
Cu-catalyzed C–N bond cleavage of 3-aminoindazoles for the C–H arylation of enamines†
Yao
Zhou
 a,
Ya
Wang
a,
Zhiyi
Song
b,
Tamaki
Nakano
b and
Qiuling
Song
*ac
a,
Ya
Wang
a,
Zhiyi
Song
b,
Tamaki
Nakano
b and
Qiuling
Song
*ac
aInstitute of Next Generation Matter Transformation, College of Materials Science & Engineering and College of Chemical Engineering at Huaqiao University, 668 Jimei Blvd, Xiamen, Fujian 361021, P. R. China. E-mail: qsong@hqu.edu.cn; Fax: +86-592-6162990
bInstitute for Catalysis (ICAT), Hokkaido university, N 21, W 10, Kita-ku, Sapporo 001-0021, Japan
cFujian University Key Laboratory of Molecule Synthesis and Function Discovery, College of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, China
First published on 1st November 2019
Abstract
The Cu-catalyzed C–H arylation of enamines via the oxidative C–N cleavage of 3-aminoindazoles is presented. A diverse array of arylated enamines were produced in decent yields with a wide substrate scope under mild conditions. Here, 3-aminoindazoles were harnessed as novel arylating agents via a radical process.
Transition-metal-catalyzed transformation of C–N bonds has inarguably been a subject of great importance in organic chemistry, organometallic chemistry and biochemistry.1 In comparison with the transformation of activated C–N bonds, for example those in diazonium salts,2 ammonium salts,3 pyridinium salts,4 triazoles5 and strained azaheterocycles,6 the transformation of unactivated C–N bonds7 has presented itself as a challenging albeit promising theme in organic synthesis, owing to the inert nature of the unreactive C–N bond. For the transformation of unactivated aromatic C–N bonds, Shi and other groups have recently demonstrated a range of excellent coupling reactions via selective cleavage of aryl C–N bonds.7n–r Although remarkable progress has been achieved in the last decades in transition-metal-catalyzed selective cleavage of C–N bonds, very few methodologies have to date been successfully established for the transformation of multifarious C–N bonds in one targeted molecule under one set of conditions. In particular, the direct disassembly of relatively stable five-membered heterocycles via selective cleavage of multiple C–N bonds in one pot is still far from being achieved.
In recent years, enamines have emerged as multifunctional building blocks in synthetic chemistry.8 A myriad of efficient methodologies have been established to produce N-heterocycles by using enamines as robust synthons.9,16 The π-donating capacity of the nitrogen atom makes the carbon–carbon double bonds of enamines relatively electron rich (compared for example to those of simple olefins), which affords a plethora of excellent transformations for these carbon–carbon double bonds. Given this unique property of enamines, the alkenyl C–H functionalization of enamines has received much attention by organic chemists for the purpose of streamlining the syntheses of multiple functionalized enamines.10 With respect to C–H arylation of enamines, a range of readily available arylating agents, such as electron-rich arenes, aryl halides, triflates, organoboranes and others, have been applied to accomplish the olefinic C–H arylation of enamines (Scheme 1a).11a–e Despite such undisputed advances, it is desirable to carry out the C–H arylation of enamines concomitant with the introduction of other functional groups under one set of conditions; the resulting products could undergo further derivatization to expand the molecular complexity of synthesized materials. The cyano group can be handily transformed to a diverse array of functional groups, which makes nitrile-derived compounds are significant building blocks in synthetic chemistry.12 Furthermore, nitrile-containing derivatives are pervasively included in natural products, drug reagents, herbicides and agrochemicals.13 As part of our continuous interest in developing new reactions of 3-aminoindazoles,14 in the current work we used an expedient approach to achieve the C–H arylation of enamines via Cu-catalyzed C–N bond activation of 3-aminoindazoles concomitant with the generation of the cyano group in situ (Scheme 1b).
