
Azidofluoromethane: synthesis, stability and reactivity in [3 + 2] cycloadditions†
Svatava
Voltrová
 a,
Josef
Filgas
ab,
Petr
Slavíček
b and
Petr
Beier
*a
a,
Josef
Filgas
ab,
Petr
Slavíček
b and
Petr
Beier
*a
aInstitute of Organic Chemistry and Biochemistry AS CR, Flemingovo nám. 2, 160 00 Prague 6, Czech Republic. E-mail: beier@uochb.cas.cz
bUniversity of Chemistry and Technology, Technická 5, 160 00 Prague 6, Czech Republic
First published on 5th November 2019
Abstract
Azidofluoromethane was prepared for the first time by the nucleophilic displacement of bromofluoromethane with sodium azide. This volatile and unstable compound was isolated by low temperature vacuum distillation with a suitable solvent and fully characterized. Theoretical calculations of its decomposition activation energies and rate constants were performed and the values were compared to those for azidomethane, azidodifluoromethane and azidotrifluoromethane. Azidofluoromethane underwent [3 + 2] cycloadditions with alkynes, 1,3-diones and β-ketoesters to furnish 1-fluoromethyl-1,2,3-triazoles.
Introduction
Organic azides are compounds of high synthetic utility. For example, their cycloaddition reaction with alkynes to form 1,2,3-triazoles is widely used in medicinal chemistry and in chemical biology. Thermal and photo decomposition give highly reactive nitrenes and the reaction with phosphines affords iminophosphoranes and eventually imines. However, small organic azides with a large proportion of nitrogen in the overall mass (such as azidomethane) are often unstable and can decompose explosively. Fluorine substituents are widely used to modify the physical and biological properties of bioactive molecules in the desired way1–4 and fluorinated azidomethanes have emerged as underdeveloped fluorinated C1 building blocks of high synthetic potential. We recently reintroduced azidodifluoromethane5 and azidotrifluoromethane6 and showed that they are stable and undergo [3 + 2] cycloadditions with dipoles. The resulting 1,2,3-triazoles transform into various nitrogen heterocycles in Rh-catalyzed transannulations7 or into novel β-enamido triflates in the reaction with triflic acid.8 Azidofluoromethane is the last unknown member of the family of fluorinated azidomethanes, and in this work, we set out to investigate its synthesis, stability and cycloaddition to form the respective 1-fluoromethyl 1,2,3-triazoles.Results and discussion
Azidofluoromethane has never been prepared and characterized. It is mentioned in a patent9 without any relevant characterization or experimental details and in two theoretical studies.10,11 Each fluorinated azidomethane requires a different synthetic approach. The synthesis of CF3N3 is based on the transfer of a nucleophilic trifluoromethyl group to an electrophilic azide source (Scheme 1A). For the synthesis of F2CHN3 a difluorocarbene precursor was used in the reaction with nucleophilic azide (Scheme 1B). To access FCH2N3 we proposed a nucleophilic displacement of the halide leaving group with an azide source (Scheme 1C).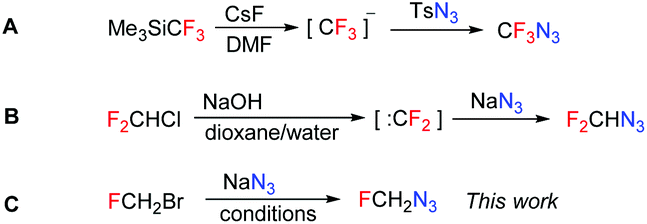 | ||
| Scheme 1 Synthetic approaches to fluorinated azidomethanes. | ||
Optimization experiments for the synthesis of azidofluoromethane are shown in Table 1. When DMF was used as the first solvent of choice to dissolve the starting bromofluoromethane and water was used to dissolve sodium azide at 0 °C, no product formation was observed. At elevated temperature, the reaction proceeded to achieve a complete conversion of the bromide, but only traces of the azide could be detected. Higher azide yields were obtained when THF was added to the reaction mixture; however, decomposition of the product occurred after an extended time. Acetone and chloroform gave negligible product yields. More promising was the experiment in DMSO with the addition of a small amount of water. It led to product formation, but decomposition took place after 3 hours. Finally, NMP was found to be the best solvent. It exhibited excellent solubility for both bromo- and azidofluoromethane, and sufficient solubility for sodium azide. With a two-fold excess of sodium azide, the product was formed with complete conversion at ambient temperature. No pressurization of the reaction vessel was observed. Isolation of the product was performed by distillation after the addition of THF as the co-solvent. Other solvents such as CDCl3, DCM or DCE can be also used for this purpose. In these experiments, we were aware of the possible low stability of azidofluoromethane and its potential to decompose explosively similarly to azidomethane.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. (°C) | Time (h) | Yielda (%) |
a ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1H NMR yield.
