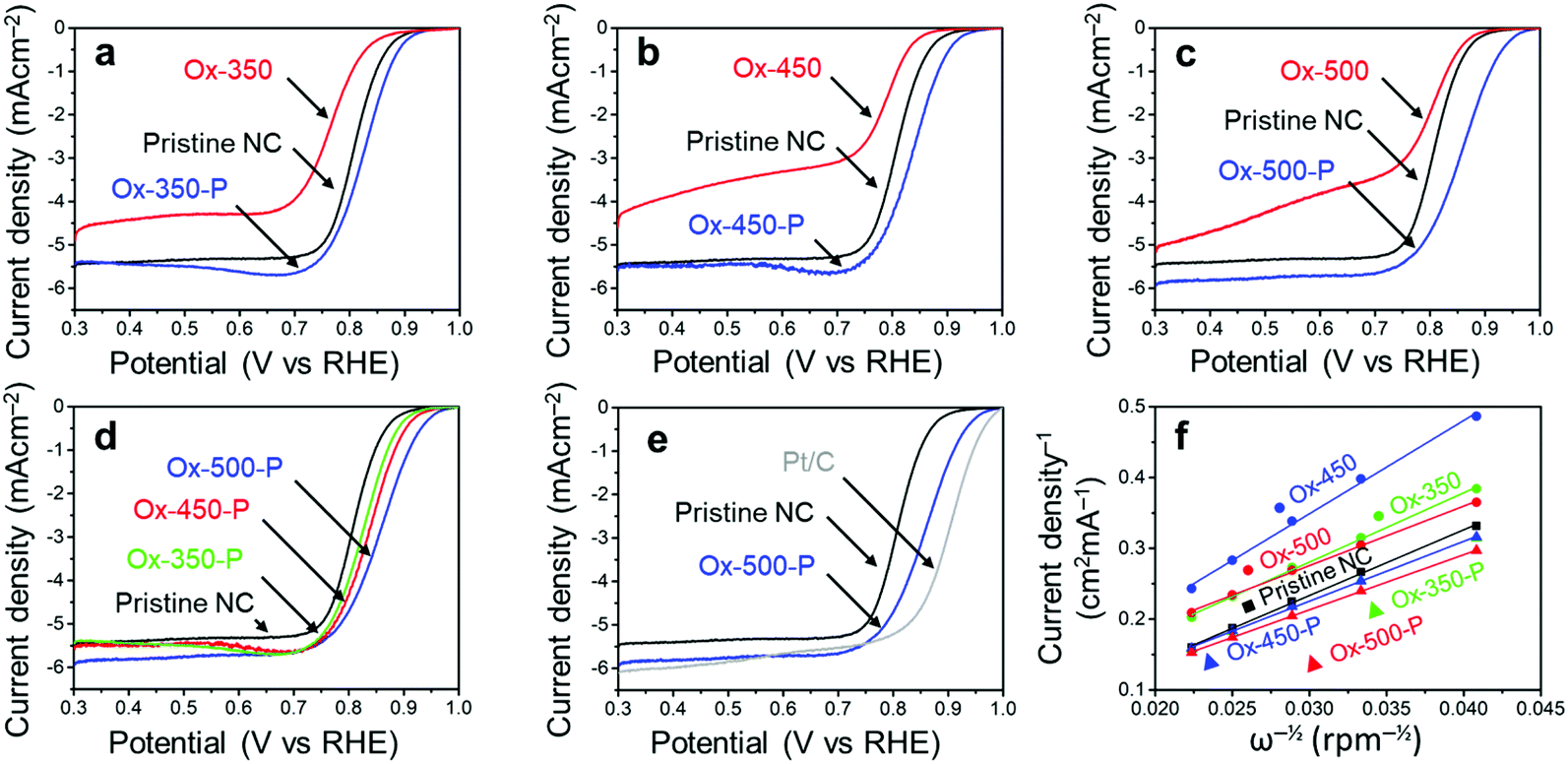 | ||
| Fig. 4 (a) LSV curves of the NCs oxidized at 350 °C. (b) LSV curves of the NCs oxidized at 450 °C. (c) LSV curves of the NCs oxidized at 500 °C. (d) LSV curves of the pyrolysed NCs, showing a trend between the ORR activity and oxidation temperature. (e) LSV curves of pristine NCs, Ox-500-P and commercial Pt/C. (f) The Koutecký–Levich plots of all the NC samples at 0.5 V. All electrochemical experiments were performed in 0.1 M KOH oxygen saturated electrolyte with a scan rate of 10 mV s−1 and 1600 rpm. | ||
| Entry | Sample | V onset (V) | V halfwave (V) | n |
|---|---|---|---|---|
| a At a faradaic current of 100 μA cm−1. b Between 0.35 and 0.65 V. c Theoretical value. | ||||
| 1 | Pristine NC | 0.91 | 0.81 | 3.0–3.1 |
| 2 | Ox-350 | 0.89 | 0.76 | 2.8–3.2 |
| 3 | Ox-350-P | 0.93 | 0.83 | 3.3–3.4 |
| 4 | Ox-450 | 0.87 | 0.79 | 2.5–1.9 |
| 5 | Ox-450-P | 0.94 | 0.84 | 3.3–3.4 |
| 6 | Ox-500 | 0.89 | 0.80 | 3.0–3.5 |
| 7 | Ox-500-P | 0.97 | 0.85 | 3.5–3.6 |
| 8 | Pt/C | 0.99 | 0.90 | 4c |
As we reported elsewhere,37 the pristine NC has a high Vonset of 0.91 V and a Vhalfwave of 0.81 V (Table 1, entry 1). After the oxidation, these values decreased to 0.89 V and 0.76 V for Ox-350 (Fig. 4a and Table 1, entry 2), indicating a negative effect of the oxidation on the ORR activity. This negative effect was even more pronounced for Ox-450 (Fig. 4b and Table 1, entry 4), with a Vonset of 0.87 V and a Vhalfwave of 0.79 V. Simultaneously, the selectivity for the 4e− pathway decreased between Ox-350 and Ox-450. However, Ox-500 did not show this trend. The Vonset and Vhalfwave (Fig. 4c and Table 1, entry 6) of this sample were closer to those of the pristine NC, as was the 4e− pathway selectivity. After the pyrolysis treatment, a positive correlation between the oxidation temperature and ORR activity was observed (Fig. 4d). Compared to the pristine NC sample, the Vonset increased by 20 mV, 30 mV and 60 mV for Ox-350-P (Table 1, entry 3), Ox-450-P (entry 5) and Ox-500-P (entry 7), respectively. A similar effect was observed for the Vhalfwave and n of these samples. In fact, the ORR activity of Ox-500-P becomes exceptionally high and comes close to the activity of commercial Pt/C (Fig. 4e and Table 1, entry 8), surpassing the state-of-the-art N-doped carbon catalysts in the literature (see the comparison in Table S2†).
