
Enantioselective synthesis of chiral multicyclic γ-lactones via dynamic kinetic resolution of racemic γ-keto carboxylic acids†
Zhichao
Xiong
a,
Jiangyan
Tian
a,
Peng
Xue
a,
Xumu
Zhang
 b and
Hui
Lv
*ac
b and
Hui
Lv
*ac
aKey Laboratory of Biomedical Polymers of Ministry of Education & College of Chemistry and Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, Sauvage Center for Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, China. E-mail: huilv@whu.edu.cn
bShenzhen Grubbs Institute and Department of Chemistry, South University of Science and Technology of China, Shenzhen, Guangdong 518055, P. R. China
cKey Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, China
First published on 25th November 2019
Abstract
Ru-Catalyzed asymmetric transfer hydrogenation of γ-keto carboxylic acids has been achieved by using the formic acid–triethylamine azeotrope as the hydrogen source, affording chiral multicyclic γ-lactones in high yields with excellent diastereo- and enantioselectivities (up to 92% yield, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99% ee). This method provides a highly efficient approach to obtain valuable multicyclic γ-lactones through a reduction/lactonization sequence. Moreover, a concise synthetic route to obtain bioactive molecules (+)-GR24 and (+)-epi-GR24 has also been developed by using this methodology as a key step.
:1 dr and 99% ee). This method provides a highly efficient approach to obtain valuable multicyclic γ-lactones through a reduction/lactonization sequence. Moreover, a concise synthetic route to obtain bioactive molecules (+)-GR24 and (+)-epi-GR24 has also been developed by using this methodology as a key step.
Multicyclic lactones are prestigious scaffolds widely occurring in natural products,1 drugs,2 and synthetic bioactive molecules,3 and have exhibited some important bioactivities.4 Particularly, strigolactones, important plant signalling molecules containing a tricyclic γ-lactone core, widely exist in the roots of plants (Fig. 1), and play important roles in the mediation of vital biological functions in plants.5 The typical bioactivities include seed germination,6 inhibition of shoot-branching,7 regulation of plant architecture and the response to abiotic factors such as nutrient availability and light,8 and root-colonization by symbiotic arbuscular mycorrhizal fungi.9 However, the natural abundance of strigolactones in plants is very low, which greatly limits the studies and applications of strigolactones. Therefore, the synthesis of strigolactones and their derivatives has attracted wide attention, and several approaches have been developed.10 Nevertheless, asymmetric synthetic routes are still rare, and the development of efficient methods for the construction of chiral molecules with a tricyclic γ-lactone core is urgent.
 | ||
| Fig. 1 Representative strigolactones and their analogues. | ||
Owing to the high efficiency in the preparation of optically pure molecules from racemic compounds, dynamic kinetic resolution (DKR) has emerged as a powerful strategy for the construction of chiral molecules.11,12 In particular, dynamic kinetic asymmetric hydrogenation of racemic α-substituted ketones has proved to be an efficient and reliable approach to obtain chiral alcohols with two contiguous stereocenters.13 In this context, the dynamic kinetic resolution of α-keto esters and β-keto esters has been widely investigated to prepare functionalized α-/β-hydroxyl esters or their derivatives.14 Among them, Johnson reported his pioneering studies on dynamic kinetic asymmetric transfer hydrogenation of the well-designed α-keto esters to synthesize multiply substituted γ-butyrolactones through a reduction/lactonization sequence (Scheme 1-1).14f,g Unfortunately, the substrate scope of this method is limited to β-aryl α-keto esters, which greatly restricted its applications in the synthesis of multicyclic γ-lactones. Recently, McErlean reported one example of dynamic kinetic resolution of cyclic γ-keto esters to synthesize tricyclic γ-lactones.10d Nevertheless, this approach needs two steps to achieve this transformation, and the additive pyridinium para-toluenesulfonate (PPTS) is essential in the lactonization step. Therefore, the direct construction of tricyclic γ-lactones by dynamic kinetic resolution is still a challenge. As alternative substrates of γ-keto esters, the dynamic kinetic resolution of γ-keto carboxylic acids provides a potential access to γ-lactones. However, in comparison with keto esters, the analogous resolution of keto carboxylic acids has remained essentially undeveloped, despite its extensive potential synthetic utility in organic synthesis. Herein, we report the highly enantioselective synthesis of chiral multicyclic γ-lactones via Ru-catalyzed asymmetric transfer hydrogenation of racemic γ-keto carboxylic acids (Scheme 1-2).