b Using NaN3 (2 equiv.).
c Isolated yield of the azide co-distilled with THF. 1H NMR yield.
b Using NaN3 (2 equiv.).
c Isolated yield of the azide co-distilled with THF.
|
||||
| 1 | DMF/water (1:1) |
0–50 | 16 | 0 |
| 2 | DMF/THF/water (1:2:1) |
35 | 1.5 | 40 |
| 3 | DMF/THF/water (1:2:1) |
35 | 3 | 45 |
| 4 | DMF/THF/water (1:2:1) |
35 | 4.5 | 12 |
| 5 | Acetone/water (1:1) |
30 | 16 | 0 |
| 6 | CHCl3/water (1:1) |
20 | 4 | 1 |
| 7 | CHCl3/water (1:1) |
40 | 4 | 7 |
| 8 | DMSO/water (100:1) |
50 | 1.5 | 80 |
| 9 | DMSO/water (100:1) |
50 | >3 | 0 |
| 10 | NMP | 20 | 1 | 90 |
| 11 | NMPb | 20 | 1 | 100 (82)c |

Stability of 1 was found to be solvent and temperature dependent. A solution of azide 1 in CDCl3 was not stable even during short-time storage (possibly due to acid-accelerated decomposition of 1). Nevertheless, we were able to record the 1H, 19F and 13C NMR (short-time experiment) and HRMS spectra of 1 in CDCl3 and thus fully characterize it. However, at room temperature, after prolonged time (>2 h), decomposition of even a diluted CDCl3 solution of azidofluoromethane was repeatedly observed (build-up of pressure in NMR tubes). Therefore, the storage of the solution of azidofluoromethane is not advisable, but rather the immediate use of the in situ formed 1 or distilled solution of 1 in THF is advisable. Azide 1 was found to be stable in THF at low temperature (below −20 °C); at room temperature, evaporation of 1 rather than decomposition was observed.
The identification of the decomposition products of 1 was of interest. In one experiment with the solution of 1 in CDCl3 in a sealed NMR tube, a non-explosive decomposition of 1 took place after storage at room temperature for 6 hours and the formation of hydrogen cyanide was observed by 13C NMR. The proposed mechanism of the decomposition of azidofluoromethane starts with nitrogen elimination. The highly reactive nitrene formed rearranges to fluoroimine which eliminates hydrogen fluoride and forms hydrogen cyanide (Scheme 2A). In contrast, azidodifluoromethane and azidotrifluoromethane are more stable (their chloroform solutions were found to be stable at 150 °C).5,6 Neat CF3N3 was reported to decompose explosively at 330 °C. High-temperature decomposition of both azides was studied and they yielded nitrogen and FCN (Scheme 2B and C).12,13
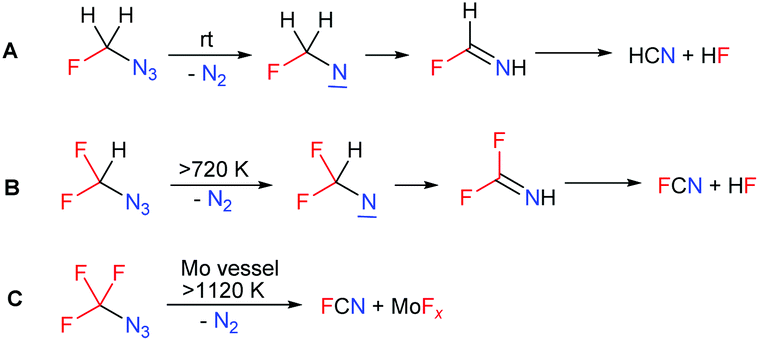 | ||
| Scheme 2 Decomposition of fluorinated azidomethanes. | ||