The above results show a positive correlation between the degree of amorphous carbon removal (oxidation temperature) and the ORR activity. Indeed, the low catalytic performance of the amorphous domains can be ascribed to the lack of electrical conductivity and the selectivity to the 2-electron-transfer ORR. However, three key questions remain: do the two domains contain rather distinguished type/amount of N moieties? How does oxidizing these amorphous domains alone cause the decrease in ORR activity? What leads to the performance increase after a subsequent pyrolysis?
A common explanation for the increased ORR activity of NCs is the changes in the morphology and SSA. Indeed, the treatments increased both the porosity and SSA, which will affect the ORR activity. However, these properties did not alter in the oxidized and pyrolyzed samples, yet the Vonset and Vhalfwave changed dramatically. Moreover, the electrochemically-active surface area (EASA) of all samples did not vary much (see the specific capacitance values in Table S1†), implying that the change in the surface area is unlikely to play a major role in the activity change. Note that the specific capacitance of both Ox-350 and Ox-450 is higher than the remaining samples. This might pertain to the generation of aromatic OCGs, increasing the pseudo-capacitance of the samples. The subsequent pyrolytic treatment removed these moieties and the capacitance thus decreased (vide infra).
To understand this, we decided to study further how the oxidation and pyrolysis treatments affect the surface structure. X-ray photoelectron spectroscopy (XPS) showed that the atomic composition of the surface changed after the oxidation treatment (Table 2). The pristine NC (entry 1) surface had only 4% oxygen, while this increased to 11% for both Ox-450 (entry 4) and Ox-500 (entry 6). Surprisingly, no change in the oxygen content was observed for Ox-350 (entry 2). According to the previous studies using diffuse reflectance infrared Fourier transform spectroscopy, a carbon with minimal sp2 characteristics, e.g. amorphous domains, will undergo full gasification during oxidation without generating oxygen functionalities on the surface.45 We therefore infer that there was a selective gasification of the amorphous domains at 350 °C. At 450 °C and higher temperature, the graphitic domains started to oxidize. But the subsequent pyrolysis removed the oxygen atoms, yielding similar oxygen content values as the pristine NC. The nitrogen content did not change during the oxidation (the fluctuations are attributed to localized heterogeneity in the material). This also reflects the fact that both domains contained similar amounts of nitrogen.