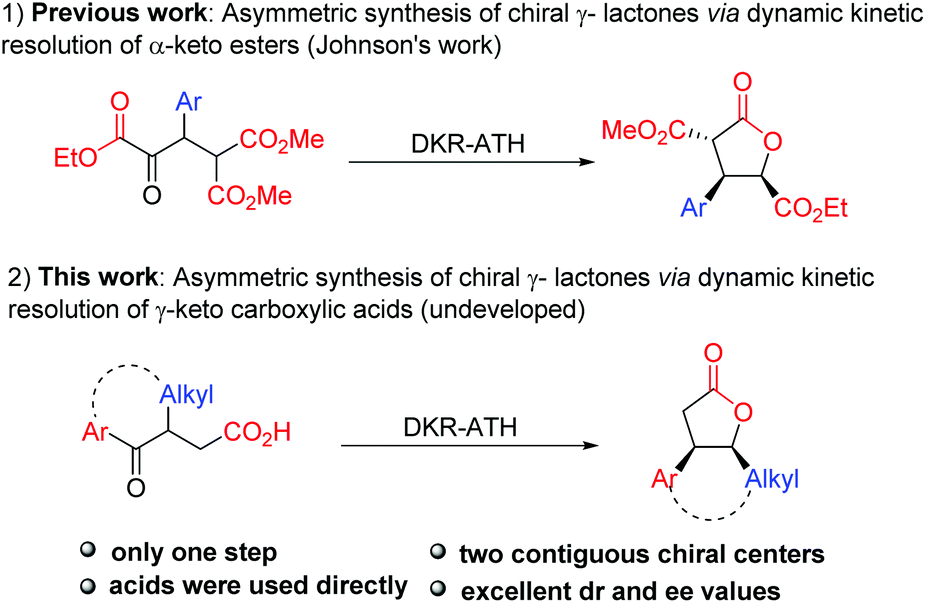 | ||
| Scheme 1 . Asymmetric synthesis of γ-lactones by dynamic kinetic resolution. | ||
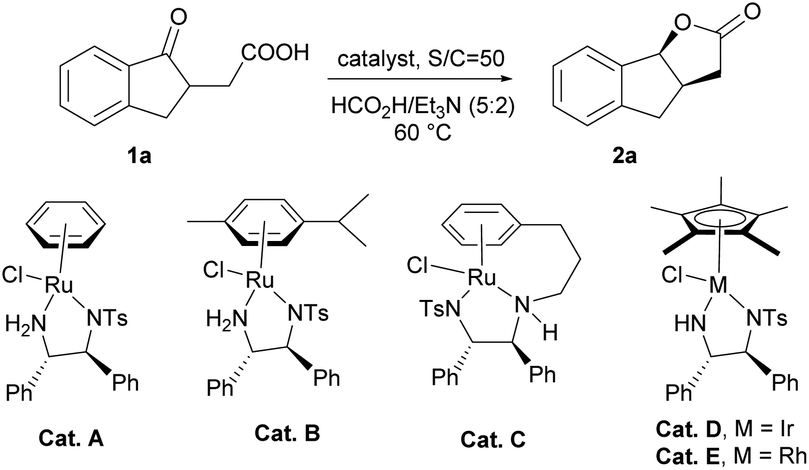
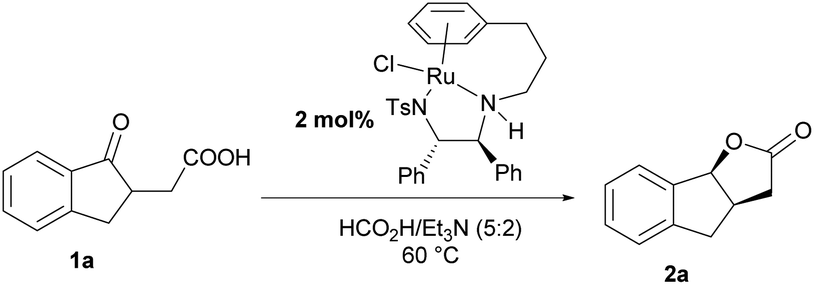
Initially, we chose asymmetric transfer hydrogenation of 2-(1-oxo-2,3-dihydro-1H-inden-2-yl)acetic acid (1a) as a model reaction to optimize the reaction conditions (Table 1). When the reaction was conducted in THF at 60 °C using the HCOOH–Et3N (5:2) azeotrope as the hydrogen source, a series of catalysts were tested. To our delight, the dynamic kinetic resolution of 1a proceeded very well in the presence of Cat. A, giving tricyclic γ-lactone 2a in moderate yield with high enantioselectivity (entry 1). Then Cat. B with substituents on the aromatic ring was tested, and the yield and enantioselectivity increased slightly (entry 2). Subsequently, Cat. C, a tethered catalyst developed by Wills’ group,15 was employed in the dynamic kinetic resolution of 1a, and the target product was obtained with stereospecific syn-selectivity in high yield with excellent enantioselectivity (entry 3). When a Rh or Ir catalyst was used instead of a Ru catalyst for this transformation, the yield decreased to some extent, suggesting that the Ru catalyst was more suitable for this kind of reduction (entries 4 and 5). We also tested the variation in the molar ratio of formic acid/trimethylamine, and the results showed that reducing the ratio of formic acid is detrimental to the yield (from 5:2 to 3:2 and 1:1).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yieldb (%) | eec(%) | drd |
|
a All reactions were carried out by using 1a (0.16 mmol), catalyst (0.0032 mmol), 5:2 HCO2H/Et3N (1 mL) under argon protection at 60 °C.