 | ||
| Scheme 1 The transition metal-catalyzed C–H arylation of enamines. | ||
To initiate our study, the commercially available 3-amino-1H-indazole (1a) and easily accessible (Z)-ethyl 3-amino-3-phenyl acrylate (2a) were selected as model substrates for the optimization experiments. To our delight, when the model reaction was carried out by using 20 mol% Cu(OAc)2·H2O as a catalyst and tert-butyl peroxybenzoate (TBPB) as an oxidant at 60 °C, the denitrogenative C–H arylation was indeed observed, and the product 3a was isolated in 58% yield (Table 1, entry 1). Subsequently, the model reaction was submitted to a round of oxidative systems (Table 1, entries 2–6). However, only marginal improvements were observed. As a follow-up optimization, a series of oxidants were investigated in the presence of Cu(OAc)2·H2O as a catalyst (Table 1, entries 7–17). TBHP was found to be the optimal oxidant, affording the product 3a in 80% yield (Table 1, entry 8). We also screened several Cu salts (Table 1, entries 18–21); Cu(OAc)2·H2O still demonstrated the best effectiveness. Hoping to enhance the reactivity, the effect of reaction temperature was also tested (Table 1, entries 22–24). When this C–H arylation reaction was carried out at room temperature instead of at 60 °C, the yield of the expected product 3a was significantly increased to 88%. The reaction was sluggish when conducted without Cu(OAc)2 or TBHP, indicating that Cu salt and oxidant were both obligatory for this denitrogenative C–H arylation (Table 1, entries 25–26).
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Oxidant | T (°C) | Yield of 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.4 mmol), [M] (20 mol%), oxidant (0.5 mmol), under air, 20 h. b GC yield. c Isolated yield. d 10 mol% RuCl3. e 5 mol% Ru(PPh)3Cl2. | ||||
| 1 | Cu(OAc)2·H2O | TBPB | 60 | 58% |
| 2 | Mn(OAc)3 | TBPB | 60 | Trace |
| 3 | Co(OAc)2 | TBPB | 60 | 18% |
| 4d | RuCl3 | TBPB | 60 | 9% |
| 5e | Ru(PPH)3Cl2 | TBPB | 60 | N.D. |
| 6 | — | TBPB | 60 | N.D. |
| 7 | Cu(OAc)2·H2O | DTBP | 60 | 35% |
| 8 | Cu(OAc)2·H2O | TBHP | 60 | 80% |
| 9 | Cu(OAc)2·H2O | O2 | 60 | 39% |
| 10 | Cu(OAc)2·H2O | K2S2O8 | 60 | 42% |
| 11 | Cu(OAc)2·H2O | Na2S2O8 | 60 | 31% |
| 12 | Cu(OAc)2·H2O | LPO | 60 | 8% |
| 13 | Cu(OAc)2·H2O | CAN | 60 | N.D. |
| 14 | Cu(OAc)2·H2O | PIDA | 60 | N.D. |
| 15 | Cu(OAc)2·H2O | PIFA | 60 | Trace |
| 16 | Cu(OAc)2·H2O | IBX | 60 | 22% |
| 17 | Cu(OAc)2·H2O | PhIO | 60 | 46% |
| 18 | Cu(OTf)2 | TBHP | 60 | 10% |
| 19 | CuI | TBHP | 60 | 63% |
| 20 | CuBr2 | TBHP | 60 | 44% |
| 21 | CuCl2 | TBHP | 60 | 51% |
| 22 | Cu(OAc)2·H2O | TBHP | 80 | 72% |
| 23 | Cu(OAc)2·H2O | TBHP | 50 | 83% |
| 24 | Cu(OAc)2·H2O | TBHP | RT | 92% (88%)c |
| 25 | — | TBHP | RT | N.D. |
| 26 | Cu(OAc)2·H2O | — | RT | Trace |

Having identified the reaction conditions for this denitrogenative C–H arylation, the generality of this Cu-catalyzed C–H arylation reaction was then assessed (Scheme 2). The scope with respect to the enamines was inspected initially. We were glad to find that a sequence of β-enamine esters bearing electron-donating or electron-withdrawing groups on the aromatic ring were well tolerated under the optimal conditions, enabling the production of the targeted C–H arylation products (3a–3g) in good to high yields. To our delight, heteroaromatic β-enamine esters (2h and 2i) also proved to be good candidates for this C–H functionalization, leading to the formation of ethyl (Z)-3-amino-2-(2-cyanophenyl)-3-(pyridin-4-yl)acrylate (3h) and ethyl (Z)-3-amino-2-(2-cyanophenyl)-3-(thiophen-3-yl)acrylate (3i) in 67% and 88% yields, respectively. In addition to the aromatic enamines, the alkyl-substituted β-enamine esters also demonstrated decent reactivity in this oxidative C–H arylation, furnishing the expected products (3j–3p) in moderate to good yields. The structure of 3n was explicitly identified using X-ray crystallographic analysis.15 N-protected enamines were amenable to this transformation as well, giving rise to the generation of corresponding products 3q and 3r in 72% and 69% yields, respectively. Subsequently, the scope with regard to a battery of 3-aminoindazoles was also investigated. As showcased in Scheme 2, a group of halo-substituted 3-aminoindazoles were suitable donors for this Cu-catalyzed denitrogenative ring-opening, providing the desired C–H arylation products 3s–3v in 57%–68% yields.
