
Asymmetric total synthesis of nodulisporiviridin E†
Yang
Ji
a,
Zhengyuan
Xin
a,
Yingbo
Shi
a,
Haibing
He
*b and
Shuanhu
Gao
 *ab
*ab
aShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, 3663N Zhongshan Road, Shanghai 200062, China. E-mail: hbhe@chem.ecnu.edu.cn; shgao@chem.ecnu.edu.cn
First published on 18th November 2019
Abstract
The asymmetric total synthesis of (+)-nodulisporiviridin E, a fungal metabolite from Nodulisporium sp., was achieved in 16 steps. The synthesis features a convergent approach from two conventional fragments. An intramolecular Heck cyclization was applied to construct the C-ring and the all-carbon quaternary center at C-10. The furan E ring was installed by means of an intramolecular oxonium trapping reaction. This approach provides an advanced Michael acceptor, which might facilitate the preparation of various analogues and derivatives for biological studies.
Furanosteroids viridin (1)1 and wortmannin (2)2 attracted considerable attention from the chemistry and biology community because they potently inhibit phosphatidylinositol 3-kinase (PI3K),3 which is associated with reduced apoptosis and enhanced survival and growth of many cancer cells (Fig. 1). Biological studies revealed that natural furanosteroids are easily degraded and highly toxic in vivo. Structural modification has been proven to be a reliable strategy to divergently prepare derivatives and analogues, which resulted in a drug candidate PX-866 in Phase II clinical trials.4 Biogenetically related furanosteroids include viridiol (3),5 demethoxyviridin (4),6 9-epi-viridiol (5)7 and 1-deoxyviridiol (6).8 In 2015, Gao and coworkers reported eight new viridin-like natural products nodulisporiviridins (7–13), which were isolated from the extract of an endolichenic fungal strain Nodulisporium sp. fermented with potato dextrose broth.9 The short-term memory assay on an Aβ transgenic drosophila model of Alzheimer's disease showed that nodulisporiviridins improved the short-term memory capacity, with potencies close to that of the positive control.9
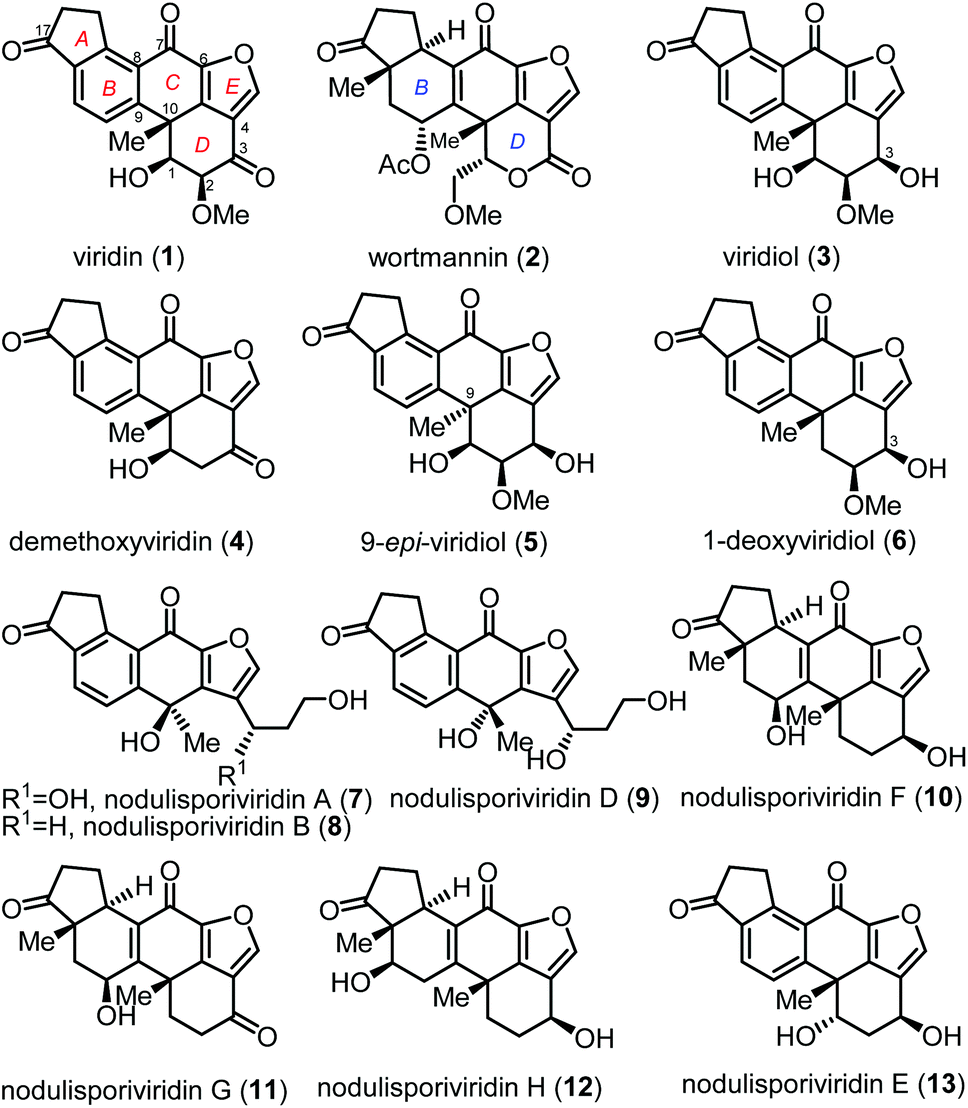 | ||
| Fig. 1 Structures of furanosteroids. | ||
The common structural feature of furanosteroids is the pentacyclic skeleton involving a strained furan ring fused to the steroid scaffold. The major structural difference of this subgroup of natural molecules relies on A and C rings due to the multiply-substituted functional groups, various oxidation levels and contiguous chiral centers. The challenging structures and biological potential of furanosteroids attracted our attention. We planned to develop new strategies for the total synthesis of structurally and biogenetically related furanosteroids for medicinal chemistry and biological studies. Recently, we reported the asymmetric total synthesis of viridin (1) and viridiol (3) using the