b Isolated yield.
c Determined by chiral HPLC analysis using a Chiralpak AD-H column.
d Determined by NMR.
e 3:2 HCO2H/Et3N was used.
f 1:1 HCO2H/Et3N was used.
|
||||
| 1 | Cat. A | 65 | 90 | >20:1 |
| 2 | Cat. B | 70 | 95 | >20:1 |
| 3 | Cat. C | 90 | 98 | >20:1 |
| 4 | Cat. D | 58 | 95 | >20:1 |
| 5 | Cat. E | 65 | 93 | >20:1 |
| 6e | Cat. C | 45 | 95 | >20:1 |
| 7f | Cat. C | 30 | 93 | >20:1 |
Subsequently, the solvent effect was investigated, and the results disclosed that the solvents have no influence on the diastereo- and enantioselectivities (Table 2), and all solvents tested in the experiments afforded the target products with excellent diastereo- and enantioselectivities (>20:1 dr, 98–99% ee). However, the yield of γ-lactone was greatly affected by the solvents. When the reaction was conducted in alcohol or ether solvents, it proceeded very smoothly, affording multicyclic lactone 2a with high yields (entries 1–4). When toluene or acetonitrile was employed as a solvent, only moderate yields were obtained (entries 5 and 6). N,N-Dimethylformamide was also a good solvent for this reaction (entry 7). Interestingly, the azeotropic mixture of HCOOH/Et3N, the hydrogen source of this reaction, was employed as a solvent, and the reaction worked very well, furnishing the desired product with high yield and excellent stereoselectivity (entry 8). Considering the reaction results and easy operation, the azeotropic mixture of HCOOH/Et3N (molar ratio = 5:2) was chosen as the best solvent. Subsequently, the reaction temperature was evaluated, and the results disclosed that the lower temperature resulted in the yield dropping significantly, even if the reaction time was prolonged.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Yieldb (%) | eec (%) | drd |
|
a Unless otherwise noted, all reactions were carried out with a substrate/catalyst ratio of 50:1 at 60 °C for 12 h.
b Isolated yield.
c Determined by HPLC analysis using a chiral stationary phase.
d Determined by 1H NMR spectroscopy.
e 1 mL HCO2H/Et3N (5:2) mixture was used.
f Room temperature, 24 h.
g 40 °C, 18 h.