| Entry | Sample | C content (%) | N content (%) | O content (%) |
|---|---|---|---|---|
| a Calculated from the areas of the XPS peaks of C 1s, N 1s and O 1s. | ||||
| 1 | Pristine NC | 92.2 | 4.2 | 3.5 |
| 2 | Ox-350 | 91.2 | 4.8 | 4.0 |
| 3 | Ox-350-P | 91.8 | 3.7 | 4.4 |
| 4 | Ox-450 | 84.7 | 4.3 | 11.0 |
| 5 | Ox-450-P | 91.5 | 3.5 | 5.0 |
| 6 | Ox-500 | 83.5 | 5.2 | 11.3 |
| 7 | Ox-500-P | 93.3 | 4.1 | 2.7 |
While the nitrogen content was unaffected by the treatments, the specific surface groups hardly interchanged either. The different nitrogen moieties, shown in Fig. 5, were characterized by deconvoluting the N 1s XPS peak (see Fig. S7 in the ESI†). The deconvolution gave four nitrogen peaks: graphitic (398.2 eV), pyrrolic/pyridonic (399.7 eV), pyridinic (401.1 eV) and oxidized nitrogen (403.2 eV). Again, the biggest change in the ratios was observed for Ox-450 (Table 3, entry 4) and Ox-500 (entry 6). At these temperatures, the pyridinic nitrogen was oxidized to the pyridonic one. This change hardly affected the ORR activity according to the previous studies, e.g., Guo showed that the pyridones, originated from the pyridines, were the intermediates during the ORR.18 The pyrolysis of the oxidized samples converted these groups back to pyridinic nitrogen.
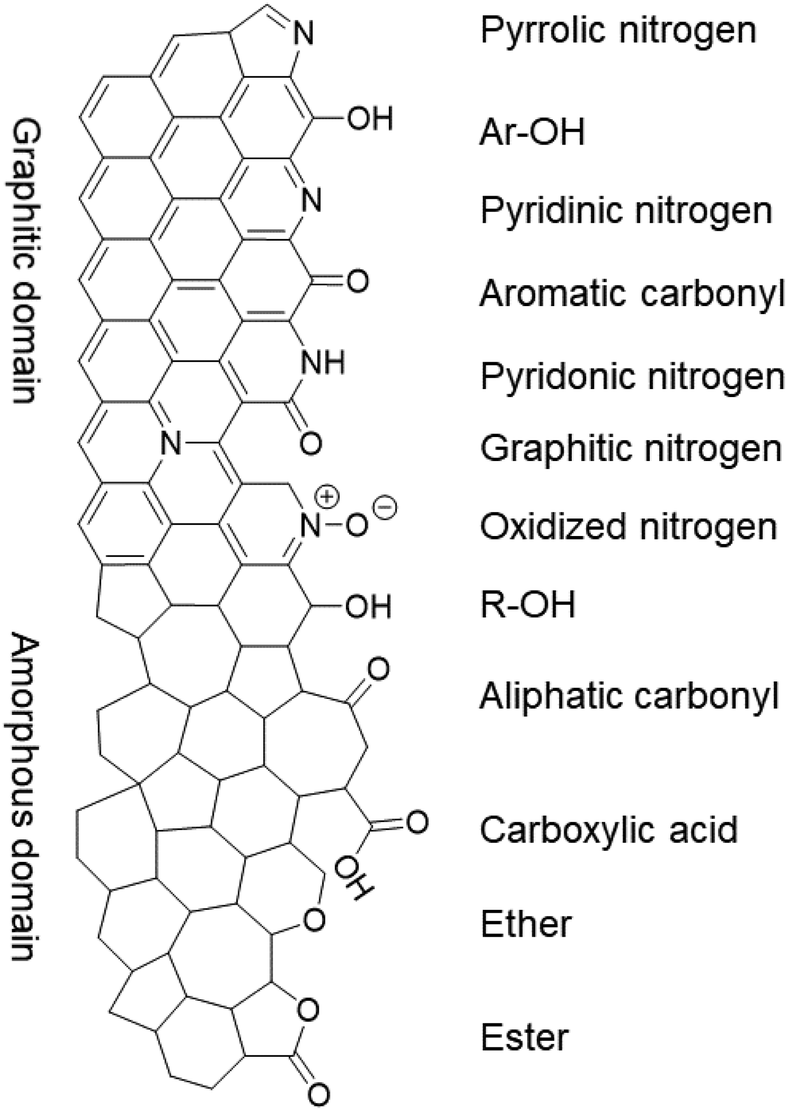 | ||
| Fig. 5 The nitrogen and oxygen functional groups which are expected in the pristine and/or oxidized form of the NC, divided over the graphitic and amorphous carbon domains. | ||
| Entry | Sample | Graphitic (%) | Pyridinic (%) | Pyrrolic/pyridonic (%) | Oxidized (%) |
|---|---|---|---|---|---|
| a Based on the deconvolution of the N 1s XPS peaks. The first number represents the absolute percentage of the nitrogen functionality in the sample, while the bracketed number represents the contribution of the functionality to the total amount of nitrogen. | |||||
| 1 | Pristine NC | 1.22 (29.1) | 1.65 (39.2) | 0.53 (12.6) | 0.80 (19.0) |
| 2 | Ox-350 | 1.33 (27.7) | 1.81 (37.8) | 0.76 (15.8) | 0.90 (18.8) |