 | ||
| Scheme 2 Scope for the denitrogenative C–H arylation of enamines. All reactions unless otherwise stated were carried out with 1 (0.2 mmol), 2 (0.4 mmol), Cu(OAc)2 (20 mol%) and TBHP (2.5 equiv.) in MeCN (1.0 mL) at RT for 18 h. The yields of the isolated products are given. | ||
The scalability of this Cu-catalyzed C–H arylation of enamines was also investigated. When 3 mmol of β-enamine ester 2c was subjected to this transformation, the desired ethyl (Z)-3-amino-2-(2-cyanophenyl)-3-(4-methoxyphenyl)acrylate (3c) was readily produced in 85% yield without a loss of the efficiency (Scheme 3a). In recent years, enamines have emerged as promising materials for producing 2H-azirines.16 Owing to the characteristic strain, 2H-azirines are versatile building blocks to streamline construction of multitudinous nitrogen heterocycles in organic synthesis.17 In addition, 2H-azirines also exhibit unique biological properties in medicinal chemistry.18 The synthetic utility of this Cu-catalyzed C–H arylation was showcased by the oxidation of β-enamine esters 3a to assemble the 2H-azirine derivative 4 in excellent yield under mild conditions (Scheme 3b). 1-Aminoisoquinoline derivatives are pervasively found as privileged core structural frameworks in a plethora of bioactive compounds.19 This C–H arylation product 3c was also effectively converted into ethyl 1-amino-4-(p-tolyl)isoquinoline-3-carboxylate 5 in 92% yield, under basic conditions (Scheme 3c).
 | ||
| Scheme 3 Synthetic applications and transformations. | ||
To gain deeper insight into the reaction mechanism of this C–H arylation, a couple of control experiments were carried out. When 3-amino-1H-indazole (1a) was subjected to the identified conditions without enamines, a trace amount of benzonitrile (6) and the dimeric compound [1,1′-biphenyl]-2,2′-dicarbonitrile (7) were detected using GC-MS (Scheme 4a). When unsubstituted, 3-iodo, and 3-carboxylic acid indazoles were submitted to this transformation, these starting materials were intact and no C–N activation products were detected (Scheme 4b), indicating that attachment of the NH2 group to the C3 position of indazole is a prerequisite for this C–N activation of the indazole ring. When 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) were added as radical inhibitors in this Cu-catalyzed C–H arylation, only a stagnant reaction was observed (Scheme 4c). And compound 8 was also detected using GC-MS when 1,1-diphenylethylene was utilized as the additive in this C–H arylation (Scheme 4d). Based on these control experiments, it was supposed that a radical-type reaction regime might be involved in this Cu-catalyzed C–H arylation.
 | ||
| Scheme 4 Control experiments. | ||
According to the above experiments, a possible reaction mechanism for this Cu-catalyzed denitrogenative C–H arylation of enamine is shown in Scheme 5. According to this proposed mechanism, firstly, the oxidation of 3-aminoindazole 1 leads to the formation of compound A. Simultaneously, an electron is transferred from the Cu(II) species to TBHP to deliver the Cu(III) species and the tert-butoxyl radical.20 Subsequently, the tert-butoxyl radical abstracts a hydrogen atom from compound A, which then extrudes one molecular nitrogen to provide the benzonitrile radical intermediate B.21 Then, radical addition of intermediate B to the enamine 2 gives rise to the production of radical intermediate C,22 which is oxidized by the Cu(III) species to afford the carbocation intermediate D. At the same time, the Cu(II) species is released to continue the next catalytic cycle. Finally, deprotonation of intermediate D furnishes the desired C–H arylation product 3.