|
||||
| 1 | MeOH | 83 | 98 | >20:1 |
| 2 | EtOH | 75 | 98 | >20:1 |
| 3 | THF | 85 | 98 | >20:1 |
| 4 | Dioxane | 82 | 98 | >20:1 |
| 5 | CH3CN | 40 | 98 | >20:1 |
| 6 | Toluene | 44 | 98 | >20:1 |
| 7 | DMF | 89 | 98 | >20:1 |
| 8e | — | 90 | 98 | >20:1 |
| 9e,f | — | 65 | 99 | >20:1 |
| 10e,g | — | 80 | 98 | >20:1 |
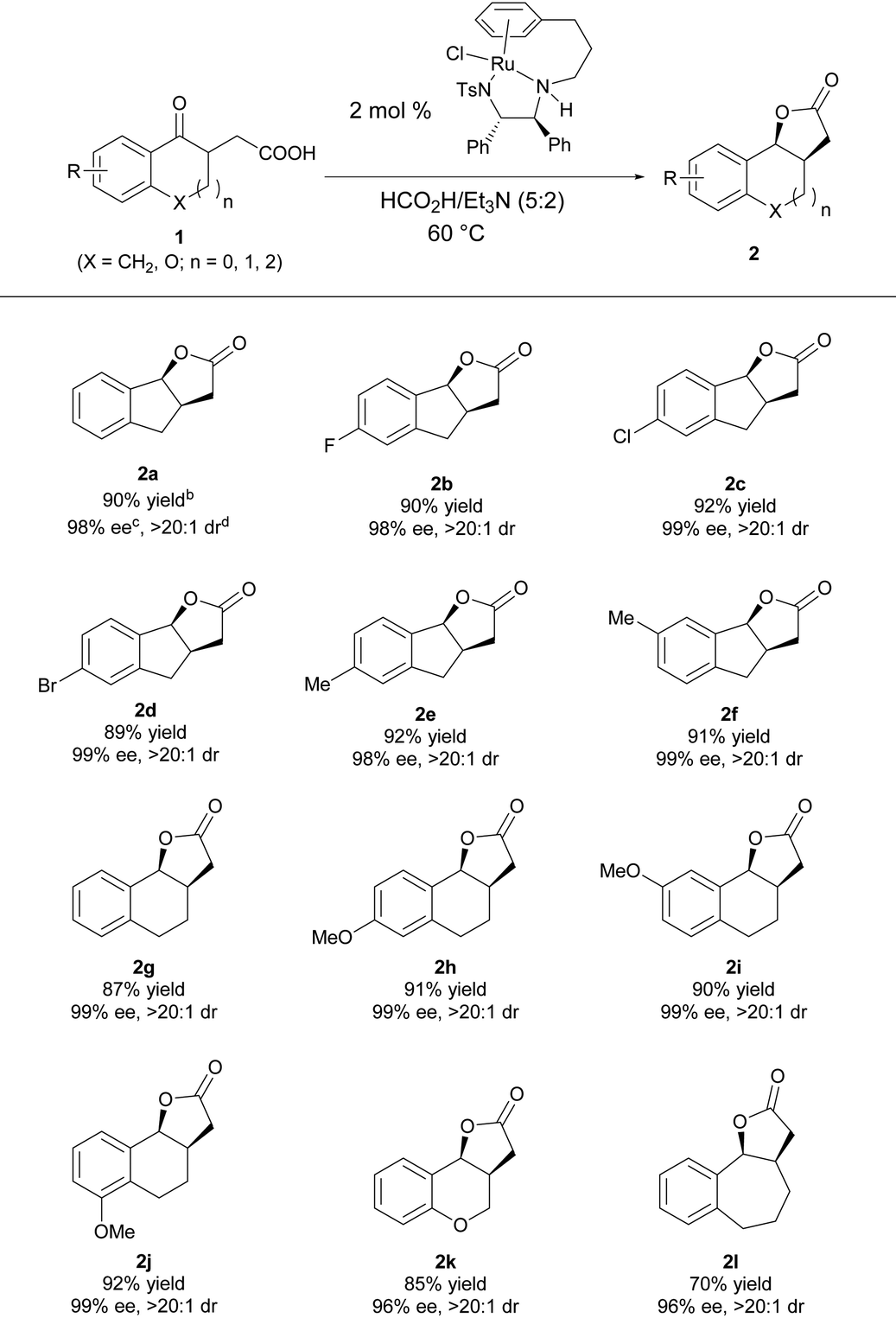
Under the optimal reaction conditions, a series of γ-keto carboxylic acids were hydrogenated to evaluate the substrate scope of this reaction. As shown in Table 3, the reaction has a good substrate generality, and the electronic properties and the position of the substituent on the benzene ring have no effect on the yield and diastereo- and enantioselectivities; all of them furnished the desired product with high yields and excellent stereoselectivities (2a–2f).16 Changing the size of the cycloketone to a six- or seven-membered ring, the reaction proceeded smoothly, giving the target products 2g–2l in good yields with excellent enantio- and diastereoselectivities.