| 3 | Ox-350-P | 1.08 (29.3) | 1.38 (37.3) | 0.53 (14.3) | 0.71 (19.1) |
| 4 | Ox-450 | 1.29 (30.0) | 1.19 (27.6) | 1.02 (23.8) | 0.80 (18.6) |
| 5 | Ox-450-P | 1.05 (30.0) | 1.05 (30.0) | 0.67 (19.1) | 0.73 (20.9) |
| 6 | Ox-500 | 1.55 (29.8) | 1.51 (29.0) | 1.31 (25.2) | 0.83 (16.0) |
| 7 | Ox-500-P | 1.07 (26.2) | 1.32 (32.3) | 0.60 (14.7) | 1.09 (26.7) |
Since no change in the nitrogen active sites among the samples was detected, the increase in oxygen functionalities was attributable to the drop of ORR activities for the oxidized samples. While OCGs are active in the ORR, the activities of each of these species is much lower than those of nitrogen moieties.46 To prove this, we analyzed the different oxygen functionalities by deconvoluting the O 1s XPS peak (Fig. S8†). Quinones, aromatic carbonyls and amides (530.7 eV), aliphatic carbonyls (532.1 eV), alcohols and ethers (532.8 eV), aromatic alcohols and esters (533.6 eV), and carboxyl/adsorbed water (534.9 eV) were used to represent the envelope of the O 1s XPS peak.
The main contribution to low oxygen content in the pristine NC came from both aromatic and aliphatic carbonyl groups (Fig. 6a and Table 4, entry 1). A small shift between the ratios of these two functional groups was observed for Ox-350 (entry 2). This resulted from the gasification of amorphous carbon, increasing the contribution of the aromatic carbonyls/quinones. These groups favor a 2e− ORR pathway and require a higher overpotential compared to nitrogen functionalities.47,48 Their content increase was reflected by the higher overpotential and low n for Ox-350 (see Fig. 4a). The oxygen content of Ox-450 and Ox-500 increased drastically, both of which had a significant amount of aromatic carbonyl/quinone functional groups (Fig. 6b and Table 4). These groups were generated by the oxidation of the edges of the graphitic domains. Because of the high number of quinones, the ORR activity was dominated by the 2e− pathway.