metal-hydride H atom transfer (MHAT) as a key step.1e As a part of this big project, we aim to prepare an advanced intermediate which might facilitate its late-stage modification and structure–activity relationship (SAR) studies. Under this consideration, a powerful intramolecular Heck reaction10 was applied to build the core tetracyclic skeleton (A–B–C–D ring) which has also been successfully used in the total synthesis of xestosaprol N and O.11 Herein, we report the first total synthesis of (+)-nodulisporiviridin E (13) which has a lower oxidation state than viridin.
As outlined in Scheme 1, we envisioned that furanosteroids, including nodulisporiviridins and their analogues, could be obtained from 14 by means of the oxidation state adjustment of the enone moiety in the advanced intermediate 15. In turn, the core pentacyclic skeleton as well as the all-carbon quaternary carbon center on C-10 could be established by an intramolecular Heck reaction of triflate 18. We believe that a convergent approach might support the efficient preparation of 18via the fragment coupling of 19 (A–B ring) and chiral aldehyde 20 (D ring). Regarding the formation of the furan E ring, an acid-promoted cyclization was designed through an intramolecular oxonium trapping reaction.
 | ||
| Scheme 1 Retrosynthetic analysis of nodulisporiviridin E. | ||
Our synthesis began with the preparation of fragment 20 (Scheme 2). The chiral enone 21 was prepared from (−)-quinic acid according to a known procedure,12 which was converted to unsaturated ketone 22 in good yield and diastereoselectivity (dr >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) through a substrate-controlled alkylation with methyl bromoacetate (BAM). A one-pot operation was developed to transform 22 into cyclohexene derivative 23 in 79% overall yield, via a reaction sequence involving selective 1,4-reduction, in situ triflation and methylation. Aldehyde 20 was then obtained through the process of the amidation reaction to Weinreb amide13 and DIBAL-H reduction. Lithium-bromide exchange of aryl bromide 19 with t-BuLi followed by the interaction with aldehyde 20 gave rise to the corresponding adduct which was oxidized to ketone 24 through Dess–Martin oxidation14 in 55% yield over two steps. Replacement of the t-butyldimethylsilyl (TBS) on the phenol group with triflate yielded the precursor of the cyclization reaction 18 in 81% yield. Then, the key Pd-catalyzed intramolecular Heck reaction was investigated. Treatment of 18 with catalytic Pd(OAc)2 in the presence of the 1,4-bis(diphenylphosphino)butane (dppb) ligand in reflux acetonitrile produced the cyclized product 17 in 89% yield. This intramolecular Heck reaction proceeded through a stereospecific process, via the intermediate 18a, and formed the all carbon quaternary center on C-10 as a single diastereomer.