To demonstrate the utility of this protocol, a gram-scale hydrogenation of 1a was conducted in the presence of 1 mol% catalyst loading, and it proceeded very efficiently, giving 2a in high yield and excellent stereoselectivity (Scheme 2a, 85% yield, >20:1 dr, 98% ee). In addition, (+)-GR24 and (+)-epi-GR24, the analogues of strigolactones but with higher germination activity and better stability than natural strigolactones, were efficiently synthesized. As shown in Scheme 2b, 2a reacted with HCOOMe smoothly in the presence of KOtBu, and then reacted with bromobutenolide 3, delivering (+)-GR24 and (+)-epi-GR24 in 35% yield respectively (Scheme 2b).
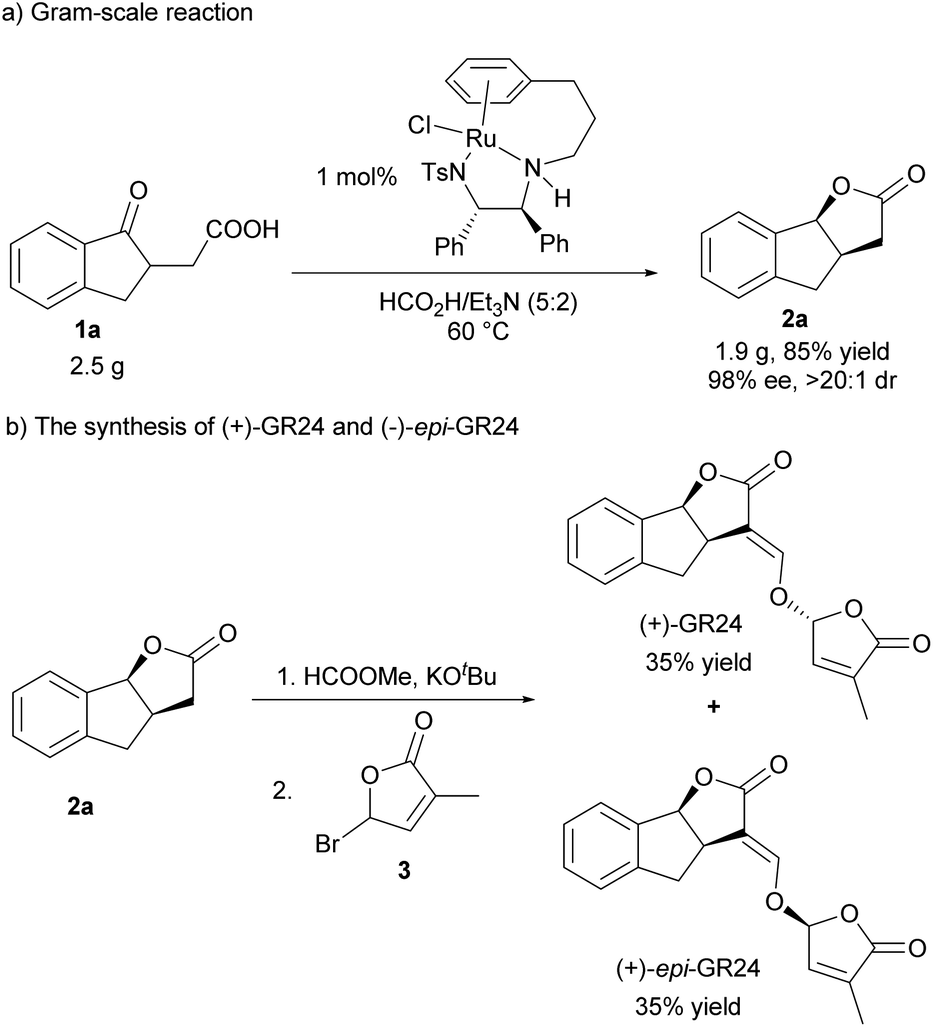 | ||
| Scheme 2 Gram-scale reaction and derivative reaction. | ||
In summary, we have developed an efficient and practical methodology for the synthesis of chiral multicyclic γ-lactones by a tethered Ru(II) complex catalyzed asymmetric transfer hydrogenation of γ-keto carboxylic acids. The reaction featured a wide substrate scope, high yields, and excellent enantio- and diastereoselectivities. Furthermore, the gram-scale reaction and efficient synthesis of (+)-GR24 and (+)-epi-GR24 disclosed that the method has potential application in the synthesis of biologically active compounds bearing a multicyclic lactone core. Further investigations on asymmetric hydrogenation initiated cascade reactions are underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21871212, 21402145), the Natural Science Foundation of Hubei Province (2018CFB430), the Open fund of CAS Key Laboratory of Molecular Recognition and Function (2017LMRF002), and the “111” Project of the Ministry of Education of China, JSGG (20160608140847864).Notes and references
- (a) X. N. K. Yoneyama, X. N. Xie, K. Yoneyama, Y. Takeuchi, Pe. Xie, K. Yoneyama and K. Yoneyama, Annu. Rev. Phytopathol., 2010, 48, 93 CrossRef PubMed; (b) K. Yoneyama, X. N. Xie, K. Yoneyama and Y. Takeuchi, Pest Manage. Sci., 2009, 65, 467 CrossRef CAS; (c) B. R. Borade, R. Nomula, R. G. Gonnade and R. Kontham, Org. Lett., 2019, 21, 2629 CrossRef CAS.