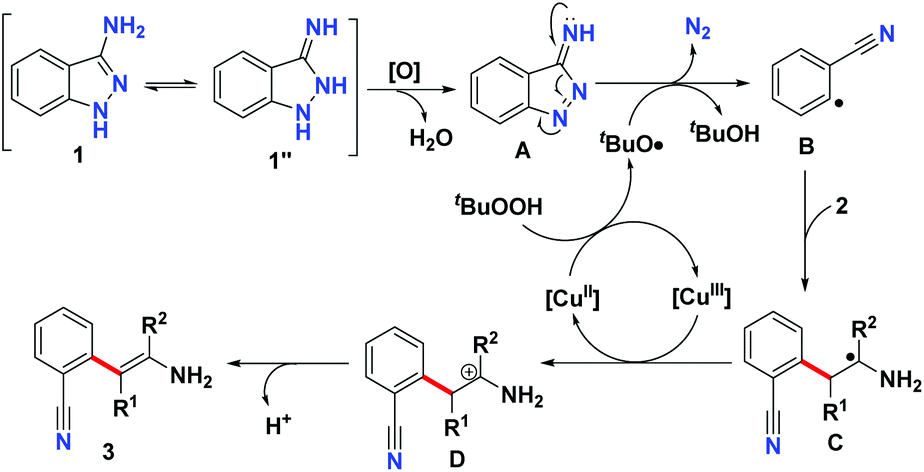 | ||
| Scheme 5 Proposed reaction mechanism. | ||
In conclusion, we have disclosed an expedient approach to achieve the C–H arylation of enamines via the Cu-catalyzed C–N bond activation of 3-aminoindazoles, concomitant with the generation of a cyano group in situ. A range of enamines could be arylated in good yields by using 3-aminoindazoles as new arylating agents. The mechanism study indicated that a radical process was involved in this Cu-catalyzed C–N bond cleavage of 3-aminoindazoles for the C–H arylation of enamines.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This research was financially supported by the National Natural Science Foundation (21772046) and the Natural Science Foundation of Fujian Province (2016J01064), which are gratefully acknowledged. We also thank the Instrumental Analysis Center of Huaqiao University for analysis support.Notes and references
- For some reviews on transition-metal-catalyzed C–N activation: (a) K. Ouyang, W. Hao, W.-X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045 CrossRef CAS; (b) Q. Wang, Y. Su, L. Li and H. Huang, Chem. Soc. Rev., 2016, 45, 1257 RSC; (c) G. Meng, S. Shi and M. Szostak, Synlett, 2016, 27, 2530 CrossRef CAS; (d) C. Liu and M. Szostak, Chem. – Eur. J., 2017, 23, 7157 CrossRef CAS.
- (a) S. Mahouche-Chergui, S. Gam-Derouich, C. Mangeney and M. M. Chehimi, Chem. Soc. Rev., 2011, 40, 4143 RSC; (b) F. Mo, D. Qiu, Y. Zhang and J. Wang, Acc. Chem. Res., 2018, 51, 496 CrossRef CAS.
- (a) L.-G. Xie and Z.-X. Wang, Angew. Chem., Int. Ed., 2011, 50, 4901 CrossRef CAS; (b) P. Maity, D. M. Shacklady-McAtee, G. P. A. Yap, E. R. Sirianni and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 280 CrossRef CAS; (c) S. Yu, S. Liu, Y. Lan, B. Wan and X. Li, J. Am. Chem. Soc., 2015, 137, 1623 CrossRef CAS; (d) C. H. Basch, K. M. Cobb and M. P. Watson, Org. Lett., 2016, 18, 136 CrossRef CAS; (e) Y.-Q.-Q. Yi, W.-C. Yang, D.-D. Zhai, X.-Y. Zhang, S.-Q. Li and B.-T. Guan, Chem. Commun., 2016, 52, 10894 RSC; (f) M. Guisan-Ceinos, V. Martín-Heras and M. Tortosa, J. Am. Chem. Soc., 2017, 139, 8448 CrossRef CAS; (g) L.-L. Liao, G.-M. Cao, J.-H. Ye, G.-Q. Sun, W.-J. Zhou, Y.-Y. Gui, S.-S. Yan, G. Shen and D.-G. Yu, J. Am. Chem. Soc., 2018, 140, 17338 CrossRef CAS.