:1) through a substrate-controlled alkylation with methyl bromoacetate (BAM). A one-pot operation was developed to transform 22 into cyclohexene derivative 23 in 79% overall yield, via a reaction sequence involving selective 1,4-reduction, in situ triflation and methylation. Aldehyde 20 was then obtained through the process of the amidation reaction to Weinreb amide13 and DIBAL-H reduction. Lithium-bromide exchange of aryl bromide 19 with t-BuLi followed by the interaction with aldehyde 20 gave rise to the corresponding adduct which was oxidized to ketone 24 through Dess–Martin oxidation14 in 55% yield over two steps. Replacement of the t-butyldimethylsilyl (TBS) on the phenol group with triflate yielded the precursor of the cyclization reaction 18 in 81% yield. Then, the key Pd-catalyzed intramolecular Heck reaction was investigated. Treatment of 18 with catalytic Pd(OAc)2 in the presence of the 1,4-bis(diphenylphosphino)butane (dppb) ligand in reflux acetonitrile produced the cyclized product 17 in 89% yield. This intramolecular Heck reaction proceeded through a stereospecific process, via the intermediate 18a, and formed the all carbon quaternary center on C-10 as a single diastereomer.
 | ||
| Scheme 2 Construction of the core tetracyclic ring through an intramolecular Heck reaction. | ||
With the basic tetracarbocyclic skeleton 17 (ring A–B–C–D) in hand, the next task is to form the furan ring E (Scheme 3). Firstly, a carbonyl group is required on C-6. After exploration of various conditions, we found that the oxidation occurred to generate product 25 in 76% yield when DMF and t-BuOH were used as a co-solvent in the presence of t-BuOK. We reasoned that oxygen in air acted as an oxidant during this process which involved the formation of a peroxide intermediate. This observation was matched with the results achieved by Harada and co-workers.1525 exists as an enol form which could be easily protected as its methoxymethyl (MOM) ether. After removal of TBS groups and Dess–Martin oxidation, the dicarbonyl compound 16 was achieved. We proposed that the methoxymethyl ether could serve as the hydroxymethyl source via an oxonium intermediated under acidic conditions. After extensive study of the reaction conditions (see Table S1 in the ESI†), we found that the reaction of 16 with boron trifluoride etherate leads to the formation of acetal compound 28 in 45% yield. It is reasonable to assume that the deprotection of the MOM group occurred first and generated formic aldehyde which was intermolecularly trapped to form 27a. After tautomerization and acetalization, 28 was produced. This was further validated by the observation that the reaction yield could be improved to 79% by adding paraformaldehyde into the reaction mixture. To our delight, treatment of 1,3-dioxane 28 with 3 N hydrochloric acid (HCl) furnished the desired pentacyclic product 30. p-Toluenesulfonic acid (PTSA) mediated dehydration of 30 forged 15 with the furan E ring in good yield. Compound 15 contains the pentacyclic core of furanosteroids and enone group on the A ring,16 which could serve as a Michael acceptor to prepare the analogues. Indeed, an oxa-Michael addition smoothly occurred under acidic conditions to generate β-hydroxyl ketone 31 as a single diastereomer in 76% yield. Stereoselective reduction of 31 with sodium triacetoxyborohydride NaB(OAc)3H gave (+)-nodulisporiviridin E (13) in 71% yield. The synthetic sample showed the same spectra of 1H and 13C NMR and high-resolution mass spectrometry as the data reported by Gao and co-workers.9
 | ||
| Scheme 3 Total synthesis of nodulisporiviridin E. | ||
Conclusions
In summary, we have achieved the first total synthesis of (+)-nodulisporiviridin E via a convergent strategy in 16 steps. In this synthesis, two coupling fragments of dihydroindenol and chiral D rings were easily prepared from the commercially available material or chiral building block. A substrate controlled intramolecular Heck reaction was applied to build the core tetracyclic ring with the quaternary carbon center. The furan E ring was installed by means of an intramolecular oxonium trapping reaction. This approach provided an advanced Michael acceptor, which might facilitate the preparation of various analogues and derivatives by using different nucleophiles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (21971068 and 21772044), the “National Young Top-Notch Talent Support Program”, Program of Shanghai Academic/Technology Research Leader (18XD1401500), the Program of Shanghai Science and Technology Committee (18JC1411303) and “the Fundamental Research Funds for the Central Universities” for generous financial support.References
- For the isolation of viridin, see: (a) P. W. Brian and J. C. McGowan, Nature, 1945, 156, 144 CrossRef CAS; (b) J. F. Grove, P. McCloskey and J. S. Moffatt, J. Chem. Soc. C, 1966, 743, 743 RSC. For the total synthesis of viridin, see: (c) E. A. Anderson, E. J. Alexanian and E. J. Sorensen, Angew. Chem., Int. Ed., 2004, 43, 1998 CrossRef CAS; (d) M. Del Bel, A. R. Abela, J. D. Ng and C. A. Guerrero, J. Am. Chem. Soc., 2017, 139, 6819 CrossRef CAS PubMed; (e) Y. Ji, Z. Xin, H. He and S. Gao, J. Am. Chem. Soc., 2019, 141, 16208 CrossRef CAS PubMed.