To correlate the observed differences in the stability of azidomethane and its fluorinated analogues, we computed their activation Gibbs energies (ΔG#) and rate constants (k) for the decomposition by using the ab initio method CCSD(T) (Table 2). The value of ΔG# somewhat decreases upon the fluorination of azidomethane and then it increases with increasing fluorine content. This trend correlates well with the qualitative observations from the experiments, showing increased kinetic stability of the azides upon fluorination. At the DFT level, the transition states for the azide decomposition are characterized by a concerted N–N dissociation and hydrogen transfer. The imine product would then be directly formed without the nitrene intermediate. The multi-reference ab initio methods14,15 support a shallow nitrene minimum. The general trend of the activation barriers remains unchanged – the value of the activation barrier is primarily determined by the dissociation of the N–N bond into the nitrene (see the ESI†). Alternatively, thermal decomposition can proceed via a spin-forbidden pathway as the nitrenes have a triplet ground state.15 The S/T crossings mediating the decomposition are somewhat lower in energy than the transition state in the singlet electronic state, with the crossing point corresponding again with the N–N dissociation. The spin-allowed pathway was shown to be largely dominating.15 More details on the topology of the potential energy surface can be found in the ESI.†
| Entry | Azide | 300 K | 473.15 K | ||
|---|---|---|---|---|---|
| ΔG# (eV) | k (s−1) | ΔG# (eV) | k (s−1) | ||
| a Experimental values for azidomethane at 473.15 K: ΔG# in the range 1.64 eV to 1.66 eV and k in the range 2.28 × 10−5 s−1 to 3.15 × 10−5 s−1.16 | |||||
| 1 | CH3N3 | 1.61 | 5.78 × 10−15 | 1.58a | 1.49 × 10−4a |
| 2 | FCH2N3 | 1.27 | 3.48 × 10−9 | 1.23 | 8.79 × 10−1 |
| 3 | F2CHN3 | 1.44 | 4.84 × 10−12 | 1.39 | 1.60 × 10−2 |
| 4 | F3CN3 | 2.16 | 3.89 × 10−24 | 2.01 | 3.89 × 10−9 |
Azidofluoromethane was used for copper-catalyzed azide–alkyne cycloaddition (CuAAC). An equimolar amount of azide was used as in situ prepared aqueous DMF solution (Method A) or THF solution after distillation (Method B) (Scheme 3). The click reaction proceeded smoothly with various aromatic or aliphatic alkynes and catalytic copper(I) 3-methylsalicylate (CuMeSal) with regioselective formation of 4-substituted triazoles 2. The known related 1-fluoromethyl-substituted benzotriazoles were prepared by electrophilic fluoromethylation using monofluoromethyl halides,17 sulfonium ylides18 or sulfoximines.19
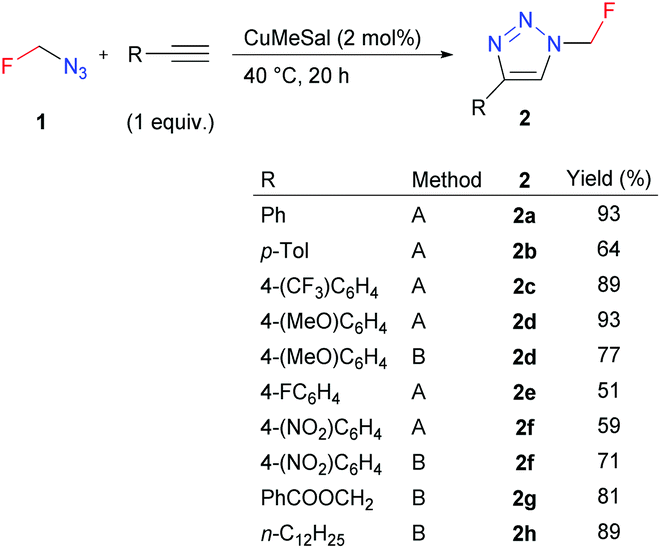 | ||
| Scheme 3 Scope of the copper-catalyzed azide–alkyne cycloaddition (CuAAC). Method A: Solution of 1 in aqueous DMF prepared in situ. Method B: Solution of 1 in THF after distillation. CuMeSal denotes copper(I) 3-methylsalicylate. | ||
Azidofluoromethane also reacted with alkyne, base, iodine monochloride, and stoichiometric amounts of CuI to form 5-iodotriazole 3, which was used in the Suzuki coupling reaction to afford 4,5-disubstituted triazole 4 (Scheme 4).