- F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943 CrossRef CAS.
- (a) B. Chattopadhyay and V. Gevorgyan, Angew. Chem., Int. Ed., 2012, 51, 862 CrossRef CAS; (b) A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 1371 CrossRef CAS; (c) H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151 RSC; (d) Y. Jiang, R. Sun, X.-Y. Tang and M. Shi, Chem. – Eur. J., 2016, 22, 17910 CrossRef CAS.
- (a) I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef CAS; (b) G. S. Singh, M. D′hooghe and N. D. Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS; (c) S. Stanković, M. D′hooghe, S. Catak, H. Eum, M. Waroquier, V. V. Speybroeck, N. D. Kimpe and H.-J. Ha, Chem. Soc. Rev., 2012, 41, 643 RSC.
- For some selected examples on transformation of unactivated C–N bond, see: (a) Y. Liu, Y. Xie, H. Wang and H. Huang, J. Am. Chem. Soc., 2016, 138, 4314 CrossRef CAS PubMed; (b) L. Li, X. Zhou, B. Yu and H. Huang, Org. Lett., 2017, 19, 4600 CrossRef CAS; (c) C. Rao, M. Shao and Q. Song, Org. Lett., 2017, 19, 4726 CrossRef CAS; (d) B. Gao and H. Huang, Org. Lett., 2017, 19, 6260 CrossRef CAS; (e) R. S. Mane and B. M. Bhanage, Adv. Synth. Catal., 2017, 359, 2621 CrossRef CAS; (f) H. Yu, B. Gao, B. Hu and H. Huang, Org. Lett., 2017, 19, 3520 CrossRef CAS; (g) Y. Zhou, S. Deng, S. Mai and Q. Song, Org. Lett., 2018, 20, 6161 CrossRef CAS; (h) Y. Fu, Q.-S. Xu, C.-Z. Shi, Z. Du and C. Xiao, Adv. Synth. Catal., 2018, 360, 3502 CrossRef CAS; (i) Y. Zhou, Y. Lou, Y. Wang and Q. Song, Org. Chem. Front., 2019, 6, 3355 RSC; (j) Y. He, Z. Zheng, Y. Liu, J. Qiao, X. Zhang and X. Fan, Org. Lett., 2019, 21, 1676 CrossRef CAS; (k) Y. Zhou, Y. Wang, Y. Lou and Q. Song, Chem. Commun., 2019, 55, 10265 RSC; (l) H.-H. Xu, X.-H. Zhang and X.-G. Zhang, J. Org. Chem., 2019, 84, 7894 CrossRef CAS; (m) K. He, P. Li, S. Zhang, Q. Chen, H. Ji, Y. Yuan and X. Jia, Chem. Commun., 2018, 54, 13232 RSC; (n) J. Hu, Y. Zhao, J. Liu, Y. Zhang and Z. Shi, Angew. Chem., Int. Ed., 2016, 55, 8718 CrossRef CAS; (o) Z.-C. Cao, X.-L. Li, Q.-Y. Luo, H. Fang and Z.-J. Shi, Org. Lett., 2018, 20, 1995 CrossRef CAS; (p) Z.-C. Cao, S.-J. Xie, H. Fang and Z.-J. Shi, J. Am. Chem. Soc., 2018, 140, 13575 CrossRef CAS; (q) Z. Zhang, D. Zheng, Y. Wan, G. Zhang, J. Bi, Q. Liu, T. Liu and L. Shi, J. Org. Chem., 2018, 83, 1369 CrossRef CAS; (r) Z.-B. Zhang, C.-L. Ji, C. Yang, J. Chen, X. Hong and J.-B. Xia, Org. Lett., 2019, 21, 1226 CrossRef CAS.