- For the isolation of wortmannin, see: (a) P. W. Brian, P. J. Curtis, H. G. Hemming and G. L. F. Norris, Trans. Br. Mycol. Soc., 1957, 40, 365 CrossRef CAS. For the total synthesis of wortmannin, see: (b) S. Sato, M. Nakada and M. Shibasaki, Tetrahedron Lett., 1996, 37, 6141 CrossRef CAS; (c) T. Mizutani, S. Honzawa, S. Tosaki and M. Shibasaki, Angew. Chem., Int. Ed., 2002, 41, 4680 CrossRef CAS PubMed; (d) H. Shigehisa, T. Mizutani, S. Tosaki, T. Ohshima and M. Shibasaki, Tetrahedron, 2005, 61, 5057 CrossRef CAS; (e) Y. Guo, T. Quan, Y. Lu and T. Luo, J. Am. Chem. Soc., 2017, 139, 6815 CrossRef CAS PubMed.
- (a) H. Fujiwara, K. Matsunaga, M. Saito, S. Hagiya, K. I. Furukawa, H. Nakamura and Y. Ohizumi, Eur. J. Pharmacol., 2001, 413, 37 CrossRef CAS PubMed; (b) P. Wipf and R. J. Halter, Org. Biomol. Chem., 2005, 3, 2053 RSC.
- (a) P. Wipf, D. J. Minion, R. J. Halter, M. I. Berggren, C. B. Ho, G. G. Chiang, L. Kirkpatrick, R. Abraham and G. Powis, Org. Biomol. Chem., 2004, 2, 1911 RSC; (b) N. T. Ihle, R. Williams, S. Chow, W. Chew, M. I. Berggren, G. Paine-Murrieta, D. J. Minion, R. J. Halter, P. Wipf, R. Abraham, L. Kirkpatrick and G. Powis, Mol. Cancer Ther., 2004, 3, 763 CAS.
- For the isolation of viridiol, see: J. S. Moffatt, J. D. Bu'Lock and T. H. Yuen, J. Chem. Soc. D, 1969, 839a RSC.
- For the isolation of demethoxyviridin and demethoxyviridiol, see: D. C. Aldridge, W. B. Turner, A. J. Geddes and B. Sheldrick, J. Chem. Soc., Perkin Trans. 1, 1975, 943 RSC.
- For the isolation of 9-epi-viridiol, see: P. Y. Phuwapraisirisan, J. Rangsanyz, P. Siripongx and S. Tip-Pyang, Nat. Prod. Res., 2006, 20, 1321 CrossRef CAS PubMed.
- P. F. Andersson, S. Bengtsson, M. Cleary, J. Stenlid and A. Broberg, Phytochemistry, 2013, 86, 195 CrossRef CAS PubMed.
- For the isolation of nodulisporiviridin E, see: Q. Zhao, G. Chen, X. Feng, Y. Yu, R. He, X. Li, Y. Huang, W. Zhou, L. Guo, Y. Zheng, X. Yao and H. Gao, J. Nat. Prod., 2015, 78, 1221 CrossRef CAS PubMed.
- (a) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS; (b) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 232 CrossRef; (c) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945 CrossRef CAS PubMed; (d) K. Du, P. Guo, Y. Chen, Z. Cao, Z. Wang and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 3033 CrossRef CAS PubMed.
- Y. Shi, Y. Ji, K. Xin and S. Gao, Org. Lett., 2018, 20, 732 CrossRef CAS PubMed.
- M. T. Barros, C. D. Maycock and M. R. Ventura, J. Chem. Soc., Perkin Trans. 1, 2001, 166 RSC.
- S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS.
- (a) D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS; (b) D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277 CrossRef CAS.
- N. Harada, T. Sugioka, H. Uda, T. Kuriki, M. Kobayashi and I. J. Kitagawa, J. Org. Chem., 1994, 59, 6606 CrossRef CAS.
- (a) J. R. Hanson, M. A. O'Leary, H. J. Wadsworth and B. L. Yeoh, J. Chem. Soc., Perkin Trans. 1, 1985, 1311 RSC; (b) Y. Lang, F. E. S. Souza, X. Xu, N. J. Taylor, A. Assoud and R. Rodrigo, J. Org. Chem., 2009, 74, 5429 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qo01278h |
| This journal is © the Partner Organisations 2020 |