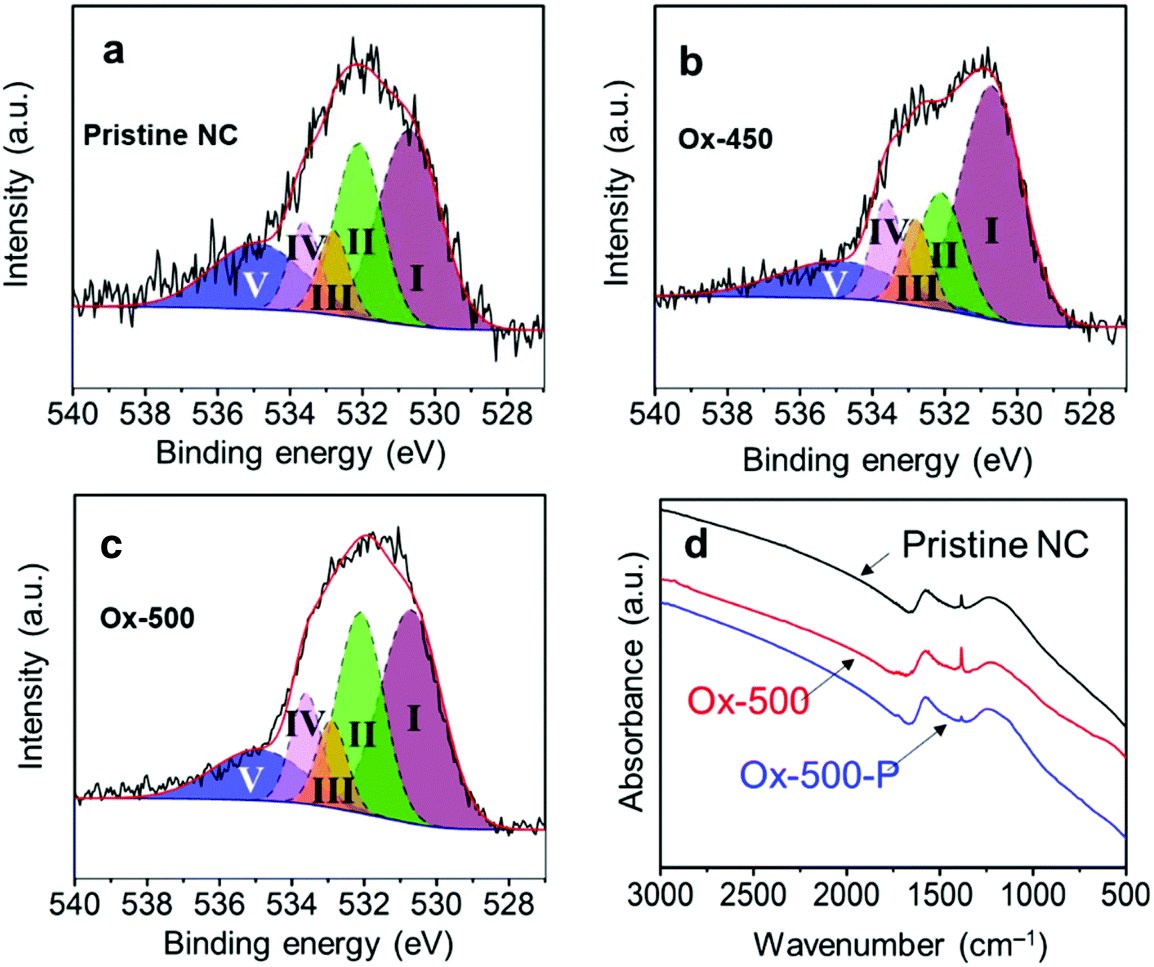 | ||
| Fig. 6 Deconvolution of the O 1s XPS spectra into I aromatic carbonyl/quinone, II aliphatic carbonyl, III R–OH and ethers, IV Ar–OH and esters and IV adsorbed water for (a) pristine NC; (b) Ox-450; (c) Ox-500. (d) The FTIR spectra comparison of pristine NC, Ox-500 and Ox-500-P. | ||
| Entry | Sample | Aromatic carbonyl/quinones (%) | Aliphatic carbonyls (%) | R–OH and C–O–C (ethers) (%) | Ar–OH and C–O–C (esters) (%) | Adsorbed water (%) |
|---|---|---|---|---|---|---|
| a Based on the deconvolution of the O 1s peak in XPS. The first number represents the absolute percentage of the nitrogen functionality in the sample, while the bracketed number represents the contribution of the functionality to the total amount of nitrogen. | ||||||
| 1 | Pristine NC | 1.33 (38.1) | 0.90 (25.6) | 0.29 (8.3) | 0.31 (8.8) | 0.67 (19.3) |
| 2 | Ox-350 | 1.70 (42.5) | 0.78 (19.5) | 0.43 (10.6) | 0.34 (8.6) | 0.75 (18.7) |
| 3 | Ox-350-P | 1.80 (41.0) | 1.14 (25.8) | 0.38 (8.7) | 0.52 (11.7) | 0.57 (12.9) |
| 4 | Ox-450 | 5.24 (47.6) | 1.97 (17.9) | 0.97 (8.9) | 1.14 (10.4) | 1.67 (15.2) |
| 5 | Ox-450-P | 2.36 (47.2) | 1.17 (23.3) | 0.39 (7.9) | 0.34 (6.8) | 0.74 (14.9) |
| 6 | Ox-500 | 4.47 (39.6) | 3.15 (27.9) | 0.92 (8.1) | 1.16 (10.3) | 1.60 (14.2) |
| 7 | Ox-500-P | 0.77 (28.4) | 0.84 (31.1) | 0.22 (8.0) | 0.32 (12.0) | 0.56 (20.6) |
After pyrolysis, the oxygen content of both Ox-450 and Ox-500 decreased back to the pristine carbon level (see Tables 2 and 4). In particular, the aromatic carbonyl/quinone content of Ox-450-P and Ox-500-P dropped pronouncedly. This is also in good agreement with the results of Fourier-transform infrared spectroscopy (FTIR) shown in Fig. 6d. The peak at 1380 ± 20 cm−1 was assigned to the stretching vibration of C–H and/or the in-plane bending vibration of C–O, which is linked to the aromatic carbonyl. Its intensity increased substantially after oxidizing pristine carbon at 500 °C. In contrast, the sequential