- (a) Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679 CrossRef CAS; (b) J. S. Liu, Y. L. Zhu, C. M. Yu, Y. Z. Zhou, Y. Y. Han, F. W. Wu and B. F. Qi, Can. J. Chem., 1986, 64, 837 CrossRef CAS.
- (a) B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed; (b) A. Albrecht, Ł. Albrecht and T. Janecki, Eur. J. Org. Chem., 2011, 2011, 2747 CrossRef; (c) B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931 CrossRef CAS PubMed; (d) M. Seitz and O. Reiser, Curr. Opin. Chem. Biol., 2005, 9, 285 CrossRef CAS PubMed; (e) C. Schneider and F. Abels, Org. Biomol. Chem., 2014, 12, 3531 RSC; (f) S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS PubMed; (g) T. Sekikawa, T. Kitaguchi, H. Kitaura, T. Minami and Y. Hatanaka, Org. Lett., 2015, 17, 3026 CrossRef CAS PubMed.
- (a) Y. S. Rao, Chem. Rev., 1964, 64, 353 CrossRef CAS; (b) H. M. R. Hoffmann and J. Rabe, Angew. Chem., Int. Ed., 1985, 24, 94 CrossRef; (c) E. Negishi and M. Kotora, Tetrahedron, 1997, 53, 6707 CrossRef CAS; (d) F. Q. Alali, X. X. Liu and J. L. McLaughlin, J. Nat. Prod., 1999, 62, 504 CrossRef CAS PubMed; (e) R. Bandichhor, B. Nosse and O. Reiser, Top. Curr. Chem., 2005, 243, 43 CrossRef CAS; (f) A. Quintavalla, Curr. Med. Chem., 2018, 25, 917 CrossRef CAS PubMed.
- (a) C. E. Cook, L. P. Whichard, B. Turner and M. E. Wall, Science, 1966, 154, 1189 CrossRef CAS; (b) E. M. Mangnus, P. L. A. Stommen and B. Zwanenburg, J. Plant Growth Regul., 1992, 11, 91 CrossRef CAS; (c) B. Zwanenburg, A. S. Mwakaboko, A. Reizelman, G. Anilkumar and D. Sethumadhavan, Pest Manage. Sci., 2009, 65, 478 CrossRef CAS.
- (a) Y. Kapulnik, P. M. Delaux, N. Resnick, E. Mayzlish-Gati, S. Wininger, C. Bhattacharya, N. Sejalon-Delmas, J. P. Combier, G. Becard, E. Belausov, T. Beeckman, E. Dor, J. Hershenhorn and H. Koltai, Planta, 2011, 233, 209 CrossRef CAS; (b) C. Ruyter-Spira, W. Kohlen, T. Charnikhova, A. van Zeijl, L. van Bezouwen, N. de Ruijter, C. Cardoso, J. A. Lopez-Raez, R. Matusova, R. Bours, F. Verstappen and H. Bouwmeester, Plant Physiol., 2011, 155, 721 CrossRef CAS.
- (a) M. Umehara, A. Hanada, S. Yoshida, K. Akiyama, T. Arite, N. Takeda-Kamiya, H. Magome, Y. Kamiya, K. Shirasu, K. Yoneyama, J. Kyozuka and S. Yamaguchi, Nature, 2008, 455, 195 CrossRef CAS; (b) V. Gomez-Roldan, S. Fermas, P. B. Brewer, V. Puech–Pagès, E. A. Dun, J. P. Pillot, F. Letisse, R. Matusova, S. Danoun, J. C. Portais, H. Bouwmeester, G. Bécard, C. A. Beveridge, C. Rameau and S. F. Rochange, Nature, 2008, 455, 189 CrossRef CAS.