- For reviews on the enamine chemistry, see: (a) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455 RSC; (b) R. Matsubara and S. Kobayashi, Acc. Chem. Res., 2008, 41, 292 CrossRef CAS PubMed; (c) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599 CrossRef CAS; (d) M.-X. Wang, Chem. Commun., 2015, 51, 6039 RSC; (e) J.-P. Wan and Y. Gao, Chem. Rec., 2016, 16, 1164 CrossRef CAS.
- (a) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 14254 CrossRef CAS; (b) J. Huang, Y. Liang, W. Pan, Y. Yang and D. Dong, Org. Lett., 2007, 9, 5345 CrossRef CAS; (c) R. Zhang, Y. J. Liang, G. Y. Zhou, K. W. Wang and D. Dong, J. Org. Chem., 2008, 73, 8089 CrossRef CAS PubMed; (d) R. Zhang, D. Zhang and D. Dong, J. Org. Chem., 2011, 76, 2880 CrossRef CAS PubMed; (e) P. Huang, N. Zhang, R. Zhang and D. Dong, Org. Lett., 2012, 14, 370 CrossRef CAS; (f) D. Xiang, X. Xin, X. Liu, R. Zhang, J. Yang and D. Dong, Org. Lett., 2012, 14, 644 CrossRef CAS; (g) P. Huang, R. Zhang, Y. Liang and D. Dong, Org. Lett., 2012, 14, 5196 CrossRef CAS; (h) C.-H. Lei, D.-X. Wang, L. Zhao, J. Zhu and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 4708 CrossRef CAS; (i) Q. Zhang, X. Liu, X. Xin, R. Zhang, Y. Liang and D. Dong, Chem. Commun., 2014, 50, 15378 RSC; (j) P. Gao, J. Wang, Z. Bai, D. Yang, M.-J. Fan and Z.-H. Guan, Chem. – Asian J., 2017, 12, 1865 CrossRef CAS; (k) Y. Zhou, Z. Tang and Q. Song, Adv. Synth. Catal., 2017, 359, 952 CrossRef CAS; (l) C. Li, J. Yuan, Q. Zhang, C. B. Rao, R. Zhang, Y. Zhao, B. Deng and D. Dong, J. Org. Chem., 2018, 83, 14999 CrossRef CAS; (m) P. Shi, S. Li, L.-M. Hu, C. Wang, T.-P. Loh and X.-H. Hu, Chem. Commun., 2019, 55, 11115 RSC; (n) S. S. Jang, J. Y. Chang, G. Y. Kang and S. W. Youn, Asian J. Org. Chem., 2019, 8, 1668 CrossRef CAS; (o) D. Cheng, Z. Deng, X. Yan, M. Wang, X. Xu and J. Yan, Adv. Synth. Catal., 2019, 361, 5025 CrossRef CAS.
- For a review, see: N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Chem. – Eur. J., 2014, 20, 7548 CrossRef CAS.
- (a) H. Zhou, Y.-H. Xu, W.-J. Chung and T.-P. Loh, Angew. Chem., Int. Ed., 2009, 48, 5355 CrossRef CAS PubMed; (b) H. Zhou, W.-J. Chung, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2009, 3472 RSC; (c) S. Pankajakshan, Y.-H. Xu, J. K. Cheng, M. T. Low and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 5701 CrossRef CAS; (d) N. Gigant, L. Chausset-Boissarie, M.-C. Belhomme, T. Poisson, X. Pannecoucke and I. Gillaizeau, Org. Lett., 2013, 15, 278 CrossRef CAS PubMed; (e) Z.-Q. Lei, J.-H. Ye, J. Sun and Z.-J. Shi, Org. Chem. Front., 2014, 1, 634 RSC; (f) N. Gigant, L. Chausset-Boissarie and I. Gillaizeau, Org. Lett., 2013, 15, 816 CrossRef CAS PubMed; (g) A. L. Hansen and T. Skrydstrup, J. Org. Chem., 2005, 70, 5997 CrossRef CAS; (h) Y. Liu, D. Li and C.-M. Park, Angew. Chem., Int. Ed., 2011, 50, 7333 CrossRef CAS; (i) H. Ge, M. J. Niphakis and G. I. Georg, J. Am. Chem. Soc., 2008, 130, 3708 CrossRef CAS; (j) J.-P. Wan, Z. Tu and Y. Wang, Chem. – Eur. J., 2019, 25, 6907 CrossRef CAS.