pyrolysis removed most of them. The lower fraction of the aromatic carbonyl/quinone in Ox-500-P compared to the pristine one also implied the fact that these functionalities were richer in amorphous domains. Therefore, all the oxidized samples after the pyrolytic treatment showed a remarkable increase in ORR activity.
Fig. 7 summarizes the effects of sequential oxidative-pyrolytic treatment on the nitrogen-doped carbon. At 350 °C, only the amorphous domains are gasified, while the graphitic domains remain unaffected. Increasing the temperature to 450 °C starts the oxidation of the graphitic domain edges and introduces oxygen functionalities onto the surface. This effect is amplified at 500 °C, where near complete removal of amorphous carbon is expected and the C–C bonds in the graphitic network break, resulting in surface amorphization in addition to the generation of OCGs. In the electrocatalytic oxygen reduction reaction, a positive correlation between the ORR activity and the degree of graphitization/oxygen functional group removal is observed. We therefore conclude that the amorphous domains and the surface OCGs are unfavored in the ORR catalysis. Rather than the lower level of the active pyridinic N moiety, such a decrease in activity is ascribed to the lower electrical conductivity and the enrichment of oxygen-containing groups. The lower level of the amorphous domain also significantly increases the corrosion resistance, contributing to a robust electrocatalyst in fuel cells.29–32
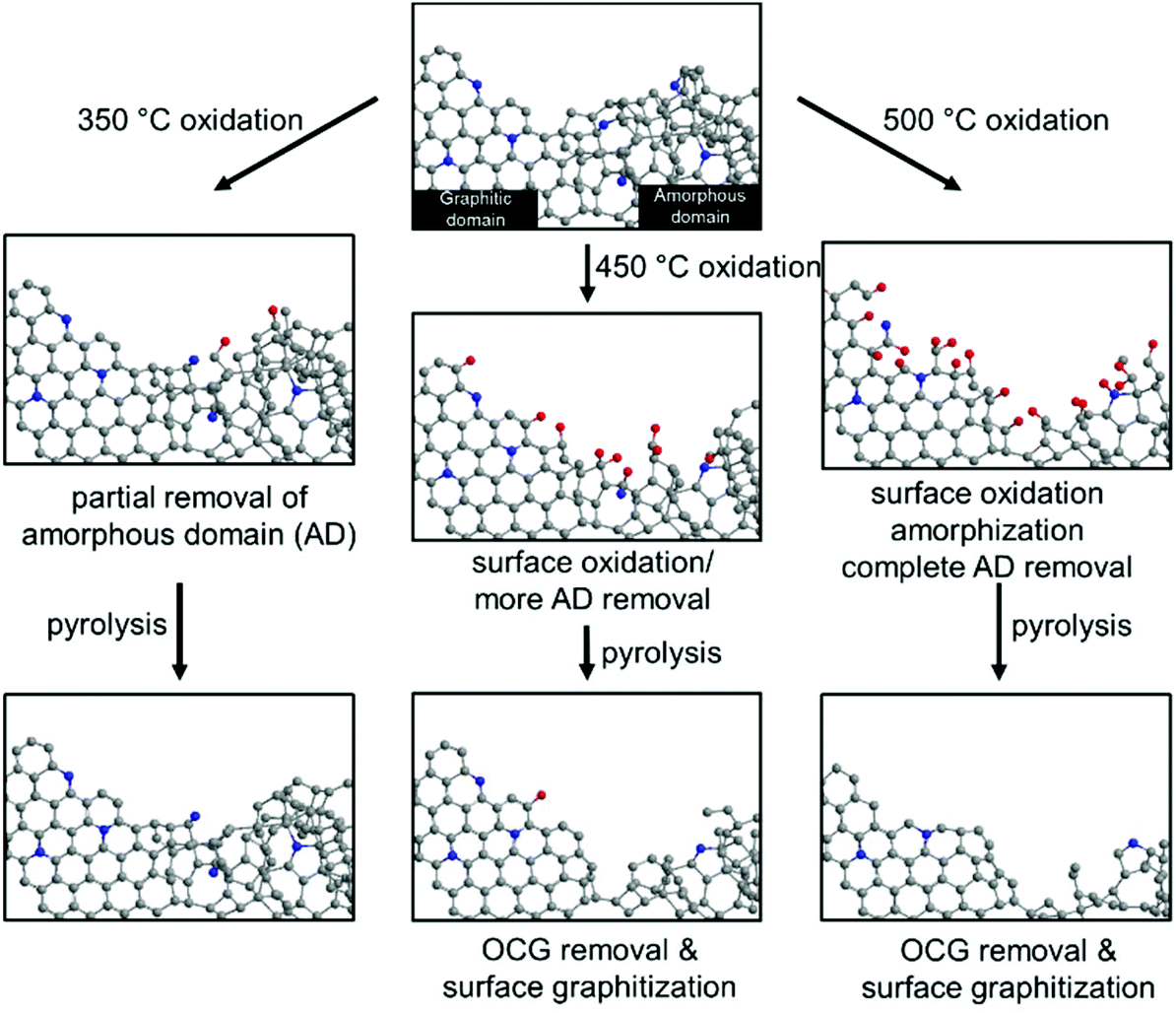 | ||
| Fig. 7 The schematic representation of the effects of oxidation on the NC at 350 °C, 450 °C and 500 °C, and the subsequent pyrolysis treatment. | ||
3. Conclusions