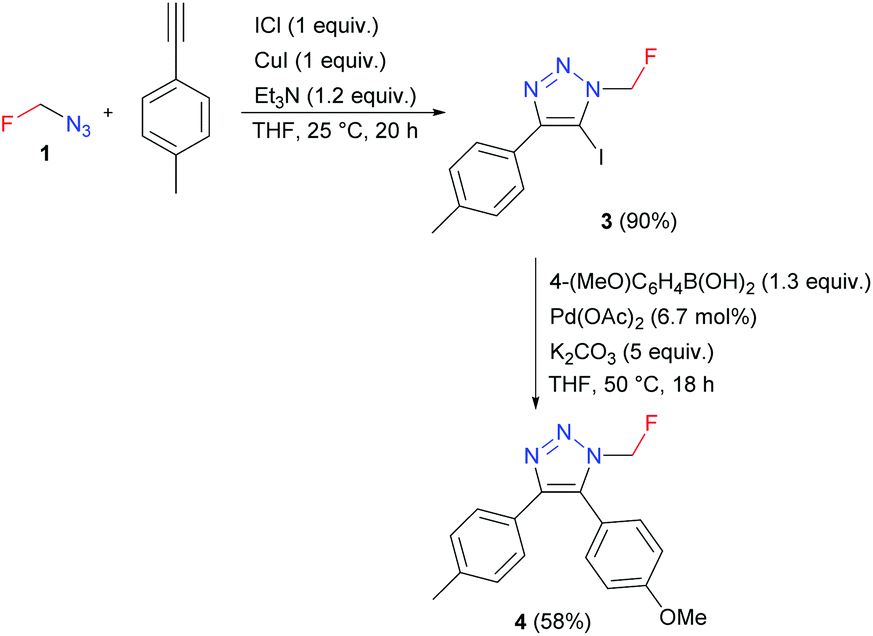 | ||
| Scheme 4 Preparation and Suzuki coupling of 5-iodotriazole 3. | ||
4,5-Disubstituted triazoles 5 were obtained in good to high yields by the organocatalyzed [3 + 2] cycloaddition of azidofluoromethane with β-ketoesters or 1,3-diones (Scheme 5). Ethyl esters 5 were subjected to alkaline hydrolysis to obtain acids 6, which thermally decarboxylated to 5-substituted triazoles 7 (Scheme 6).
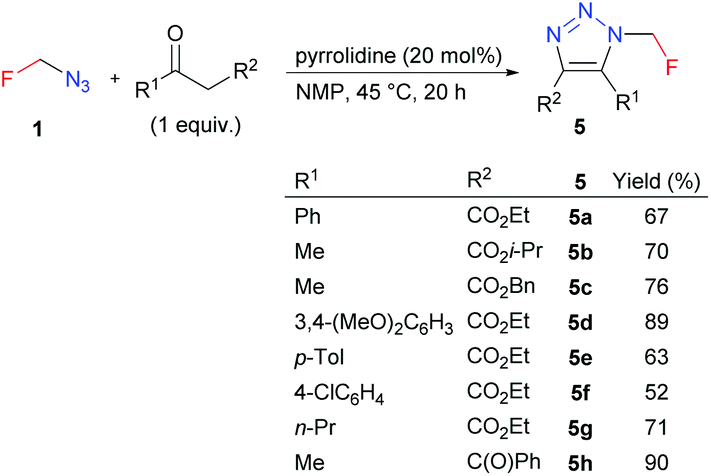 | ||
| Scheme 5 Azidofluoromethane-ketone [3 + 2] cycloadditions. | ||
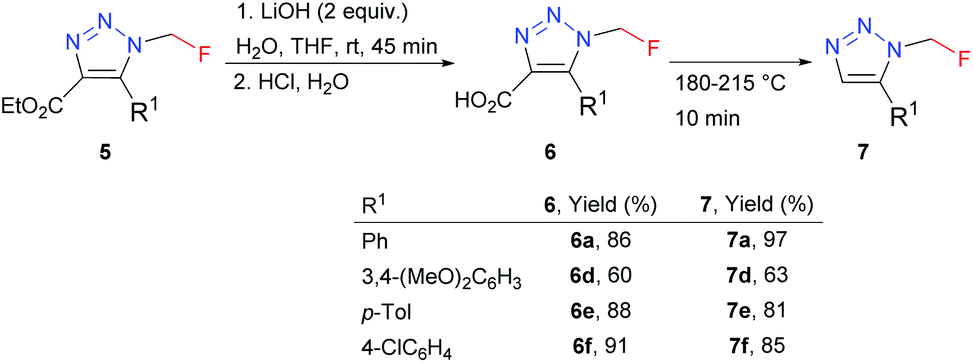 | ||
| Scheme 6 Preparation of 1,5-disubstituted triazoles. | ||
Experimental
Synthetic procedure for the preparation of azidofluoromethane 1
Bromofluoromethane (826 mg, 7.31 mmol) was dissolved in degassed NMP (2.558 g) and cooled to 10 °C. Sodium azide (951 mg, 14.62 mmol) was suspended in NMP (3.141 g) in a 10 mL round-bottom flask with a glass stopper and stirred at rt. A safety shield and a metal safety clamp were used. The cooled bromide solution was added at once to the sodium azide suspension, the reaction vessel was closed and the mixture was stirred to complete conversion (NMR control) over 2 h. Then, THF (5 mL) was added and distilled together with the product (rt, 4 Torr) into a cooled trap (−70 °C). The distillate contained azide 1 (6.17 mmol, 84% yield) in THF. Its full characterization is given in the ESI.†Calculations
Optimal geometries and transition states were calculated at the PBE0/6-31+g* level. The activation energies were then re-calculated at the CCSD(T) level with the aug-cc-pVQZ basis set. These values were then corrected for thermal and zero point energies at the PBE0/6-31+g* level, yielding the final values of activation Gibbs energies ΔG#. The rate constants were then calculated using the Eyring–Polanyi formula. The calculations were performed in the gas phase, considering that the solvent within the dielectric models had no effect on the calculated quantities. All calculations were done using the Gaussian 09 code.20Conclusions
The last remaining unknown fluorinated azidomethane – azidofluoromethane – was prepared from bromofluoromethane by the substitution of bromide with sodium azide and fully characterized in a THF or CDCl3 solution. The azide was found to be unstable at room temperature in chloroform and decomposed to hydrogen fluoride and hydrogen cyanide. Nevertheless, the azidofluoromethane solution underwent [3 + 2] cycloaddition with various dipoles to form 1-fluoromethyl 1,2,3-triazoles. With terminal alkynes, Cu(I)-catalyzed cycloaddition afforded 4-substituted triazoles. With an equimolar amount of CuI and iodine monochloride, 4-substituted 5-iodo