- K. Yoneyama, X. N. Xie, H. Sekimoto, Y. Takeuchi, S. Ogasawara, K. Akiyama, H. Hayashi and K. Yoneyama, New Phytol., 2008, 179, 484 CrossRef CAS.
- (a) K. Akiyama, K. I. Matsuzaki and H. Hayashi, Nature, 2005, 435, 824 CrossRef CAS; (b) H. J. Bouwmeester, C. Roux, J. A. Lopez-Raez and G. Bécard, Trends Plant Sci., 2007, 12, 224 CrossRef CAS; (c) A. Besserer, V. Puech-Pagès, P. Kiefer, V. Gomez-Roldan, A. Jauneua, S. Roy, J.-C. Portais, C. Roux, G. Bécard and N. Séjalon-Delmas, PLoS Biol., 2006, 41, 1239 Search PubMed; (d) K. Akiyama, S. Ogasawara, S. Ito and H. Hayashi, Plant Cell Physiol., 2010, 51, 1104 CrossRef CAS.
- (a) A. W. Johnson, G. Roseberry and C. Parker, Weed Res., 1976, 16, 23 CrossRef; (b) A. W. Johnson, G. Gowda, A. Hassanali, J. Knox, S. Monaco, Z. Razawi and G. Roseberry, J. Chem. Soc., Perkin Trans. 1, 1981, 1734 RSC; (c) B. Zwanenburg, A. S. Mwakaboko, A. Reizelman, G. Anilkumar and D. Sethu-madhavan, Pest Manage. Sci., 2009, 65, 478 CrossRef CAS; (d) L. J. Bromhead, J. Visser and C. S. P. McErlean, J. Org. Chem., 2014, 79, 1516 CrossRef CAS; (e) Y. Sugimoto, S. C. M. Wigchert, J. W. J. F. Thuring and B. Zwanenburg, J. Org. Chem., 1998, 63, 1259 CrossRef CAS; (f) A. Reizelman, M. Scheren, G. H. L. Nefkens and B. Zwanenburg, Synthesis, 2000, 1944 CrossRef CAS; (g) L. J. Bromhead, J. Smith and C. S. P. McErlean, Aust. J. Chem., 2015, 68, 1221 CrossRef CAS.
- Selected reviews on dynamic kinetic resolution, see: (a) V. Bhat, E. R. Welin, X. Guo and B. M. Stoltz, Chem. Rev., 2017, 117, 4528–4561 CrossRef CAS; (b) P.-G. Echeverria, T. Ayad, P. Phansavath and V. Ratovelomanana-Vidal, Synthesis, 2016, 48, 2523 CrossRef CAS; (c) H. Pellissier, Tetrahedron, 2016, 72, 3133 CrossRef CAS; (d) H. Pellissier, Tetrahedron, 2011, 67, 3769 CrossRef CAS; (e) H. Pellissier, Adv. Synth. Catal., 2011, 353, 659 CrossRef CAS; (f) B. Martin-Matute and J.-E. Bäckvall, Curr. Opin. Chem. Biol., 2007, 11, 226–232 CrossRef CAS; (g) R. Noyori, M. Tokunaga and M. Kitamura, Bull. Chem. Soc. Jpn., 1995, 68, 36 CrossRef CAS; (h) R. S. Ward, Tetrahedron: Asymmetry, 1995, 6, 1475 CrossRef CAS.
- For selected examples on dynamic kinetic resolution, see: (a) L. Rasu, J. M. John, E. Stephenson, R. Endean, S. Kalapugama, R. Clément and S. H. Bergens, J. Am. Chem. Soc., 2017, 139, 3065 CrossRef CAS; (b) K. M.-H. Lim and T. Hayashi, J. Am. Chem. Soc., 2017, 139, 8122 CrossRef CAS; (c) X. Chen, J. Z. M. Fong, J. Xu, C. Mou, Y. Lu, S. Yang, B. A. Song and Y. R. Chi, J. Am. Chem. Soc., 2016, 138, 7212 CrossRef CAS; (d) J. Paetzold and J. E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620 CrossRef CAS; (e) W. Bhat, S. Wang, B. M. Stoltz and S. C. Virgil, J. Am. Chem. Soc., 2013, 135, 16829 CrossRef; (f) S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149 CrossRef CAS; (g) J.-H. Xie, Z.-T. Zhou, W.-L. Kong and Q.- L. Zhou, J. Am. Chem. Soc., 2007, 129, 1868 CrossRef CAS.