By simply applying a sequential oxidative-pyrolytic treatment of nitrogen doped carbon, we managed to selectively remove the amorphous domains and the surface oxygen-containing groups. Via a control experiment, we understand that the graphitic domain in nitrogen-doped carbons is more favoured in ORR catalysis than the amorphous ones. The higher degree of graphitization, high conductivity, and less OCGs render superior ORR activity and corrosion resistance of the “upgraded” nitrogen-doped carbon. Since most of the heteroatom-doped carbon materials today contain both domains, our approach and understanding might open new possibilities of optimizing their catalytic performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the financial support from the Netherlands Organization for Scientific Research (NWO) NWO-GDST Advanced Materials program (project no. 729.001.022). We also thank Mr T. Slot (UvA) for the Raman analysis, Dr Y. Liu (Wuhan University) for the XPS measurements, and Ms X. H. Wang (Thermo Fisher Scientific China) for the FTIR analysis. This work is part of the Research Priority Area Sustainable Chemistry of the UvA (http://suschem.uva.nl).References
- R. Schlögl, ChemSusChem, 2010, 3, 209–222 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef.
- G. W. Crabtree, M. S. Dresselhaus and M. V. Buchanan, Phys. Today, 2004, 57, 39–44 CrossRef CAS.
- B. Wang, J. Power Sources, 2005, 152, 1–15 CrossRef CAS.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS.
- K. Yamamoto, T. Imaoka, W.-J. Chun, O. Enoki, H. Katoh, M. Takenaga and A. Sonoi, Nat. Chem., 2009, 1, 397–402 CrossRef CAS.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS.
- J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132–2135 CrossRef CAS PubMed.
- M. Cao, D. Wu and R. Cao, ChemCatChem, 2014, 6, 26–45 CrossRef CAS.
- M. Zhang and L. Dai, Nano Energy, 2012, 1, 514–517 CrossRef CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167 RSC.
- J. Shui, M. Wang, F. Du and L. Dai, Sci. Adv., 2015, 1, e1400129 CrossRef.
- D. Eisenberg, W. Stroek, N. J. Geels, S. Tanase, M. Ferbinteanu, S. J. Teat, P. Mettraux, N. Yan and G. Rothenberg, Phys. Chem. Chem. Phys., 2016, 18, 20778–20783 RSC.
- D.-S. Yang, S. Chaudhari, K. P. Rajesh and J.-S. Yu, ChemCatChem, 2014, 6, 1236–1244 CAS.