triazole was formed which subsequently underwent a cross-coupling reaction with arylboronic acid. Cycloaddition of azidofluoromethane with β-ketoester or 1,3-diones afforded 4,5-disubstituted triazoles. Finally, hydrolysis and decarboxylation of the ethoxycarbonyl group led to 5-substituted triazoles.Ab initio calculations of the activation energies for the decomposition of (fluorinated) azidomethanes confirmed the observed strongly positive effect of fluorine substitution on the stability of the substituted azidomethane.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Academy of Sciences (RVO: 61388963) and by the Ministry of Education, Youth and Sports of the Czech Republic (LTAUSA 18037).References
- I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, 2009 Search PubMed.
- V. Gouverneur and K. Müller, Fluorine in Pharmaceutical and Medicinal Chemistry, Imperial College Press, 2012 Search PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS.
- H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. – Eur. J., 2019, 25, 11797–11819 CrossRef CAS.
- S. Voltrová, I. Putovný, V. Matoušek, B. Klepetářová and P. Beier, Eur. J. Org. Chem., 2018, 5087–5090 CrossRef.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Angew. Chem., Int. Ed., 2017, 56, 346–349 CrossRef CAS.
- V. Motornov, A. Markos and P. Beier, Chem. Commun., 2018, 54, 3258–3261 RSC.
- A. Markos, S. Voltrová, V. Motornov, D. Tichý, B. Klepetářová and P. Beier, Chem. – Eur. J., 2019, 25, 7640–7644 CAS.
- E. O. Aboagye, G. Smith, Q.-D. Nguyen, E. Arstad and M. E. Glaser, PCT Int. Appl, WO 2010026388A1, 2010 Search PubMed.
- C. Glidewell and H. D. Holden, J. Mol. Struct.: THEOCHEM, 1982, 89, 325–332 CrossRef.
- M. H. Palmer and A. D. Nelson, J. Mol. Struct., 2004, 689, 161–173 CrossRef.
- H. Bock and R. Dammel, Angew. Chem., Int. Ed. Engl., 1987, 26, 504–526 CrossRef.
- C. Wentrup, Chem. Rev., 2017, 117, 4562–4623 CrossRef CAS.
- J. F. Arenas, J. I. Marcos, J. C. Otero, A. Sánchez-Gálvez and J. Soto, J. Chem. Phys., 1999, 111, 551–561 CrossRef CAS.
- M. Besora and J. N. Harvey, J. Chem. Phys., 2008, 129, 044303 CrossRef.
- M. S. O'Dell and B. de B. Darwent, Can. J. Chem., 1970, 48, 1140–1147 CrossRef.
- W. Zhang, L. Zhu and J. Hu, Tetrahedron, 2007, 63, 10569–10575 CrossRef CAS.
- Y. Liu, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2017, 56, 9930–9934 CrossRef CAS.
- X. Shen, M. Zhou, C. Ni, W. Zhang and J. Hu, Chem. Sci., 2014, 5, 117–122 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental and synthetic procedures, characterization and copies of NMR spectra of all synthesized compounds, and calculation methods. See DOI: 10.1039/c9qo01295h |
| This journal is © the Partner Organisations 2020 |