- (a) J. H. Xie, S. Liu, W.-L. Kong, W.-J. Bai, X.-C. Wang, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2009, 131, 4222 CrossRef CAS; (b) K. Makino, Y. Hiroki and Y. Hamada, J. Am. Chem. Soc., 2005, 127, 5784–5785 CrossRef CAS; (c) M. T. Corbett and J. S. Johnson, J. Am. Chem. Soc., 2013, 135, 594 CrossRef CAS; (d) A. Lei, S. Wu, S. He and X. Zhang, J. Am. Chem. Soc., 2004, 126, 1626–1627 CrossRef CAS; (e) W. Wu, C. You, C. Yin, Y. Liu, X.-Q. Dong and X. Zhang, Org. Lett., 2017, 19, 2548 CrossRef CAS; (f) J. Limanto, S. W. Krska, B. T. Dorner, E. Vazquez, N. Yoshikawa and L. Tan, Org. Lett., 2010, 12, 512 CrossRef CAS; (g) D. Bao, X. S. Gu, J.-H. Xie and Q.-L. Zhou, Org. Lett., 2017, 19, 118 CrossRef CAS; (h) X. Tao, W. Li, X. Li, X. Xie and Z. Zhang, Org. Lett., 2013, 15, 72 CrossRef CAS; (i) A. Ros, A. Magriz, H. Dietrich, R. Fernández, E. Alvarez and J. M. Lassaletta, Org. Lett., 2006, 8, 127 CrossRef CAS; (j) A. E. Cotman, B. Modec and B. Mohar, Org. Lett., 2018, 20, 2921 CrossRef CAS; (k) Z. Xiong, C. Pei, P. Xue, H. Lv and X. Zhang, Chem. Commun., 2018, 54, 3883 RSC; (l) F. Eustache, P. I. Dalko and J. Cossy, Org. Lett., 2002, 4, 1263 CrossRef CAS.
- (a) D. Cartigny, K. Püntener, T. Ayad, M. Scalone and V. Ratovelomanana-Vidal, Org. Lett., 2010, 12, 3788–3791 CrossRef CAS; (b) B. Seashore-Ludlow, F. Saint-Dizier and P. Somfai, Org. Lett., 2012, 14, 6334 CrossRef CAS; (c) C. G. Goodman, D. T. Do and J. S. Johnson, Org. Lett., 2013, 15, 2446 CrossRef CAS; (d) G. Gu, J. Lu, O. Yu, J. Wen, Q. Yin and X. Zhang, Org. Lett., 2018, 20, 1888 CrossRef CAS; (e) D. Lavergne, C. Mordant, V. Ratovelomanana-Vidal and J. P. Genet, Org. Lett., 2001, 3, 1909 CrossRef CAS; (f) K. M. Steward, E. C. Gentry and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 7329 CrossRef CAS; (g) K. M. Steward, M. T. Corbett, C. G. Goodman and J. S. Johnson, J. Am. Chem. Soc., 2012, 134, 20197 CrossRef CAS.
- (a) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318 CrossRef CAS; (b) D. J. Morris, A. M. Hayes and M. Wills, J. Org. Chem., 2006, 71, 7035 CrossRef CAS; (c) R. Soni, J.-M. Collinson, G. C. Clarkson and M. Wills, Org. Lett., 2011, 13, 4304 CrossRef CAS; (d) K. E. Jolley, A. Zanotti-Gerosa, F. Hancock, A. Dyke, D. M. Grainger, J. A. Medlock, H. G. Nedden, J. J. M. Le Paih, S. J. Roseblade, A. Seger, V. Sivakumar, I. Prokes, D. J. Morris and M. Wills, Adv. Synth. Catal., 2012, 354, 2545 CrossRef CAS; (e) Z. Fang and M. Wills, J. Org. Chem., 2013, 78, 8594 CrossRef CAS; (f) R. Soni, K. E. Jolley, G. J. Clarkson and M. Wills, Org. Lett., 2013, 15, 5110 CrossRef CAS PubMed; (g) H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2619 CrossRef CAS PubMed.
- The absolute configuration of compound 2a has been established by comparison with the literature data (for 2a in ref. 10d: ([α]20D = 110 (c 0.985, CHCl3), 99% ee, our experiment result of 2a: [α]20D = 100 (c = 0.985, CHCl3) (98% ee)). All the other configurations are uncertain and based on the assumption that the configuration follows that of 2a.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra of compounds. See DOI: 10.1039/c9qo01047e |
| This journal is © the Partner Organisations 2020 |