- C. Du, X. Liu, G. Ye, X. Gao, Z. Zhuang, P. Li, D. Xiang, X. Li, A. Z. Clayborne and X. Zhou, et al. , ChemSusChem, 2019, 12, 1017–1025 CrossRef CAS.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361–365 CrossRef CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936–7942 RSC.
- C. V. Rao, C. R. Cabrera and Y. Ishikawa, J. Phys. Chem. Lett., 2010, 1, 2622–2627 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- Y. Okamoto, Appl. Surf. Sci., 2009, 256, 335–341 CrossRef CAS.
- L. Yu, X. Pan, X. Cao, P. Hu and X. Bao, J. Catal., 2011, 282, 183–190 CrossRef CAS.
- M. Li, L. Zhang, Q. Xu, J. Niu and Z. Xia, J. Catal., 2014, 314, 66–72 CrossRef CAS.
- L. Zhang and Z. Xia, J. Phys. Chem. C, 2011, 115, 11170–11176 CrossRef CAS.
- J. Chen, X. Wang, X. Cui, G. Yang and W. Zheng, Chem. Commun., 2014, 50, 557–559 RSC.
- L. Cao, Z. Lin, J. Huang, X. Yu, X. Wu, B. Zhang, Y. Zhan, F. Xie, W. Zhang and J. Chen, et al. , Int. J. Hydrogen Energy, 2017, 42, 876–885 CrossRef CAS.
- K. Waki, R. A. Wong, H. S. Oktaviano, T. Fujio, T. Nagai, K. Kimoto and K. Yamada, Energy Environ. Sci., 2014, 7, 1950–1958 RSC.
- W. Ng, Y. Yang, K. van der Veen, G. Rothenberg and N. Yan, Carbon, 2018, 129, 293–300 CrossRef CAS.
- D. von Deak, E. J. Biddinger and U. S. Ozkan, J. Appl. Electrochem., 2011, 41, 757–763 CrossRef.
- P. N. Ross, J. Electrochem. Soc., 1988, 135, 1464 CrossRef CAS.
- L. Li and Y. Xing, J. Power Sources, 2008, 178, 75–79 CrossRef CAS.
- Y. Shao, G. Yin, J. Zhang and Y. Gao, Electrochim. Acta, 2006, 51, 5853–5857 CrossRef CAS.
- Y. Shao, J. Sui, G. Yin and Y. Gao, Appl. Catal., B, 2008, 79, 89–99 CrossRef CAS.
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- D. Eisenberg, W. Stroek, N. J. Geels, C. S. Sandu, A. Heller, N. Yan and G. Rothenberg, Chem. – Eur. J., 2016, 22, 501–505 CrossRef CAS.
- C. Li and T. C. Brown, Carbon, 2001, 39, 725–732 CrossRef CAS.
- L. R. Radović, P. L. Walker and R. G. Jenkins, Fuel, 1983, 62, 849–856 CrossRef.
- E. Illeková and K. Csomorová, J. Therm. Anal. Calorim., 2005, 80, 103–108 CrossRef.
- M. Pawlyta, J.-N. Rouzaud and S. Duber, Carbon, 2015, 84, 479–490 CrossRef CAS.
- A. Sadezky, H. Muckenhuber, H. Grothe, R. Niessner and U. Pöschl, Carbon, 2005, 43, 1731–1742 CrossRef CAS.
- D. Eisenberg, P. Prinsen, N. J. Geels, W. Stroek, N. Yan, B. Hua, J.-L. Luo and G. Rothenberg, RSC Adv., 2016, 6, 80398–80407 RSC.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2001 Search PubMed.
- P. E. Fanning and M. A. Vannice, Carbon, 1993, 31, 721–730 CrossRef CAS.
- R.-S. Zhong, Y.-H. Qin, D.-F. Niu, J.-W. Tian, X.-S. Zhang, X.-G. Zhou, S.-G. Sun and W.-K. Yuan, J. Power Sources, 2013, 225, 192–199 CrossRef CAS.
- G. S. Calabrese, R. M. Buchanan and M. S. Wrighton, J. Am. Chem. Soc., 1983, 105, 5594–5600 CrossRef CAS.
- G. Jürmann, D. J. Schiffrin and K. Tammeveski, Electrochim. Acta, 2007, 53, 390–399 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. See DOI: 10.1039/c9qi00983c |
| This journal is © the Partner Organisations 2020 |