- (a) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappoport, Wiley-VCH, New York, NY, 1983 Search PubMed. For some reviews on the synthesis of nitriles, see: (b) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC; (c) G. Yan, Y. Zhang and J. Wang, Adv. Synth. Catal., 2018, 359, 4068 CrossRef.
- (a) A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substance: Synthesis, Patents, Applications, Georg Thieme, Stuttgart, 4th edn, 2001 Search PubMed; (b) L. H. Jones, N. W. Summerhill, N. A. Swain and J. E. Mills, MedChemComm, 2010, 1, 309 RSC.
- (a) Y. Zhou and Q. Song, Org. Chem. Front., 2018, 5, 3245 RSC; (b) Y. Zhou, Y. Wang, Y. Lou and Q. Song, Org. Lett., 2018, 20, 6494 CrossRef CAS; (c) W. Kong, Y. Zhou and Q. Song, Adv. Synth. Catal., 2018, 360, 1943 CrossRef CAS; (d) Y. Zhou, L. Lin, Y. Wang, J. Zhu and Q. Song, Org. Lett., 2019, 21, 7630 CrossRef CAS; (e) Y. Zhou, Y. Wang, Y. Lou and Q. Song, Org. Lett., 2019 DOI:10.1021/acs.orglett.9b02288.
- CCDC 1954088 (3n)† contains the supplementary data for this communication.
- (a) X. Li, Y. Du, Z. Liang, X. Li, Y. Pan and K. Zhao, Org. Lett., 2009, 11, 2643 CrossRef CAS; (b) X. Sun, Y. Lyu, D. Zhang-Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 6222 CrossRef CAS; (c) X. Duan, X. Kong, X. Zhao, K. Yang, H. Zhou, D. Zhou, Y. Zhang, J. Liu, J. Ma, N. Liu and Z. Wang, Tetrahedron Lett., 2016, 57, 1446 CrossRef CAS; (d) Y. Zhang, X. Zhao, C. Zhuang, S. Wang, D. Zhang-Negrerie and Y. Du, Adv. Synth. Catal., 2018, 360, 2107 CrossRef CAS; (e) M. Wang, J. Hou, W. Yu and J. Chang, J. Org. Chem., 2018, 83, 14954 CrossRef CAS.
- For a review on 2H-azirine chemistry, see: A. F. Khlebnikov, M. S. Novikov and N. V. Rostovskii, Tetrahedron, 2019, 75, 2555 CrossRef CAS.
- (a) T. F. Molinski and C. M. Ireland, J. Org. Chem., 1988, 53, 2103 CrossRef CAS; (b) C. K. Skepper and T. F. Molinski, J. Org. Chem., 2008, 73, 2592 CrossRef CAS; (c) J. L. Keffer, A. Plaza and C. A. Bewley, Org. Lett., 2009, 11, 1087 CrossRef CAS.
- (a) A. Smith, F. DeMorin, N. Paras, Q. Huang, K. Petkus, E. Doherty, T. Nixey, J. Kim, D. Whittington, L. Epstein, M. Lee, M. Rose, C. Babij, M. Fernando, K. Hess, Q. Le, P. Beltran and J. Carnahanz, J. Med. Chem., 2009, 52, 6189 CrossRef CAS; (b) S. Yang, H. Van, T. Le, D. Khadka, S. Cho, K. Lee, H. Chung, S. Lee, C. Ahn, Y. Lee and W. Cho, Bioorg. Med. Chem. Lett., 2010, 20, 5277 CrossRef CAS.
- J. Wang, C. Liu, J. Yuan and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 2256 CrossRef CAS.
- This denitrogenation of compound A probably attributes to the formation of thermodynamic stable cyano group, which could also explain the result of Scheme 4b (the NH2 group installed on the C3 position of indazoles is indispensable for this ring-opening of indazole ring).
- (a) Y. Gao, Y. Liu and J.-P. Wan, J. Org. Chem., 2019, 84, 2243 CrossRef CAS; (b) Y. Guo, Y. Xiang, L. Wei and J.-P. Wan, Org. Lett., 2018, 20, 3971 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1954088. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo01177c |
| This journal is © the Partner Organisations 2020 |